A trifluoroacetic acid adduct of a trifluoroacetate-bridged μ4-oxo-tetranuclear zinc cluster, Zn4(OCOCF3)6O·CF3CO2H: synthesis under mild conditions and catalytic transesterification and oxazoline formation†
Yukiko
Hayashi
a,
Takashi
Ohshima
*b,
Yuka
Fujii
a,
Yoshimasa
Matsushima
c and
Kazushi
Mashima
*a
aDepartment of Chemistry, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: mashima@chem.es.osaka-u.ac.jp; Fax: +81 6-6850-6249; Tel: +81 6-6850-6246
bGraduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi Higashi-ku, Fukuoka 812-8582, Japan. E-mail: ohshima@phar.kyushu-u.ac.jp; Fax: +81 92-642-6650; Tel: +81 92-642-6650
cCorporate Research and Development Division, Takasago International Corporation, 1-4-11 Nishi-yawata, Hiratsuka, Kanagawa, 254-0073, Japan
First published on 14th February 2011
Abstract
Synthesis of a trifluoroacetate-bridged tetranuclear zinc cluster and its trifluoroacetic acid (TFA) adduct under mild conditions (∼110 °C) was achieved. The catalytic activity and functional group tolerance of the TFA adduct in transesterification and oxazoline formation were almost identical to those of the original zinc cluster.
Transesterification is one of the most straightforward organic transformations,1 and various catalyst systems have been developed.2–5 Recently, we reported that a trifluoroacetate-bridged tetranuclear zinc cluster Zn4(OCOCF3)6O (1a) serves as an efficient catalyst with broad substrate generality for the transesterification of various methyl esters with various alcohols,6a,bcatalytic acetylation in ethyl acetate,6c and catalytic deacetylation in methanol,6d as well as the direct conversion of esters, lactones, and carboxylic acids to oxazolines.6e Because these catalyses proceed under neutral conditions, various functional groups, such as THP ether, TES ether, acetal, as well as N-Boc, N-Cbz, and N-Fmoc groups, remain intact, and undesirable side reactions, such as elimination, isomerization, and epimerization, are suppressed. Further, a unique hydroxy group-selective acylation in the presence of much more nucleophilic primary and secondary aliphatic amino groups is achieved by 1a.6a,c
The requirements for any suitable catalyst are easy preparation, easy handling, and ready generation of catalytically active species under desired conditions. Zinc cluster 1a, the best catalyst for transesterification, was originally synthesized by heating a zinc trifluoroacetate hydrate Zn(OCOCF3)2·xH2O (2a) at 360 °C under high vacuum (<0.02 mmHg) to produce solidified zinc cluster 1a on a glass wall, followed by collection of the resulting white solid under argon atmosphere, avoiding contact with moisture, to give pure 1a in 84% yield.6c,7 This preparation method could be performed in gram-scale. This synthetic methodology was based on the first synthesis of the acetate-bridged tetranuclear zinc cluster Zn4(OCOCH3)6O (1b), which was obtained by vacuum distillation of zinc acetate hydrate Zn(OCOCH3)2·xH2O (2b) by Auger and Robin in 1924,8 and its μ4-oxo structure was later established by X-ray crystallographic analysis by Wyart9a and Koyama and Saito9b (Fig. 1). Other tetranuclear zinc clusters with different carboxylate ligands, such as Zn4(OCOR)6O (R = Et, n-Pr, t-Bu, Ph, etc.), were subsequently prepared using this synthetic route or similar pyrolytic methods.10 Although such sublimation provides highly pure zinc clusters, the high temperature synthesis and moisture-free handling required for 1a decrease its accessibility to organic chemists, and thus the development of a more practical synthetic method of 1a is in high demand. Herein, we report the novel synthesis and catalytic performance of a TFA adduct of 1a, namely Zn4(OCOCF3)6O·CF3CO2H (3), which was practically synthesized under toluene refluxed conditions (∼110 °C) in greater-than kilogram scale. The catalytic activity and functional group tolerance of the TFA adduct 3 were almost identical to those of 1a. Another great advantage of 3 compared with 1a is its easy handling due to its much less hygroscopic nature.
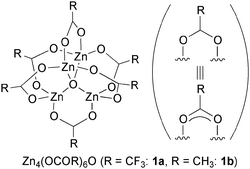 | ||
| Fig. 1 Structure of μ4-oxo tetranuclear zinc clusters 1. | ||
Intensive studies to evaluate the reaction temperature indicated that even at a much lower temperature (<160 °C) the assembly of the requisite zinc ions, trifluoroacetates, and μ-oxo ligand proceeded smoothly to form the μ4-oxo tetranuclear zinc carboxylate motif. Heating the zinc trifluoroacetate hydrate 2a11 at 120 °C under high vacuum conditions for 1.5 h, and then gradually rising the temperature to 160 °C produced a white solid A by sublimation of the reaction product (Fig. 2, path A). Electrospray ionization (ESI) mass analysis of this white solid A (negative mode) showed a peak that was assigned to [Zn4(OCOCF3)6O·CF3CO2]− (Fig. 3). In addition, 19F-NMR and 13C-NMR spectra of solid A were quite similar to those of the zinc cluster 1a, though the 1H-NMR spectrum of A displayed an additional signal at around 10 ppm, which was assigned to an acidic proton. Based on these results, solid A is considered to be a TFA adduct of 1a, that is, Zn4(OCOCF3)6O·CF3CO2H (3).12 To obtain more information about the composition of solid A, we investigated the zinc content of A by titration. The theoretical zinc content of zinc cluster 1a and TFA adduct 3 was 27.4% and 24.5%, respectively, and the observed zinc content of A was 25.2%, suggesting that solid A was a 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶72 mixture of 1a and 3. Thus, the yield of A based on the zinc content was determined to be 98%. Re-sublimation of A at 170 °C with generation of TFA increased the zinc content to 26.6%, suggesting that the ratio of 1a increased (1a∶3 = 75∶25). These experimental results indicated that high temperature is essentially required, not for the construction of the μ4-oxo tetranuclear zinc carboxylate motif, but for the dissociation of the coordinated TFA in 3. To confirm this hypothesis, we performed a density functional theory calculation at the B3LYP13/(LANL2DZ for Zn and 6-31G(d,p) for others) level and found that the energy of the TFA adduct 3 is 5.18 kcal mol−1 lower than that of 1a + TFA.7
∶72 mixture of 1a and 3. Thus, the yield of A based on the zinc content was determined to be 98%. Re-sublimation of A at 170 °C with generation of TFA increased the zinc content to 26.6%, suggesting that the ratio of 1a increased (1a∶3 = 75∶25). These experimental results indicated that high temperature is essentially required, not for the construction of the μ4-oxo tetranuclear zinc carboxylate motif, but for the dissociation of the coordinated TFA in 3. To confirm this hypothesis, we performed a density functional theory calculation at the B3LYP13/(LANL2DZ for Zn and 6-31G(d,p) for others) level and found that the energy of the TFA adduct 3 is 5.18 kcal mol−1 lower than that of 1a + TFA.7
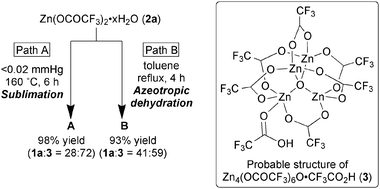 | ||
| Fig. 2 Synthesis of zinc clusters and probable structure of Zn4(OCOCF3)6O·CF3CO2H (3). | ||
![(a) Negative ion ESI mass spectrum of the obtained compound A and (b) calculated isotope pattern for [Zn4(OCOCF3)6O·CF3CO2]−.](/image/article/2011/CY/c0cy00048e/c0cy00048e-f3.gif) | ||
| Fig. 3 (a) Negative ion ESI mass spectrum of the obtained compound A and (b) calculated isotope pattern for [Zn4(OCOCF3)6O·CF3CO2]−. | ||
The observation that the μ4-oxo tetranuclear zinc carboxylate motif is formed at a relatively low temperature (∼160 °C) led us to develop a more practical and scalable method for preparing the zinc cluster. After screening various conditions, we successfully developed a new azeotropic dehydration method as a highly efficient method in industrial preparation of the zinc cluster (Fig. 2, path B). Heating at 60 °C in toluene dissolved 2a completely, and then refluxing the resulting solution for 4 h generated white precipitates together with the formation of an azeotropic mixture (pH ≈ 1) of toluene, water, and TFA. The resulting precipitates were filtered, leaving a white solid B.14 The spectroscopic data of B were identical to those of A, except for a zinc content of 25.6%, suggesting that the white solid B was a mixture of 1a and 3 in a ratio of 41∶59 and thus the yield of B, based on zinc content, was 93%. This reaction protocol was applicable in kilogram scale with the same efficiency. Solids A and B were much less hygroscopic than 1a, probably because the additional TFA stabilized the Lewis acidic coordination site of 1a.
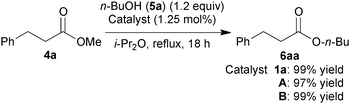
To determine whether the solids A and B could be alternative catalysts to 1a, we first examined the catalytic activities of A and B for the transesterification of methyl 3-phenylpropionate (4a) with 1.2 equiv. of n-butanol (5a) (eqn (1)).6b Under optimized conditions, A and B efficiently catalyzed the reaction to afford the product 6aa in excellent yield with the same efficiency as 1a, suggesting that the coordinated TFA in A and B readily dissociated to generate the same active species as generated using 1a under the reaction conditions.
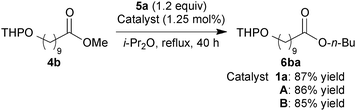
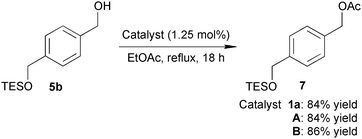
We next examined the effects of additional TFA in A and B on functional group tolerability using substrates with acid-sensitive functional groups.6b Reactions of methyl 10-(tetrahydro-2H-pyran-2-yloxy)decanoate (4b) with 5a using 1.25 mol% of either A or B provided the corresponding product 6ba in high yield without decomposition of the acid-sensitive THP ether (eqn (2)). Moreover, catalysts A and B were successfully applied to the acetylation of alcohol 5b,6c which has a highly acid-sensitive TES ether, to afford the product 7 in 84% and 86% yields, respectively (eqn (3)).
 | (1) |
 | (2) |
 | (3) |
The transesterification of various N-protected amino esters 8a–c with 5a proceeded smoothly to give the corresponding n-butyl esters 9 in high yield without O-alkyl transesterification of carbamates (Table 1). Notably, even in the case of the phenylglycine derivative 8c, no epimerization was observed (entries 7–9).

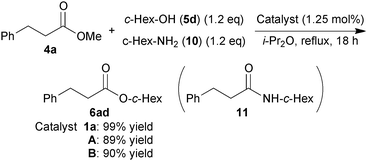
The most attractive feature of 1a is its high hydroxy group selectivity over a more nucleophilic amino group in an acylation reaction.6a,c We thus performed chemoselective acylation of cyclohexanol (5d) and cyclohexylamine (10) using catalyst A or B (eqn (4)). In both cases, transesterification product 6ad was obtained in excellent yield along with recovery of 4a (ca. 8%), and cyclohexylamide 11 was not detected, consistent with the chemoselectivity of zinc cluster 1a.
 | (4) |
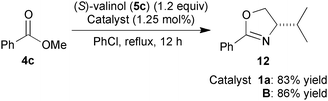
Finally, we investigated the catalytic activities of the zinc clusters in the direct conversion of ester to oxazoline (eqn (5)). The reaction of methyl benzoate (4c) with (S)-valinol (5c) using catalyst B afforded oxazoline 12 in 86% yield, which was almost identical to the 83% yield produced using zinc cluster 1a.
 | (5) |
Overall, catalysts A and B exhibited almost identical catalytic activity and functional group tolerability to 1a.
In conclusion, we revealed that construction of the μ4-oxo tetranuclear zinc trifluoroacetate motif proceeded smoothly even at 110 °C, a much milder condition than that used in the preceding synthetic methods (sublimation at 360 °C). Heating zinc trifluoroacetate hydrate 2a under toluene reflux conditions and simple filtration of the resulting precipitates afforded a white solid B, which was a mixture of trifluoroacetate-bridged tetranuclear zinc cluster Zn4(OCOCF3)6O (1a) and its TFA adduct Zn4(OCOCF3)6O·CF3CO2H (3). Because the present method does not require high temperature conditions or a sublimation process, kilogram scale synthesis of the zinc cluster proceeded with the same efficiency. The catalytic activity and functional group tolerance of the TFA adduct 3 in transesterification and oxazoline formation were almost identical to those of 1a. Another advantage of 3 is its low hygroscopic nature, which allows for handling without the concern to introducing moisture. Industrial application of 3-catalyzed reactions is under investigation.
This work was supported by a Research for Promoting Technological Seeds and Core Research for Evolutional Science and Technology from Japan Science and Technology Agency. Y.H. express her special thanks for Global COE Program ‘Global Education and Research Center for Bio-Environmental Chemistry’ of Osaka University and JSPS Research Fellowship for Young Scientists.
Notes and references
- For general reviews, see: (a) R. Larock, Comprehensive Organic Transformations, Wiley-VCH, New York, 2nd edn, 1996 Search PubMed; (b) J. Mulzer, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1992, vol. 6 Search PubMed; (c) J. Otera, Esterification, Wiley-VCH, Weinheim, 2003 Search PubMed; (d) J. Otera, Chem. Rev., 1993, 93, 1449 CrossRef CAS; (e) H. E. Hoydonckx, D. E. De Vos, S. A. Chavan and P. A. Jacobs, Top. Catal., 2004, 27, 83 CrossRef CAS; (f) G. A. Grasa, R. Singh and S. P. Nolan, Synthesis, 2004, 971 CAS.
- For the use of excess amounts of either of the reactant, see: (a) W. Pereira, V. A. Close, W. Patton and B. Halpern, J. Org. Chem., 1969, 34, 2032 CrossRef CAS; (b) Y. Misaki, N. Tanaka and T. Miura, Chem. Lett., 1997, 55 CAS; (c) B. C. Ranu, P. Dutta and A. Sarkar, J. Org. Chem., 1998, 63, 6027 CrossRef CAS; (d) B. C. Ranu, P. Dutta and A. Sarkar, J. Chem. Soc., Perkin Trans. 1, 2000, 2223 RSC; (e) P. Baumhof, R. Mazitschek and A. Giannis, Angew. Chem., Int. Ed., 2001, 40, 3672 CrossRef CAS; (f) K. Ramalinga, P. Vijayalakshmi and T. N. B. Kaimal, Tetrahedron Lett., 2002, 43, 879 CrossRef CAS; (g) J. Xiang, S. Toyoshima, A. Orita and J. Otera, Angew. Chem., Int. Ed., 2001, 40, 3670 CrossRef CAS.
- For the use of enol ester as a reactant, see: (a) Y. Ishii, M. Takeno, Y. Kawasaki, A. Muromachi, Y. Nishiyama and S. Sakaguchi, J. Org. Chem., 1996, 61, 3088 CrossRef CAS; (b) A. Orita, A. Mitsutome and J. Otera, J. Org. Chem., 1998, 63, 2420 CrossRef CAS; (c) P. Ilankumaran and J. G. Verkade, J. Org. Chem., 1999, 64, 9063 CrossRef CAS; (d) Y. Shirae, T. Mino, T. Hasegawa, M. Sakamoto and T. Fujita, Tetrahedron Lett., 2005, 46, 5877 CrossRef CAS; (e) J. W. J. Bosco, A. Agrahari and A. K. Saikia, Tetrahedron Lett., 2006, 47, 4065 CrossRef CAS; (f) J. W. J. Bosco and A. K. Saikia, Chem. Commun., 2004, 1116 RSC.
- For removal of the resulting lower alcohol by molecular sieves, see: (a) G. W. Nyce, J. A. Lamboy, E. F. Connor, R. M. Waymouth and J. L. Hedrick, Org. Lett., 2002, 4, 3587 CrossRef; (b) G. A. Grasa, T. Gueveli, R. Singh and S. P. Nolan, J. Org. Chem., 2003, 68, 2812 CrossRef CAS; (c) R. Singh, R. M. Kissling, M.-A. Letellier and S. P. Nolan, J. Org. Chem., 2004, 69, 209 CrossRef CAS.
- For removal of the resulting lower alcohol by continuous distillation, see: (a) D. Seebach, E. Hungerbuehler, R. Naef, P. Schnurrenberger, B. Weidmann and M. Zueger, Synthesis, 1982, 138 CrossRef CAS; (b) P. Schultheiss-Reimann and H. Kuntz, Angew. Chem., Int. Ed. Engl., 1983, 22, 63; (c) J. Otera, T. Yano, A. Kawabata and H. Nozaki, Tetrahedron Lett., 1986, 27, 2383 CrossRef CAS; (d) H. Waldmann and H. Kunz, J. Org. Chem., 1988, 53, 4172 CrossRef CAS; (e) A. M. Klibanov, Acc. Chem. Res., 1990, 23, 114 CrossRef CAS; (f) A. L. Gutman, E. Shkolnik and M. Shapira, Tetrahedron, 1992, 48, 8775 CrossRef CAS; (g) C. Blandy, J.-L. Pellegatta and P. Cassoux, Catal. Lett., 1997, 43, 139 CrossRef CAS; (h) D. E. Ponde, V. H. Deshpande, V. J. Bulbule, A. Sudalai and A. S. Gajare, J. Org. Chem., 1998, 63, 1058 CrossRef; (i) P. Krasik, Tetrahedron Lett., 1998, 39, 4223 CrossRef CAS; (j) D. E. Ponde, V. H. Deshpande, V. J. Bulbule, A. Sudalai and A. S. Gajare, J. Org. Chem., 1998, 63, 1058 CrossRef; (k) K. Wakasugi, T. Misaki, K. Yamada and Y. Tanabe, Tetrahedron Lett., 2000, 41, 5249 CrossRef CAS; (l) J. Xiang, S. Toyoshima, A. Orita and J. Otera, Angew. Chem., Int. Ed., 2001, 40, 3670 CrossRef CAS; (m) J. Xiang, A. Orita and J. Otera, Adv. Synth. Catal., 2002, 344, 84 CrossRef CAS; (n) S. P. Chavan, K. Shivasankar, R. Sivappa and R. Kale, Tetrahedron Lett., 2002, 43, 8583 CrossRef CAS; (o) J. Otera, Acc. Chem. Res., 2004, 37, 288 CrossRef CAS; (p) D.-W. Yoo, J.-H. Han, S. H. Nam, H. J. Kim, C. Kim and J.-K. Lee, Inorg. Chem. Commun., 2006, 9, 654 CrossRef CAS; (q) T. Funatomi, K. Wakasugi, T. Misaki and Y. Tanabe, Green Chem., 2006, 8, 1022 RSC; (r) T. Shinada, M. Hamada, K. Miyoshi, M. Higashino, T. Umezawa and Y. Ohfune, Synlett, 2010, 2141 CrossRef CAS; (s) Y. M. Lee, Y. J. Song, J. I. Poong, S. H. Kima, H. G. Koo, J. A. Lee, C. Kima, S.-J. Kimb and Y. Kimb, Inorg. Chem. Commun., 2010, 13, 101 CrossRef CAS.
- (a) T. Ohshima, T. Iwasaki, Y. Maegawa, A. Yoshiyama and K. Mashima, J. Am. Chem. Soc., 2008, 130, 2944 CrossRef CAS; (b) T. Iwasaki, Y. Maegawa, Y. Hayashi, T. Ohshima and K. Mashima, J. Org. Chem., 2008, 73, 5147 CrossRef CAS; (c) T. Iwasaki, Y. Maegawa, Y. Hayashi, T. Ohshima and K. Mashima, Synlett, 2009, 1659 CAS; (d) T. Iwasaki, K. Agura, Y. Maegawa, Y. Hayashi, T. Ohshima and K. Mashima, Chem. Eur. J., 2010, 16, 11567 CrossRef CAS; (e) T. Ohshima, T. Iwasaki and K. Mashima, Chem. Commun., 2006, 2711 RSC.
- For details, see ESI†.
- V. Auger and I. Robin, C. R. Hebd. Seances Acad. Sci., 1924, 178, 1546 Search PubMed.
- (a) J. Wyart, Bull. Soc. Fr. Mineral., 1926, 49, 148 Search PubMed; (b) H. Koyama and Y. Saito, Bull. Chem. Soc. Jpn., 1954, 27, 112 CAS.
- (a) R. M. Gordon and H. B. Silver, Can. J. Chem., 1983, 61, 1218 CAS; (b) W. Clegg, D. R. Harbron, C. D. Homan, P. A. Hunt, J. R. Little and B. P. Straughan, Inorg. Chim. Acta, 1991, 186, 51 CrossRef CAS; (c) Y. Ming-cai, M. Chi-wei, A. Chang-chun, Y. Liang-jie and S. Ju-tang, Wuhan Univ. J. Nat. Sci., 2004, 9, 939 CrossRef; (d) S. Hermes, T. Witte, T. Hkov, D. Zacher, S. Bahnmüller, G. Langstein, K. Huber and R. A. Fischer, J. Am. Chem. Soc., 2007, 129, 5324 CrossRef CAS; (e) O. Berkesi, I. Dreveni and A. Andor, Inorg. Chim. Acta, 1991, 181, 285 CrossRef CAS.
- The use of zinc trifluoroacetate anhydrous instead of hydrate gave an unsatisfactorily low yield of the zinc cluster. Based on analysis with gas chromatography, no hexafluoroacetone was generated during the reaction.
- We confirmed that the addition of TFA to 1a gave 3 by 1H NMR analysis.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- White solid B prepared via path B is commercially available as ZnTAC24TM from Takasago International Corp. and Strem Chemicals Inc.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data. See DOI: 10.1039/c0cy00048e |
| This journal is © The Royal Society of Chemistry 2011 |