Retracted article: Towards near zero-sulfur liquid fuels: a perspective review
Barbara Pawelec, Rufino M. Navarro, José Miguel Campos-Martin and José L. G. Fierro*
Sustainable Energy and Chemistry Group, Institute of Catalysis and Petrochemistry, CSIC, Marie Curie 2, Cantoblanco, 28049 Madrid, Spain. E-mail: jlgfierro@icp.csic.es; Web: http://www.icp.csic.es/eqs/
First published on 4th February 2011
Abstract
We, the named authors, hereby wholly retract this Catalysis Science & Technology article, due to significant similarity with previously published work. Signed: B. Pawelec, R. M. Navarro, J. M. Campos-Martin and J. L. G. Fierro, October 2012. Retraction endorsed by Jamie Humphrey, Editor, Catalysis Science & Technology.
Introduction
With the growing awareness that oil is a finite resource, it is a very important task to try to utilize it as effectively as possible whilst limiting its environmental impact, for example, by reducing the sulfur and aromatic levels in the various hydrocarbonfuels. The problem of the CO2 emissions intrinsically associated with hydrocarboncombustion and its climatic consequences must be taken seriously. In the overall context, fuel quality and fuel economy are important parameters that the refining and automotive industries will have to improve continuously, prompted by ever-stricter environmental legislation. In addition, last century's hike in oil production now manifests itself in a degeneration of the average crude quality, as the remaining contents of the known reservoirs tend to be of heavier and sourer composition. New wells are constantly being sought out; nowadays, prospectors often have to explore remote and hostile off-shore areas, yet major discoveries are encountered increasingly seldom. Although the necessity of alternative energy solutions is widely recognized, oil and hydrocarbonfuels are predicted to prevail for many years to come.1,2Fossil fuels are used in the day-to-day production of electricity and the propulsion of vehicles. In particular, motorised transport is over 95% dependent on oil and accounts for almost half its global use. This made road transport a significant source of air pollution in the last century, and car engines are still a major producer of toxic emissions, with subsequent adverse effects on human health.3
The combustion of fossil fuels releases harmful emissions into the atmosphere that impact on living organisms, as well as cause the greenhouse effect. Major emissions of NOx, SOx, CO2 and particulate matter are the cause of most concern for pollution in our environment. Emissions of NOx, together with hydrocarbons, increase the ozone levels in the troposphere while SOx gases cause acid rain. The greenhouse gases (GHGs) most closely identified with transportation are carbon dioxide (CO2), nitrous oxide (N2O), and methane (CH4). Other vehicle-related pollutants also contribute to global warming, although their quantification is more difficult; these include CO, non-methane hydrocarbons (NMHCs), nitrogen dioxide (NO2), and ozone (O3). Black carbon (soot) emitted from diesel vehicles is another emerging GHG concern.4Sulfur-containing compounds and polyaromatic hydrocarbons present in diesel fuel are responsible for the particulates or soot in diesel.5,6 Traces of sulfur present in diesel fuels also poison the oxidationcatalysts in the emission control system and reduce their effectiveness for the oxidation of harmful carbon monoxide, hydrocarbons and volatile organic matter.7
Environmental regulations have been introduced in many countries around the world to reduce the sulfur content of diesel fuel to ultra low levels (10 ppm) with the aim of lowering the diesel engine's harmful exhaust emissions and improving air quality.8 Meanwhile, the demand for transportation fuels has been increasing in developed countries over the past two decades. Basically, all these motorfuels are derived from oil, with the sole exception of coal (and natural gas)-derived fuels produced in South Africa by Fischer–Tropsch synthesis. The global demand for energy and, in particular, transportation fuels keeps growing. Global energy consumption in 2007 was about 12.0 billion toe (toe = tonnes of oil equivalent) and is expected to grow until 13.3 billion toe by 2015 (Fig. 1) Oil consumption rose from around 70 million barrels per day in 1995 (1 barrel = 159 L) to over 80 in 2005, and it is projected to grow to over 90 million barrels per day in 2020.9 While a significant part of the demand is located in North America, the largest growth is observed in the Asia-Pacific region. The future growth of energy demand in the Asia-Pacific area has recently seen a series of calls for new grass-root refineries, mainly to be located in the Middle East and Asia-Pacific. Current predictions suggest that only 60% of all the new projects announced will actually materialize. This means that new refineries imply new hydroprocessing units and further growth of the overall market for hydroprocessing catalysts. This is just one reason for the industry now needing hydrocracking units and catalysts to process the “bottom of the barrel”.
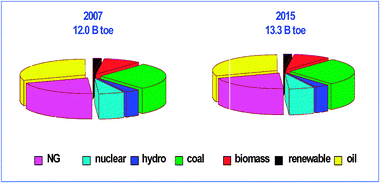 | ||
| Fig. 1 Global energy demand by type in billion tonnes of oil equivalent in 2007 (left) and estimates for 2015 (right). Adapted by British Petroleum from the World Energy Outlook 2009. | ||
In addition, the new environmental regulations that limit the S-levels of transportation fuels, and specifically diesel fraction, are beneficial from an environmental point of view, and meeting their stringent requirements is a major challenge for the petroleum refining industry.10–14 The very low S-levels to be achieved in motorfuels necessarily imply the ultra-deep desulfurisation of the diesel feed stream. The shift from normal to ultra-deep desulfurisation is a very complicated issue. Factors such as feedstock source,15,16catalysts,17–20 reactivity of S-compounds,21,22 inhibition effects of H2S,23–25nitrogen compounds,26,27 aromatics,28,29 and process parameters,30 among others, have a significant bearing on the degree of desulfurisation of feeds.
The increasing attention being paid worldwide to the chemistry of diesel fuel processing is related to both thermal efficiency and environmental aspects, which include both pollutants and greenhouse gas emissions. As a general trend, diesel fuel demand is expected to increase significantly over the next two decades.31 In addition, interest in ultra-clean fuels also stems from the use of new emission control technologies for internal combustion engines (especially diesel fuels), and for using the on-board or on-site reforming of hydrocarbonfuels to supply the H2 required to feed the future generation of electric cars. Since the general chemistry of diesel fuels is well known,32 the objective of this Perspective Article is to provide a selective overview of new design approaches and the associated catalysis and chemistry, as well as of processes for the deep desulfurisation and deep hydrogenation of hydrocarbonfuels, particularly diesel fuels. In a first instance, the major drivers of the demand for new and better hydroprocessing catalysts are examined, and then the challenges faced in catalyst development are considered.
2. Drivers of ultra low sulfurfuel production
2.1 Environmental legislation on diesel fuel specification
The SOxoxides, particulates and other harmful pollutants present in exhaust engines are the main reasons for targeting sulfur content in fuel, mainly in the diesel fraction, and decreasing it to ever lower levels in many countries worldwide. It has been shown that low S-levels in fuels and the use of appropriate particulate filters can reduce particulate matter by as much as 90%.33 In the USA, the acceptable level of sulfur in highway diesel was first reduced from 2000 ppm to 500 ppm by the Clean Air Act (CAA) amendments in the nineties, then to 350 ppm, 50 ppm and 15 ppm in the years 2000, 2005, and 2006, respectively (Fig. 2).34 In the EU, Germany was the first country to adopt the 10 ppm sulfur limit for diesel as from January 2003. Other EU countries and Japan introduced diesel fuel with 10 ppm into the market from the year 2008 (Fig. 3).35,36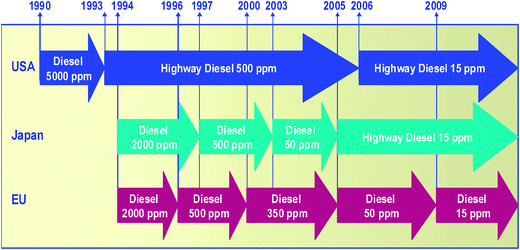
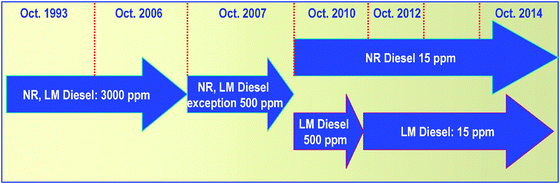 | ||
| Fig. 3 Trends in sulfur specifications for non-road diesel (NR: non-road, and LM: locomotive and marine diesel). | ||
Similar ultra low sulfur specifications are also targeted in many other countries, and will be predominant worldwide during the next decade. The specifications proposed for clean diesel by the Worldwide Fuel Charter (WWFC), which reflects the view of car/engine manufacturers concerning the fuel qualities required for engines in use and for those yet to be developed, require a higher cetane index, significant reduction of polyaromatic hydrocarbons (PAH), and a lower T95distillation temperature in addition to ultra low sulfur levels (Table 1). Car manufacturers have concluded that substantial reductions in both gasoline and diesel fuelsulfur levels to quasi S-free levels are essential to enable future vehicle technologies to meet stringent vehicle emission control requirements and reduce fuel consumption. Similar tighter sulfur specifications have also been introduced for non-road diesel37 (Fig. 3). In 2004, the US Environmental Agency (USEPA) issued the clean air-non-road-Tier 4 final rule which mandates that starting from 2007, the fuelsulfur level in non-road diesel fuel should be reduced to 500 ppm from its current 3000 ppm level. This includes fuels used in locomotive and marine applications (except marine residual fuel used by very large engines in ocean-going tankers). In 2010, fuelsulfur levels in most non-road diesel fuel have been reduced to 15 ppm, making it possible for engine manufacturers to use advanced emission control systems that significantly reduce harmful fumes.
2.2 Heavier crudes
Oil refiners are increasingly processing heavier and worse quality feedstocks, while the specifications for middle distillates and gasoline/diesel engines are becoming more stringent.38 There is a continuous trend within the refining industry whereby crudes are becoming heavier (higher density and viscosity) and sourer, which means a larger amount of sulfur. A prediction by Hart is that over the next 15 years an extra 0.15 wt% of sulfur will be recorded in crude, giving an average level of 1.28 wt%.39 In addition, the shift in crude diversity and source will continue. For example, more Russian and Caspian crude import is expected in Western Europe, and these crudes are heavier and contain high heteroatom amounts of contaminant compounds, such as arsenic compounds, for example, compared to traditional North Sea and Middle East crudes. The effects from heavier crude supply are thus twofold. Since the demand for light products is increasing at the expense of heavier fuels (e.g. heavy fuel oil), more crude conversion will be needed to upgrade the heavy crudes to provide the desired product mix. Secondly, the higher level of metal-containing micelles in the feed will require more effective guard bed catalysts and systems.A very exciting and emerging field is the production of syncrudes (synthetic oils) in Canada and Venezuela. Canadian syncrudes are produced from tar sands by a process of extraction and bitumen upgrading.40 The upgrading of these tar sands, via bitumen, to synthetic crude oil is most challenging with respect to hydrotreating, since the bitumen contains two to four times as much sulfur, nitrogen and aromatic compounds as standard crudes. The levels of arsenic- and silicon-containing compounds can be quite high, which creates its own problems in such processing schemes. The operating conditions for the hydroprocessing of these types of crude are much more severe compared to the processing of conventional crude fractions. Consequently, the experience with conventional hydrotreating catalysts operating under these sorts of conditions is limited, and it can be assumed that opportunities will open up with the development of bespoke catalysts for this area.
3. Deep HDS
3.1 Reactivities of sulfur compounds
The sulfur compounds can be classified into four groups according to their HDS reactivities that are described by the pseudo-first-order rate constants: (i) sulfur compounds dominated by alkyl benzothiophenes (BTs); (ii) dibenzothiophenes (DBT) and alkyl dibenzothiophenes (DBTs) without alkyl substituents at the 4- and 6-positions; (iii) alkyl DBTs with only one alkyl substituent at either the 4- or 6-position; and (iv) alkyl substituents at the 4- and 6-positions (as shown in Fig. 4).12,41,42 The latter species are termed refractory sulfur compounds and both steric hindrance and electronic factors are claimed to be responsible for their low reactivity.42–46 The relative rate constants of HDS, assuming pseudo first-order kinetics, for some alkyl-substituted dimethyl DBTs are displayed in Fig. 5.44 According to these values, the relative reactor volume requirements for various degrees of sulfur removal by conventional single-stage HDS of diesel fuels have been calculated. The estimation of volume requirements is based on the results from HDS kinetics studies using a commercial CoMo/Al2O3catalyst,44 assuming 1.0 wt% S in feed.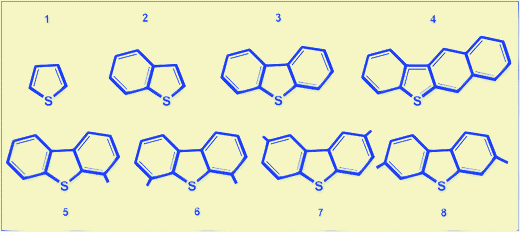 | ||
| Fig. 4 Structures of refractory sulfur compounds. (1) thiophene; (2) benzothiophene; (3) dibenzothiophene; (4) benzo(b)naphtha 2,3-thiophene; (5) 4-methyl dibenzothiophene; (6) 4,6-dimethyl dibenzothiophene; (7) 2,8-dimethyl dibenzothiophene; and (8) 3,7-dimethyl dibenzothiophene. | ||
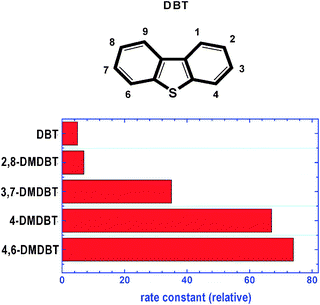 | ||
| Fig. 5 Relative values of the rate constants (kthiophene/kDMDBTs) for the desulfurization of different dimethyl substituted dibenzothiophenes. | ||
Sulfur removal in gasoline is relatively straightforward by current catalytic HDS processes. The challenge in the deep desulfurisation of FCCnaphtha involves the selective conversion of sulfur compounds without saturating olefinic compounds, which contribute to octane number enhancement. For the straight-run kerosene used for making jet fuels, sulfur removal by HDS is more difficult than that from naphtha, but less difficult compared to that from gas oil. Recent papers have reported that the sulfur compounds remaining in diesel fuels at a sulfur level below 500 ppm are the DBTs with alkyl substituents at the 4- and/or 6-position, because of their lower HDS reactivity.10,41–44 When the total S-content is reduced to 30 ppmw, the sulfur compounds remaining in the hydrotreated oil belong solely to the fourth group, indicating that the lower the sulfur content, the lower the HDS reactivity, which has been discussed by Whitehurst et al. in their review paper.45 More recent studies using various straight-run gas oils from different crude oils have confirmed the differences in reactivity between different sulfur compounds.46
Based on conventional approaches to the standard HDS processing of diesel fuels when reducing the sulfur level from the current 500 to 15 ppmw (the regulation in 2006), the volume of catalyst bed will have to be increased to 3.2 times that of the current HDS catalyst bed. When reducing the sulfur level to 0.1 ppm by conventional HDS processing for fuelcell application, the volume of catalyst bed will have to be increased by about sevenfold. There is no doubt that the use of high temperature and high pressure renders the process prohibitive. In other words, using current commercial HDS processes without changing the reactor volume means catalyst activity will have to be increased by a factor of 3.2 and 7 to meet the new regulation and fuelcell applications, respectively. It might be difficult to meet this requirement by making small incremental improvements to existing hydrotreating catalysts that have been developed over the past 50 years.
The petroleum refining industry is therefore facing a major challenge to meet the new stricter sulfur specifications in the early 21st century, when the quality of crude oils continues to decline in terms of increased sulfur content and decreased API gravity.
3.2 Reaction pathways
Important progress has been made in the fundamental understanding and practical application of hydrotreating catalysis and metal sulfide-based catalysts for HDS, as discussed in several excellent reviews.47–50 The following discussion focuses on the deep desulfurisation of polycyclic sulfur compounds.The hydrodesulfurisation of DBT and alkyl DBTs proceeds mainly via two parallel routes, as shown in Fig. 6. The first route involves direct desulfurisation (DDS) leading to the formation of biphenyl, while the second route involves the hydrogenation (HYD) of one of the benzene rings of the DBT, producing tetrahydrodibenzothiophene in the first step, which is further desulfurised to cyclohexylbenzene.51,52 It has been shown that the HDS of unsubstituted DBT occurs preferentially via the direct sulfurextraction (DDS) route. The alkyl substituents affect the HDS of DBT in two ways: (i) they reduce their HDS reactivity; and (ii) they change the ratio between the rates of the two routes. The HYD route becomes dominant with the introduction of alkyl substituents in the 4- and/or 6-positions of the DBT molecules.48,50 It has been documented that partial saturation changes the spatial configuration of the molecule,50 making the sterically hindered sulfur more accessible for adsorption on the active site prior to reaction.
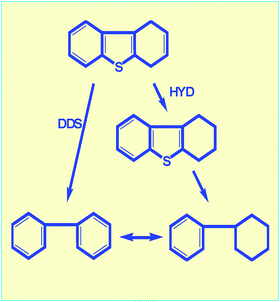 | ||
| Fig. 6 Direct and hydrogenation reaction pathways for hydrodesulfurization of 4,6-dimethyl dibenzothiophene. | ||
The DDS pathway is severely inhibited, while the HYD pathway is hardly affected by the presence of alkyl groups in the 4- and 6-positions of DBT. It has been suggested that the alkyl groups in the 4,6-DMDBT do not play a significant role in the reactivity of 4,6-DMDBT along the HYD pathway, and the difference in reactivity between DBT and 4,6-DMDBT lies essentially in the selective promoting effect on the DDS pathway.53
A common dihydro-dibenzothiophene intermediate was suggested for the DDS and HYD reaction routes. The orientation of the reaction toward one or the other of the two possible pathways is due to the difference in reactivity of the common dihydro intermediate in further hydrogenation or in the C–S bond cleavage through elimination. The hindrance of the C–S bond cleavage in the partially hydrogenated intermediate could thus be the most likely reason for the low reactivity of alkyl DBTs with the alkyl substituent in the 4 and 6 positions.
The proposal of a common dihydrodibenzothiophene intermediate for both DDS and HYD reaction pathways poses certain doubts. It has been proposed that the two HDS pathways do not have a common intermediate and are determined by the conformation of the adsorbed DBT molecule. DDS reaction starts with the π adsorption of the DBT molecule via the S-atom, whereas the HYD reaction starts with σ adsorption of the reactant via the aromatic system. This would require different sites and different adsorption constants for the DDS and HYD pathways. The main reason for this difference is the aromaticity of the DBT structure, making π adsorption more likely than σ adsorption. It has been shown that methyl groups hinder the perpendicular σ adsorption of DBT, but hardly affect flat adsorption via the aromatic π system. It is suggested that in the HYD route, 1,2,3,4-tetrahydrothiophene is formed as a partially hydrogenated intermediate, and the final removal of the S-atom from tetrahydrothiophene could occur by hydrogenolysis (through σ adsorption). Therefore, the methyl groups suppress not only the σ adsorption of the reactant, but also of the partially hydrogenated intermediate. The DDS pathway is considerably slower than the sulfur removal via the HYD pathway due to the presence of the methyl groups. This may be due to the lower flexibility in DBT and 4,6-DMDBT than in their partly hydrogenated intermediates, making adsorption easier for the latter molecule.
3.3 Inhibition effects
In the production of ultra low sulfur diesel fuel (<10 ppm S), more than 99% of the S-containing compounds, including the traces of least reactive alkyl DBTs present in the feedstock, have to be removed during catalytic hydrotreating. The main problem in the HDS of the sterically hindered alkyl DBTs is the inhibiting effects of different poisons, such as N-containing compounds, H2S and aromatic molecules, on their reactivity under reaction conditions imposed for deep desulfurisation. As shown in the previous section, sulfur removal from DBT and alkyl substituted DBTs can take place along DDS and HYD routes. The former involves direct sulfurextraction, while the latter involves two steps: hydrogenation of one of the aromatic rings in the first step, followed by S-removal as H2S in the second step. The poisoning effects of the inhibitors (H2S, nitrogen compounds and aromatics) are different for the two routes and are discussed in some detail in several papers.13,54–56 In general, the inhibition order is as follows: nitrogen compounds > organic sulfur compounds > polyaromatics ≈ oxygen compounds ≈ H2S > monoaromatics. The inhibition is not only for HDS but also between inter- and intra-molecular reactions, as well as its intermediate reaction products.57–594. Catalyst development for deep HDS
Catalysts consisting of molybdenum supported on alumina and promoted with cobalt or nickel are traditionally used in hydrotreating processes. The role of the catalyst is to enhance the removal of S, N and aromatics present in the refinery streams by promoting HDS, hydrodenitrogenation (HDN) and hydrogenation (HYD) reactions. Conventional CoMo- and NiMo-based catalysts, however, do not have sufficient activity to desulfurise diesel feed streams to ultra low sulfur levels under normal operating conditions. They require severe operating conditions, such as high temperature, low space velocity and high hydrogen partial pressure. Such severe processing conditions generally lead to rapid catalyst deactivation, shorter cycle times and reduced throughput. The development and application of more active and stable catalysts are among the most promising options for reducing the S-content of diesel to ultra low levels by deep desulfurisation.60,61Studies have shown that around 4 times more active catalysts are required to reduce the S-content of diesel fuel from 500 to 50 ppm. Further improvement in catalyst activity is necessary when the sulfur content is to be reduced to ultra low levels (<15 ppm). Intensive efforts have been devoted worldwide to the development of highly active hydrotreating catalysts for the deep desulfurisation of diesel to ultra low sulfur levels,62–65 and new generation catalysts with very high HDS activity are being continuously developed and introduced to the market. The development of improved hydrotreating catalysts has been possible through a clear understanding of the key properties, namely, the nature of the active sites and their structure, support effects and the textural characteristics of supports, which have significant influence on catalyst performance.66–68 The scientific basis for the high activity of the new generation hydrotreating catalysts is presented and discussed in detail in the following sections.
4.1 Conventional catalysts
The nature of the active phase in unpromoted and promoted MoS2catalysts has been widely studied and reported in several reviews.69,70 For unpromoted molybdenum sulfidecatalysts, it has been proposed that coordinatively unsaturated (CUS) sites or exposed Mo ions with S-vacancies at the edges and corners of MoS2 structures are active in hydrogenation and hydrogenolysis reactions. Basal planes are inactive in the adsorption of molecules and are probably unimportant in hydrotreating reactions. For Co- or Ni-promoted catalysts, several different structural models, such as monolayer model, intercalation model, contact synergy model, Co(Ni)–Mo–S phase model, and catalyticCo site model, have been proposed to explain the role of the promoter and its location in the catalyst. Among these, the Co(Ni)–Mo–S model proposed by Topsøe69 is now widely accepted. The Co(Ni)–Mo–S structures are small MoS2 nano-crystals with the Co- (or Ni)-atoms located at the edges of the MoS2 layers on the same plane of MoS2 layers (Fig. 7). The relative amount of Co-atoms present as the Co–Mo–S phase was found to correlate linearly with HDS activity. Further studies on the structure–activity correlation of these catalyst systems allowed the identification of two types of Co–Mo–S structures, one type having substantially higher activity than the other. The high and low activity forms of Co–Mo–S were termed types II and I Co–Mo–S, respectively. The type I Co–Mo–S structures were deemed to be incompletely sulfided and have some remaining Mo–O–Al linkages to the support. The presence of such linkages was related to the interaction that occurs in the calcined state between Mo and surface alumina OH groups leading to oxygen bridged monolayer-type structures that are difficult to sulfide completely. In type II Co(Ni)–Mo–S phase, the support interactions are weaker and fully sulfided. The underlying MoS2 in type II Co–Mo–S phases is less disperse, consisting of multiple slabs not linked to the support. Further studies have shown that the degree of stacking in MoS2 layers and Co–Mo–S structures can be controlled by tailoring support properties and preparation parameters. These will have a high MoS2 edge dispersion that can accommodate more Co-atoms to form higher active single slab type II Co–Mo–S structures.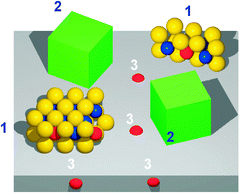 | ||
| Fig. 7 Schematic representation of the CoMoS model under reaction conditions. Co is present in three different phases. (1) The active CoMoS nanoparticles; (2) a thermodynamically stable cobalt sulfide (Co9S8); (3) Co dissolved in the Al2O3 support. Only the CoMoS particles are catalytically active. Taken from ref. 69. | ||
Harris and Chianelli72 posited that the specific and unique activity of the cobalt (or nickel) promoted MoS2 phase is related to an electron donation from Co to Mo decreasing the Mo–S bond strength to an optimum range for HDS activity. These electron donations will increase the electron density in the highest occupied molecular orbital (HOMO) of Mo, occupying anti-bonding orbitals and then somewhat weakening the Mo–S bond energy. This electron transfer will also decrease the occupancy of anti-bonding orbitals for Co, strengthening the Co–S bond. The S-atom shared between Co and Mo will then present intermediate metal–S bond strength. It was then assumed that this intermediate strength would be in an optimum range for HDS activity. This weakening effect of Co(Ni) promotion on the metal–S bond strength is now well accepted and was confirmed using theoretical calculations as well as experimental techniques.73,74 Based on S-bond energy calculation and experimental work, Thomazeau et al.75 used DFT calculations to predict that Ni promoted trimetallic NiMoW sulfidecatalysts are better than bimetallic NiWS and NiMoS (or CoMoS and CoWS) catalysts, as shown in Fig. 8.
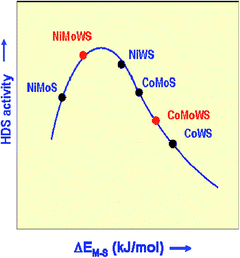 | ||
| Fig. 8 Schematic representation of the relative positions of the Co(Ni)Mo(W)S systems on the volcano curve and explanation of the new synergy effects for NiMoWS systems (DEMS, energy variation associated to a catalytic system). Adapted from ref. 75. | ||
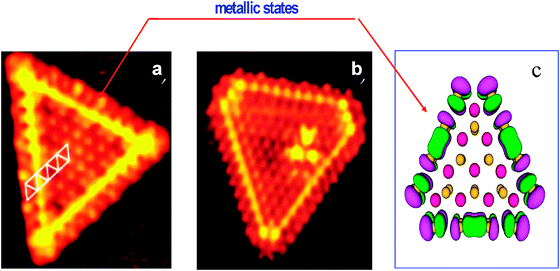
Further insight into atomic structure, morphology of MoS2 and Co(Ni)–Mo–S phases was forthcoming using a combination of advanced experimental and theoretical techniques, such as scanning tunnelling microscopy (STM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and density functional theory (DFT). STEM showed atom-resolved images of the catalytically active edges of MoS2 and Co(Ni)–Mo–S nano-clusters.76,77 The Mo-edge was found to have a special electronic edge state identified as brim sites76 (Fig. 9). DFT calculations revealed the metallic nature of the brim sites. The metallic nature of these brim sites means they may bind S-containing molecules, and when hydrogen is available at the neighbouring edge sites in the form of SHgroups, hydrogen transfer and hydrogenation reactions can take place. Therefore, these brim sites are catalytically active for hydrogenation reactions, whereas direct sulfur removal can take place at both edges.
4.2 Preparation of ultra deep HDS catalysts
It has been well established that in CoMo- and NiMo-based hydrotreating, type II Co(Ni)–Mo–S catalyst sites are highly active for HDS. Therefore, intensive efforts have been made over the past decade to improve the HDS activity of alumina-supported CoMo and NiMocatalysts by maximizing the concentration of the type II sites by a variety of methods. To achieve this, approaches such as modified supports, the development of new carriers, improvements in catalyst impregnation and preparation techniques, and the use of certain additives, have been considered and discussed in excellent reviews.11,78,79 As mentioned above, the deep HDS of sterically hindered sulfur compounds in diesel fuel will require the enhancement of hydrogenation activity to hydrogenate the aromatic rings of the S-containing compounds. The use of supports with high acidity can also improve the HDS of refractory sulfur compounds by promoting isomerization and dealkylation reactions.53,80,81The active components in hydrotreating catalysts are mixed sulfides of Mo and Co(Ni) as Co(Ni)–Mo–S phases deposited on a carrier. The carrier or support material usually provides a high surface area to maximize active phase dispersion. The support also gives the catalyst mechanical strength. Alumina is the most widely used support material in hydrotreating catalysts because it combines almost all the above characteristics. It is highly stable, contains acidic and basic sites, has a reasonably high surface area and porosity, can be easily formed into desired shapes, and is relatively inexpensive. Despite these advantages, great interest has still been shown in new supports for HDS catalysts due to the need to develop better ones.82,83 A large number of new materials with high surface area and other properties suitable for support applications have been developed and tested. These include TiO2, ZrO2, MgO, carbon, SiO2, zeolites, etc.84–87 Attempts have also been made to modify the alumina by mixing it with zeolites and other metal oxides such as SiO2, TiO2, ZrO2, etc. to exploit the favourable properties of both carriers. As these mixed oxides have acid–base properties, they promote the desulfurisation of alkyl DBTs.88 Differences in catalytic performance may arise as a result of variations in metal–support interactions that may influence the dispersion and morphology of active phases. The process involving the formation of the Co(Ni) –Mo–S active phases of the catalyst and their dispersion on the support surface is strongly influenced by the interaction with the support.
Support interaction also influences the degree of stacking of MoS2 and Co–Mo–S structures. Very weak support interaction resulted in the multi-stacking of type II Co–Mo–S phases. It has been reported that the degree of stacking in MoS2 and Co–Mo–S phases can be controlled by tailoring support properties. Stable, single-slab MoS2 has been observed on the alumina substrate. These slabs with a high MoS2-edge dispersion accommodate more Co-atoms to form higher active single-slab type II Co–Mo–S structures. A few studies have focused on the acidic and basic natures of the support and its effect on the nature of interaction with the active metal. TiO2 mixed oxide has received relatively higher attention because it records higher activity. NiMo/Ti–Al–O catalysts were more active than NiMo/Al2O3catalysts for the model compound HDS of DBT, 4-MDBT and 4,6-DMDBT.69 HDS tests of straight-run distillate gas oil showed that sulfidecatalysts supported on Ti–Al–O composite (11 mol%) reduce the sulfur level of diesel fuel from 500 to 50 ppm under conventional HDS conditions.82,83Ti-containing mixed oxide support not only enhanced light molecule HDS but also clearly promoted crude oil HDS.89
Based on theoretical calculations and experiments, it has been shown that both DBT and 4,6-DMDBT adsorbed horizontally on the catalyst surface and, therefore, the low reactivity of 4,6-DMDBT compared with DBT came from the reduced access of hydrogen to the C–S–C bridge after adsorption.90 In addition, zeolites and amorphous silica–alumina (ASA) supports have been widely used for the HDS of S-containing compounds. For the sulfide phases deposited on these substrates the specific rate constant for the HDS of refractory sulfur compounds such as 4,6-DMDBT was found to be substantially higher than on the conventional alumina-supported catalysts. For instance, when using ASA mixed with a NiMoaluminacatalyst, a 2.5-fold increase in the 4,6-DMDBT HDS was found.91 As both ASA and zeolite substrates show Brønsted acidity, the enhancement in the performance was associated to the Brønsted acidity-induced isomerization of 4,6-DMDBT to 3,6-DMDBT on the acid sites of these substrates. It is likely that the methyl substituents in the 3- and 6-positions do not sterically hinder the vertical adsorption that leads to direct desulfurization, in which the S-atom interacts directly with the surface of sulfide phases.
However, zeolites do not permit a good dispersion of the active phase. Zeolites as cation exchangers should have intrinsic difficulties to disperse molybdate salt precursors which are anionic. In addition, the size of the zeolitespores is too small (<1 nm) to accommodate the active phase and ensure the access of bulky reactants like 4,6-DMDBT. Unlike zeolites, the pore size of ASA is not a limiting factor for active phase dispersion and for the diffusion of the large reactant molecules to the active sites located within the pores. Recently, mesostructured systems such as MCM-41, Al-MCM-41, SBA-15 and SB-16 have received considerable attention as supports for Co(Ni) and promoted Mo(W) sulfidecatalysts.92–94 The high surface area of these nanoporous systems (pore size 5–10 nm) allows high CoMo or NiMo loading without the restricted access of the zeolite structure to bulky molecules.94 Thus, the performance of these catalysts is expected to be high due to their defined textural properties (pore size 5–30 nm, wall thickness 3–5 nm) and high dispersion of nano-sized MoS2, as shown in Fig. 10. Substantially more active catalysts than alumina-supported ones can be obtained by using these materials as support. The surface acidity of these materials can be modified by the incorporation of additives such as Al, Ti, Zr, etc. into the silica framework during synthesis.88,94,95
 | ||
| Fig. 10 TEM image of the mesoporous SBA-15 (a) showing a well-ordered hexagonal array of mesopores when the electron beam is parallel to the main axis of the cylindrical pores. HRTEM image of the sulfideCoMo/SBA-15 catalyst showing the slabs of the CoMoS phase. | ||
Carbon-supported catalysts have also been found to record high HDS activity with reduced deactivation by coke deposition due to the lower acidity of the carbon support.96,97 Rapid sintering of the active phase under reaction conditions and its unsuitability for regeneration (after deactivation) are the major disadvantages of using this support.
Unsupported transition metal sulfides have also been studied in recent years as potential new catalysts for hydrotreating reactions. The catalysts tested include sulfides of the type RuS2, V2S3, Rh2S3, NbS3, and mixed sulfides of Co–Mo, Ni–Mo, Ni–W, Ni–Mo–W, Fe–W, Fe–Mo and Ni–Ru. Among them, RuS2, Rh2S3 and NbS3 were found to exhibit improved HDS performance over the traditional Co–Ni and Mo–W sulfides.98 Unsupported Ni–Mo–W sulfidecatalysts exhibited at least 3 times higher activity than the conventional alumina-supported NiMo and CoMocatalysts.2Unsupported Ni–Mo–W sulfide composition is commercially used for deep HDS of diesel under the commercial trade name NEBULA.99 Apart from the unsupported transition metal sulfides, the hydrotreating activities of carbides and nitrides of Mo and W have also been investigated. These materials are stable under the hydroprocessing reaction conditions, which form surface sulfide and remain active in HDS reactions with time-on-stream.100
Unsupported metal phosphides have also received much attention due to their high activity for hydrodesulfurization (HDS) and hydrodenitrogenation (HDN) of petroleum feedstocks.101–103 It has been shown that MoP and WP have moderate activity and Ni2P has excellent activity in hydroprocessing. The overall activity was found to be in the order: Fe2P < CoP < MoP < WP < Ni2P in the simultaneous HDS of dibenzothiophene and HDN of quinoline.104
5. ‘Non-HDS’ based desulfurisation alternatives
Significant advances have been made over the past few years on developing new alternatives to the conventional catalytic HDS reactions for removing sulfur from liquid fuels to ultra low levels. Higher temperatures and pressures, more active catalysts, longer residence times, and/or additional reactors are required to increase the effectiveness of the HDS process. In view of this, efforts have been also made to develop new processes for desulfurisation, particularly to remove those highly refractory sterically hindered S-compounds under mild operating conditions. Among these, oxidative desulfurisation (ODS),105–111 biodesulfurisation (BDS),112–117 and S-extraction using solvents118–125 and ionic liquids126–130 have been investigated. The fundamentals and the advantages and limitations of these methodologies are given in the sub-sections below.5.1 Shifting the boiling point by alkylation
A simple procedure for removing organosulfur compounds from oil fractions is to increase their boiling temperature. These heavier compounds can then be separated from light fractions by distillation and concentrated in the heavy boiling part of refinery streams. British Petroleum used this concept in an advanced process for desulfurising FCC gasoline streams called olefinic alkylation of thiophenic sulfur (OATS).131 This process consists in the alkylation of thiophenic compounds via a reaction with the olefins present in the stream (Fig. 11). The boiling temperature of the resulting S-containing alkylate such as 3-hexylthiophene (221 °C) is much higher than that of thiophene (ca. 85 °C). This simple approach means it can be easily separated from the main gasoline stream by distillation. The high-boiling compounds produced can be blended into the diesel pool and desulfurised by conventional hydrotreating, as the octane number is not important for diesel.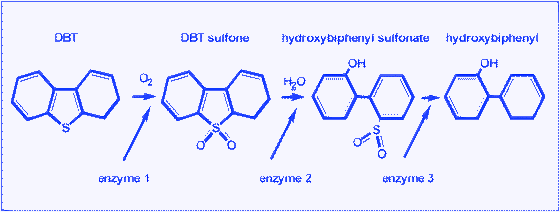 | ||
| Fig. 11 Schematic diagram of alkylation of thiophene with 1-olefins to obtain alkylthiophenes with much higher boiling points. | ||
In the OATS process, there are two basic factors to be tackled: (i) the selection of the alkylation agent, as a suitable carbochain can raise the boiling points of the alkylated thiophenes to an appropriate level; and (ii) the choice of the catalyst would have less influence if a gasoline feed is employed because olefins are present. Several alcohols and alkenes, such as methanol, propene/propanol, butene and propene, have been chosen as the alkylation agents.132 The alkylationcatalysts include supported BF3, AlCl3, ZnCl2, or SbCl5, phosphoric acid, Hβ zeolite and mesoporous materials.133,134 In most cases, OATS studies have focused on the conversion of thiophenes. Some insights into the mechanism of the reaction and certain detailed functions of the catalysts have also been illustrated. Besides improving the activities of the catalysts for thiophenealkylation, the most important problem is to prevent alkene oligomerization and aromatics alkylation, as these parallel reactions decrease the efficiency of the OATS and increase the losses in gasoline yield. As shown by previous studies,135thiophenealkylation, aromatics alkylation and hexene oligomerization coexist through the carbocation mechanism, and the only difference between them is the extent of their reaction. The reactivity is known to be associated with the acidity of the catalysts. In other words, variation in catalyst acidity would lead to a change in the activity of all three reactions.
Alkylation is carried out in an OATS reactor and the alkylate is then sent to a conventional distillation column where it is separated into a light S-free naphtha and a heavy S-rich stream. The light naphtha is sent directly to the gasoline pool and the heavy stream is preferably hydrotreated. By means of OATS technology, over 99.5% of the sulfur can be removed from the gasoline stream.136 It has been reported that sulfur can be reduced in gasoline from 2330 ppm to fewer than 20 ppm with a loss of only two octane.136 Another advantage of the OATS process is that it consumes less hydrogen because only a relatively low volume of the FCC gasoline stream is hydrotreated. Fortunately, under the conditions used, the alkylation of the S-containing compounds is much faster than that of aromatics. One of the disadvantages of the OATS process is that the alkylated sulfur compounds produced require more severe hydrotreating conditions to eliminate sulfur.
5.2 Desulfurisation by adsorption
The organosulfur compounds present in liquid fuels can be removed by adsorption on a solid sorbent. Based on the mechanism of S-compound interaction with the surface of the sorbent, the adsorption can be physical or chemical: physical adsorption is usually called adsorptive adsorption, whereas chemical adsorption is called reactive adsorption desulfurisation. Adsorptive desulfurisation is based on the interaction of the organosulfur compounds on the solid sorbent surface by weak (van der Waals) forces. Therefore, the sorbent can be easily regenerated, usually by flushing the spent sorbent with an inert gas, resulting in a high organosulfur compound concentration flow. On the other hand, reactive adsorption desulfurisation occurs through the formation of true chemical bonds of the S-containing compounds and the sorbent. Sulfur is fixed in the sorbent, usually as sulfide, and the S-free hydrocarbon is released into the purified fuel stream. Regeneration of the spent sorbent is usually conducted in the presence of air, which oxidizes the sulfide into SOx. The efficiency of the desulfurisation is determined mainly by the sorbent adsorption capacity, selectivity for organosulfur compounds, durability and regenerability.It has recently been shown145,146 that metal–organic frameworks (MOFs) are excellent candidates for eliminating the refractory dibenzothiophene (DBT) and alkyldibenzo-thiophenes that are predominantly present in the feed after hydrotreatment. Experimental results have shown that the extent of DBT adsorption at temperatures close to ambient (31 °C) is about nine times higher on the Basolite C300 (Cu3(C9H3O6)2) MOF system than on the benchmarked Y-type zeolite and activated carbons. Experiments designed for this comparison were conducted with DBT over a wide concentration range (10–1700 ppmw of sulfur) and the temperature was kept constant and close to ambient (31 °C). Under these conditions, the amounts of sulfur retained at equilibrium, expressed as g of sulfur per kg of sorbent, are shown in Fig. 12. From this figure, it is evident that the extent of DBT adsorption is much higher on the MOF sample than on the benchmarked Y-type zeolite.145
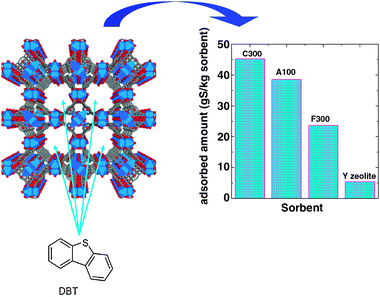 | ||
| Fig. 12 Structure of the C300 metal oxide framework (MOF) (left) and extent of DBT adsorption at constant temperature (31 °C) for different MOF sorbents. Y-zeolite is also included for comparison (right). 1 g sorbent per 100 g solution, initial S concentration 0.9908% DBT (1724 ppm). | ||
An adsorptive desulfurisation process called IRVAD was developed and targeted to remove a wide spectrum of organosulfur compounds from various refinery streams including FCC gasoline.147 The process is based on moving bed technology and uses a solid sorbent, which is counter-currently brought into contact with an S-rich hydrocarbon stream. The desulfurised hydrocarbon stream is produced at the top of the adsorber, whereas the spent sorbent is withdrawn at the bottom. The spent sorbent is circulated into the reactivation reactor where organosulfur compounds and some adsorbed hydrocarbons are desorbed from the sorbent surface. The regenerated sorbent is recirculated back to the adsorber (Fig. 13). Alumina is used as the sorbent and operates up to 240 °C at low pressure with a hydrocarbon/sorbent weight ratio of around 1.4. Under these conditions, the efficiency of the IRVAD process has been proven in pilot plant experiments for FCC feedstock (1276 ppm S) and coker naphtha (2935 ppm), providing at least a 90% reduction in sulfur content.148
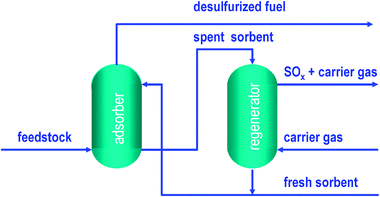 | ||
| Fig. 13 Simplified adsorptive desulfurization process flow. | ||
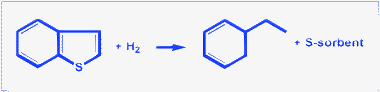 | ||
| Fig. 14 Scheme for the reactive adsorption desulfurization. | ||
Conoco-Phillips Petroleum Co. developed a technology called S-Zorb for the deep desulfurisation of diesel by reactive adsorption.152 In this process, the diesel stream is contacted with the sorbent in a fluid bed reactor in the presence of hydrogen. The sulfur-loaded sorbent is regenerated in a separate reactor and reused. Sulfur is typically reduced to about 5 ppm from feedstocks containing 500 ppm sulfur. Feedstocks containing greater than 2000 ppm could also be deeply desulfurised to ultra low sulfur levels by this process. Depending upon the feedstock, the process operating conditions can vary in the range: pressure = 20–35 bar; temperature about 350 °C. For an optimized composition of the Ni/ZnO sorbent equal to 13 wt% Ni, it has been reported that fewer than 0.03 ppm of sulfur were present in the effluent for one year for a kerosene feed containing 51 ppm of sulfur when working at a liquid space hourly velocity (LSHV) of 0.27 h−1.
The principal limitation of the reactive adsorption desulfurisation process is the rapid overloading of the sorbent in the case of refinery streams with high S-content. High S-content requires either a large amount of sorbent or a suitable process configuration based on fast kinetics of the deactivation–regeneration reactions. Both sorbent capacity and sorbent performance can be optimized by an appropriate composition of the sorbent applied. For low S-containing streams (usually involving the removal of the last ppm S) reactive adsorption requires the treatment of large volumes of sulfur diluted reactant. This results in a high energy penalty due to the pumping of the hydrocarbons through the reactor. To minimize this effect, it seems appropriate to apply structured low-pressure drop reactors (i.e. monolith reactors), and also to combine reactive adsorption desulfurisation with other processes that pre-concentrate sulfur.
5.3 S-removal by extraction
The concept of extractive desulfurisation means that organosulfur compounds are more soluble than hydrocarbons in an appropriate solvent. As shown by the process flow (Fig. 15), the oil and solvent are first mixed in a tank where the sulfur compounds are transferred from the oil phase into the solvent due to their higher solubility in the solvent. The solvent–fuel mixture is then fed into a separator in which the desulfurised oil is separated from the solvent. The desulfurised hydrocarbon stream can then be used either as a component to be blended into the final product or as a feed for further transformations. The organosulfur compounds are separated by distillation and the solvent is recycled. The most attractive feature of extractive desulfurisation is its application at low temperature and low pressure. To make the process efficient, the solvent must be carefully selected to satisfy a number of requirements. The organosulfur compounds must be highly soluble in the solvent whose boiling temperature is quite different from that of the sulfur-containing compounds, and it must be inexpensive for process viability.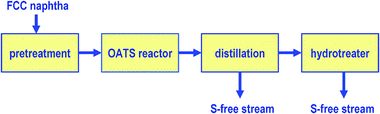 | ||
| Fig. 15 General process flow of extractive desulfurization. | ||
It has been reported that the sulfur content of a hydrotreated middle distillate can be reduced sevenfold (from 0.2% to 0.029%) and the aromatics content threefold (from 27.1% to 8%) by extraction with solvents such as methanol, furfural and ethylene glycol.153 Other solvents commonly used for sulfurextraction are acetonitrile, lactones like gamma butyrolactone, DMF, N-containing solvents like amines and pyrrolidones, or sulfur-containing solvents like DMSO and sulfolane.
As stated, the efficiency of extractive desulfurisation is limited mainly by the solubility of the organosulfur compounds in the solvent. Solubility can be enhanced by choosing an appropriate solvent taking into account the nature of the sulfur compounds to be removed. This is usually achieved by preparing mixtures of two or more solvents. Preparation of such mixtures of solvents is a complex task and intrinsically non-efficient since its composition depends markedly on the spectrum of the organosulfur compounds present in the feed stream. Solubility can also be enhanced by transforming the organosulfur compounds to increase their solubility in a polar solvent. One way to do this is by selectively oxidizing the organic sulfur compound (thiophene, BTs, DBTs) to sulfones possessing higher polarity.
In a study on the ability of 1,3-dimethylimidazolium dimethyl phosphate [MMIM][DMP], 1-ethyl-3-methylimidazolium diethyl phosphate [EMIM][DEP] and 1-butyl-3-methylimidazolium dibutyl phosphate[BMIM][DBP] to extract sulfur from diesel fuel over a wide range of sulfur concentrations, it was revealed that the solubility of DBT and BT in aqueous solutions of ILs at 25 ° varies in the following order: [BMIM][DBP] > [EMIM][DEP] > [MMIM][DMP].156 The most suitable IL for the desulfurisation of diesel fuel was found to be [EMIM][DEP], which is fairly highly reactive toward sulfur, poorly soluble in fuel, and affects only slightly other properties of the fuel. The ILs synthesized from organic acids (formic, acetic, and benzoic) and N-containing bases (aniline, piperidine, and diethylamine) seem quite interesting.154 After the threefold extraction of catalytic cracking gasoline with the aforementioned ILs, the sulfur content of the latter decreased from 240 to 30 ppm, and the content of aromatic hydrocarbons, from 26 to 14%. Ionic liquids can be regenerated by treating the extract with an excess of low-boiling paraffins and repeatedly used for desulfurisation. A high efficiency of ILs in the desulfurisation of diesel fuel was proven by the example of ILs containing 1-butyl-3-methylimidazolium as cation and tetrafluoroborate, hexafluorophosphate, octyl sulfate, ethyl sulfate, and dimethyl phosphate.126 The suggested scheme involving the steps of extraction and regeneration of ILs reduced the sulfur content of diesel fuel from 500 to 10 ppm.
In the absence of oxidants, the ILs themselves fail to provide a high degree of sulfur removal.154,157,158 For example, the ionic liquid [HMIm]BF4 removes only 6% of the sulfur, but in the presence of hydrogen peroxide it recorded a sulfur removal of 65, 70, and 93% at 70, 80, and 90 °C, respectively.157 However, a very high H2O2/S ratio is necessary to reach these sulfur removal levels. This drawback can be avoided by using catalysts: the tungsten and molybdenum peroxo complexes,159Fenton-like catalyst160 or phosphotungstic acid161 can remove the bulk of sulfur with a low hydrogen peroxide sulfur ratio. This finding provides evidence in favour of combining catalyticoxidation and extraction.
In a recent study, the extractive desulfurisation performance of low viscosity ILs based on the dicyanamide ([N(CN)2]−) anion was reported.162 Interestingly, these systems present much shorter extraction equilibrium times than other previously reported ILs, that is, less than 5 min for all the ILs at room temperature, which is important for generating a high production yield at industrial scale. Compared with cyclic thiophenium [S2][N(CN)2] and tetrahedral trialkylsulfonium [EtMe2S][N(CN)2], the aromatic imidazoliums [BMI][N(CN)2] and [EMI][N(CN)2] were more efficient. For [BMI][N(CN)2], the S-removals from gasoline and diesel fuel were 48.5 and 68.7%, respectively, in a single extraction at 25 °C, IL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶oil = 1∶1 (w/w), 5 min; negligible S-content could be obtained after 3–5 extraction cycles. The insensitivity of the extraction efficiency to temperature and initial S-content, along with easy regeneration and good reusability, are also useful for industrial application.
∶oil = 1∶1 (w/w), 5 min; negligible S-content could be obtained after 3–5 extraction cycles. The insensitivity of the extraction efficiency to temperature and initial S-content, along with easy regeneration and good reusability, are also useful for industrial application.
In addition, a new approach for the desulfurization was recently proposed using supported ionic liquid phase (SILP) systems and a comparison of extraction was made between biphasic and SILP phase. These SILP systems were prepared by dispersing the ionic liquid as a thin film on highly porous silica, which exhibited a significantly higher extraction performance owing to their large surface area, reducing the sulfur content to less than 100 ppm in one stage. Multistage extraction with this SILP technology offers very efficient utilization of ionic liquids and circumvents mass transfer limitations. However, leaching of the small quantity of ionic liquid from the support should be avoided and hence clever engineering of the ionic liquid loading and solid support is required.163
One of the principal challenges nowadays to be addressed before ILs can be used routinely is cost. In general, ILs are more expensive in comparison to common organic solvents. However, because ionic liquids can be recycled and their cost is decreasing drastically along the last few years, a wider application is expected for the removal of refractory S-containing compounds present in transportation fuels.
5.4 Oxydesulfurisation (ODS)
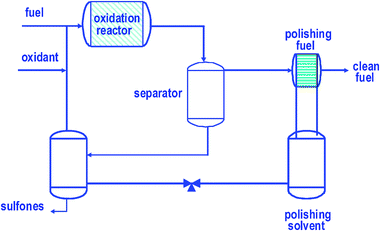
Oxidative desulfurisation (ODS) is a relatively new technology for the deep desulfurisation of light oil. This desulfurisation process includes two stages: (i) oxidation in a first step; and (ii) liquid extraction at the end. It is evident that the greatest advantages of the ODS process are low reaction temperature and pressure, and that expensive hydrogen is not used in the process. Another feature of ODS is that the refractory S-containing compounds in ODS are easily converted by oxidation. Therefore, ODS has great potential as a complementary process to traditional HDS for producing deeply desulfurised light oil.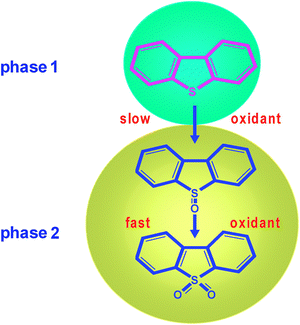 | ||
| Fig. 16 Simplified scheme showing the oxidation of DBT. The oxidant donates O-atoms to the DBT molecule present in the organic phase to form first sulfoxide and then sulfones (these oxidized compounds accumulate mainly in the polar phase). | ||
 | ||
| Fig. 17 Schematic biphasic simultaneous oxidation/extraction ODS reaction of DBT. | ||
The second step of this process involves removing the oxidized compounds by contacting the oil with a selective extractionsolvent. A simple L–L extraction technique using water-soluble polar solvents (i.e.acetonitrile, DMF and DMSO) is usually employed. Acetonitrile is particularly suited mainly because it has a relatively low boiling point (82 °C) and is easily separated from the sulfones by distillation. When acetonitrile is contacted with light oil, a large quantity of aromatics is extracted simultaneously with the sulfones. The addition of water, however, suppresses the extraction of the sulfones. The solvents should therefore be sufficiently polar to be selective for polar compounds in the process of extraction. Methanol, for instance, has sufficient polarity, but its density, 0.79 g cm−3, is about the same as that of standard light oil, making separation very difficult. Other properties to be considered include boiling point, freezing point, and surface tension.
The first ODS process was proposed in 1974 by using NO2 as oxidant followed by extraction with methanol to remove both S- and N-containing compounds from oil fractions.164 Later, in 1988, a process was described for purifying hydrocarbon aqueous oils containing both S- and N-containing compounds, such as shale oils, by first reacting the oil with an oxidizing gas containing NO2 and then extracting the oxidized oil with solvents in two stages (amines and formic acid).165 The oxidized products consist of a liquid phase and a by-product that is a semisolid-like residue with high S-content. However, interest has substantially increased in the early 2000s, in parallel with more restrictive limitations on sulfur concentration in fuels, and the need to remove refractory organosulfur compounds.
Organic hydroperoxides
Organic hydroperoxides, and most specifically tert-butyl hydroperoxide (TBHP) have been widely employed for sulfur removal from oil fractions. An ODS process based on this oxidant was first patented by EniChem/UOP166,167 and then by Lyondell Chemicals.168,169 EniChem/UOP removes sulfones by adsorption, whereas the Lyondell process uses extraction for sulfone separation. The major drawbacks of TBHP oxidant are: (i) the high cost of tert-butyl hydroperoxide, and the waste treatment of the tert-butyl alcohol by-product generated, as well as sulfone waste treatment, and (ii) the low amount of active oxygen by mass unit (17–10%), for this reason the transportation of this compound is very expensive, and it is therefore recommendable to install a hydrocarbonoxidation unit on-site to produce hydroperoxides, with an ensuing increase in capital costs. The oxidation of sulfur compounds with organic hydroperoxides takes place in the presence of catalysts. The active centre of these catalysts is a transition metal in a high oxidation state with Lewis acidity, such as Mo(IV), Ti(IV), V(V), W(IV), among which molybdenumcatalysts remain prominent.170–175 However, molybdenum tends to be leached to the reaction medium, where the catalyst is not very stable and the main part of the catalytic activity is due to solubilized molybdenum. Other much more stable catalysts, such as Co/Mn,171Ti–silica169 and Ti-MCM-41176catalysts, have been proposed. The best results have been obtained with Ti-based systems, as their silylation improves catalyst activity and life.176Organic peracids
Peracids are powerful oxidizing agents, and can oxidize sulfur compounds. These compounds are highly reactive and corrosive, and hence they are produced in situ by reacting hydrogen peroxide with a carboxylic acid, usually formic acid or acetic acid. The peracids are powerful oxidizing agents, and are able to oxidize sulfur compounds. These compounds are highly reactive and corrosive, for this reason they are produced in situ by reaction of hydrogen peroxide and a carboxylic acid, usually formic acid or acetic acid. The first studies have been based on the direct use of peracids in the oxidation of sulfur compounds.177–179 However, the amount of oxidizing agent used is very high in comparison to the sulfur removed. For this reason, it was imperative to incorporate catalysts capable of oxidising the sulfur compound more efficiently. The most common option is the use of transition metal salts, and specifically tungsten salts180 which are very active and selective in the oxidation of sulfur compounds with peracids. Modified active carbons are also catalysts for this reaction.181,182 In the presence of active carbon with carboxylic groups, more sulfur is removed (>95%) from the fuel than in their absence (60%).181 A second option is the use of transition metal salts that can form radicals from peracids (R–COOO), like Co183 or Fe.184 These radicals are very reactive and with a high oxidation capacity. The peracids of short carboxylic acids used in these reactions are highly polar and form two immiscible liquid phases in the reactor, so there are diffusional limitations between the fuel and polar phase with the oxidizing agent. Accordingly, studies have been conducted on the use of the phase transfer compound. The presence of the phase transfer compound facilitates the transfer of products and reagents at the polar–apolar interface, substantially increasing the mass transfer along the interface.185Hydrogen peroxide
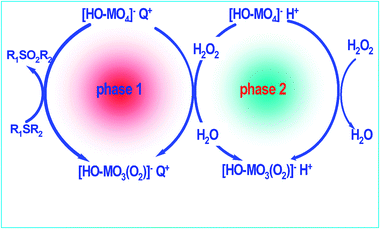
Hydrogen peroxide is the best candidate as oxidizing agent because it presents a high amount of active oxygen by mass unit (47%); it is a commercial product often used at industrial level; and its by-product is only water.186 A catalyst is required to ensure hydrogen peroxide efficiently oxidizes the sulfur compounds. The first reports on ODS with hydrogen peroxide are related to the use of photosensitizers,187,188 these compounds are first excited under light radiation and then activate the sulfur compound, and these excited compounds are oxidized by the hydrogen peroxide, with benzophenone being especially effective.188 Under these conditions, the desulfurisation of commercial light oil was also achieved and the sulfur content was reduced from 0.2 to 0.05 wt% by 48 h of photoirradiation. This long reaction time renders this process unattractive for industrial application.The second group of catalysts is based on transition metal salts in a high oxidation state with Lewis acidity.188–191 These systems allow the oxidation of DBT in short reaction time,192 however the complete oxidation of substituted DBT is slower, and then long residence times are necessary for use in refinery schemes. The main problem lies in the presence of two reaction phases—an oil phase with the sulfur compound and a polar phase that contains the oxidant agent (H2O2), which is not soluble in the oil phase. To avoid this difficulty, phase transfer catalysis has been proposed.110,111,193–195 The presence of the phase transfer agent facilitates the transfer of products and reagents at the polar–apolar interface, significantly increasing the mass transfer along the interface. Such a biphasic oxidation reaction follows the cycle illustrated in Fig. 18. In the polar phase, the catalyst precursor is rapidly oxidized by H2O2. The resulting peroxo-compound is transferred to the apolar phase by H+–Q+ ion exchange with the phase transfer agent (Q+). The peroxo-compound in the apolar phase oxidizes thioethers into sulfones, which can then be regenerated at the L–L interface with H2O2 or transferred to the polar phase where it reacts with hydrogen peroxide. The sulfones obtained are then transferred to the polar phase due to the solubility of sulfones in a polar solution, with the subsequent production of a sulfur-free polar phase. Mass transfer limitations still remain, which make this reaction slow for industrial application. Some interesting approaches have been studied, such as the use of microemulsions196 and microstructured reactors.197 In both cases the distances between bulk and interface are massively reduced, and diffusional limitations are therefore minimized, if not avoided.
The ideal catalytic systems belong to the class of heterogeneous catalysts. Consequently, efforts are being devoted to the development of processes using highly robust and recyclable catalysts that provide higher atom use and minimized pollution levels using greener ingredients. In recent years, the selective oxidation of S-containing compounds by H2O2 has been carried out using a large number of redox solid catalysts such as Ti,107,198–203 V,204–206 W,207–210Mo,106,210Co/Mn,211Ag,212Au,213Re.214
Improvement of catalytic activity at low H2O2/S ratios
Many catalytic systems are very active in the oxidation of sulfur compounds. However, only some catalysts can oxidize in short reaction times (<15 min) operating with a hydrogen peroxide sulfur molar ratio close to the stoichiometric one (H2O2/S = 2). A competitive commercial process requires working with an amount of oxidant that is as small as possible. Liquid fuels are very complex mixtures containing alkenes and aromatics, and these compounds can be also oxidized consuming part of the oxidant and degrading the quality of the fuel. These unwanted oxidation reactions are evident at temperatures about 80–90 °C for this short reaction time (<15 min). For this reason, the reaction has to be conducted at temperatures lower than 80 °C and with a short reaction time for real fuels.Another important area for improvement is the relative reactivity with different sulfur compounds. The relative oxidation reactivity of small peracids (formic or acetic) is 4,6-DMDBT > 4-MDBT > DBT97,207 which is the opposite to HDS and makes a very good complement to HDS. When a catalyst is used, irrespectively if it is homogeneous or heterogeneous, the relative oxidation reactivity is different: DBT > 4-MDBT > 4,6-DMDBT > BT.105,110,203,210 The relative reactivities of DBT and substituted DBT compounds appear to be related to the steric hindrance of the ethyl and methyl groups at positions 4 and 6 in the DBT molecule, hindering the formation of reaction intermediates prior to oxidisation, whereas it is more difficult to oxidize in terms of S–C bond stabilization in the BT molecule.105,110,215 Consequently, the activity in the oxidation of substituted DBT compounds must be improved because these compounds are present in the treated diesel stream, making it difficult to comply with very restrictive environmental regulations.
Increasing mass transfer in the reactor
The reaction is conducted in the presence of two liquid reaction phases: an oil phase with the sulfur compound and a polar phase that contains the oxidant agent (H2O2), which is not soluble in the oil phase. Consequently, the overall sulfur removal rate is limited by the mass transfer between phases with very active catalysts. For this reason, an increase in mass transfer clearly improves the overall sulfur removal rate. Some attempts have been made using microemulsions216,217 and microstructured reactors.218 Microemulsions are interesting from a conceptual point of view but need intense development if they are to be applied in a refinery process. A very interesting concept is the use of microstructured reactors. Chemical microstructured reactors (MSR) are devices containing open paths for fluid flow with dimensions in the sub-millimetre range. Most MSR are designed as multiple parallel channels with diameters between ten and several hundred micrometres where the chemical transformation occurs.219 This gives a high specific surface area in the range of 10000 to 50000 m2 m−3219,220 and allows an effective mass and heat transfer when compared to more traditional chemical reactors of ≈100 m2 m−3. These reactor systems improve mass transfer (100 times).Process-engineering research should focus on decreasing the volumetric reaction rate (polar solvent/oil ratio). The solvent/oil volume ratio is among the most important technical bottlenecks in the development of ODS processes, and in order to reduce the operational costs associated with the handling, separation and disposal of water, the volume ratios of solvent/oil should ideally be minimized.
Enhancing the post-treatment of sulfones
The concentrations of sulfur compounds are very low, but the refinery stream to be treated is huge, so a large amount of sulfones is produced. Some sulfones can be used as a chemical intermediate but in a real fuel the variety of sulfur compounds is very high, and therefore the number of different sulfones is also high and very difficult to separate. For this reason, an adequate post-treatment of sulfones is very important. The amount of fuel lost in the ODS process depends on the initial sulfur concentration and the amount of sulfur removed; for instance, 1000 ppm of S removed implies a decrease of around 5–6% in fuel mass. It is therefore highly expedient to recover, at least in part, the hydrocarbon in the oxidized sulfur compounds.The first option when treating these sulfones is to eliminate the sulfur and produce an S-free fuel. The oxidised sulfur-containing hydrocarbon is contacted with a metal to form a metal–sulfur containing compound. This process therefore relies on the adsorption of oxidised sulfur compounds from the hydrocarbon using a metal capable of forming a metal sulfide. The metal is selected from the group consisting of Ni, Mo, Co, W, Fe, Zn, V, Cu, Mn, Hg, and mixtures thereof. This process is different from conventional hydrodesulfurisation in that the sulfur is immobilized in the form of a metallic sulfur compound (e.g. a metal sulfide) rather than converted to hydrogen sulfide. For this reason, the addition of free molecular hydrogen, as required in hydrodesulfurisation, is overcome. The second option is the catalyticdecomposition on solid acids like zeolites or silica–alumina, or basic catalysts like MgO or hydrotalcite.221,222 This catalytic conversion takes place under relatively mild conditions, without the use of a hydrogen atmosphere. However, these processes need to be improved.
6. Biodesulfurisation (BDS)
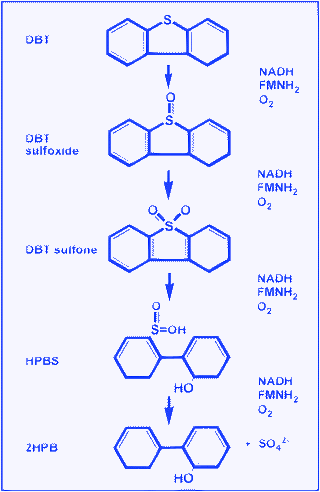
One of the alternative options for removing sulfur from fossil fuel is by biological methods. Sulfur atoms form 0.5–1% of the dry weight of bacterial cells. Microorganisms require sulfur for their growth and biological activities. Sulfur generally occurs in the structure of some enzymecofactors (such as Coenzyme A, thiamine and biotin), amino acids and proteins (cysteine, methionine, and disulfur bonds).223 Energy BioSystems Corporation (EBC) was initially the only commercial venture dedicated to the development of biodesulfurisation technology. EBC's concept for a biodesulfurisation process was to treat diesel, but also to produce a value-added surfactant by-product to achieve a more economical process.113,115 A number of reviews have recently been published in this field,115,224 so we will focus on a general description of the process and on some improvements necessary for developing a BDS process.Depending on their enzymes and metabolic pathways, microorganisms may have the ability to obtain the sulfur they need from different sources. Some microorganisms consume the sulfur in thiophenic compounds such as DBT and reduce the sulfur content in fuel. In terms of DBT utilization, two main pathways have been reported: ring-destructive (degradation) and sulfur-specific (desulfurisation) pathways. To date, two ring-destructive pathways for DBTmetabolism have been recognized.
A very attractive, green option is to utilize microorganisms to do the oxidation chemistry of sulfur species as illustrated in Fig. 19. The reaction takes place in the presence of water and oxygen under ambient conditions, i.e., ambient temperature and pressure. Bacteria converting dibenzothiophene and alkyl sulfides are relatively well known while fewer bacteria are found for benzothiophene, and only a few bacteria detected for thiophene.223 In general, many reports of bio-desulfurization do not show very deep desulfurization, down to 50–200 ppm sulfur at best. The reason for this lies in the fact that higher bacterial activity occurs in the higher concentration range. Therefore the potential of biodesulfurization for achieving the 10 ppm S level might be not so great. In addition, another important aspect to be considered in the bio-system is the competitive reactions caused by other bacteria. A successful H2S biological process like the one from ThioPaque224 has the great advantage that only target type bacteria seem to survive in a very noxious environment of H2S, oxidizing H2S to elemental sulfur efficiently. In that respect, crude and diesel desulfurization would need an additional effort to promote the working of target bacteria. Occasionally, reports appear on development of new bacteria for oil desulfurization.225,226
 | ||
| Fig. 19 The pathway of biological desulfurization of DBT relies on biocatalysts for specificity. NADH is reduced nicotinamide adenosine dinucleotide; FMN is flavin mononucleotide; DSZA, DSZB, DSZC and DSZD are the catalyticgene products of dszA, dszB, dszC and dszD, respectively. | ||
Microbial sulfur-specific transformations have been shown to selectively desulfurize organic S-containing molecules.227 Discoveries related to biodesulfurization mechanisms may lead to commercial application of biodesulfurization through engineering recombinant strains for over-expression of biodesulfurization genes and combining relevant industrial and environmental traits with improvement in bioprocess design.226 Key improvements lie in the enhancement of biocatalyst stability, faster kinetics, minimization of mass transfer limitations, and temperature and solvent tolerance. Once these advances can be achieved biocatalysis may be a cost-effective approach to obtain low sulfur gasoline. Indeed, the challenge shall be to reach such improvements by the time the regulations for low sulfurfuels go into effect with the objective to be competitive with emerging non-biological approaches.
6.1 Destructive biodesulfurisation
The most common pathway for DBTdegradation, known as the ‘Kodama pathway’ (Fig. 20), is analogous to that of naphthalenedegradation.228 In this pathway, initial dioxygenation is carried out at the peripheral aromatic ring of DBT, followed by cleavage of the ring. This process leads to the accumulation of 3-hydroxy-2-formylbenzothiophene as a water-soluble end product, with lower carbon content than DBT. No desulfurisation of the organosulfur substrate occurs in this pathway. Denome et al.229 cloned and sequenced a 9.8 kb DNA fragment from Pseudomonas strain C18 that encoded DBT-degrading enzymes. Nine ORFs were identified and designated doxABDEFGHIJ; collectively, these genes were referred to as the DOX (DBToxidation) pathway. The results indicated that a single genetic pathway controls the metabolism of DBT, naphthalene and phenanthrene in strain C18.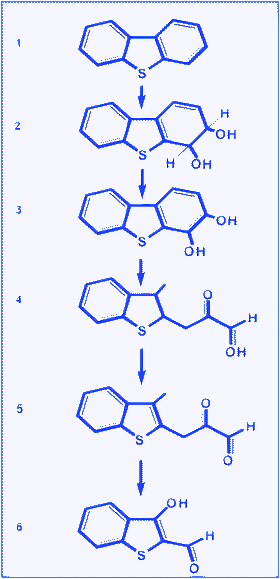 | ||
| Fig. 20 Kodama pathway of DBTdegradation. | ||
Another ring-destructive pathway that results in mineralization of DBT is the one proposed by van Afferden et al.230 They isolated Brevibacterium sp. DO, capable of using DBT for growth as the sole source of carbon, sulfur and energy. During DBTmineralization three metabolites were identified: DBT sulfoxide (DBTO), DBT sulfone (DBTO2) and benzoate. This pathway resulted in the complete mineralization of DBT, with the release of the sulfur atom as sulfite stoichiometrically, which then oxidized to sulfate. There are no detailed studies of the enzymology or molecular biology of DBTdegradation by this strain. This ring-destructive pathway may be valuable in biodegradation of DBT in the environment.
The above ring-destructive pathways are not commercially useful for the petroleum industry, because use of the carbon skeleton of S-containing compounds by the bacteria reduces the fuel's calorific value.
7. Concluding remarks
Concerns for cleaner air and increasingly more stringent regulations on sulfur contents in transportation fuels will make desulfurisation more and more important. The sulfur problem is becoming more serious in general, particularly for diesel fuels, as the regulated sulfur content is recording a lower order of magnitude, while the sulfur contents of crude oils refined in the US are becoming higher. The chemistry of diesel fuel processing has evolved significantly around the central issue of how to produce cleaner diesel fuels in a more efficient and eco-friendly manner. New design approaches are required for producing affordable ultra-clean diesel and gasoline.Research on the deep desulfurisation of diesel fuel to ultra low sulfur levels has recently gained considerable importance, and over the past seven years a growing number of articles have been published in the scientific literature concerning various aspects of ULSD production by academic research institutions and industrial research laboratories. The renewed interest in ULSD research is driven by the need to have a thorough understanding of the various factors influencing the deep desulfurisation of diesel to ultra low levels as well as to find cost-effective ways to produce ULSD, which are expected to soon dominate the global distillate fuels market. ULSDfuel will enable the use of advanced emission control devices in diesel engines for lowering harmful exhaust emissions. Remarkable progress has been made in recent years in our understanding of the key factors, such as catalytic sites, inhibitory effects, feed quality, hydrogen partial pressure and other process variables, kinetic and thermodynamic effects, etc. influencing the desulfurisation of the least reactive sterically hindered alkyl DBTs (e.g.4,6-DMDBT). Significant improvements in HDS catalysts and reactor design have been made, and optimum operating strategies have been developed to minimize the inhibition effects of H2S and N-compounds and enhance the removal of the last traces of refractory sulfur compounds. The conventional diesel hydrotreating units for ULSD production have been revamped based on the results of this research and development. In addition, new hydrotreating process concepts and technologies and alternative non-hydrogenation routes have been developed for ULSD production.
Refurbishing existing hydrotreaters designed in the 1990s has been undertaken by refineries to produce low sulfur diesel with 500 ppm S. The general trend in the future is for an increasing global demand for ULSD, whereby refiners will be required to upgrade poor quality feedstocks such as LCO, heavy gas oils, etc. to produce the additional volume of ULSD. Furthermore, future ULSD specifications may require improvements in other diesel properties such as higher cetane number, lower aromatics content, density reduction, etc. The production of such high quality ULSD from low quality feeds is a tough challenge, but the economic incentive is high.
Deep S-removal means high hydrogen consumption, which is a serious problem affecting the process economics in ULSD production. The use of non-hydrogenation processes such as ODS will also gain increasing importance for ULSD production in the future. The refractory sulfur species (e.g.4,6-DMDBT) are preferentially attacked and desulfurised in the ODS process. Because of the mild process conditions with no hydrogen requirement, the capital and operating costs of the ODS process will be substantially lower, and it could be used in combination with existing conventional hydrotreaters for desulfurising moderately hydrotreated low sulfur diesel.
We must begin to contemplate the use of zero-sulfur transportation fuels in the near future. One particular case, which presents a major challenge for the ultra-deep desulfurisation of liquid hydrocarbonfuels, is a fuel processor for proton-exchange membranefuelcells, which basically require zero-sulfurhydrogen streams generated by reforming liquid hydrocarbonfuels.
Acknowledgements
We are grateful to many of our colleagues for stimulating discussions on diesel fuel desulfurization and hydrogenation. We would like to acknowledge the MICINN sponsor under research grants ENE2007-07345-C03-01/ALT, ENE2010-21198-C04-01 and CTQ2010-19157-C03-01.References
- M. Booth, J. G. Buglass and J. F. Unsworth, Top. Catal., 2001, 16/17, 39–46 CrossRef.
- H. Hoshi and H. Hayashi, Toyota Tech. Rev., 2004, 53, 10–15 Search PubMed.
- R. N. Colvile, E. J. Hutchinson, J. S. Mindell and R. F. Warren, Atmos. Environ., 2001, 35, 1537–1565 CrossRef CAS.
- V. Ramanathan and G. Carmichael, Nat. Geosci., 2008, 1, 221–227 Search PubMed.
- J. R. Katzer, M. P. Ramage and A. V. Sapre, Chem. Eng. Prog., 2000, 96, 41–51 Search PubMed.
- S. F. Venner, Hydrocarbon Process., Int. Ed., 2000, 5, 51–52 Search PubMed.
- R. B. Miller, A. Macris and A. R. Gentry, Pet. Technol. Q., 2001, 2, 69–73 Search PubMed.
- US EPA, Diesel Fuel Quality: Advance Notice of Proposed Rulemaking, EPA420-F-99-011, Office of Mobile Sources, May 1999.
- BP Statistical Review, 2006, http://www.bp.com/.
- P. T. Vasudevan and J. L. G. Fierro, Catal. Rev., 1996, 38, 161–188 CrossRef CAS.
- I. V. Babich and J. A. Moulijn, Fuel, 2003, 82, 607–631 CrossRef CAS.
- C. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- M. Breysse, G. Djega-Mariadassou, S. Pessayre, C. Geantet, M. Vrinat, G. Perot and M. Lemaire, Catal. Today, 2003, 84, 129–138 CrossRef CAS.
- B. H. Cooper and K. G. Knudsen, in Practical Advances in Petroleum Processing, 2006, ch. 10, p. 297 Search PubMed.
- T. C. Ho, Appl. Catal., A, 2003, 244, 115–128 CrossRef CAS.
- A. Marafi, A. Al-Hindi and A. Stanislaus, Fuel Process. Technol., 2007, 88, 905–911 CrossRef CAS.
- H. Topsøe, B. Hinnemann, J. K. Nørskov, J. V. Lauritsen, F. Besenbacher, P. L. Hansen, G. Hytoft, R. G. Egeberg and K. G. Knudsen, Catal. Today, 2005, 107–108, 12 CrossRef CAS.
- T. Fujikawa, Top. Catal., 2009, 52, 872–879 CrossRef CAS.
- M. Breysse, C. Geantet, P. Afanasiev, J. Blanchard and M. Vrinat, Catal. Today, 2008, 130, 3–13 CrossRef CAS.
- S. T. Oyama, T. Gott, H. Zhao and Y.-K. Lee, Catal. Today, 2009, 143, 94–107 CrossRef CAS.
- H. Schulz, W. Bohringer, P. Waller and F. Ousmanov, Catal. Today, 1999, 49, 87–97 CrossRef CAS.
- T. C. Ho, Catal. Today, 2004, 98, 3–18 CrossRef CAS.
- M. Egorova and R. Prins, J. Catal., 2004, 225, 417–427 CrossRef CAS.
- V. Rabarihoela-Rakotovao, S. Brunet, G. Perot and F. Diehl, Appl. Catal., A, 2006, 306, 34–44 CrossRef CAS.
- H. Farag, Appl. Catal., A, 2007, 331, 51–59 CrossRef CAS.
- K. H. Choi, Y. Korai, I. Mochida, I. W. Ryu and W. Min, Appl. Catal., B, 2004, 50, 9–16 CrossRef CAS.
- V. Rabarihoela-Rakotovao, F. Diehl and S. Brunet, Catal. Lett., 2008, 129, 50–56.
- T. Song, Z. Zhang, J. Chen, Z. Ring, H. Yang and Y. Zheng, Energy Fuels, 2006, 20, 2344–2349 CrossRef CAS.
- Z. Liu, Q. Zhang, Y. Zheng and J. Chen, Energy Fuels, 2008, 22, 860–866 CrossRef CAS.
- R. R. Bharvani and R. S. Henderson, Hydrocarbon Process., Int. Ed., 2002, 81, 61–64 Search PubMed.
- World Energy Outlook 2009. OECD/IEA 2009, p. 622.
- Chemistry of Diesel Fuels, ed. C. Song, S. Hsu, I. Mochida, Taylor & Francis, New York, 2000 Search PubMed.
- Diesel Emission Control-Sulphur Effects (DECSE) Program, Final Report, The US Department of Energy, Engine Manufactures Association, 2001 Search PubMed.
- Z. C. Mester, in Meeting Sulphur Specifications for 2000 and Beyond, San Francisco, 2000, March 26–29 Search PubMed.
- EU Fuel Regulations: http://www.dieselnet.com/standards/eu/fuel.php.
- Japan Fuel Regulations: http://www.dieselnet.com/standards/jp/fuel.php.
- EIA, Official Energy Statistics from the US Government, http://www.eia.doe.gov/oiaf/aeo/otheranalysis/aeo2005analysispapers/candr.html.
- M. Gürü, U. Karakaya, D. Altiparmak and A. Alicilar, Energy Convers. Manage., 2002, 43, 1021–1025 CrossRef CAS.
- Hart World Refining and Fuel Service, http://www.hartwrfs.com/.
- S. Patel, Hydrocarbon Process., Int. Ed., 2007, 86(2), 65–68 Search PubMed.
- M. J. Girgis and B. C. Gates, Ind. Eng. Chem. Res., 1991, 30, 2021–2058 CrossRef CAS.
- T. A. Zepeda, B. Pawelec, J. L. G. Fierro and T. Halachev, Appl. Catal., B, 2007, 71(3–4), 223–236 CrossRef CAS.
- X. Ma, K. Sakanishi, T. Isoda and I. Mochida, Ind. Eng. Chem. Res., 1995, 34, 748–754 CrossRef CAS.
- X. Ma, K. Sakanishi and I. Mochida, Ind. Eng. Chem. Res., 1994, 33, 218–222 CrossRef CAS.
- D. D. Whitehurst, H. Farag, T. Nagamatsu, K. Sakanishi and I. Mochida, Catal. Today, 1998, 45, 299–305 CrossRef CAS.
- H. Schulz, W. Bohringer, F. Ousmanov and F. Waller, Fuel Process. Technol., 1999, 61, 5–42 CrossRef CAS.
- M. Daage and R. R. Chianelli, J. Catal., 1994, 149, 414–427 CrossRef CAS.
- H. Topsøe, B. S. Clausen, F. E. Massoth, Hydrotreating: Catalysis, Science and Technology, Springer, Berlin, 1996, pp. 310 Search PubMed.
- D. D. Whitehurst, T. Isoda and I. Mochida, Adv. Catal., 42, 345–471 Search PubMed.
- K. G. Knudsen, B. H. Cooper and H. Topsoe, Appl. Catal., A, 1999, 189, 205–215 CrossRef CAS.
- B. C. Gates and H. Topsøe, Polyhedron, 1997, 16, 3213–3217 CrossRef CAS.
- R. Shafi and G. J. Hutchings, Catal. Today, 2000, 59, 423–442 CrossRef CAS.
- G. Perot, Catal. Today, 2003, 86, 111–128 CrossRef CAS.
- E. J. M. Hensen, V. H. J. de Beer, J. A. R. van Veen and R. A. van Santen, J. Catal., 2003, 215, 353–357 CrossRef CAS.
- H. Farag and K. Sakanishi, J. Catal., 2004, 225, 531–535 CrossRef CAS.
- H. Topsøe, B. Hinnemann, J. K. Nørskov, J. V. Lauritsen, F. Besenbacher, P. L. Hansen, G. Hytoft, R. G. Egeberg and K. G. Knudsen, Catal. Today, 2005, 107–108, 12–22 CrossRef CAS.
- A. Logadottir, P. G. Moses, B. Hinnemann, N. Y. Topsøe, K. G. Knudsen, H. Topsøe and J. K. Nørskov, Catal. Today, 2006, 111, 44–51 CrossRef CAS.
- F. Besenbacher, M. Brorson, B. S. Clausen, S. Helveg, B. Hinnemann, J. Kibsgaard, J. V. Lauritsen, P. G. Moses, J. K. Nørskov and H. Topsøe, Catal. Today, 2008, 130, 86–96 CrossRef CAS.
- H. Wang and R. Prins, J. Catal., 2009, 264, 31–43 CrossRef CAS.
- T. Fujikawa, J. Jpn. Pet. Inst., 2007, 50, 249–261 Search PubMed.
- D. S. Stratiev, I. Shishkova, T. Tzingov and P. Zeuthen, Ind. Eng. Chem. Res., 2009, 48, 10253–10261 CrossRef CAS.
- T. A. Zepeda, B. Pawelec, J. L. G. Fierro and T. Halachev, J. Catal., 2006, 242(2), 254–269 CrossRef CAS.
- W. Pang, Y. Z. Zhang, K. H. Choi, J. K. Lee, S. H. Yoon, I. Mochida and K. Nakano, Petroleum Science and Technology, 2009, 27, 1349–1359 Search PubMed.
- G. F. Wan, A. Duan, Y. Zhang, Z. Zhao, G. Jiang, D. Zhang, Z. Gao, J. Liu and K. H. Chung, Energy Fuels, 2009, 23, 3846–3852 CrossRef CAS.
- R. Ismagilov, S. A. Yashnik, A. N. Startsev, A. I. Boronin, A. I. Stadnichenko, V. V. Kriventsov, S. Kasztelan, D. Guillaume, M. Makkee and J. A. Moulijn, Catal. Today, 2009, 144, 235–250 CrossRef.
- H. Topsøe, B. S. Clausen, N. Y. Topsøe and P. Zeuthen, Stud. Surf. Sci. Catal., 53, 77–102 Search PubMed.
- B. Delmon, Stud. Surf. Sci. Catal., 53, 1–40 Search PubMed.
- E. J. M. Hensen, P. J. Kooyman, Y. van der Meer, A. M. van der Kraan, V. H. J. de Beer, J. A. R. van Veen and R. A. van Santen, J. Catal., 2001, 199, 224–235 CrossRef CAS.
- H. Topsøe, Appl. Catal., A, 2007, 322, 3–8 CrossRef CAS.
- S. Eijsbouts, S. W. Mayo and K. Fujita, Appl. Catal., A, 2007, 322, 58–66 CrossRef CAS.
- J. K. Nøreskov, B. S. Clausen and H. Topsøe, Catal. Lett., 1992, 13, 1–8 CrossRef.
- S. Harris and R. R. Chianelli, J. Catal., 1986, 98, 17–31 CrossRef CAS.
- P. Raybaud, J. Hafner, G. Kresse, S. Kasztelan and H. Toulhoat, J. Catal., 2000, 190, 128–143 CrossRef CAS.
- R. R. Chianelli, Oil Gas Sci. Technol., 2006, 61, 503–513 CrossRef CAS.
- C. Thomazeau, C. Geantet, M. Lacroix, M. Danot, V. Harlec and P. Raybaud, Appl. Catal., A, 2007, 322, 92–97 CrossRef CAS.
- J. Kibsgaard, J. V. Lauritsen, B. S. Clausen, H. Topsøe and F. Besenbacher, J. Am. Chem. Soc., 2006, 128, 13950–13958 CrossRef CAS.
- J. V. Lauritsen, J. Kibsgaard, G. H. Olesen, P. G. Moses, B. Hinnemann, J. K. Stig Helveg, B. S. Nørskov, H. Clausen, E. Topsøe, E. Lægsgaard and F. Besenbacher, J. Catal., 2007, 249, 220–233 CrossRef CAS.
- C. Song and X. Ma, Appl. Catal., B, 2003, 41, 207–238 CrossRef CAS.
- A. Stanislaus, A. Marafi and M. S. Rana, Catal. Today, 2010, 153, 1–68 CrossRef CAS.
- B. Pawelec, S. Damyanova, R. Mariscal, J. L. G. Fierro, I. Sobrados, J. Sanz and L. Petrov, J. Catal., 2004, 223, 86–97 CrossRef CAS.
- T. A. Zepeda, B. Pawelec, J. L. G. Fierro and T. Halachev, Appl. Catal., B, 2006, 71, 223–236.
- M. Breysse, P. Afanasiev, C. Geantet and M. Vrinat, Catal. Today, 2003, 86, 5–16 CrossRef CAS.
- G. Muralidhar, B. N. Srinivas, M. S. Rana, M. Kumar and S. K. Maity, Catal. Today, 2003, 86, 45–60 CrossRef.
- A. Carati, G. Ferraris, M. Guidotti, G. Moretti, R. Psaro and C. Rizzo, Catal. Today, 2003, 77, 315–323 CrossRef CAS.
- R. Tian, B. Shen, F. Wang, C. Lu and C. Xu, Energy Fuels, 2009, 23, 55–59 CrossRef CAS.
- T. D. Tang, C. Yin, L. Wang, Y. Ji and F. S. Xiao, J. Catal., 2008, 257, 125–133 CrossRef CAS.
- Y. Zhao, B. Shen, W. Zhang, R. Tian, Z. Zhang and J. Gao, Fuel, 2008, 87, 2343–2346 CrossRef CAS.
- A. J. Duan, R. L. Li, G. Y. Jiang, J. Gao, Z. Zhao, G. Wan, D. Zhang and C. Keng, Catal. Today, 2009, 140, 187–191 CrossRef CAS.
- M. S. Rana, J. Ancheyta and P. Rayo, Catal. Today, 2005, 109, 24–32 CrossRef CAS.
- V. Meille, E. Schulz, L. Lemaire and M. Vrinat, J. Catal., 1997, 170, 29–37 CrossRef CAS.
- P. Michaud, J. L. Lemberton and G. Perot, Appl. Catal., A, 1998, 169, 343–353 CrossRef CAS.
- J. Ren, A. Wang, X. Li, Y. Chen, H. Liu and Y. Hu, Appl. Catal., A, 2008, 344, 175–182 CrossRef CAS.
- O. Y. Gutierrez, F. Perez, G. A. Fuentes, X. Bokhimi and T. Klimova, Catal. Today, 2008, 130, 292–301 CrossRef CAS.
- T. A. Zepeda, B. Pawelec, J. L. G. Fierro, A. Montesinos, A. Olivas, S. Fuentes and T. Halachev, Microporous Mesoporous Mater., 2008, 111(1–3), 493–506 CrossRef CAS.
- R. S. Araújo, D. A. S. Maia, D. C. S. Azevedo, C. L. Cavalcante, Jr., E. Rodríguez-Castellón and A. Jimenez-López, Appl. Surf. Sci., 2009, 255, 6205–6209 CrossRef CAS.
- B. Pawelec, R. Mariscal, J. L. G. Fierro, A. Greenwood and P. T. Vasudevan, Appl. Catal., A, 2001, 206, 295–307 CrossRef CAS.
- Z. Vít, Catal. Lett., 1992, 13, 131–136 CrossRef CAS.
- N. Hermann, M. Brorson and H. Topsøe, Catal. Lett., 2000, 65, 169–174 CrossRef CAS.
- F. L. Plantenga, R. Cefortain, S. Eijsbouts, F. van Houtert, S. L. Soled, S. Miseo, R. Krycak, G. Anderson and K. Fujita, Stud. Surf. Sci. Catal., 145, 407–410 Search PubMed.
- D. J. Sajkowski and S. T. Oyama, Appl. Catal., A, 1996, 134, 339–349 CrossRef CAS.
- W. Li, B. Dhandapani and S. T. Oyama, Chem. Lett., 1998, 207–208 CrossRef CAS.
- C. Stinner, R. Prins and T. Weber, J. Catal., 2000, 191, 438–444 CrossRef CAS.
- S. J. Sawhill, K. A. Layman, D. R. Van Wyk, M. H. Engelhard, C. Wang and M. E. Bussell, J. Catal., 2005, 231, 300–313 CrossRef CAS.
- P. Clark, X. Wang and S. T. Oyama, J. Catal., 2002, 207, 256–265 CrossRef CAS.
- M. Te, C. Fairbridge and Z. Ring, Appl. Catal., A, 2001, 219, 267–280 CrossRef CAS.
- J. L. Garcia-Gutierrez, G. A. Fuentes, M. E. Hemandez-Teran, P. Garcia, F. Murrieta-Guevara and F. Jimenez-Cruz, Appl. Catal., A, 2008, 334, 366–373 CrossRef CAS.
- V. Hulea, F. Fajula and J. Bousquet, J. Catal., 2001, 198, 179–186 CrossRef CAS.
- A. V. Anisimov, E. V. Fedorova, A. Z. Lesnugin, V. M. Senyavin, L. A. Aslanov, V. B. Rybakov and A. V. Tarakanova, Catal. Today, 2003, 78, 319–325 CrossRef CAS.
- J. Palomeque, J. M. Clacens and F. Figueras, J. Catal., 2002, 211, 103–108 CrossRef CAS.
- J. M. Campos-Martin, M. C. Capel-Sanchez and J. L. G. Fierro, Green Chem., 2004, 6, 557–562 RSC.
- X. Jiang, H. Li, W. Zhu, L. He, H. Shu and J. Lu, Fuel, 2009, 88, 431–436 CrossRef CAS.
- J. M. Campos-Martin, M. C. Capel-Sanchez, P. Perez-Presas and J. L. G. Fierro, J. Chem. Technol. Biotechnol., 2010, 85, 879–890 CrossRef CAS.
- D. J. Monticello, Curr. Opin. Biotechnol., 2000, 11, 540–546 CrossRef CAS.
- J. Kilbane II, Curr. Opin. Biotechnol., 2006, 17, 305–314 CrossRef.
- M. Soleimani, A. Bassi and A. Margaritis, Biotechnol. Adv., 2007, 25, 570–596 CrossRef CAS.
- B. Yu, P. Xu, Q. Shi and C. Ma, Appl. Environ. Microbiol., 2006, 72, 54–58 CrossRef CAS.
- G. Mohebali and A. S. Ball, Microbiology, 2008, 154, 2169–2183 CrossRef CAS.
- J. G. Park, C. H. Ko, K. B. Yi, J. H. Park, S. S. Han, S. H. Cho and J. N. Kim, Appl. Catal., B, 2008, 81, 244–250 CrossRef CAS.
- X. Ma, S. Velu, I. H. Kim and C. Song, Appl. Catal., B, 2005, 56, 137–147 CrossRef CAS.
- J. H. Kim, X. Ma, A. Zhou and C. Song, Catal. Today, 2006, 111, 74–83 CrossRef CAS.
- S. Ve1u, X. Ma, C. Song, M. Namazian, S. Sethuraman and G. Venkataraman, Energy Fuels, 2005, 19, 1116–1125 CrossRef.
- X. Ma, L. Sun and C. Song, Catal. Today, 2002, 77, 107–116 CrossRef CAS.
- A. J. Hernandez-Maldonado and R. T. Yang, Catal. Rev., 2004, 46, 111–150 CrossRef.
- V. Selvavathi, V. Chidambaram, A. Meenakshisundaram, B. Saíram and B. Sivasankar, Catal. Today, 2009, 141, 99–102 CrossRef CAS.
- Y. Sano, K. Sugahara, K. H. Choi, Y. Korai and I. Mochida, Fuel, 2005, 84, 903–910 CrossRef CAS.
- J. Eber, P. Wasserscheid and A. Jess, Green Chem., 2004, 6, 316–322 RSC.
- S. Zhang, Q. Zhang and C. Zhang, Ind. Eng. Chem. Res., 2004, 43, 614–622 CrossRef CAS.
- J. D. Holbrey, I. Lopez-Martin, G. Rothenberg, K. R. Seddon, G. Silvero and X. Zheng, Green Chem., 2008, 10, 87–92 RSC.
- D. Liu, J. Gui, L. Song, X. Zhang and Z. Sun, Petroleum Science and Technology, 2008, 26, 973–982 Search PubMed.
- C. C. Cassoll, A. P. Umpierre, G. Ebeling, B. Ferrera, S. S. X. Chiaro and J. Dupont, Int. J. Mol. Sci., 2007, 8, 593–605 CrossRef CAS.
- G. A. Huff, O. S. Owen, B. D. Alexander, D. N. Rundel, W. J. Reagen and J. S. Yoo, WO 9909117, 1999 Search PubMed.
- V. Bellière, C. Geantet, M. Vrinat, Y. Ben-Taarit and Y. Yoshimura, Energy Fuels, 2004, 18, 1806–1813 CrossRef CAS.
- B. D. Alexander, G. A. Huff, V. R. Pradhan, W. J. Reagan and R. H. Cayton, US Patent 6,024,865, 2000 Search PubMed.
- Z. Zhang, S. Liu, X. Zhu, Q. Wang and L. Xu, Fuel Process. Technol., 2008, 89, 103–110 CrossRef CAS.
- D. Richardeau, G. Joly, C. Canaff, P. Magnoux, M. Guisnet, M. Thomas and A. Nicolaos, Appl. Catal., A, 2004, 263, 49–61 CrossRef CAS.
- P. T. Burnett, G. A. Huff, V. R. Pradhan, M. Hodges, J. A. Glassett, S. G. McDaniel, P. Hurst, The European Refining Technology Conference, Rome, Italy, November 13–15, 2000 Search PubMed.
- R. T. Yang, A. J. Hernández-Maldonado and F. H. Yang, Science, 2003, 301, 79–81 CrossRef CAS.
- M. Dastanian and F. Seyedeyn-Azad, Ind. Eng. Chem. Res., 2010, 49, 11254–11259 CrossRef CAS.
- C. H. Ko, J. G. Park, J. C. Park, H. J. Song, S. S. Han and J. S. Kim, Appl. Surf. Sci., 2007, 253, 5864–5867 CrossRef CAS.
- A. Zhou, X. Ma and C. S. Song, J. Phys. Chem. B, 2006, 110, 4699–4707 CrossRef CAS.
- E. Deliyanni, M. Seredych and T. J. Bandosz, Langmuir, 2009, 25, 9302–9312 CrossRef CAS.
- Y. Sano, K. H. Choi, Y. Korai and I. Mochida, Appl. Catal., B, 2004, 49, 219–225 CrossRef CAS.
- H. J. Jeon, C. H. Ko and S. H. Kim, Energy Fuels, 2009, 23(5), 2537–2543 CrossRef CAS.
- A. B. S. H. Salem and H. S. Hamid, Chem. Eng. Technol., 1997, 20, 342–347 CAS.
- G. Blanco-Brieva, J. M. Campos-Martin, S. M. Al-Zahrani and J. L. G. Fierro, Fuel, 2011, 90, 190–197 CrossRef CAS.
- K. A. Cychosz, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 6938–6943 CrossRef CAS.
- R. L. Irvine and D. M. Varraveto, Pet. Technol. Q., 1999, 3, 37–42 Search PubMed.
- R. L. Irvine, US Patent 5,730,860, 1998 Search PubMed.
- C. H. Ko, J. G. Park, S. S. Han, J. H. Park, S. H. Cho and J. N. Kim, Stud. Surf. Sci. Catal., 165, 881–884 Search PubMed.
- C. H. Ko, J. G. Park, J. C. Park, H. Song, S. S. Han and J. N. Kim, Appl. Surf. Sci., 2007, 253, 5864–5867 CrossRef CAS.
- M. V. Landau, M. Herskowitz, R. Agnihotri and J. E. Kegerreis, Ind. Eng. Chem. Res., 2008, 47, 6904–6916 CrossRef CAS.
- P. Slater, B. Johnson and D. Kidd, in NPRA Annual Meeting Papers, 2002, p. 9 Search PubMed.
- P. Petkov, J. Tasheva and D. Stratiev, Pet. Coal, 2004, 46, 13–18 Search PubMed.
- P. S. Kulkarni and C. A. M. Afonso, Green Chem., 2010, 12, 1139–1149 RSC.
- C. Huang, B. Chen, J. Zhang, Z. Liu and Y. Li, Energy Fuels, 2004, 18, 1862–1864 CrossRef CAS.
- Y. Nie, C. Li, A. Sun, H. Meng and Z. Wang, Energy Fuels, 2006, 20, 2083–2087 CrossRef CAS.
- L. Lu, S. F. Cheng, J. B. Gao, G. H. Gao and M. Y. He, Energy Fuels, 2007, 21, 383–384 CrossRef CAS.
- E. Lissner, W. F. de Souza, B. Ferrera and J. Dupont, ChemSusChem, 2009, 2, 962–964 CrossRef.
- W. S. Zhu, H. M. Li, X. Hang, Y. S. Yan, H. D. Lu and J. X. Xia, Energy Fuels, 2007, 21, 2514–2516 CrossRef CAS.
- J. Zhang, W. Zhu, H. Li, W. Jiang, L. Jiang, W. Huang and Y. Yan, Green Chem., 2009, 11, 1801–1807 RSC.
- H. Li, X. Jiang, W. Zhu, J. Lu, H. Shu and Y. Yan, Ind. Eng. Chem. Res., 2009, 48, 9034–9039 CrossRef CAS.
- C. Asumana, G. Yu, X. Li, J. Zhao, G. Liu and X. Chen, Green Chem., 2010, 12, 2030–2037 RSC.
- E. Kuhlmann, M. Haumann, A. Jess, A. Seeberger and P. Wasserschild, ChemSusChem, 2009, 2, 969–977 CrossRef CAS.
- P. S. Tam, J. R. Kittrell and J. W. Eldridge, Ind. Eng. Chem. Res., 1990, 29, 321–324 CrossRef CAS.
- S. T. Darian and S. H. Arabshshi, US Patent 4,746,420, 1988 Search PubMed.
- J. A. Kocal, US Patent 6277271, 2001 Search PubMed.
- J. A. Kocal and T. A. Branvold, US Patent 6368495, 2002 Search PubMed.
- L. J. Karas, Y. Z. Han and D. W. Leyshon, US Patent 2004/178122, 2004 Search PubMed.
- L. J. Karas, R. A. Grey and M. W. Lynch, US Patent 2008/047875, 2008 Search PubMed.
- A. Ishihara, D. H. Wang, F. Dumeignil, H. Amano, E. W. H. Qian and T. Kabe, Appl. Catal., A, 2005, 279, 279–287 CrossRef CAS.
- A. Chica, G. Gatti, B. Moden, L. Marchese and E. Iglesia, Chem.–Eur. J., 2006, 12, 1960–1967 CrossRef CAS.
- X. R. Zhou, C. X. Zhao, J. Z. Yang and S. F. Zhang, Energy Fuels, 2007, 21, 7–10 CrossRef CAS.
- L. Kuznetsova, L. Detusheva, N. Kuznetsov, V. Duplyakin and V. Likholobov, Kinet. Catal., 2008, 49, 644–652 CrossRef CAS.
- V. V. D. N. Prasad, K. E. Jeong, H. J. Chae, C. U. Kim and S. Y. Jeong, Catal. Commun., 2008, 9, 1966–1969 CrossRef CAS.
- O. González-García and L. Cedeño-Caero, Catal. Today, 2009, 148, 42–48 CrossRef CAS.
- A. Chica, A. Corma and M. E. Domine, J. Catal., 2006, 242, 299–308 CrossRef CAS.
- P. de Filippis and M. Scarsella, Energy Fuels, 2003, 17, 1452–1455 CrossRef CAS.
- K. Yazu, M. Makino and K. Ukegawa, Chem. Lett., 2004, 33, 1306–1307 CrossRef CAS.
- A. M. Dehkordi, M. A. Sobati and M. A. Nazem, Chin. J. Chem. Eng., 2009, 17, 869–874 Search PubMed.
- F. Al-Shahrani, T. C. Xiao, S. A. Llewellyn, S. Barri, Z. Jiang, H. H. Shi, G. Martinie and M. L. H. Green, Appl. Catal., B, 2007, 73, 311–316 CrossRef CAS.
- G. X. Yu, S. X. Lu, H. Chen and Z. N. Zhu, Carbon, 2005, 43, 2285–2294 CrossRef CAS.
- X. Zhou, Q. Tan, G. Yu, L. Chen, J. Wang and O. Novaro, Kinet. Catal., 2009, 50, 543–549 CrossRef CAS.
- L. J. Chen, S. H. Guo and D. S. Zhao, Chin. J. Chem. Eng., 2007, 15, 520–523 Search PubMed.
- W. F. de Souza, I. R. Guimarães, M. C. Guerreiro and L. C. A. Oliveira, Appl. Catal., A, 2009, 360, 205–209 CrossRef CAS.
- D. Zhao, H. Ren, J. Wang, Y. Yang and Y. Zhao, Energy Fuels, 2007, 21, 2543–2547 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS.
- T. Hirai, K. Ogawa and I. Komasawa, Ind. Eng. Chem. Res., 1996, 35, 586 CrossRef CAS.
- Y. Shiraishi, H. Hara, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 1999, 38, 1589–1595 CrossRef CAS.
- B. Zapata, F. Pedraza and M. A. Valenzuela, Catal. Today, 2005, 106, 219–221 CrossRef CAS.
- J. L. Lai and G. X. Luo, Pet. Sci. Technol., 2006, 24, 1357–1362 CrossRef CAS.
- S. Mondal, Y. Hangun-Balkir, L. Alexandrova, D. Link, B. Howard, P. Zandhuis, A. Cugini, C. P. Horwitz and T. J. Collins, Catal. Today, 2006, 116, 554–561 CrossRef CAS.
- F. Figueras, J. Palomeque, S. Loridant, C. Fèche, N. Essayem and G. Gelbard, J. Catal., 2004, 226, 25–31 CrossRef CAS.
- D. Huang, Y. J. Wang, L. M. Yang and G. S. Luo, Ind. Eng. Chem. Res., 2006, 45, 1880–1885 CrossRef CAS.
- O. Etemadi and T. F. Yen, Energy Fuels, 2007, 21, 2250–2257 CrossRef CAS.
- D. Huang, Z. Zhai, Y. C. Lu, L. M. Yang and G. S. Luo, Ind. Eng. Chem. Res., 2007, 46, 1447–1451 CrossRef CAS.
- H. Y. Lu, J. B. Gao, Z. X. Jiang, F. Jing, Y. X. Yano, G. Wang and C. Li, J. Catal., 2006, 239, 369–375 CrossRef.
- D. Huang, Y. C. Lu, Y. J. Wang, L. M. Yang and G. S. Luo, Ind. Eng. Chem. Res., 2008, 47, 3870–3875 CrossRef CAS.
- Y. Shiraishi, T. Hirai and I. Komasawa, J. Chem. Eng. Jpn., 2002, 35, 1305–1311 CrossRef CAS.
- L. Y. Kong, G. Li and X. S. Wang, Catal. Lett., 2004, 92, 163–167 CrossRef CAS.
- Y. Wang, G. Li, X. S. Wang and C. Z. Jin, Energy Fuels, 2007, 21, 1415–1419 CrossRef CAS.
- Y. Jia, G. Li, G. Ning and C. Jin, Catal. Today, 2009, 140, 192–196 CrossRef CAS.
- T. Napanang and T. Sooknoi, Catal. Commun., 2009, 11, 1–6 CrossRef CAS.
- M. C. Capel-Sanchez, J. M. Campos-Martin and J. L. G. Fierro, Energy Environ. Sci., 2010, 3, 328–333 RSC.
- L. Cedeno-Caero, H. Gomez-Bernal, A. Fraustro-Cuevas, H. D. Guerra-Gomez and R. Cuevas-Garcia, Catal. Today, 2008, 133–135, 244–254 CrossRef CAS.
- F. Gregori, I. Nobili, F. Bigi, R. Maggi, G. Predieri and G. Sartori, J. Mol. Catal. A: Chem., 2008, 286, 124–127 CrossRef CAS.
- H. Gómez-Bernal, L. Cedeño-Caero and A. Gutiérrez-Alejandre, Catal. Today, 2009, 142, 227 CrossRef CAS.
- D. Huang, Y. J. Wang, Y. C. Cui and G. S. Luo, Microporous Mesoporous Mater., 2008, 116, 378–385 CrossRef CAS.
- G. Rodriguez-Gattorno, A. Galano and E. Torres-García, Appl. Catal., B, 2009, 92, 1–8 CrossRef CAS.
- X. M. Yan, P. Mei, J. Lei, Y. Mi, L. Xiong and L. Guo, J. Mol. Catal. A: Chem., 2009, 304, 52–57 CrossRef CAS.
- M. C. Capel-Sanchez, P. Perez-Presas, J. M. Campos-Martin and J. L. G. Fierro, Catal. Today, 2010, 157, 328–333.
- J. T. Sampanthar, H. Xiao, H. Dou, T. Y. Nah, X. Rong and W. P. Kwan, Appl. Catal., B, 2006, 63, 85–93 CrossRef.
- L. Y. Kong, G. Li, X. S. Wang and B. Wu, Energy Fuels, 2006, 20, 896–902 CrossRef CAS.
- X. Si, S. Cheng, Y. Lu, G. Gao and M. Y. He, Catal. Lett., 2007, 122, 321–324.
- A. di Giuseppe, M. Crucianelli, F. de Angelis, C. Crestini and R. Saladino, Appl. Catal., B, 2009, 89, 239–245 CrossRef CAS.
- S. Otsuki, T. Nonaka, N. Takashima, W. H. Qian, A. Ishihara, T. Imai and T. Kabe, Energy Fuels, 2000, 14, 1232–1239 CrossRef CAS.
- O. Etemadi and T. F. Yen, Energy Fuels, 2007, 21, 2250–2257 CrossRef CAS.
- D. Huang, Z. Zhai, Y. C. Lu, L. M. Yang and G. S. Luo, Ind. Eng. Chem. Res., 2007, 46, 1447–1451 CrossRef CAS.
- X. Jiang, H. Li, W. Zhu, L. He, H. Shu and J. Lu, Fuel, 2009, 88, 431–436 CrossRef CAS.
- K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Angew. Chem., Int. Ed., 2004, 43, 406–446 CrossRef.
- L. Kiwi-Minsker and A. Renken, Catal. Today, 2005, 110, 2–14 CrossRef CAS.
- M. A. Kertesz, FEMS Microbiol. Rev., 1999, 24, 135–175.
- G. Mohebali and B. S. Andrew, Microbiology, 2008, 154, 2169–2183 CrossRef CAS.
- D. J. Boron, W. R. Deever, R. M. Atlas, B. L. McFarland, J. A. Meyer, A. R. Johnson, NPRA Meeting, San Antonio, 1999, AM–99–54 Search PubMed.
- W. E. Kleinjan, A. de Keizer and A. J. H. Janssen, in Biologically produced sulphur, ed. R. Steudel, Top. Curr. Chem., 2003, vol. 230, p. 167 Search PubMed.
- S. Maghsoudi, M. Vossoughi, A. Heirolomoom, E. Tanaka and S. Katoh, Biochem. Eng. J., 2001, 8, 151 CrossRef CAS.
- L. Mingfang, G. Zhongxuan, X. Jianmin, L. Huizhou and C. Jiayong, J. Chem. Technol. Biotechnol., 2003, 78, 873 CrossRef.
- B. L. McFarland, D. J. Boron, W. Deever, J. A. Meyer, A. R. Johnson and R. M. Atlas, Crit. Rev. Microbiol., 1998, 24, 99–147 CAS.
- K. Kodama, K. Umehara, K. Shimizu, S. Nakatani, Y. Minoda and K. Yamada, Agric. Biol. Chem., 1973, 37, 45–50 CAS.
- S. Denome, D. C. Stanley, E. S. Olson and K. D. Young, J. Bacteriol., 1993, 175, 6890–6901 CAS.
- M. van Afferden, D. Tappe, M. Beyer, H. G. Truper and J. Klein, Fuel, 1993, 72, 635–643.
| This journal is © The Royal Society of Chemistry 2011 |