Superparamagnetic nanoparticles for asymmetric catalysis—a perfect match
Kalluri V. S.
Ranganath
and
Frank
Glorius
*
Westfälische Wilhelms-Universität Münster, Organisch-Chemisches Institut, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de; Fax: +49 251-83-33202
First published on 31st January 2011
Abstract
The aim of this perspective is to highlight the potential application of (superpara)magnetic nanoparticles in asymmetric catalysis. The unique combination of high enantioselectivity and enhanced reactivity combined with its recyclability and ease of separation makes this chiral nanotechnology one of the most promising strategies for the formation of enantiomerically enriched compounds on an industrial scale. This perspective focuses on the most representative examples of this young and emerging research field and highlights recent achievements and future prospects of magnetic nanoparticles in asymmetric catalysis.
 Kalluri V. S. Ranganath | Kalluri V. S. Ranganath obtained his PhD from IICT, Hyderabad, India under the supervision of Dr B. M. Choudary. Later he joined the laboratory of Prof. Junji Inanaga, Kyushu University, as a JSPS-Postdoctoral Fellow. Thereafter, he moved to the University of Münster as an Alexander von Humboldt Research Fellow in the group of Prof. Frank Glorius. His research interests are asymmetric heterogeneous catalysis, nanoparticles, surface chemistry and materials science. |
 Frank Glorius | Frank Glorius was educated in chemistry at the Universität Hannover, Stanford University (Prof. Paul A. Wender), Max-Planck-Institut für Kohlenforschung and Universität Basel (Prof. Andreas Pfaltz) and Harvard University (Prof. David A. Evans). He began his independent career at the Max-Planck-Institut für Kohlenforschung (mentor: Prof. Alois Fürstner) in 2001 and was appointed Associate Prof. at the Philipps-Universität Marburg in 2004. Since 2007 he is a Full Professor of Organic Chemistry at the Westfälische Wilhelms-Universität Münster. His research program focuses on the development of new concepts for catalysis and their implementation in organic synthesis. |
1. Introduction
Catalysis is key for the efficient and sustainable synthesis of organic compounds, representing one of the most economically and ecologically impacting technologies of our days. Driven by the growing demand for enantiopure compounds in life sciences, asymmetric catalysis is becoming more and more important.1Homogeneous asymmetric catalysis has made tremendous progress in the last years. In this field, mechanistic and structural understanding of the often well-defined molecular catalysts has allowed the optimization and fine-tuning of the catalyst systems, resulting in high levels of enantioselectivity and reactivity. On the other hand, several distinct disadvantages limit their applicability on an industrial scale: chiral homogeneous catalysts are often expensive and neither very robust nor broad in scope. In addition, the handling, separation and recycling of the catalysts is troublesome, often resulting in undesired (transition-metal) contaminations of the final product. Thus, most reported homogeneous asymmetric catalyst systems are not suitable for a commercial application on an industrial scale.2In around 80% of the industrially relevant catalytic processes, heterogeneous catalysts are employed.3 Indeed, heterogeneous catalysts are greatly preferred as they allow easy recovery of the catalyst from the reaction products. Furthermore, high turnover numbers (TONs) and turnover frequencies (TOFs) make heterogeneous catalysis very cost efficient. Thus, supported by advances in the field of solid state chemistry and improved analytical tools, various methods have been developed and applied to immobilize privileged chiral homogeneous catalysts.4 Since the resulting heterogeneous catalysts possess the same modes of action as their homogeneous counterparts, these catalysts can be applied in the same reactions, with similar levels of selectivity and reactivity. In some cases, selectivity and reactivity are reduced as a direct consequence of steric and mass transport effects, since a large proportion of the active sites are buried deep inside the supporting matrix. Thus, the reactants have limited access to the catalytic site, which is in stark contrast to readily accessible homogeneous catalysts. Another potential disadvantage of these immobilized catalysts is the additional cost incurred in the immobilization process. This is especially true, when the often observed low stability of the catalytically active entity of the immobilized catalyst limits repetitive recycling of the catalyst.
Nanoparticles (NPs) range from 1 to 100 nm in size, with an increasing relative surface area, the smaller they get. Thus, chemically active nanoclusters are often between 1–10 nm, because in this range the proportion of surface atoms is especially large. For example, in an NP consisting of 13 atoms, 92% of the atoms are surface atoms (whereas in an NP of 55 atoms, only 76% of those are surface atoms; 147 (63%); 309 (52%); 1415 (35%)). Similarly, in an NP with a diameter of 1 nm, around 50% of the atoms are surface atoms, with the percentage of surface atoms exponentially decreasing with an increasing diameter (10 nm diameter–10% surface atoms). These particles often exhibit physical and chemical properties that are between those of their smallest components (such as metal atoms or metal oxide units) and those of bulk materials, where only a few percent of the atoms lie on the surface.5–7 NPs can either be employed in a quasi-homogeneous phase or serve as precursors for heterogeneous catalysts.8,9 Based on their unique properties, NPs are considered to be a bridge between homogeneous and heterogeneous supports.
The large relative surface areas of NPs are comparable to those of porous materials, but without problems of mass transport, because the active sites are highly accessible. Thus, NPs are well suited for the preparation of heterogeneous asymmetric catalysts. Two different strategies have to be distinguished (Fig. 1):
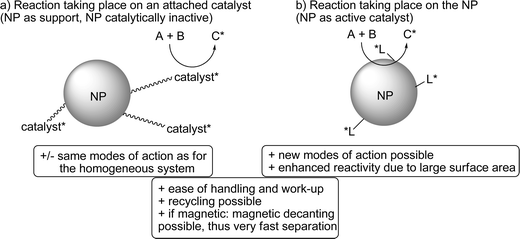 | ||
| Fig. 1 Applications of NPs in asymmetric catalysis: as support for a chiral catalyst (a) or as catalytically active entity (b); NP = nanoparticle; L* = chiral modifier; A, B = achiral substrates; C* = chiral product. | ||
(a) NPs can be used for the immobilization of privileged chiral homogeneous catalysts. In this case, the NP is chemically inactive and, thus, the attached/immobilized catalyst follows its standard mode of action. Most often, levels of activity and selectivity are comparable to the homogeneous counterparts.
(b) NPs can also be utilized as the catalytically active part of the resulting chiral catalyst system. Generally, the mode of enantioinduction has no equivalent in homogeneous asymmetric catalysis, which renders these systems especially challenging but also especially valuable.
As the particle size decreased, a large fraction of active metal atoms are exposed to the surface, and NPs of transition metals exhibit activity comparable to molecular catalysts. In particular, metal NPs, such as Pd, Pt, Rh and Au, have been evaluated for a variety of reactions including C–C coupling, oxidations and hydrogenations.10,11 Recently, the development of functionalized metal NPs for application in asymmetric catalysis has been a subject of interest.12 However, the small size of NPs requires special methods for their separation like centrifugation, nanofiltration or precipitation–flocculation. Recently, magnetite and other superparamagnetic NPs have been extensively employed as alternative catalyst supports, in view of their high surface area, robustness and easy recycling,13 as separation by magnetic decantation can greatly facilitate the recovery of these catalysts.
Nanocrystalline metal oxides like MgO, CaO, SrO, BaO, Fe3O4, Al2O3, ZnO or CeO have attracted great attention due to their unusual optical, physical, surface and catalytic properties.14–16Oxides of many metals possess very high lattice energies and melting points because of their highly ionic nature and because surfaces of these oxides exhibit both Lewis base and Lewis acid character. These nanostructured metal oxides display good catalytic activity, due to their high surface area, inherent adsorptive properties and accessibility of their active sites.17 For example, nanocrystallites of MgO were used in Claisen–Schmidt condensations, Wittig and benzylation reactions.18 NPs of barium oxide prepared with superior chemical homogeneity by using reverse-microemulsion exhibited excellent catalyticmethane combustion activity compared to other conventionally prepared samples.19 The TiO2 and ZnO NPs contained in suntan lotion are excellent examples of the optical transparency of thin layers of NPs when the size of NPs is well below the wavelength of visible light (380–780 nm).20 NPs of lanthanum–BINOL of 50 nm in size are also used in the asymmetric epoxidation of unsaturated ketones.21
Superparamagnetic magnetite NPs are of great interest for different disciplines, including catalysis, biotechnology/biomedicine, magnetic resonance imaging (MRI), data storage and environmental remediation and as magnetic fluids.22,23 In superparamagnetic nanoparticles, magnetization can randomly flip direction and their magnetization appears to be zero (superparamagnetic state). However, an external magnetic field can magnetize these nanoparticles, with the magnetic susceptibility being much larger than the one of paramagnets. Functionalized magnetic NPs (MNPs) with good stability are very promising candidates for application in catalysis, especially in liquid phase catalytic reactions;24–26 such small and magnetically separable particles may be useful as quasi homogeneous systems that combine advantages of high dispersion, high reactivity and easy separation. Magnetically driven separations make the recovery of catalysts in a liquid-phase reaction much easier than by cross-flow filtration and centrifugation, especially when the size of the catalysts is in the sub-micrometre range. In many catalytic reactions, the catalysts were separated by a simple magnetic separation process and re-used successfully without any loss of catalytic activity and selectivity. Arguably, one of the highest numbers of recycles reported for a catalytic reaction is the electrocatalytic oxidation of ammonia borane over a Fe@Pt catalyst (>100 recycles).27 Using these NPs offers advantages for “clean and green” chemistry, in addition to being readily recovered, non-toxic and widely accessible.28 In particular these applications are possible mainly as a result of the unique size distribution of NPs. Moreover, research applications using related iron oxide based nano-materials are found in medicinal and biotechnology such as targeted drug delivery, magnetic resonance imaging, cell isolation and hyperthermia. Since the size of NPs resembles that of biomolecules, such as cells, viruses and proteins, there is ample opportunity for interesting interactions among these species.29,30
Among all iron oxide NPs, magnetite (Fe3O4) exhibits the most interesting properties because the presence of non-equivalent iron cations in two valence states, Fe2+ and Fe3+, in the crystal structure gives rise to a unique magnetic structure. Magnetite is a cubic mineral found in abundance in nature and can also be readily synthesized.31 It has an inverse spinel structure (Fig. 2) following the formula Fe3O4 = (Fe3+)A(Fe3+Fe2+)BO4, where A and B symbolize the iron cations, distributed on tetrahedral and octahedral interstitial sites, respectively.
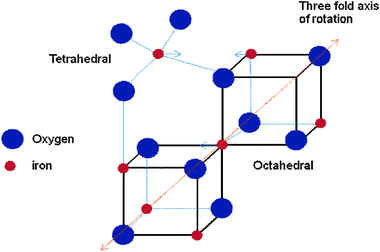 | ||
| Fig. 2 Spinel structure of Fe3O4.31 | ||
2. Applications of NPs in asymmetric catalysis:
(a) NPs as support only
An excellent and representative example for the use of MNPs as support for well-known privileged catalysts is the work of Lin et al.32 After having prepared a 4-phosphonic acid substituted (R)-BINAP derivative, Lin et al. immobilized the corresponding ruthenium(II) complex, [Ru(4-P(O)(OH)2-(R)-binap-(dpen)Cl2], with the help of the phosphonate group onto the surface of magnetite NPs (NP1, Scheme 1). These magnetite nanocomposites were easily dispersible in common organic solvents and recoverable by the application of an external magnet.32NP1 catalyzed the asymmetric hydrogenation of aromatic ketones with remarkably high activity and enantioselectivity in a heterogeneous fashion. Significantly higher enantioselectivities were obtained throughout when the iron oxide NP employed was prepared through the co-precipitation technique. Eight different aromatic ketones were reduced quantitatively to alcohols at room temperature with only 0.1 mol% catalyst loading providing very good ees from 77 to 98%. These ee values are significantly higher than or comparable to those of the parent homogeneous catalyst. Furthermore, the heterogenized catalyst was recycled up to 14 times without loss of activity and enantioselectivity. The supernatant (before and after hydrogenation) is not active for hydrogenation of aromatic ketones, indicating the true heterogeneous nature of the magnetite NP-supported catalytic system. The catalyst exhibited a saturation magnetization (σs) of 50 emu g−1, which is smaller than that of bulk magnetite of 92 emu g−1.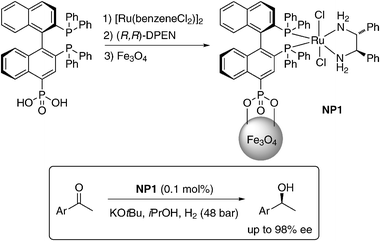 | ||
| Scheme 1 Asymmetric hydrogenation of ketones using Ru–BINAP supported on Fe3O4 NPs utilizing phosphonic acid as a linker.32 | ||
This example represents an anchored homogeneous catalyst that, unlike its homogenous counterpart, can easily be separated and recycled. At the same time, its catalytic properties are closely related to the homogeneous system. Despite these attractive properties, the increased cost of the phosphonic acid modified BINAP derivative as compared to BINAP itself presents a significant downside of this process.
The interaction between the catalyst and the NP surface can also be detrimental to activity and selectivity. Thus, surface-capping methods were introduced not only as simple approaches to stabilize and functionalize the MNPs, but also to prevent undesired interactions between NP and the organocatalyst. Silanes are frequently used to coat the surface of magnetite NPs because of the strong affinity of iron oxide towards silica. By covering iron oxide with other materials like silica, the attractive magnetic properties of iron oxide can also be utilized by other materials. Magnetic Fe2O3 NPs contained in a mesoporous silica foam have been used to immobilize Ru–TsDPEN (TsDPEN = N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine) complexes grafted on the silica matrix by Liet al. (Scheme 2). The resulting heterogeneous system has been used as a catalyst for asymmetric transfer hydrogenation of imines (up to 95% ee) and ketones (up to 97% ee). The catalyst could be recycled up to nine times without loss of activity.33
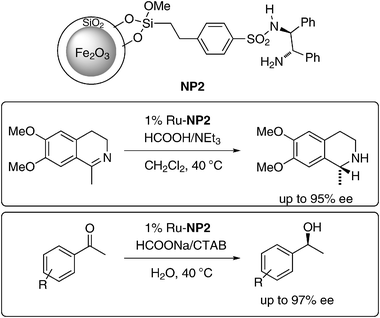 | ||
| Scheme 2 Asymmetric transfer hydrogenation of imines and ketones with TsDPEN–Ru supported on mesoporous silica foam.33 | ||
Long chain carboxylic acids are well known ligands and stabilizers of cobalt NPs. Pericàs and co-workers prepared long chain alkyl carboxylic acids functionalized with an amino alcohol group and attached them to the surface of cobalt NPs (Scheme 3). Whereas the carboxylate group binds to the metal surface, the aminoalcohol group acts as the ligand for the mononuclear catalytically active site (NP3). These NPs have been used as magnetically decantable ligands in the ruthenium catalyzed asymmetric transfer hydrogenation of ketones, providing ee's up to 87% (Scheme 3). The main drawback of this process is the surprisingly poor reusability caused by leaching of the carboxylate ligands.34
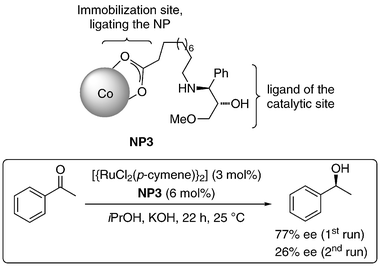 | ||
| Scheme 3 Asymmetric transfer hydrogenation of ketones with NP3–Ru catalyst.34 | ||
Kantam et al. used copper ferrite NPs in the asymmetric hydrosilylation of ketones using commercially available (S)-BINAP as the chiral modifier (Scheme 4). They observed that monoaryl silanes such as phenylsilane afforded excellent yields with good ees up to 99% at room temperature. Ketoesters were also reduced under the reaction conditions with up to 83% ee. The catalyst could be reused up to three times without significant loss of ee and it was characterized by atomic absorption spectroscopy (AAS) to determine the copper content, where 27.32% of copper was found. Leaching of copper after the first cycle was also determined using AAS to be at a low level of 0.045%. The NP size was found to be 10–12 nm and remained unchanged even after three cycles as determined by TEM.35
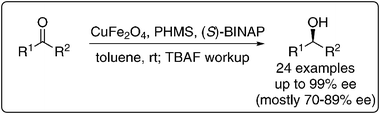 | ||
| Scheme 4 Asymmetric hydrosilylation of ketones with copper ferrite NPs using BINAP as the modifier.35 | ||
Very recently, Reiser and co-workers reported two different azide functionalized magnetic silica coated NPs for the immobilization of azabis(oxazoline) copper(II) complexes.36Azabisoxazoline ligands are particularly attractive candidates for immobilization due to greatly reduced tendency for metal leaching from their complexes, in contrast to the corresponding metal–bisoxazoline complexes. The heterogenized catalyst (NP4) was prepared utilizing azide linkage through click chemistry. These silica coated magnetic NPs (MNPs) demonstrated good activity and selectivity (up to 98% ee; Scheme 5) for the asymmetric benzoylation of racemic 1,2-diols, being only slightly less selective than the homogeneous catalyst (99% ee) and being superior to common polymer resin supports, such as MeOPEG (up to 82% ee) or Merryfield resin (up to 67% ee).36 The catalyst could be recycled for three times and high activity and ee was maintained for all three cycles.
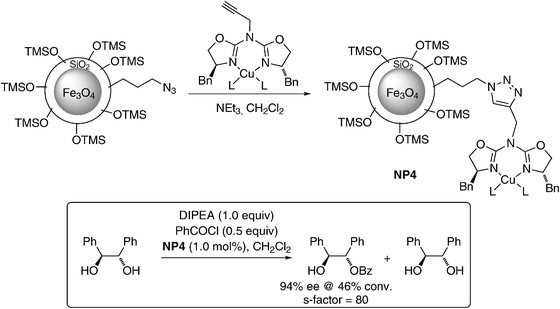 | ||
| Scheme 5 Formation of NP4 and its application in the asymmetric benzoylation of 1,2-diols.36 | ||
Hyeon et al. reported another catalyst utilizing embedded MNPs. They prepared a new type of heterogeneous chiral catalyst system on the hierarchically ordered mesoporous silica (HMMS), using [DHQ-PHAL-DHQ-S-(CH2)5CH2OH] as the chiral ligand. The immobilized system was used for asymmetric dihydroxylation reactions and was recycled eight times without significant loss of activity.37
Organocatalysis has made spectacular advances in the last years.38 However, high catalyst loadings often distract from the attractiveness of these processes. An efficient method for catalyst separation and recycling may solve this problem, and the immobilization of privileged organocatalysts on MNPs appears to be a valuable solution. The MNPs supported chiral diamine catalysts (loading: 0.39 mmol g−1) were successfully used for the aldol reaction of acetone or cyclohexanone with a variety of aldehydes (Scheme 6). MNP supported catalysts showed similar activity and selectivity as homogeneous catalysts. A strong acid, such as triflic acid, is necessary to accelerate the reaction.
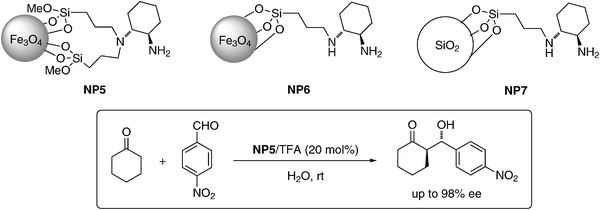 | ||
| Scheme 6 Asymmetric aldol reaction catalyzed by amine supported on Fe3O4 NPs.39 | ||
After the reaction, the MNP supported catalyst was conveniently separated with the help of an external magnet and reused for 11 cycles without loss of activity, and only a slight loss of selectivity. To compare the activity, different supported catalysts were prepared (NP5 and NP6). It was observed that tertiary/primary amine catalyst NP5 supported on magnetite was better than secondary/primary amine supported catalyst NP6. Luo et al. also observed that MNP supported catalyst NP5 exhibits superior activity compared to the silica supported catalyst NP7.39
In addition to the silane ligands, enediol ligands such as catechols are known to have a high affinity for under-coordinated surface sites of metal oxide nanoparticles.40a Therefore, dopamine has attracted some attention since it features an additional amine moiety that allows immobilization of metal centers or further covalent modification. The first chiral-N,N′-dimethylaminopyridine (DMAP) derivatives supported on MNPs was reported by Connon and co-workers (NP8, Scheme 7). The catalyst was prepared by anchoring the N-methyldopamine hydrochloride onto MNPs, followed by a nucleophilic substitution reaction with a chiral chloropyridine in toluene.40b
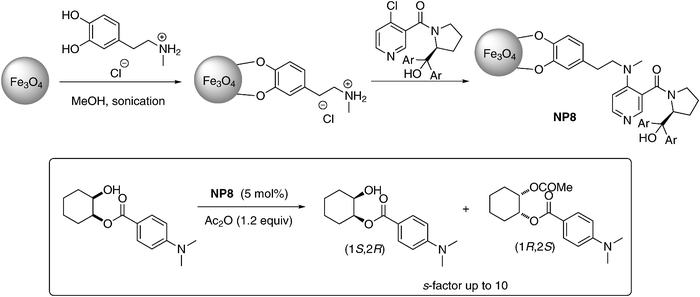
NP8 was used in acylative kinetic resolution of monoprotected cis-diols. Using 5 mol% of the catalyst enabled secondary alcohols to be resolved with 99% ee at 72% conversion. The catalyst could be reused up to 32 cycles without significant loss of activity. Furthermore, NPs did not undergo any degradation as observed by TEM analysis even after 32 cycles.40
MNPs have also emerged as a new type of support for the immobilization of enzymes. Because of high price and sensitivity to environmental changes, the focus of biocatalysts is on the regeneration of the catalyst without loss of activity.41 Tsang and coworkers reported the use of stable silica coated MNPs as a biocatalyst carrier.42Glutaraldehyde was used to link β-lactamase to the amine-functionalized silica surface. An assay using phenoxymethylpenicillin (penicillin V) as a substrate showed that these modified MNPs contained 54% of active enzyme, much higher than its carbon nano-material-supported counterpart, which only had a 16% active content. This nicely demonstrates the advantages of this immobilization technique: site (enzyme) isolation, good accessibility, recovery and reusability. In another interesting approach, Chen and Lee reported a silica encapsulated enzyme on MNP. In this process, a dual-fusion enzyme was recovered and immobilized by affinity to Cu(II)–iminodiacetic acid modified MNPs.43 Finally, a biosilicification provided the silica encapsulation of the enzyme, protecting the enzyme and also stabilizing its tertiary structure and preserving its activity.
Recently, Li and coworkers reported the immobilization of chloroperoxidase (CPO) on magnetite NPs coated with a polymeric shell. The resulting nanobio catalyst was successfully applied for the oxidation of thioanisole using H2O2, exhibiting good activity and a selectivity of up to 99% ee, comparable to the free enzyme. The polymer shell coated catalyst could be recovered and recycled for up to 11 times and is more stable than when non-coated iron oxide NPs were used as support.44 Compared with enzymes immobilized on other supports, a nanobio catalyst on MNPs could achieve a much higher enzyme loading capacity and exhibits enhanced mass transfer efficiency. Hyeon et al. reported a stable cross-linked enzyme cluster (CEC) and covalently attached enzymes (CA) on magnetite supports.45 They observed that the activities of the CEC, which are normalized per unit weight of enzyme, are significantly higher than those of CAs. Gao et al. reported that lipase immobilized on magnetite NPs demonstrated high stereoselectivity in the kinetic resolution of racemic carboxylic acids and even improved stability compared to its parent free enzyme.46 The enzyme was immobilized on magnetite NPs through the 3-aminopropyltriethoxysilane linker (7.5 μg of lipase protein per mg of NP, Scheme 8, NP9). The activity of the supported lipase was determined by kinetic resolution of three racemic carboxylates.46 The ee values of isolated carboxylates and esters were usually higher than 99%, comparable to those reported for non-immobilized free lipase. In addition, the long-term stability of this catalyst system was demonstrated by three recycles providing 29%, 21% and 21% yield, respectively, with each cycle taking 7 days. In comparison, the free lipase demonstrated much higher enzymatic activity in the first run, providing 55% conversion. However, only 7% yield was obtained in the second cycle, and a complete loss of activity was observed in the third run.
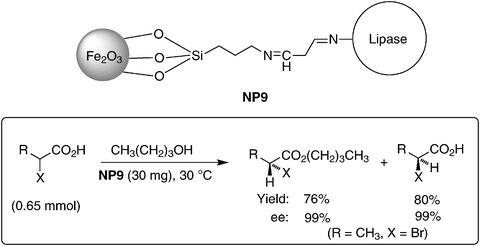 | ||
| Scheme 8 Schematic representation of immobilization of lipase on the surface of magnetite NPs through linker (NP9) and resolution of racemic carboxylates using lipase supported on MNPs (NP9).46 | ||
Even though not a catalytic application, a recent report on “enantioselective fishing”, an enantioselective separation technique utilizing chirally modified MNPs, is noteworthy. Choi and Hyun demonstrated that silica coated magnetite NPs together with an appropriate chiral “selector” (ligand) can be utilized for the separation of enantiomers of a racemic mixture.47MNPs coated with silica were used to anchor either (R)- or (S)-N-(2,2-dimethyl-5-ethoxydimethylsilylpentanoyl)-proline-3,5-dimethylanilide.47 The resulting MNPs were used for the separation of two enantiomers of N-(3,5-dinitrobenzoyl)-α-amino acid N-propylamides. By utilizing this NP technique, N-(3,5-dinitrobenzoyl)alanine, valine and leucine N-propylamides could be enriched enantiomerically. Furthermore, the quality of the separation was improved by using both enantiomers of the selector, one immobilized on the magnetic and the other one on non-magnetic NPs, resulting in ees of up to 80%.
(b) NPs as catalytically active part
The modification of a surface with a strongly adsorbing chiral agent (chiral modifier) to form chiral heterogeneous catalysts is an intellectually intriguing method and of great interest.4,48 Currently, there are two different directions in catalysis at chiral metal surfaces. Great effort has contributed to the discovery of new synthetic applications but the role of the solid surface in these reactions remains to be determined. On the other hand, surface scientists focus mainly on the fundamental aspects of idealized chiral surfaces and less on new catalytic applications. Chirally modified metal surfaces of Pd, Pt and Ni are very well known in various asymmetric transformations, in particular for hydrogenation reactions. The chiral modifier system is not only limited to transition metal surfaces, but also extended to alkaline earth NPs. Choudary and co-workers have used nanocrystals of MgO successfully in asymmetric epoxidations, Henry and Michael reactions with various chiral modifiers.48b Baiker and co-workers reported cinchonidine modified Pt immobilized on silica coated MNPs for the enantioselective hydrogenation of activated ketones.49 The cinchonidine modified platinum NP (4.4 nm diameter) on MNP showed activity and selectivity related to that of the established heterogeneous Pt/Al2O3 catalyst for the enantioselective hydrogenation of α-ketoesters and α,α,α-trifluoroacetophenone (Scheme 9). The catalyst could be recycled with little variation in reactivity up to eight times. The enantioselectivity decreased slightly, from 57% to 52% ee after the eighth cycle.49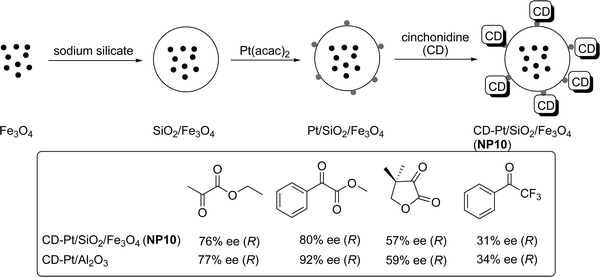 | ||
| Scheme 9 Asymmetric hydrogenation of ketones to the corresponding secondary alcohols (under 100 bar H2 pressure; absolute configuration of the products given in brackets), comparing CD-modified Fe3O4 NPs with CD-modified Pt/Al2O3.49 | ||
Core and shell type NPs have potential applications in catalysis and also in medicine. FePd NPs with a Fe-core and Pd-shell have been modified with chiral 2,2′-bis(diphenylphosphino)-1,1′-binaphthalene (BINAP) through a simple exchange procedure, creating a platform for Pd-based asymmetric catalytic reactions. FexOy rich core and Pd shell NPs were prepared by thermal decomposition of iron carbonyl complexes followed by reduction of Pd(acac)2 in the presence of oleic acid and oleylamine. Thus, these core and shell NPs were modified with BINAP to form the asymmetric heterogeneous catalyst. The crystallite sizes of Fe3O4 core and Pd shells were found to be 4.7 nm and 0.9 nm, respectively, using X-ray diffraction (XRD). TEM images show that the average diameter is 5.6 nm and the composition of iron and Pd remained unchanged after modification with BINAP. The structural properties of the catalyst were well characterized by SQUID and XANES. The bimetallic catalyst was successfully used in a challenging asymmetric Suzuki cross-coupling reaction at room temperature giving 48% ee at 99% conversion; only one example was given (Scheme 10). The heterogeneous catalytic system could be recycled once without further addition of chiral modifier.50 Furthermore, a hot filtration test confirmed that the reaction proceeded on the surface of chiral NPs only. The Fe core and Pd shell possess attractive features compared to Pd based systems. First, the replacement of the interior of precious Pd NPs with inexpensive core metals lowers the cost of production. Second, the magnetic core allows convenient separation of the nanocatalyst after the reaction by applying a magnetic field.
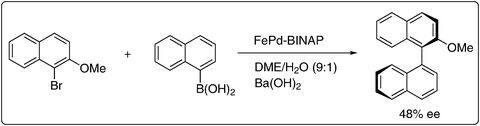 | ||
| Scheme 10 Asymmetric Suzuki coupling catalyzed by FePd NPs using BINAP as modifier.50 | ||
N-Heterocyclic carbenes (NHCs) have found a wide range of applications in organometallic chemistry and catalysis.51,52 Heterogenization of NHC–metal complexes through the binding of the silane group, carboxylic group, or pyrene moiety is a common strategy.53 Very recently, Glorius and co-workers reported the first successful application of NHCs as chiral modifiers of NP in asymmetric catalysis.54 This was achieved by the preparation of Pd NPs on magnetite NPs and subsequent modification with a chiral NHC to generate an asymmetric catalytic surface without any additional linker (NP11, Scheme 11). The chiral NHC modified catalyst was characterized using XPS and Pd is shown to exhibit zero oxidation state even after surface modification. The use of an NHC is ideal due to its distinct properties: a strong metal–NHC bond prevents leaching and its electron-richness results in the transfer of electron-density onto the metal. This should create reactive hot spots around the NHC ligands. In addition, structurally diverse NHC architectures exist and related projects utilizing NHCs as chiral modifiers can be expected in the near future.
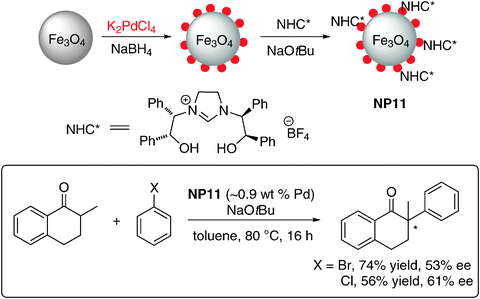 | ||
| Scheme 11 Asymmetric arylation of 2-methyl-1-tetralone and halo benzene using NHC as chiral modifier on the surface of Pd/Fe3O4 NPs.54 | ||
Pd-mediated asymmetric cross-coupling reactions to generate a quaternary stereocenter have received considerable attention. Glorius et al. demonstrated the competency of NP11 as a chiral nanocatalyst in the asymmetric α-arylation of 2-methyl-1-tetralone with bromobenzene to afford the corresponding α-arylated products in moderate to good ee's (Scheme 11), with up to 61% ee for an intermolecular and 85% ee for an intramolecular α-arylation reaction. Several experiments indicate the heterogeneous nature of this catalyst system (filtration test; mercury drop test; trace metal analysis). Intriguingly, homogeneous Pd complexes of this chiral NHC ligand resulted in the formation of a racemic product in the intermolecular α-arylation reaction (Scheme 11), another indication for the heterogeneous nature and new mode of action of NP11 catalyzed α-arylation. The catalyst was characterized by ICP-OES after the reaction, which showed the loss of only 0.04% of Pd. The yields and selectivities are comparable to that reported for the homogeneous system utilizing BINAP as the chiral ligand.55
3. Summary and outlook
For a long time, immobilization of homogeneous chiral catalysts had been accompanied by a loss of activity and selectivity. With the advent of reliable and easily reproducible protocols for NP formation, the use of NPs has received increasing attention from the scientific community. Choosing NPs or MNPs as support for catalysts can provide similar or even enhanced selectivity and reactivity compared to the homogeneous parent system. The attractive properties of NP change its role from an inevitable appendage to a well-defined material that favorably influences the outcome of a catalyzed reaction. Furthermore, it allows facile separation by magnetic decantation. Even more exciting is the development of catalytically active NP with chiral modifiers as unprecedented heterogeneous catalysts. These catalyst systems possess new modes of action and are highly valuable. However, more mechanistic and structural insight on the interactions between the modifier and the NP is needed in order to develop more efficient catalytic systems. In addition, synthesis and stabilization of mono-disperse NPs remains a challenge. Many spectacular advances can be expected in the field of asymmetric nanocatalysis in the coming years.Acknowledgements
The Alexander von Humboldt-Foundation (K.V.S.R.), the Deutsche Forschungsgemeinschaft and the Alfried Krupp von Bohlen and Halbach Foundation are gratefully acknowledged. In addition, we thank Fan Liu for careful proofreading of the manuscript.Notes and references
-
Comprehensive Asymmetric Catalysis I–III, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999 Search PubMed
.
-
Asymmetric Catalysis on Industrial Scale, ed. H. U. Blaser and E. Schmidt, Wiley-VCH, Weinheim, 2004 Search PubMed
-
(a)
Fine Chemicals Through Heterogeneous Catalysis, ed. R. A. Sheldon and H. van Bekkum, Wiley-VCH, Weinheim, 2001 Search PubMed
-
(a) M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS
-
(a)
Nanoparticles and Catalysis, ed. D. Astruc, Wiley-VCH, Weinheim, 2008 Search PubMed
-
(a) G. Schmidt, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS
-
(a) J. Grunes, J. Zhu and G. A. Somorjai, Chem. Commun., 2003, 2257–2260 RSC
-
(a)
Clusters and Colloids, ed. G. Schmidt, Wiley-VCH, Weinheim, 1994 Search PubMed
-
(a) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS
-
(a) M. Beller, H. Fischer, K. Kuehlein, C.-P. Reisinger and W. A. Herrmann, J. Organomet. Chem., 1996, 520, 257–259 CrossRef CAS
- D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS
-
(a) S. Jansat, M. Gómez, K. Philippot, G. Muller, E. Guiu, C. Claver, S. Castillon and B. Chaudret, J. Am. Chem. Soc., 2004, 126, 1592–1593 CrossRef CAS
-
(a) A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS
- K. J. Klabunde, J. Stark, O. Koper, C. Mohs, D. G. Park, S. Decker, Y. Jiang, I. Lagadic and D. Zhang, J. Phys. Chem., 1996, 100, 12142–12143 CrossRef CAS
- R. Richards, W. Li, S. Decker, C. Davidson, O. Koper, V. Zaikovski, A. Volodin, T. Rieker and K. J. Klabunde, J. Am. Chem. Soc., 2000, 122, 4921–4925 CrossRef CAS
- M. Utiyama, H. Hattori and K. Tanabe, J. Catal., 1978, 53, 237 CrossRef CAS
- A. G. Pelmenschikov, G. Morosi and A. Gamba, J. Phys. Chem., 1995, 99, 15018–15022 CrossRef CAS
-
(a) B. M. Choudary, M. L. Kantam, K. V. S. Ranganath, K. Mahendar and B. Sreedhar, J. Am. Chem. Soc., 2004, 126, 3396–3397 CrossRef CAS
- A. J. Zarur and J. Y. Ying, Nature, 2000, 403, 65–67 CrossRef CAS
-
Light Scattering by Small Particles, ed. H. C. van de Hulst, Dover Publications, New York, 1981 Search PubMed
- H. Furuno, S. Onitsuka and J. Inanaga, J. Synth. Org. Chem., Jpn., 2007, 65, 977–988 CAS
-
(a) D. E. Speliotis, J. Magn. Magn. Mater., 1999, 193, 29–35 CrossRef CAS
-
(a)
The Iron Oxides Structure, Properties, Reactions, Occurrences and Uses, ed. R. M. Cornell and U. Schwertmann, Wiley-VCH, Weinheim, 1996 Search PubMed
-
(a) S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1192 CrossRef CAS
-
(a) P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 4435–4437 RSC
-
(a) G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9212–9228 CrossRef CAS
- X. B. Zhang, J. M. Yan, S. Han, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2009, 131, 2778–2779 CrossRef CAS
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC
- K. Naka, A. Narita, H. Tanaka, Y. Chujo, M. Morita, T. Inubushi, I. Nishimura, J. Hiruta, H. Shibayama, M. Koga, S. Ishibashi, J. Seki, S. Kizaka-Kondoh and M. Hiraoka, Polym. Adv. Technol., 2008, 19, 1421–1429 CrossRef CAS
-
Viruses and Nanotechnology, ed. M. Manchester and N. F. Steinmetz, Springer, Berlin, 2008 Search PubMed
-
(a)
Iron Oxides in the Laboratory, ed. R. M. Cornell and U. Schwertmann, VCH, Weinheim, Germany, 1991 Search PubMed
- A. Hu, G. T. Yee and W. Lin, J. Am. Chem. Soc., 2005, 127, 12486–12487 CrossRef CAS
- J. Li, Y. Zhang, D. Han, Q. Gao and C. Li, J. Mol. Catal. A: Chem., 2009, 298, 31–35 CrossRef CAS
- F. Michalek, A. Lagunas, C. Jimeno and M. A. Pericàs, J. Mater. Chem., 2008, 18, 4692–4697 RSC
- M. L. Kantam, J. Yadav, S. Laha, P. Srinivas, B. Sreedhar and F. Figueras, J. Org. Chem., 2009, 74, 4608–4611 CrossRef CAS
- A. Schätz, M. Hager and O. Reiser, Adv. Funct. Mater., 2009, 19, 2109–2115 CrossRef
- D. Lee, J. Lee, H. Lee, S. Jin, T. Hyeon and B. M. Kim, Adv. Synth. Catal., 2006, 348, 41–46 CrossRef CAS
-
(a)
A. Berkessel and H. Groeger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, 2005 Search PubMed
- S. Luo, X. Zheng and J.-P. Cheng, Chem. Commun., 2008, 5719–5721 RSC
-
(a) T. Rajh, L. X. Chen, K. Lukas, T. Liu, M. C. Thurnauer and D. M. Tiede, J. Phys. Chem. B, 2002, 106, 10543–10552 CrossRef CAS
- K. S. Lee, M. H. Woo, H. S. Kim, E. Y. Lee and I. S. Lee, Chem. Commun., 2009, 3780–3782 RSC
- X. Gao, K. M. K. Yu, K. Y. Tam and S. C. Tsang, Chem. Commun., 2003, 2998–2999 RSC
- L. Chen and C. Lee, Biotechnol. Bioeng., 2008, 100, 223–230 CrossRef
- W. Wang, Y. Xu, D. I. C. Wang and Z. Li, J. Am. Chem. Soc., 2009, 131, 12892–12893 CrossRef CAS
- J. Lee, Y. Lee, J. K. Youn, H. B. Na, T. Yu, H. Kim, S. M. Lee, Y.-M. Koo, J. H. Kwak, H. G. Park, H. N. Chang, M. Hwang, J.-G. Park, J. Kim and T. Hyeon, Small, 2008, 4, 143–152 CrossRef CAS
- H. M. R. Gardimalla, D. Mandal, P. D. Stevens, M. Yen and Y. Gao, Chem. Commun., 2005, 4432–4434 RSC
- H. J. Choi and M. H. Hyun, Chem. Commun., 2009, 6454–6456 RSC
-
(a) T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS
- B. Panella, A. Vargas and A. Baiker, J. Catal., 2009, 261, 88–93 CrossRef CAS
- K. Mori, Y. Kondo and H. Yamashita, Phys. Chem. Chem. Phys., 2009, 11, 8949–8954 RSC
- Reviews on NHCs in catalysis:
(a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS
-
L. Gade, in N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin, 2007, p. 117 Search PubMed
-
(a) P. D. Stevens, G. Li, J. Fan, M. Yen and Y. Gao, Chem. Commun., 2005, 4435–4437 RSC
-
(a) K. V. S. Ranganath, J. Kloesges, A. Schäfer and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7786–7789 CrossRef CAS
-
(a) J. Åhman, J. P. Wolfe, M. V. Troutman, M. Palucki and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 1918–1919 CrossRef
| This journal is © The Royal Society of Chemistry 2011 |