Catalytic activity and extra-large pores of germanosilicate UTL zeolite demonstrated with decane test reaction
Nataliia
Kasian
ab,
Gina
Vanbutsele
a,
Kristof
Houthoofd
a,
Tamas I.
Koranyi
a,
Johan A.
Martens
a and
Christine E. A.
Kirschhock
*a
aCentre for Surface Chemistry and Catalysis, K. U. Leuven, Kasteelpark Arenberg 23, B-3001 Heverlee, Belgium. E-mail: Christine.Kirschhock@biw.kuleuven.be; Fax: +32 16321998; Tel: +32 16321610
bL. V. Pisarzhevsky Institute of Physical Chemistry, National Academy of Sciences of Ukraine, Pr. Nauky 31, 03028, Kyiv, Ukraine. Fax: +38 0445256216; Tel: +38 0445251190
First published on 10th February 2011
Abstract
Pyridine adsorption monitored by FTIR spectroscopy revealed germanosilicate UTL type zeolite to contain intrinsic Brønsted acidity required for catalytic activity. Germanosilicate UTL type zeolite was converted into a bifunctional catalyst and evaluated in n-decane isomerization and hydrocracking. To assess the stability of the framework during catalyst preparation and use, three different strategies of converting the zeolite into bifunctional catalyst were followed: incipient wetness impregnation with Pt(NH3)4Cl2 solution before and after evacuation of the template by calcination, and physical mixing of calcined UTL with Pt-containing amorphous silica. All three samples showed catalytic activity but to very different degrees. The possibility of structure degradation during catalyst preparation and catalysis was investigated by XRD and 29Si MAS NMR. The impregnation of as-made germanosilicate UTL zeolite with platinum and pretreatment in the reactor led to superior stability and activity. The n-decane test previously was used to probe zeolite micropore architectures with 8-, 10- and 12-membered rings. Its validity is now extended to the extra-large pore zeolites. The linear trend of increasing ethyloctanevs.methylnonane selectivity with increasing pore size has now been confirmed also for UTL with 14 membered rings.
Introduction
Aluminosilicate zeolites are robust catalysts profusely applied in industry.1 The commercially available aluminosilicate zeolites contain pore-windows delineated with 12 membered rings (12-MR) or less, which prevents large molecules from entering the catalyst pores. One way to create zeolite frameworks with larger pores—the so called extra-large-pore zeolites—involves the introduction of heteroatoms such as Ge into the framework.2–5Recently, catalytic applications of extra-large-pore zeolites including Ge-containing structures were reviewed.6 The UTL germanosilicate zeolite contains 14-MR channels crossing 12-MR channels.7,8 The catalytic benefit of this bi-directional extra large channel system with respect to 1-D extra-large pore framework types has been demonstrated in the dealkylation reaction of tri- and di-isopropylbenzene.7 It was also predicted that the spacious UTL pore system should be ideal for catalytic hydrocarbon conversion, provided catalytic activity could be introduced. The germanosilicate composition was considered as inactive and isomorphic substitution of trivalent elements such as Al3+ was assumed necessary to provide cation exchange capacity and acidity.9 The catalytic potential of the UTL germanosilicate zeolite itself without further element substitution has not yet been assessed.
It is known that the use of germanium as a heteroelement to generate low framework density Ge-rich zeolites is paid for by limited to low stability of the obtained materials. Literature reported that the heteroatom-containing germanosilicates were rather sensitive to humidity and slowly degraded with time.5,10
It is generally assumed that the hydrothermal stability of Ge-zeolites is a function of Ge content. Materials with increasing Si/Ge ratio should show increased stability. However, calculations have shown different bond lengths and angles of Ge–O–Si compared to silicates make some frameworks poor candidates for wide ranges of Si/Ge ratios.11 Especially the UTL structure was demonstrated to be unfeasible as pure silica or germania end composition may only be synthesized at intermediate Si, Ge ranges.
Attempts were made to stabilize the framework of Ge-rich zeolitesvia post-synthesis alumination of the material.12 It was proposed that Al partially replaced Ge preventing a change of 4-fold to 6-fold coordination of these sites. Curiously, in a previous investigation13 we observed that the introduction of heteroatoms (Al and B) into the framework during crystallization had catastrophic effects on the stability. The germanosilicate UTL structure turned out as much more robust in a reactor under the conditions of the n-decane hydroconversion reaction compared to Al-UTL and B-UTL.
In the literature it is assumed that Ge does not contain acid centers other than Ge–OH and possible defects in the structure. Incorporation of Ge appeared to increase the number of defects and hydroxyl groups while having only a small effect on the strength of the acid sites.14 In the early 2000s a series of publications described synthesis and characterization of Ge-substituted zeolites in an attempt to alter their acidic properties.14–16 It was found that the Brønsted acid strength increased in the following order: silicalite-1 = Ge-ZSM-5 < B-ZSM-5 < Fe-ZSM-5 < Ga-ZSM-5 < Al-ZSM-5.17 The catalytic activities of substituted ZSM-5 zeolites in acid-catalyzed reactions increased in the same order, corresponding to their acid strength. Other authors15 observed increased activity of Ge-ZSM-5 (compared to ZSM-5) in a series of acid-catalyzed test reactions. As explanation for this difference the presence of considerable additional meso- and macroporosity in Ge-ZSM-5 was proposed. At the same time, structural distortions may have been introduced into the framework by the presence of Ge, which may also have changed the catalytic properties of the zeolite.14 For example, the catalytic activity of Ge-TS-1 in the epoxidation of propene was higher compared to the parent material TS-1.18 Zicovich-Wilson et al.19 calculated water adsorption energies for the Ge and Si compounds and showed that despite small observed energy differences, factors such as acid softness and polarizability are important. Based on these considerations a detailed analysis of the activity of UTL in the absence of other heteroatoms than Ge seemed feasible.
Here we demonstrate that the germanosilicate UTL zeolite is active and exhibits extra-large-pore properties in hydroconversion of n-decane without any heteroelement besides the presence of Ge. Pt was introduced in three different manners, with various degrees of exposure to aqueous solutions during introduction of the noble metal function into the catalyst: by the classical incipient wetness impregnation of platinum precursor on as-made and calcined UTL samples, and via dry mixing of the calcined UTL zeolite powder with Pt dispersed on an inert amorphous silica support. The decane test reaction has been frequently used for the determination of zeolite pore structures.20–22 It is known that for an “ideal” catalyst with well balanced acid and metal functions the selectivity of formation of skeletal isomers and the hydrocracking pattern are rather insensitive to differences in acidity and dispersion of the Pt metal, but very sensitive to the pore size and dimensionality.23,24 Previously, another extra-large pore zeolite with SFH topology and 1D 14-MR channels was evaluated in this reaction but the selectivity was found comparable to the values for FAU zeolite with 12-MR pores only.25
Experimental
Preparation of the UTL zeolite samples
UTL zeolite samples were synthesized following a previously published method, which allowed synthesis of highly crystalline UTL phases with a Si/Ge ratio of 4.2.26,27 Preparation of the structure directing agent (SDA) (6R,10S)-6,10-dimethyl-5-azoniaspiro-[4.5]decane hydroxide was carried out as in ref. 28. The reaction gel was prepared by dissolving amorphous germanium oxide (Aldrich) in the solution of the SDA. After that, TEOS was added to the solution and the mixture was stirred at room temperature for 30 min. The resulting fluid gel was charged into Teflon-lined stainless steel autoclaves and heated at 175 °C for 6 days under agitation. The solid product was recovered by filtration, thoroughly washed with distilled water, and dried overnight at 60 °C. To remove the SDA, the as-synthesized zeolite was calcined in air at 550 °C for 6 h with a temperature ramp of 2 °C min−1. The UTL zeolite crystallized in the shape of thin sheets of 2–5 μm in size. The micropore volume was 0.22 cm3 g−1.Characterization techniques
Transmission X-ray powder diffraction data were recorded on a STOE Stadi P diffractometer with CuKα1 radiation in transmission. Nitrogen adsorption isotherms at −196 °C were recorded on a Tristar instrument (Micromeritics, USA). Before the measurements, samples were outgassed for 12 h at 250 °C. The pore volume was determined using t-plot analysis. Thermal gravimetric analysis was performed on a Q500 thermogravimetric instrument performed in N2 atmosphere at a heating rate of 10 °C min−1. Particle size and morphology were analyzed by scanning electron microscopy (SEM) with a Philips XL-30 FEG equipped with a tungsten filament on gold-plated samples. 29Si MAS NMR spectra were recorded on a Bruker AMX300 spectrometer (7.0 T). A total of 4000 scans were accumulated with a recycle delay of 60 s. The samples were packed in 4 mm Zirconia rotors; the spinning frequency of the rotor was 5000 Hz. Tetramethylsilane was used as chemical shift reference. The spectra were deconvoluted using the program Igor pro, assuming Gaussian shape of the NMR signals.Characterization of the acid sites viaFTIR spectroscopy was carried out using a Nicolet 6700 spectrometer. Calcined samples were compressed into self-supporting wafers of 10 mg cm–2 and mounted in an in situcell. Samples were outgassed at 400 °C for 1 h, were cooled to 50 °C and saturated by pyridine vapor at a pressure of 24 mbar. After equilibration, samples were outgassed stepwise from 50 to 350 °C, and the FTIR spectra recorded at each temperature. The acid site concentration was quantified using published molar extinction coefficients.29
Precautions were taken for characterization of the samples after catalysis. To avoid exposure to ambient leading to possible structure deterioration the samples were kept in nitrogen atmosphere. Packing of rotor for 29Si MAS NMR and preparation of self-supporting wafers for FTIR spectroscopy characterizations were performed in a nitrogen glove box.
Catalytic experiments
The UTL samples were transformed into bifunctional catalysts by three different manners.As-made + Pt and calc + Pt samples. In the first case Pt was introduced by incipient wetness impregnation at room temperature of an aqueous solution of Pt(NH3)4Cl2·H2O to as-made and freshly calcined UTL samples to obtain a loading of 0.5 wt%. 15 wt% template and water content, determined by TG, was taken into account in the case of as-made UTL. The impregnated samples were dried at 60 °C for one day in an oven. Catalyst pellets were obtained by compressing the powder into flakes, crushing the flakes, and sieving to obtain a pellet size of 125–250 μm. 50 mg of these pellets were charged in parallel microreactor tubes of a high-throughput reactor unit.30 The catalyst activation involved calcination under flowing oxygen at 400 °C for 1 h, which also removed the template from the as-made + Pt sample, followed first by flushing with nitrogen and then by reduction in hydrogen at 400 °C for 1 h. Literature reported this procedure to provide homogeneous dispersions of metal clusters throughout the zeolites.23,31,32
Calc + Pt / silica sample. The third method was chosen to reduce exposure of the framework to an aqueous solution. Here, the noble metal phase was introduced on a separate carrier material. 50 mg of calcined UTL sample was mixed with 50 mg of silica gel with 0.5 wt% Pt loading and an 81.3% Pt dispersion in a mortar under ambient. The catalytic activity of the Pt-loaded silica gel in the decane hydroconversion was verified and found to be negligible in the temperature range of interest. Catalyst pellets were obtained as described above.
All catalytic tests occurred in the same reactor setup with hydrogen to hydrocarbon molar ratio of 214. A fixed contact time of 1400 mol kg−1s−1 was used. Product analysis was performed using on-line GC and a CP Sil-5 capillary column (Chrompack). Commercial USY zeolites (CBV-712, Si/Al = 5.8, and CBV-760, Si/Al = 30, PQ Corporation) with FAU topology loaded with 0.5 wt% platinumvia incipient wetness impregnation were selected as activity reference.
Results and discussion
XRD
Preliminary tests to assess the stability of the framework revealed the UTL structure did not collapse even after calcination at 1000 °C, in agreement with the literature.26,27 However, when exposed to ambient air, UTL material, like many other Ge-rich zeolites, slowly degraded after calcination, while the as-made samples still containing the organic template remained perfectly stable during long periods of storage. It could even be demonstrated that the UTL structure withstood hydrothermal conditions when the SDA molecules remained present inside the pores and protected the structure (Fig. 1). | ||
| Fig. 1 XRD patterns of as-made and calcined UTL zeolite samples after hydrothermal treatment at 95 °C in water (pH = 7) for 3 h. | ||
Thermogravimetric analysis (TG) revealed the adsorbed water and organic matter content in as-synthesized zeolite to amount to 3 wt% and 12 wt%, respectively (Fig. 2). This organic matter content corresponded to approximately 5 to 6 template molecules per unit cell. Provided the molecules distribute evenly among 14- and 12-membered channels this would mean both are occupied with template molecules with average distances of 6 and 7 Å. Comparison with the size of the template molecule which has lengths of approximately 7 Å along its longest dimensions indicated a rather tight filling of the cavities (Fig. 3). The still included template preserves the framework structure. Given the low water content of the as-synthesized zeolites (3 wt%, see higher) only few, if any, additional water molecules could enter the pores during hydrothermal treatment. The template prevents exposure of the pore walls to moisture from ambient air or from solution. This observation prompted the attempt to impregnate the sample before the template was removed to increase the stability of the bifunctional catalyst during preparation and use. In an incipient wetness impregnation of as-synthesized zeolite there will be limited penetration of the solution into the zeolite micropores and mainly interstitial voids between the zeolite particles will be filled thus minimizing potential damage.
 | ||
| Fig. 2 TG analysis of as-synthesized UTL sample. | ||
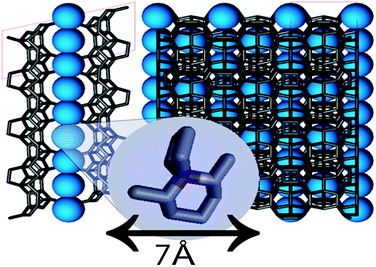 | ||
| Fig. 3 Schematic representation of even filling with 6 template molecules per unit cell of UTL along 12- and 14 ring channels. Template molecules are represented with blue spheres. | ||
The XRD patterns of UTL zeolite as-synthesized and calcined at 550 °C are shown in Fig. 4. From a comparison with published data,26 it can be deduced that the synthesized samples were of good crystallinity and did not contain amorphous or crystalline impurities in significant amounts. Shortly after impregnation of calcined zeolite with aqueous solution of Pt(NH3)4Cl2 the XRD pattern was still intact without significant changes of the width of the reflections, indicating that no serious loss of crystallinity took place. The evolution of the intensity of the Bragg reflections at low angles confirmed that the UTL contained template in the pores which was evacuated upon calcination. The reflections at low angles corresponding to the largest observed d-values can give a clear indication of porosity as especially these are strongly affected by the electron density of the framework in comparison with the electron density within the pores stemming from any adsorbed species.33 After impregnation of the calcined material the pores again were partially filled with the impregnating solution, and drying did not fully evacuate them.
 | ||
| Fig. 4 XRD patterns of as-made and freshly calcined UTL zeolite samples before and after Pt impregnation and drying at 60 °C. | ||
The integrity of the UTL samples after catalysis (vide infra) was analyzed by XRD (Fig 5). After 3 catalysis runs as-made + Pt preserved its framework structure without significant changes of the width of the reflections, indicating that no serious loss of crystallinity occurred. This was in agreement with NMR results which did not show any changes in Q4-nGe species (vide infra). After already one run of catalysis the two other samples, calc + Pt and calc + Pt/silica, showed broadened reflections indicating serious structural damage.
 | ||
| Fig. 5 XRD patterns of calcined UTL and samples taken after catalysis. | ||
NMR spectroscopy
29Si MAS NMR spectra of as-synthesized and calcined samples of germanosilicate UTL before and after catalysis are presented in Fig. 6. The spectra were typical examples of Ge-containing zeolites showing Q4silicon atoms with 4 Si neighbors and chemical shifts in the range −110 to −120 ppm (Q4-4Si), Q4silicon with one or more Ge neighbors (Q4-nGe −100 to −110 ppm) and Q3silicon atoms with one silanol group (chemical shift region −90 to −100 ppm).34,35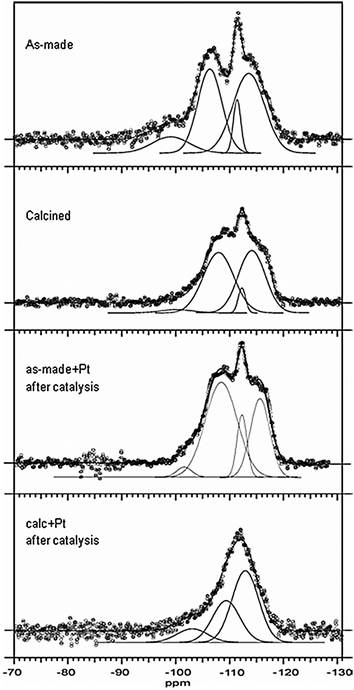 | ||
| Fig. 6 29Si MAS NMR spectra of as-made UTL zeolite, calcined UTL zeolite and sample after catalysis—as-made + Pt, calc + Pt. | ||
To semi-quantitatively assess the amount of different Si environments the spectra were fitted with Gaussians accounting for two types of Q4-4Si, one type of Q4-nGe and a broad component representing Q3silicon. The spectra did not permit further, more detailed, assignments, though the presence of at least two different types of Q4-4Si could be discerned.
The best resolved spectrum corresponded to the as synthesized sample. As-made + Pt taken after catalysis also allowed clear distinction of the different types of silicon, while calc + Pt showed significant broadening of all signals after catalysis. This was found in agreement with the XRD results which indicated loss of crystallinity for calc + Pt (Fig. 4) but preservation of the UTL structure for as-made + Pt after catalysis. Most interestingly, the intensity of the Q4-4Si signals (56–57% of all T-atoms) remained almost constant for all four samples. Neither calcination nor catalysis changed the amount of 4-coordinated Si with four Si-neighbors. However, calcination led to a significant decrease of silanol groups in favor of Q4-nGe species (from 7% in as-made to 2% in calcined). This indicated that after synthesis especially Si–O–Ge bonds were not fully formed and only dehydration and calcination led to condensation of these remaining open connections. This conclusion also is in agreement with the literature.36 Most noteworthy was the observation that after catalysis especially the number of these connections decreased to 33% for calc + Pt, again forming a large number of Q3-Si. Apparently, especially Si–O–Ge sites were affected during catalyst preparation and catalysis in calc + Pt. At the same time, the intensity of Q4-nGe for as-made + Pt, taken after catalysis, was comparable with the calcined UTL sample (39 and 42%) and catalysis did not significantly increase the number of silanols in the structure. The UTL framework is a combination of layers mostly consisting of silica and connected with D4R units preferentially occupied by Ge. The observed NMR spectra clearly indicate that especially the connection between the D4R units and the siliceous layers is the most sensitive part of the structure as after synthesis these connections are not yet fully established and might also be attacked and broken during catalyst preparation and use. To determine what exactly caused the calc + Pt structure to break down in the catalytic reactor, freshly impregnated and dried calcined UTL was heated to 400 °C and activated in H2 but not used for catalysis. The NMR spectrum closely resembled the spectrum recorded after catalysis, showing strongly broadened signals and significantly reduced numbers of Q4-nGe and increased Q3silicon species. This clearly indicated that breakdown occurred during the heating step, as also was confirmed by XRD. In all probability the structure deterioration was caused by the presence of residual water in the channels after drying at 60 °C overnight. In Table 1 we compared the peak positions and intensities of UTL samples before and after catalysis with those of Ge-MFI zeolite and ITQ-40 germanosilicate.
| Sample | Q3 | Q4-nGe | Q4-4Si |
|---|---|---|---|
| The number in parentheses corresponds to signal area, %. | |||
| As-made UTL | −99 ppm (7) | −106 ppm (36) | −114 ppm (34) |
| −111 ppm (23) | |||
| (57) | |||
| Calcined UTL | −100 ppm (2) | −107 ppm (42) | −115 ppm (39) |
| −112 ppm (17) | |||
| (56) | |||
| As-made + Pt after catalysis | −100 (4) | −107 (39) | −115.7 (32) |
| −112 (25) | |||
| (57) | |||
| Calc + Pt after catalysis | −100 ppm (11) | −109 ppm (33) | −113 ppm (56) |
| Ge-MFI 34 | −110 ppm | −113 ppm | |
| −116 ppm | |||
| ITQ-40 36 | −90 – | −105 – | |
| −105 ppm | −120 ppm | ||
FT-IR spectroscopy
The acid properties of as-made + Pt UTL zeolite before and after 3 runs of catalysis were studied by pyridine adsorption monitored by IR spectroscopy (Fig. 7 and 8). Despite the low content of silanol groups as indicated by the Q3 content based on 29Si MAS NMR (Fig. 6), FT-IR of the calcined and then outgassed (400 °C) parent germanosilicate UTL zeolite showed clear signals in the hydroxyl region of Si next to germanol species. According to the literature two intense bands in the stretching OH region (Fig. 7) at 3720 and 3649 cm−1 were assigned to the presence of terminal Si–OH and Ge–OH groups, respectively.34 The temperature dependent desorption profiles of pyridine on calcined UTL zeolite indicated the presence of Brønsted acid centers (IR bands at 1543 and 1636 cm–1) and Lewis acid sites (IR bands at 1454 and 1620 cm–1)10,29 (Fig. 8). A band at 1610 cm–1 was attributed to pyridine coordinated on Ge species which acted as weak Lewis acid sites.10,37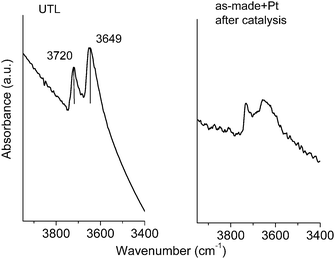 | ||
| Fig. 7 FTIR spectra in the hydroxyl stretching region of parent calcined germanosilicate UTL zeolite and as-made + Pt sample after catalysis and evacuation at 400 °C under vacuum. | ||
 | ||
| Fig. 8 FTIR spectra of pyridine adsorbed at 150 °C on parent germanosilicate UTL zeolite calcined in situ in the FTIR spectrometer and on as-made + Pt sample after catalysis. | ||
The concentration of pyridine adsorbed on acid sites was 0.33 Brønsted (B) and 0.15 Lewis (L) acid sites per unit cell of UTL sample after pyridine desorption at 150 °C.
The inherent acidity of germanosilicate UTL is lower compared to aluminosilicates, but sufficient for hydroisomerization and hydrocracking of alkanes. After 3 runs of catalytic testing the UTL as-made + Pt sample showed significantly lower Brønsted acidity (0.04 acid sites per unit cell) but no changes in Lewis acidity (0.16 acid sites per unit cell) according to pyridine adsorption at 150 °C. Closer inspection revealed small bands and shoulders in the intensity of the IR spectrum after catalysis at 1472, 1460, 1421, 1409, 1399 and 1385 cm−1. Literature assigns these bands to aliphatic C–H bands in coke.38 The presence of small amounts of coke also was confirmed by TG and may very well have caused the observed decrease in acidity and activity after three consecutive catalytic runs.
Decane hydroconversion experiments
In the catalytic experiment the feed composition and contact time were kept constant and the reaction temperature increased stepwise (Fig. 9). The conversion of n-decane started about 140 °C on calc + Pt, 186 °C on as-made + Pt and 197 °C on calc + Pt/silica. This is the temperature range of the onset of catalytic activity on reference ultrastable Y zeolites CBV-712 and CBV-760, with Si/Al ratios of 5.8 and 30, respectively, which are used as industrial hydrocracking catalysts.23,24 On the as-made + Pt and ultrastable Y zeolites, the activity increased much more rapidly with temperature compared to calc + Pt and calc + Pt/silica. This indicated that the apparent activation energies on the latter were much lower, or else that the catalyst degraded during catalytic testing. Especially calc + Pt suffered from low apparent activation energy barriers. On a well balanced bifunctional catalyst the apparent activation energy is high, reflecting the intrinsic reactions of carbocations and the increasingly favorable alkane dehydrogenation with rising temperature. | ||
| Fig. 9 Decane conversion vs. temperature on the samples CBV-712 (■), CBV-760 (●), as-made + Pt (○), calc + Pt (▲) and calc + Pt/silica (◆). | ||
Low apparent activation energies can be caused by many effects, such as intragranular or extragranular diffusion limitation, imperfect balance of catalytic functions, but a more likely interpretation in this instance is catalyst deactivation by loss of acid sites. The as-made + Pt catalyst was less active than the USY zeolites but showed a similar steep increase of n-decane conversion with increasing reaction temperature.
The promising results for as-made + Pt catalyst prompted a study of its long-term stability and re-usability. Three consecutive runs of n-decane test at increasing temperatures have been carried out to monitor its activity. After the first run of catalysis the sample was cooled down inside the reactor unit and reaction was started again under the same conditions. This procedure was repeated a third time. At no time the material was exposed to ambient. The results given in Fig. 10 showed that the catalyst can be reused and that activity and isomerization yield are only slightly lower in the second and third run. The maximum n-decane skeletal isomerization yields were 42% (1st run), 29% (2nd run) and 24% (3rd run) (Fig. 10).
 | ||
| Fig. 10 n-Decane conversion vs. temperature and yield of skeletal isomerization in the conversion of n-decane on the as-made + Pt catalyst. | ||
Taking into account the decreased Brønsted acidity on as-made + Pt sample after one catalysis run already discussed the shift of the conversion curve to higher temperatures in the second and third run likely is due to the observed decrease of acidity. This may have been caused at least in part by carbon residues accumulating in the framework as detected by FT-IR and TG.
The maximum yield of mono- and dibranched isomers was about 42% on as-made + Pt catalyst (Fig. 11), compared to ca. 60% on ultrastable Y zeolites (data not shown). A high isomerization yield as observed for as-made + Pt is typical for bifunctional catalysts with well balanced acid and metal function.
 | ||
| Fig. 11 Yield of total isomers (■) and cracking products (●) in the conversion of decane against temperature with platinum loaded catalysts. | ||
At high conversion hydrocracking predominated. The sequence of isomerization succeeded by cracking can generally be explained by the increasing sensitivity to cracking of increasingly branched isomers, which are formed with rising conversion. Some skeletal isomerization of n-decane to branched isoalkanes was also obtained at low conversion on calc + Pt/silica sample (ca. 10%). On calc + Pt the n-decane isomerization yield was very small (ca. 3%) and cracking was the prevailing reaction. According to NMR and XRD the calc + Pt underwent structural degradation in the reactor. The low isomerization yield indicated serious imbalance of the acid function on the zeolite and metal function generated by Pt impregnation or by mixing with Pt/silica.
The close to ideal bifunctional behavior of as-made + Pt catalyst enabled a detailed analysis of the skeletal isomers and assessment of shape selectivity. Several independent criteria serve for the characterization of the intracrystalline void space of unknown zeolites.20,23 The relative contribution of ethyloctane isomers (EC8) to monobranched isomers at 5% isomerization conversion is considered to be very sensitive to the diameter of the accessible channels. Diffusion and formation of ethyloctane molecules (EC8) with sizes around 5–6 × 9 Å are sterically hindered in small pores with diameters below 6 Å. These isomers are formed in small amounts on 10-MR zeolites. Among 12-MR zeolites the highest EC8 amount appears on FAU and MAZ topologies 12.5 and 13.2% (Table 2) respectively.39 The amount of EC8 formed on as-made + Pt UTL zeolite (15.9%) at 5% isomerization yield clearly exceeded EC8 for known large and medium pore zeolite structures (Fig. 12). The calc + Pt and calc + Pt/silica UTL catalysts did not strictly qualify for the test because of their low isomerization yield. However, at the lower isomerization yield of 3%, the ethyloctane content of the monobranched isomers was ca. 16% on calc + Pt and reached even 19.7% on calc + Pt/silica.
 | ||
| Fig. 12 Content of ethyloctane (EC8) against methylnonane (MC9) isomers in monobranched isodecane reaction product fraction on as-made + Pt sample, obtained at 5% n-decane isomerization conversion. | ||
Another criterion, the ratio of 3-ethyl to 4-ethyloctane (3E/4EC8) formation at 5% n-decane isomerization confirmed the observed trends. Compared with all other structures, as-made + Pt catalyst showed the 3E/4EC8 value around 0.50 (Table 2) close to the internal thermodynamic equilibrium composition as similarly also observed for the FAU topology presenting the widest pores among the 12-MR zeolites.23
The yield of hydrocracked products according to carbon numbers at 35% cracking conversion is reported in Fig. 13. As-made + Pt showed symmetrical distribution of the hydrocracked products with negligible amounts of C1, C2 as well as C8 and C9. This suggests that the predominating reaction mechanism is bifunctional and that hydrogenolysis, namely hydrocracking at the noble metal leading predominantly to methane, ethane and complimentary fragments, is minimal.23 The domination of branched isomers in the C4–C5–C6 cracked product fractions indicated that hydrocracking started from highly branched isodecane intermediates and reflected the large space available in the channels of the 2-D extra-large pore UTL structure. The asymmetric shape of the hydrocracked products distribution obtained with calc + Pt catalyst might be explained by occurrence of a contribution of secondary cracking, while the high content of C1 on calc + Pt/silica may be caused by hydrogenolysis over the platinum metal in the absence of sufficient acidity to balance the metal function.23
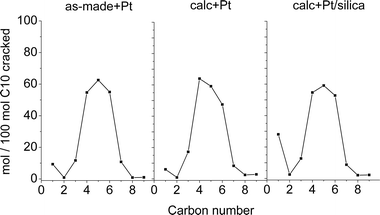 | ||
| Fig. 13 Molar yields of cracked products according to carbon number at 35% hydrocracking conversion of decane. | ||
Central cracking of the C10 feed molecule into C5 fragments was the main cracking pathway (Fig. 13). The molar yield of isopentane formed from 100 moles of C10 cracked (iC5 mol/100 mol) at 35% cracking conversion was 44.2 and 40.5 for as-made + Pt and calc + Pt/silica (Table 2) which is high and typical for a large pore zeolite. Nevertheless, this isopentane yield is lower than typically obtained with Y type zeolites, for which 50–56 mol/100 mol n-decane cracked are obtained. High isopentane yields point at cracking of tribranched isomers.40 On the UTL zeolite apparently the skeletal isomers reach a lower degree of branching before undergoing cracking. The lower iC5 yield on calc + Pt/silica sample compared to as-made + Pt is ascribed to hydrogenolysis on Pt (Table 2). The slightly decreasing iC5 yield observed in consecutive runs of as-made + Pt most probably is the result of a gradual loss of acid sites, partly due to deposition of coke residues. Hydrogenolysis may take over part of the cracking function when acidity is weakening.
Conclusions
Our results showed that preparation of bifunctional catalysts by classical incipient wetness impregnation of Pt on calcined zeolite is not suitable for germanosilicate UTL zeolite. High reaction temperatures and wet conditions of sample preparation give rise to structure collapse. Germanium is dislodged from the structure as was confirmed by NMR. Partial loss of crystallinity, acidity and catalytic activity are the consequence.To avoid contact with the aqueous solution during Pt impregnation we used mechanical mixing of calcined UTL zeolite with Pt dispersed on amorphous silica. We obtained slightly higher yields of isomerisation but the sample still barely qualified for bifunctional catalysis and showed poor stability during catalytic testing.
Our last approach was to use an as-made germanosilicate UTL zeolite sample for impregnation with precursor solution for the noble metal function. Our previous results showed that as-made samples were stable during hydrothermal treatment while calcined samples inevitably collapsed, even in the presence of only small amounts of water such as present in the atmosphere. Due to this observation we used the incipient wetness impregnation of Pt on an uncalcined sample, still containing the template in the pores. Because of the addition of platinum the organic template molecules catalytically are oxidized during catalyst pretreatment at 400 °C under flowing oxygen. The results showed that this UTL zeolite sample presented sufficient acidity for bifunctional catalysis and was stable even after 3 runs of testing. This discovery clearly indicates that to preserve the structure and activity of Ge-containing extra-large pore zeolites the inside of the pores must not be exposed to even traces of water, once the template is removed. The combination of its bi-directional channel system (with pores formed by 14- and 12-MR) together with extra-large pores (7.1 × 9.5 Å) makes UTL special in n-decane hydroconversion, widening the spectrum of accessible pores and allowing to extend the trend of increasing ethyloctanevs.methylnonane selectivity with increasing pore width. These observations also confirmed the n-decane test as a reliable tool to also assess topologies of extra-large pore zeolites.
Acknowledgements
N.K. acknowledges the BELSPO for the Research Fellowship. C.E.A.K. and J.A.M. acknowledge financial support by ESA and the Belgian Prodex office. The work was supported by the Belgian government through the IAP–PAI network and by the Flemish government through long term structural funding to J.A.M. (Methusalem). The support of Dr E. Gobechiya (capillar XRD measurements) and Els Verraedt (N2 adsorption) is gratefully acknowledged.Notes and references
- W. Vermeiren and J.-P. Gilson, Top. Catal., 2009, 52, 1131 CrossRef CAS.
- J. Sun, C. Bonneau, Á. Cantín, A. Corma, M. J. Díaz-Cabañas, M. Moliner, D. Zhang, M. Li and X. Zou, Nature, 2009, 458, 1154 CrossRef CAS.
- Y. Lorgouilloux, M. Dodin, J.-L. Paillaud, P. Caullet, L. Michelin, L. Josien, O. Ersen and N. Bats, J. Solid State Chem., 2009, 182, 622 CrossRef CAS.
- M. Dodin, J.-L. Paillaud, Y. Lorgouilloux, P. Caullet, E. Elkam and N. Bats, J. Am. Chem. Soc., 2010, 132, 10221 CrossRef CAS.
- J. Jiang, J. L. Jorda, M. J. Diaz-Cabanas, J. Yu and A. Corma, Angew. Chem., 2010, 122, 5106 CrossRef.
- J. Jiang, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120 CrossRef CAS.
- A. Corma, M. J. Díaz-Cabañas, F. Rey, S. Nicolopoulus and Kh. Boulahya, Chem. Commun., 2004, 1356 RSC.
- J.-L. Paillaud, B. Harbuzaru, J. Patarin and N. Bats, Science, 2004, 304, 990 CrossRef.
- L. Kanga, W. Dengb, T. Zhangc, Z. Liuc and K.-L. Han, Microporous Mesoporous Mater., 2008, 115, 261 CrossRef.
- M. Moliner, M. J. Díaz-Cabañas, V. Fornés, C. Martínez and A. Corma, J. Catal., 2008, 254, 101 CrossRef CAS.
- G. Sastre and A. Korma, J. Phys. Chem. C, 2010, 114, 1667 CrossRef CAS.
- F. Gao, M. Jaber, K. Bozhilov, A. Vicente, C. Fernandez and V. Valtchev, J. Am. Chem. Soc., 2009, 131, 16580 CrossRef CAS.
- N. Kasian, T. I. Koranyi, G. Vanbutsele, K. Houthoofd, J. A. Martens and C. E. A. Kirschhock, Top. Catal., 2010, 53, 1374 CrossRef CAS.
- L. G. A. van de Water, J. C. van der Waal, J. C. Jansen, M. Cadoni, L. Marchese and T. Maschmeyer, J. Phys. Chem. B, 2003, 107, 10423 CrossRef CAS.
- L. G. A. van de Water, J. C. van der Waal, J. C. Jansen and T. Maschmeyer, J. Catal., 2004, 223, 170 CrossRef CAS.
- V. A. Tuan, J. L. Falconer and R. D. Noble, Microporous Mesoporous Mater., 2000, 41, 269 CrossRef CAS.
- R. Szostak, Molecular Sieves, van Nostrand Reinhold, New York, 1989 Search PubMed.
- M. K. Faraj, U.S. Patent, 5,977,009, 1999 Search PubMed.
- C. M. Zicovich-Wilson and A. Corma, J. Phys. Chem. B, 2000, 104, 4134 CrossRef CAS.
- J. A. Martens and P. A. Jacobs, Zeolites, 1986, 6, 334 CrossRef CAS.
- A. Corma, C. Corell, F. Llopis, A. Martinez and J. Perez-Pariente, Appl. Catal., A, 1994, 115, 121 CrossRef CAS.
- A. Corma, A. Chica, J. M. Guil, F. J. Llopis, G. Mabilon, J. A. Perdigón-Melón and S. Valencia, J. Catal., 2000, 189, 382 CrossRef CAS.
- J. A. Martens, M. Tielen, P. A. Jacobs and J. Weitkamp, Zeolites, 1984, 4, 98 CAS.
- J. Weitkamp, Erdoel Kohle, Erdgas, Petrochem., 1978, 31, 13 Search PubMed.
- S. Tontisirin and S. Ernst, Angew. Chem., Int. Ed., 2007, 46, 7304 CrossRef CAS.
- O. V. Shvets, A. Zukal, N. Kasian, N. Žilková and J. Čejka, Chem.–Eur. J., 2008, 14, 10134 CrossRef CAS.
- O. V. Shvets, N. Kasian, A. Zukal, J. Pinkas and J. Čejka, Chem. Mater., 2010, 22, 3482 CrossRef CAS.
- Electronic supplementary information http://www.rsc.org/suppdata/cc/b4/b406572g.
- C. A. Emeis, J. Catal., 1993, 141, 347 CrossRef CAS.
- W. Huybrechts, J. Mijoin, P. A. Jacobs and J. A. Martens, Appl. Catal., A, 2003, 243, 1 CrossRef CAS.
- S. Bhatia, Zeolite Catalysts: Principles and Applications, CRC Press, Boca Raton, 1990 Search PubMed.
- B. C. Gagea, Y. Lorgouilloux, Y. Altintas, P. A. Jacobs and J. A. Martens, J. Catal., 2009, 265, 99 CrossRef CAS.
- R. W. Grosse-Kunstleve, L. B. McCusker and Ch. Baerlocher, J. Appl. Crystallogr., 1999, 32, 536 CrossRef CAS.
- H. Kosslick, V. A. Tuan, R. Fricke, C. Peuker, W. Pilz and W. Store, J. Phys. Chem., 1993, 97, 5678 CrossRef CAS.
- T. Blasco, A. Corma, M. J. Díaz-Cabañas, F. Rey, J. A. Vidal-Moya and C. M. Zicovich-Wilsonm, J. Phys. Chem. B, 2002, 106, 2634 CrossRef CAS.
- A. Corma, M. J. Díaz-Cabañas, J. Jiang, M. Afeworki, D. L. Dorset, S. L. Soled and K. G. Strohmaier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13997 CrossRef CAS.
- S. Leiva, M. J. Sabater, S. Valencia, G. Sastre, V. Fornes, F. Rey and A. Corma, C. R. Chim., 2005, 8, 369 CrossRef CAS.
- J. V. Ibarra, C. Royo, A. Monzon and J. Santamariamm, Vib. Spectrosc., 1995, 9, 191 CrossRef CAS.
- J. A. Martens, E. Benazzi, J. Brendlé, S. Lacombe and R. Le Dred, Stud. Surf. Sci. Catal., 2000, 130, 293 CrossRef.
- J. A. Martens, P. A. Jacobs and J. Weitkamp, Appl. Catal., 1986, 20, 239 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |