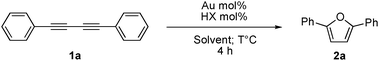
Gold(I)-catalyzed synthesis of furans and pyrroles via alkyne hydration†
Pierrick
Nun
a,
Stéphanie
Dupuy
a,
Sylvain
Gaillard
ab,
Albert
Poater
c,
Luigi
Cavallo
d and
Steven P.
Nolan
*a
aEaStCHEM School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK. E-mail: snolan@st-andrews.ac.uk
bLaboratoire de Chimie Moléculaire et Thio-organique, UMR 6507, ENSICAEN, 6 Boulevard Maréchal Juin, 14050 Caen, France
cCatalan Institute for Water Research (ICRA), H2O Building, Scientific and Technological Park of the University of Girona, Emili Grahit 101, E-17003 Girona, Spain
dDipartimento di Chimica, Università di Salerno, Via ponte don Melillo, 84084, Fisciano, Italy
First published on 31st January 2011
Abstract
Furans and pyrroles were prepared via the gold(I)-catalyzed alkyne hydration of diynes. The use of [Au(IPr)OH] as precatalyst in a silver-free protocol permits low catalyst loadings and in situ generation of the active cationic gold species. A detailed computational study confirmed the experimental results and supports the proposed mechanism.
Heterocycles, and amongst them furans and pyrroles, are structural motifs of significant importance in pharmaceutical and agrochemical compounds.1 These are also commonly employed as building blocks in organic synthesis.2 As a consequence, a large number of strategies have been developed to synthesize such compounds since the seminal work of Paal and Knorr.3 Amongst synthons leading to heterocyclic compounds, alkynes have appeared as particularly versatile substrates with various examples where these are transformed with the help of transition metals such as copper,4 titanium,5 ruthenium6 and rhodium.7
In the course of the last 10 years or so, gold complexes have emerged as powerful catalytic tools enabling numerous synthetic transformations.8 As part of our ongoing programme focusing on the use of gold(I)-catalyzed reactions, we have recently developed a new precatalyst, [Au(IPr)OH] (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene),9 which has efficiently been employed to generate the ubiquitous [Au(IPr)]+ catalytically active species by the simple protonolysis using a Brønsted acid.10 This method permits to circumvent the use of silver salts of type AgX (X = BF4, SbF6, OTf, PF6, NTf2) which are known to be light- and moisture-sensitive, as well as costly compounds. The use of gold has proven successful in pyrroles and furans syntheses. These routes have made use of alkynes as substrates, via amino-Claisen rearrangement,11 dehydrative cyclization,12 a domino approach,13 and an intramolecular acetylenic Schmidt reaction.14 More recently, Skrydstrup et al. reported access to 2,5-diaminopyrroles and 2,5-diaminofurans via hydroamination or hydration of 1,3-diynes.15 This method proved to be very efficient starting from dimerized terminal ynamides with [Au(PPh3)NTf2] (1–2 mol%) as a catalyst but when diaryl/alkyl diynes were used, [Au(SPhos)NTf2] (5 mol%) had to be used and harsher conditions (60–80 °C, 24 h) were then required. Taking advantage of our recent very promising results in gold-mediated alkyne hydration,16 we investigated whether the furan synthesis under silver-free conditions and at low catalyst loading could be achieved.
To optimize the reaction conditions, 1,4-diphenylbuta-1,3-diyne ( 1a) was chosen as the model substrate and was reacted under alkyne hydration conditions (Table 1). As expected, the reaction carried out with [Au(IPr)Cl] or [Au(IPr)OH] ( 3) alone did not yield the desired 1,4-diphenylfuran (entries 1 and 2). When 3 was activated by a Brønsted acid (HX) in order to generate [Au(IPr)]X (entries 3–8), yields leading to the furan 2a appear to be strongly dependent on the nature of the X counterion. Whereas HSbF6 and HOTf (entries 3 and 4) lead to almost no reaction, the use of HBF4 and HPF6 gave better results with, respectively, 37% and 39% yields (entries 5 and 6). However, when HNTf2 is used, generating [Au(IPr)NTf2] ( 4) in situ, the furan could be obtained in 77% yield after only 4 h (entry 7). Other reaction media were tested (entries 8–10) and although comparable results were obtained with THF/H2O (entry 8), only poor conversion to the furan was observed in DCE/H2O and MeOH/H2O reaction media (entries 9 and 10). Unsurprisingly, in the absence of gold there is no formation of 2a (entry 11). Under these conditions, the active species is formed in situ and for comparison purposes, diyne 1a was reacted in the presence of the well-defined complex17 [Au(IPr)NTf2] ( 4) (entries 12 and 13). At 80 °C and at 120 °C, the observed yield is not as good as with the in situ formed catalyst. This reactivity difference might be caused by a competition for inner sphere coordination (after acid deprotonation) between substrate 1a and counterion NTf2.18
| Entry | Catalytic systema | Solvent (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1) ∶1) |
T/°C | Yieldb (%) |
|---|---|---|---|---|
| a 1 mol% gold complex unless otherwise stated. b 1H NMR yield determined with p-nitrobenzaldehyde as an internal standard. c 5 mol% of 3 used. | ||||
| 1 | [Au(IPr)Cl] | Dioxane/H2O | 80 | 0 |
| 2 | [Au(IPr)OH] ( 3) | Dioxane/H2O | 80 | 0 |
| 3 | 3 | Dioxane/H2O | 80 | 0 |
| HOTf (1.5 mol%) | ||||
| 4 | 3 | Dioxane/H2O | 80 | 5 |
| HSbF6 (1.5 mol%) | ||||
| 5 | 3 | Dioxane/H2O | 80 | 37 |
| HBF4 (1.5 mol%) | ||||
| 6 | 3 | Dioxane/H2O | 80 | 39 |
| HPF6 (1.5 mol%) | ||||
| 7 | 3 | Dioxane/H2O | 80 | 77 |
| HNTf2 (1.5 mol%) | ||||
| 8 | 3 | THF/H2O | 80 | 65 |
| HNTf2 (1.5 mol%) | ||||
| 9 | 3 | 1,2-DCE/H2O | 80 | 40 |
| HNTf2 (1.5 mol%) | ||||
| 10 | 3 | MeOH/H2O | 80 | 4 |
| HNTf2 (1.5 mol%) | ||||
| 11 | HNTf2 (1.5 mol%) | Dioxane/H2O | 80 | 0 |
| 12 | [Au(IPr)NTf2] ( 4) | Dioxane/H2O | 80 | 25 |
| 13 | 4 | Dioxane/H2O | 120 | 62 |
| 14 | 3 | Dioxane/H2O | RT | 18 |
| HNTf2 (7.5 mol%) |
Finally, the possibility of reducing the temperature was investigated at higher catalyst loading (entry 14) but under these conditions only low conversions were obtained. Higher temperatures appear to be necessary to enable the transformation.
A scope of symmetrical and unsymmetrical diynes was then submitted to the optimised conditions obtained from Table 1 to explore the scope of this atom-economical catalytic transformation.
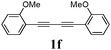
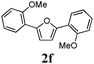
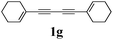
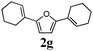
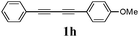
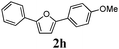
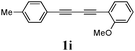
Our first efforts focused on the synthesis of symmetrical furans, starting from symmetrically substituted buta-1,3-diynes (Table 2). Aromatic diynes 1a–f were reacted under optimized conditions and in every case the corresponding furan was obtained in good yields (entries 1–7). Various substitutions at the para and ortho positions showed no real effect on the performance of the cyclisation. Even the more hindered system, 1f, with two ortho-anisoles gave good yields. Nevertheless, we were surprisingly unable to prepare furans starting from meta-substituted diynes under these conditions. A non-fully conjugated system was also prepared using 1,4-dicyclohexenylbuta-1,3-diyne with comparable results (entry 7). In the study reported by Skrydstrup et al.,15 only one example of asymmetric furan or pyrrole was described and the yield achieved was moderate. Using recently published methods to prepare asymmetric diynes,19 the reactivity of these substrates towards cyclisation was examined using our conditions (entries 8–12). As expected, comparable results were obtained with fully conjugated systems ( 2h and i) and substitutions on the phenyl ring resulted in no effect on the conversion rate (entries 8 and 9). Replacing an aromatic ring by a cyclohexenyl functionality ( 1j) proved the usefulness of these catalytic conditions to the synthesis of asymmetric non-fully conjugated furans (entry 10). To further explore this observation, buta-1,3-diynes bearing aliphatic sidechains ( 1k and l) were also prepared. In both cases, with one n-butyl or tert-butyl group and a phenyl moiety, the expected furans could be obtained in acceptable yields (entries 11 and 12). Nevertheless, when two aliphatic groups are bound to the same diyne, we were unable to obtain more than traces of the cyclized product.


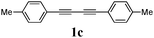
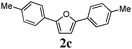
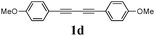
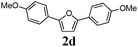
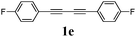


Two mechanistic hypotheses were considered when furans are formed via the alkyne hydration reaction (Fig. 1).
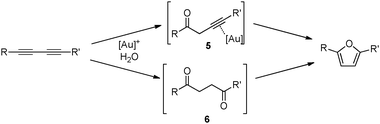 | ||
| Fig. 1 Two possible reaction pathways leading to furans. | ||
The first proceeds through a single alkyne hydration to provide the ketone 5 that cyclizes after activation of the triple-bond by the cationic gold species. The second hypothesis involves the hydration of both alkyne functions leading to the diketone 6 that cyclizes in a Robinson–Gabriel dehydrative cyclization.20 Our experimental results suggested that the first pathway might prove most probable as no diketone could be observed in NMR experiments performed during the course of the reaction. In order to fully understand the exact mechanistic picture at play in the present transformation, the commercially available diketone 1,4-diphenylbutane-1,4-dione ( 6a) was subjected to our conditions and, as expected, no cyclization occurred (Scheme 1).
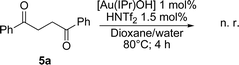
This observation supports the hypothesis where the furans are formed after cyclization of the ketone or the enol on the activated alkyne to form the 5-membered ring.
DFT calculations were undertaken to shed light on mechanistic details. Since experiments excluded the diketone pathway, we focused on the Au-catalyzed cyclisation of the monoketone.21 Calculations indicate that after metal coordination, a rapid keto–enol equilibrium, assisted by a water molecule, can be established. The keto form Ik is 3.1 kcal mol−1 more stable, and the barrier to convert Ik into the enol Ie is only 9.2 kcal mol−1, see Fig. 2. Cyclisation from Ik, through transition state (TS) Ik–IIk, requires to overcome a negligible barrier of 0.8 kcal mol−1, leading to IIk, from which the Au-dissociated furan product is obtained via a 1,2 H-shift and TS IIk–2a sits at 6.9 kcal mol−1. Cyclisation from the enol Ie also presents reasonably low energy TSs, Ie–IIe and IIe–2a are at 16.5 and 14.9 kcal mol−1, respectively, but comparison between the rate limiting IIk–2a and Ie–IIe TSs indicates that the keto pathway is favored by 9.6 kcal mol−1.
![Energy profiles (in kcal mol−1) for the Au-catalyzed formation of 2a from the Au-coordinated monoketone intermediate. [Au]+ = [Au(IPr)]+.](/image/article/2011/CY/c0cy00055h/c0cy00055h-f2.gif) | ||
| Fig. 2 Energy profiles (in kcal mol−1) for the Au-catalyzed formation of 2a from the Au-coordinated monoketone intermediate. [Au]+ = [Au(IPr)]+. | ||
Although the hydroamination reaction has already been demonstrated to proceed less easily with such alkyne substrates,22 the possibility of preparing pyrroles using the present protocol by replacing water by anilines as a nucleophile was investigated.
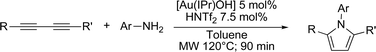
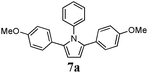
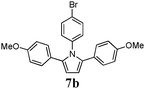
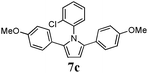
Optimum reaction conditions were determined after a short screening and resulted in the selection of toluene as the solvent and microwave irradiation as the method of heating. In this manner, the reaction times could be reduced from 24 h to only 90 min. An increased catalyst loading (5 mol%) was also necessary (Table 3). Pyrroles were obtained in all cases in moderate, but comparable, yields to those obtained by Skrydstrup et al.15 Diyne 1d proved to be the most reactive substrate. Other anilines were used as nucleophiles with no measurable effect of the diverse substitutions on yields (entries 1–4). A non-fully conjugated diyne was also reacted with aniline and the corresponding pyrrole 7e was isolated in a moderate 33% yield.
In conclusion, we have shown [Au(IPr)OH] ( 3) to be a very convenient pre-catalyst for heterocycle synthesis. Furans were prepared in high yields and under milder conditions than with other gold(I) catalysts. Pyrroles showed a lower reactivity under the optimized reaction conditions. Synergistic experimental and computational work has shed light on the reaction pathway leading to furans and pyrroles.
The ERC (Advanced Investigator Award to SPN) and the EPSRC are gratefully acknowledged for support of this work. Umicore AG are thanked for their generous gifts of materials. LC thanks the HPC team of Enea (www.enea.it) for using the ENEA-GRID and the HPC facilities CRESCO (www.cresco.enea.it) in Portici, Italy. AP thanks the Spanish MICINN for a Ramón y Cajal contract. SPN is a Royal Society-Wolfson Research Merit Award holder.
Notes and references
- (a) X.-L. Hou, Z. Yang and H. N. C. Wong, Comprehensive Heterocyclic Chemistry, Pergamon, Oxford, UK, 2003 Search PubMed; (b) G. Yin, Z. Wang, A. Chen, M. Gao, A. Wu and Y. Pan, J. Org. Chem., 2008, 73, 3377–3383 CrossRef.
- (a) N. A. Trofimov and N. A. Nedolya, Comprehensive Heterocyclic Chemistry III, Elsevier Ltd, Oxford, UK, 2008 Search PubMed; (b) H. N. C. Wong, K.-S. Yeung and Z. Yang, Comprehensive Heterocyclic Chemistry III, Elsevier Ltd, Oxford, UK, 2008 Search PubMed.
- For original report, see: (a) L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 1635–1642 CrossRef; (b) C. Paal, Ber. Dtsch. Chem. Ges., 1885, 18, 367–371 CrossRef and for more references to this strategy, see: ; (c) G. Balme, D. Bouyssi and N. Monteiro, Heterocycles, 2007, 73, 87–124 CrossRef CAS; (d) X.-L. Hou, Z. Yang, K.-S. Yeung and H. N. C. Wong, Prog. Heterocycl. Chem., 2009, 21, 179–223 Search PubMed; (e) J. S. Russel, E. T. Pelkey and S. J. P. Yoon-Miller, Prog. Heterocycl. Chem., 2009, 21, 145–178 Search PubMed.
- J. Barluenga, L. Riesgo, R. Vicente, A. Lopez and M. Tomas, J. Am. Chem. Soc., 2008, 130, 13528–13529 CrossRef CAS.
- L. Ackermann and L. T. Kaspar, J. Org. Chem., 2007, 72, 6149–6153 CrossRef CAS.
- E. Bustelo and P. H. Dixneuf, Adv. Synth. Catal., 2007, 349, 933–942 CrossRef CAS.
- J. Dheur, M. Sauthier, Y. Castanet and A. Mortreux, Adv. Synth. Catal., 2010, 352, 557–561 CrossRef CAS.
- (a) A. Arcadi, Chem. Rev., 2008, 108, 3266–3325 CrossRef CAS; (b) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208–3221 RSC; (c) D. J. Gorin, F. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351–3378 CrossRef CAS; (d) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef; (e) E. Jiménez-Nunez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326–3350 CrossRef CAS; (f) S. P. Nolan, Acc. Chem. Res., 2010 DOI:10.1021/ar1000764; (g) R. Skouta and C.-J. Li, Tetrahedron, 2008, 4917–4938 CrossRef CAS; (h) T. W. Hudnall, A. G. Tennyson and C. W. Bielawski, Organometallics, 2010, 29, 4569–4578 CrossRef CAS.
- (a) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef CAS; (b) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776–1782 RSC; (c) S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC.
- S. Gaillard, J. Bosson, R. S. Ramón, P. Nun, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2010, 16, 13729–13740 CrossRef CAS.
- (a) A. Saito, T. Konishi and Y. Hanzawa, Org. Lett., 2010, 12, 372–374 CrossRef CAS; (b) F. M. Istrate and F. Gagosz, Org. Lett., 2007, 9, 3181–3184 CrossRef CAS.
- A. Aponick, C.-Y. Li, J. Malinge and E. Finco Marques, Org. Lett., 2009, 11, 4624–4627 CrossRef CAS.
- J. T. Binder and S. F. Kirsch, Org. Lett., 2006, 8, 2151–2153 CrossRef CAS.
- D. J. Gorin, N. R. Davis and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260–11261 CrossRef CAS.
- S. Kramer, J. L. H. Madsen, M. Rottländer and T. Skrydstrup, Org. Lett., 2010, 12, 2758–2761 CrossRef CAS.
- (a) N. Marion, R. S. Ramon and S. P. Nolan, J. Am. Chem. Soc., 2009, 131, 448–449 CrossRef CAS; (b) P. Nun, R. S. Ramón, S. Gaillard and S. P. Nolan, J. Organomet. Chem., 2011, 696, 7–11 CrossRef CAS.
- L. Ricard and F. Gagosz, Organometallics, 2007, 26, 4704–4707 CrossRef CAS.
- For well-defined 4, NTf2 is required to leave the Au coordination to lead to the active species which is more energetically demanding than simple binding of substrate present in excess at the deprotonation step..
- (a) K. Balaraman and V. Kesavan, Synthesis, 2010, 3461–3466 CAS; (b) W. Yin, C. He, M. Chen, H. Zhang and A. Lei, Org. Lett., 2009, 11, 709–712 CrossRef CAS.
- K. C. Nicolaou, J. Hao, M. V. Reddy, B. R. Paraselli, R. Gerasimos, S. A. Snyder, X. Huang, D. Y.-K. Chen, W. E. Brenzovich, N. Giuseppone, P. Giannakakou and A. O'Brate, J. Am. Chem. Soc., 2004, 126, 12897–12906 CrossRef CAS.
- The GGA DFT calculations were performed with the Gaussian09 package. The BP86 functional in connection with the SVP basis set on main group atoms and the quasi-relativistic Stuttgart/Dresden ECP with the associated triple-ζ basis set on valence shells were used. The reported energies include solvent effects (H2O) estimated with PCM.
- R. A. Widenhoefer and H. Han, Eur. J. Org. Chem., 2006, 4555–4563.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details are included. See DOI: 10.1039/c0cy00055h |
| This journal is © The Royal Society of Chemistry 2011 |