A straightforward zinc-catalysed reduction of sulfoxides to sulfides†
Stephan
Enthaler
*
Technische Universität Berlin, Department of Chemistry, Cluster of Excellence “Unifying Concepts in Catalysis”, Straße des 17. Juni 135, D-10623 Berlin, Germany. E-mail: stephan.enthaler@tu.berlin.de; Fax: +49 30314 29732; Tel: +49 30314 22039
First published on 8th February 2011
Abstract
In the present study, the zinc-catalysed reduction of a variety of sulfoxides with silanes as reductant to the corresponding sulfide has been examined in detail. With the straightforward and commercially available zinc(II) triflate as pre-catalyst, excellent yields and chemoselectivities were feasible. After studying the reaction conditions and the scope and limitations several attempts were undertaken to shed light on the reaction mechanism.
Introduction
The development of sustainable, efficient and selective procedures to access organic compounds with higher value is one of the fundamental research goals in modern chemistry.1 In this regard, excellent performances have been exhibited by application of homogeneous metal-based catalysts.2 Obviously, most of the applied metals displayed difficulties by their high price and toxicity. Hence, today's research is focusing on the replacement by cheaper and low toxic metals.3 Here, the use of zinc is of great interest, due to abundance and biological relevance. Among the established strategies, reduction processes have been intensively investigated and are demonstrating excellent yields and remarkable selectivities (e.g.reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN and amides).4 However, the reduction of sulfoxides to sulfides in the presence of homogeneous zinc-catalysts has been so far unreported.5,6 The biological relevance of the deoxygenation of sulfoxides has been discussed with respect of avoidance of oxidative damage of cells.7 Furthermore, the importance of sulfides for organic synthesis are evident.8 Hence, new protocols are highly desired. Herein, we portray the successful application of an easy-to-adopt catalyst based on zinc for the homogeneous reduction of a variety of aromatic and aliphatic sulfoxides to access sulfides.
O, CN and amides).4 However, the reduction of sulfoxides to sulfides in the presence of homogeneous zinc-catalysts has been so far unreported.5,6 The biological relevance of the deoxygenation of sulfoxides has been discussed with respect of avoidance of oxidative damage of cells.7 Furthermore, the importance of sulfides for organic synthesis are evident.8 Hence, new protocols are highly desired. Herein, we portray the successful application of an easy-to-adopt catalyst based on zinc for the homogeneous reduction of a variety of aromatic and aliphatic sulfoxides to access sulfides.
Results and discussion
Initially, the reduction of p-tolyl sulfoxide (1) with silanes in toluene was studied as a model reaction to explore appropriate conditions and to examine the influence of various reaction parameters (Table 1). As expected when applying silanes (PhSiH3 or Et3SiH) in the absence of zinc sources only marginal amounts of p-tolyl sulfide (1a) after 12 h were monitored (Table 1, entry 1). However, when performing the reduction with 5.0 mol% of commercially available Zn(OTf)2 and phenylsilane as reducing reagent, an excellent yield (>99%) and chemoselectivity (>99%) was realized after 12 h under non-inert conditions (Table 1, entry 2). In addition, the effect of various silanes on the product formation was explored. Increasing the number of phenyl substituent at the silicon resulted in a decreased yield of product 1a, while an increased number of alkyl substituent led to an excellent performance (Table 1, entry 6). Next other zinc sources, such as ZnF2, ZnCl2, ZnBr2, Zn(BF4)2, Zn(OAc)2, Zn(acac)2, were tested in combination with triethylsilane. Best performance was still observed for zinc(II) triflate. On the other hand, good results were detected for zinc(II) halides, apart from ZnF2 (Table 1, entries 10–12). An increased yield was monitored for heavier halides (ZnF2 < ZnCl2 < ZnBr2). Moreover, with other zinc-based Lewis acids no product at all was attained. Besides, the loading of Zn(OTf)2 was decreased to 1.0 mol%, resulting in a diminish of product 1a (20%) even after elongation of the reaction time (20 h). Additionally, the influence of the reaction temperature was studied. Here, the amount of p-tolyl sulfide (1a) is decreased at lower temperature (80 °C: yield 20%), while the reaction in the range of (25–60 °C) is hampered. Besides toluene as solvent excellent results were obtained applying THF (Table 1, entry 18), while the reaction in PhCN and PhCOOMe proceeded smoothly (Table 1, entries 20 and 21). It is worth noting that no reduction of the solvent was observed. In addition, the zinc(II) triflate was modified by nitrogen containing ligands. However, the addition of tmeda [N,N,N′,N′-tetramethylethylenediamine], 2,2′-bipyridine, and imidazole resulted in no improvement of the reaction outcome (yields: <5%).
| Entry | Zn-source | Silane (equiv.) | Solvent | Yield (%)b |
|---|---|---|---|---|
| a Reaction conditions: 1 (0.72 mmol), Zn-source (5 mol%), silane (1.5–2.0 equiv), solvent (2.0 mL), 100 °C, 12 h. b Determined by GC methods using biphenyl as an internal standard. c Run for 24 h. d Run for 1 h at 100 °C with 1.0 mol% Zn(OTf)2. e Run for 24 h at r.t. f Run for 24 h at 60 °C. g Run for 12 h at 60 °C in 0.6 mL THF-d8. h Up-scaling: 1 (2.4 mmol), solvent (6.0 mL). | ||||
| 1c | — | PhSiH3 (1.5) | Toluene | <3 |
| 2 | Zn(OTf)2 | PhSiH3 (1.5) | Toluene | >99 |
| 3 | Zn(OTf)2 | Ph2SiH2 (1.5) | Toluene | 50 |
| 4 | Zn(OTf)2 | Ph3SiH (2.0) | Toluene | 25 |
| 5 | Zn(OTf)2 | PhMe2SiH (2.0) | Toluene | 42 |
| 6 | Zn(OTf)2 | Et3SiH (2.0) | Toluene | >99 |
| 7 | Zn(OTf)2 | (EtO)3SiH (2.0) | Toluene | 51 |
| 8 | Zn(OTf)2 | PMHS (2.0) | Toluene | 13 |
| 9 | Zn(OAc)2 | Et3SiH (2.0) | Toluene | <1 |
| 10 | ZnF2 | Et3SiH (2.0) | Toluene | <1 |
| 11 | ZnCl2 | Et3SiH (2.0) | Toluene | 41 |
| 12 | ZnBr2 | Et3SiH (2.0) | Toluene | 75 |
| 13 | Zn(BF4)2 | Et3SiH (2.0) | Toluene | <1 |
| 14 | Zn(acac)2 | Et3SiH (2.0) | Toluene | 3 |
| 15d | Zn(OTf)2 | Et3SiH (2.0) | Toluene | 20 |
| 16e | Zn(OTf)2 | Et3SiH (2.0) | Toluene | <1 |
| 17f | Zn(OTf)2 | Et3SiH (2.0) | Toluene | 20 |
| 18g | Zn(OTf)2 | Et3SiH (2.0) | THF-d8 | >99 |
| 19h | Zn(OTf)2 | Et3SiH (2.0) | Toluene | >99 |
| 20 | Zn(OTf)2 | Et3SiH (2.0) | PhCN | 63 |
| 21 | Zn(OTf)2 | Et3SiH (2.0) | PhCOOMe | 53 |
Once the optimized reaction conditions were established, the scope and limitations of the zinc-catalysed deoxygenation of sulfoxides applying Et3SiH or PhSiH3 as reducing reagent were investigated. A number of sulfoxides, including aromatic and aliphatic sulfoxides were reduced to the corresponding sulfides (Table 2). In general, a better performance was detected for PhSiH3 compared to Et3SiH. In most cases excellent yields and selectivities were obtained after 24 h at 100 °C. In case of 9 and 19 no product was observed probably due to low solubility of the starting materials. In order to study the selectivity of the process different substrates containing functional groups sensitive to reduction or a combination of the sulfoxide 1 with an additional substrate were reacted with phenylsilane in the presence of zinc(II) triflate (Table 2, entries 6–8, 12, 20 and Table 3). Excellent selectivity (>99%) for the reduction of SO bond was observed in the presence of NO2, CN, esters, and sulfonyl, while alkynyl, alkenyl, CO and amide groups were reduced. Noteworthy, if the functional group (keto- and alkenyl group, Table 2, entries 12 and 20) is closely connected to the sulfoxide to some extend reduction took place, probably due to the neighbourhood to the active centre, while in case of separation of the two functional groups (Table 3, entries 2 and 3) resulted in selective reduction of the sulfoxide to the corresponding sulfide.
| Entry | Substrate | Yield (%)b |
|---|---|---|
| a Reaction conditions: sulfoxide (0.72 mmol), Zn(OTf)2 (5 mol%), silane (1.1 mmol PhSiH3), toluene (2.0 mL), 24 h, 100° C. b Determined by GC–MS and 1H NMR. In parenthesis the isolated yield is stated. c Et3SiH. d As main product PhSEt was observed. e Only traces of product were detected, probably due to poor solubility. Reaction was also carried out PhCN and PhCOOMe with similar results. f As side reaction the reduction of the carbonyl group was observed. g Due to the odour and the volatility the reaction was carried out in C6D6 and the products were not isolated. h Full conversion with respect to the sulfoxide was detected, while 15% are reduced to the corresponding alkene and 9% are reduced to the alkane. | ||
| 1c |

|
1a: >99 (91) |
| 2 |

|
2a: >99 (96) |
| 3 |

|
3a: >99 (93) |
| 4 |

|
4a: 78 (69) |
| 5 |
|
5a: >99 (92) |
| 6 |

|
6a: >99 (95) |
| 7 |

|
7a: >99 (95) |
| 8 |

|
8a: >99d |
| 9 |

|
9a: <1e |
| 10 |

|
10a: >99 (95) |
| 11 |

|
11a: >99 (91) |
| 12 |

|
12a: 87f |
| 13g |

|
13a: 94 |
| 14g |

|
14a: 57 |
| 15g |

|
15a: 40 |
| 16g |
|
16a: >99 |
| 17g |

|
17a: >99 |
| 18g |

|
18a: >99 |
| 19 |

|
19a: <1e |
| 20 |

|
20a: 76h |







For preliminary mechanistic investigations, the reduction of 1 with PhSiH3 was performed in the presence of stoichiometric amounts of the persistent radical TEMPO (2,2,6,6-tetramethylpiperidinyloxyl) as a scavenger. However, no negative effect was observed on the yield of product 1a, hence a radical based mechanism could be excluded.9 Applying 1H NMR spectroscopy on the reduction of 1 to 1a with Et3SiH in C6D6 and using the para-methyl group as probe only the decrease of 1 and the increase of 1a were monitored, while no intermediates were observed on the NMR time scale.
In addition, with 29Si NMR spectroscopy (Et3Si)2O (δ = 9.2 ppm) was found as main product. We exclude the formation of (Et3Si)2Ovia condensation of Et3SiH and Et3SiOH, which could be formed by elimination from R2S+(H−)OSiEt3, because no Et3SiOH was monitored by 29Si NMR during the course of reaction. Aside, the condensation reaction was separately studied in the presence and absence of catalytic amounts of Zn(OTf)2.10 Here, no formation of R3SiOSiR3 was observed. However, it was detected that two equivalents of hydride per sulfoxide are required for the reduction to the sulfide.
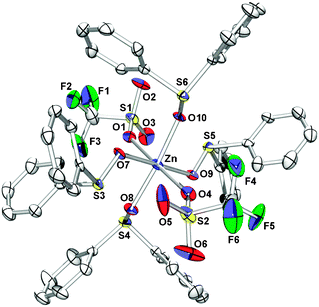
Afterwards, the interaction of Zn(OTf)2 with the silane was studied. Zn(OTf)2 was reacted with Et3SiH (10 equiv.) in THF-d8 for 1 h at 80 °C. The signal for Et3SiH was unchanged in 1H NMR as well as 29Si NMR. Only a slight shift in the 19F NMR spectra was observed, −80.7 ppm (Zn(OTf)2, Et3SiH in THF-d8)11 which is slightly shifted compared to Zn(OTf)2(THF)4 (δ = −79.1 ppm) probably caused by solvation or complexation.4r,12 Based on this result, the formation of a zinc-hydride species can be probably excluded on the NMR time scale. On the other hand the interaction of zinc with sulfoxide was investigated. A solution of Zn(OTf)2 in C6D6 was reacted first with methyl phenyl sulfoxide (5) for 1 h at 60 °C. The recorded 19F NMR spectra showed a signal at −80.8 ppm. Indeed, after one hour at room temperature colourless crystals, which were suitable for single-crystal X-ray diffraction analysis, were collected from this solution. The obtained solid structure Zn(5)6(OTf)2 (29) showed an octahedral complexation of six sulfoxides molecules to the cationic zinc centre (Fig. 1).13 Under same conditions the reaction of Zn(OTf)2 with compound 2 (10 equiv.) led to the formation of complex Zn(2)4(OTf)2 (30) (Fig. 2). In comparison to complex 29 four sulfoxides are coordinated to the metal centre. In addition the triflate anions are attached to the metal, while in 29 the triflate anions are not fixed to the metal.
 | ||
| Fig. 1 Molecular structure of Zn(5)6(OTf)2 (29). Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected distances (Å): Zn–O(1,2,3,4,5,6): 2.0817(14), O–S: 1.4265(18). | ||
 | ||
| Fig. 2 Molecular structure of Zn(2)4(OTf)2 (30). Hydrogen atoms and a C6D6 solvent molecule are omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected distances (Å): Zn–O(7,8,9,10): 2.041–2.067(3), Zn–O(1,4): 2.124–2.223(3), (S(3,4,5,6)–O(3,4,5,6): 1.517–1.522(3). | ||
Later on, after isolation of the crystals of complex 29PhSiH3 (10 equiv.) was added at room temperature. Here, no significant change in the chemical shift was observed (19F NMRδ = 80.8 ppm), while a shift to 80.3 ppm was found after heating for 1 h at 60 °C. In the 1H NMR spectra the formation of the corresponding sulfide 5a was monitored. Continuing heating for additional 23 h exhibited a shift towards −79.0 ppm, while in the 1H NMR nearly full conversion of 5 was detected. The shift in the 19F NMR spectra is probably caused by complexation of the corresponding sulfide 5a. Noteworthy, the reaction of the sulfoxide with the silane in the absence of Zn(OTf)2 resulted in no change of the 1H NMR or 29Si NMR spectra. With respect to the obtained results we excluded the formation of LxZnO species, while for various metals the abstraction of the oxide of the sulfoxide to yield metal oxides has been proposed.14 Based on our findings we assumed that the zinc-catalyst acts as a Lewis acid (Scheme 1). First the sulfoxide coordinates to the zinc (intermediate A), thus an activation of the SO bond occurs and increases the susceptibility of the sulfur for reduction.6a,15 Next, the silane reacts with the intermediate A. An interaction of the hydride with the sulfur and an interaction of the silicon with the oxygen of the triflate groupvia a six-membered transition state are assumed (intermediate B). Subsequently, a sulfonium salt analogue is formed (intermediate C), which undergoes elimination of (R3Si)2O, hydrogen and the sulfide.16 Similar processes are assumed for the reduction of sulfoxides with boranes as reductant.5q However, the precise mechanism is currently uncertain and will be the subject of ongoing studies.
 | ||
| Scheme 1 Proposed catalytic cycle for the reduction of sulfoxides. | ||
Conclusions
In summary, we have set up for the first time an efficient protocol for the reduction of sulfoxides to the corresponding sulfide with silanes as hydride source in the presence of straightforward zinc-salts under mild reaction conditions. Furthermore, mechanistic investigations indicated, that Zn(II) triflate acts a Lewis acid catalyst. Future studies will focus on the development of ligand modified zinc catalysts for improvement of the catalyst performance and chemoselectivity.Experimental
General
All compounds were used as received without further purification. THF and toluene were dried applying standard procedures. 1H, 19F and 13C NMR spectra were recorded on a Bruker AFM 200 spectrometer (1H: 200.13 MHz; 13C: 50.32 MHz; 19F: 188.31 MHz) using the proton signals of the deuterated solvents as reference. Single-crystal X-ray diffraction measurements were recorded on an Oxford Diffraction Xcalibur S Saphire spectrometer. GC–MS measurements were carried out on a Shimadzu GC-2010 gas chromatograph (30 m Rxi-5ms column) linked with a Shimadzu GCMA-QP 2010 Plus mass spectrometer.General procedure for the deoxygenation of sulfoxides
A pressure tube was charged with an appropriate amount of Zn(OTf)2 (0.036 mmol, 5.0 mol%) and the corresponding sulfoxide (0.72 mmol) and PhSiH3 (1.1 mmol) was added. After addition of toluene (2.0 mL) the reaction mixture was stirred in a pre-heated oil bath at 100 °C for 24 h. The mixture was cooled on an ice bath and biphenyl (internal standard) was added. The solution was diluted with dichloromethane and an aliquot was taken for GC-analysis (30 m Rxi-5ms column, 40–300 °C). The solvent was removed and the residue was purified by column chromatography. The analytical properties of the corresponding sulfides are in agreement with literature.15,17Di(p-methylphenyl)sulfide (1a)17
Column chromatography (n-hexane–ethyl acetate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5). Rf 0.79. 1H NMR (CDCl3, 200 MHz) δ = 7.06–7.22 (m, 8 H), 2.30 (6 H, CH3) ppm; 13C NMR (CDCl3, 50 MHz) δ = 137.1, 132.8, 131.3, 130.1, 21.3 ppm; MS (ESI) m/z = 214 (100, M+), 199 (34), 184 (20), 105 (18), 91 (33), 65 (19).
:5). Rf 0.79. 1H NMR (CDCl3, 200 MHz) δ = 7.06–7.22 (m, 8 H), 2.30 (6 H, CH3) ppm; 13C NMR (CDCl3, 50 MHz) δ = 137.1, 132.8, 131.3, 130.1, 21.3 ppm; MS (ESI) m/z = 214 (100, M+), 199 (34), 184 (20), 105 (18), 91 (33), 65 (19).
Diphenylsulfide (2a)17
Column chromatography (n-hexane–ethyl acetate 1:5). Rf 0.75. 1H NMR (CDCl3, 200 MHz) δ = 7.26–7.47 (m, 10 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 135.8, 131.0, 129.2, 127.0 ppm; MS (ESI) m/z = 186 (100, M+), 152 (11), 92 (19), 77 (27), 65 (20), 51 (43).
Di(p-chlorophenyl)sulfide (3a)15
Column chromatography (n-hexane–ethyl acetate 1:10). Rf 0.67. 1H NMR (CDCl3, 200 MHz) δ = 7.23–7.33 (m, 8 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 133.9, 133.4, 132.3, 129.4 ppm; MS (ESI) m/z = 270 (M+, not detected), 219 (33), 184 (100), 139 (11), 108 (29), 91 (18), 75 (24).
Phenyl 2-chloroethylsulfide (4a)15
Column chromatography (n-hexane–ethyl acetate 1:5). Rf 0.88. 1H NMR (CDCl3, 200 MHz) δ = 7.22–7.45 (m, 5 H), 3.59–3.68 (m, 2 H), 3.19–3.28 (m, 2 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 134.2, 130.4, 129.2, 127.1, 43.3, 36.1 ppm; MS (ESI) m/z = 172 (43, M+), 123 (100), 109 (21), 65 (17), 45 (31).
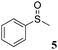
Phenyl methylsulfide (5a)15
Column chromatography (n-hexane–ethyl acetate 1:5). Rf 0.88. 1H NMR (CDCl3, 200 MHz) δ = 7.19–7.35 (m, 5 H), 2.53 (s, 3 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 138.3, 128.7, 126.5, 124.9, 15.7 ppm; MS (ESI) m/z = 124 (100, M+), 109 (51), 91 (49), 78 (51), 85 (21).
p-Cyanophenyl methylsulfide (6a)17
Column chromatography (acetone–CH2Cl2 1:9). Rf 0.90. 1H NMR (CDCl3, 200 MHz) δ = 7.48–7.53 (m, 2 H), 7.21–7.27 (m, 2 H), 2.49 (s, 3 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 146.0, 132.0, 125.3, 118.8, 107.4, 14.5 ppm; MS (ESI) m/z = 149 (100, M+), 134 (32), 116 (60), 104 (23).
p-Nitrophenyl methylsulfide (7a)17
Column chromatography (acetone–CH2Cl2 1:9). Rf 0.90. 1H NMR (CDCl3, 200 MHz) δ = 8.07–8.12 (m, 2 H), 7.24–7.29 (m, 2 H), 2.53 (s, 3 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 148.8, 144.5, 124.8, 123.7, 14.8 ppm; MS (ESI) m/z = 169 (100, M+), 139 (50), 123 (13), 111 (16), 108 (33), 82 (14), 77 (38).
Dibenzothiophene (10a)17
Column chromatography (n-hexane–ethyl acetate 1:5). Rf 0.78. 1H NMR (CDCl3, 200 MHz) δ = 8.16–8.21 (m, 2 H), 7.84–7.91 (m, 2 H), 7.44–7.53 (m, 4 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 139.0, 135.5, 126.8, 124.3, 122.8, 121.5 ppm; MS (ESI) m/z = 184 (100, M+), 152 (14), 139 (21), 92 (18), 79 (12).
Dibenzylsulfide (11a)17
Column chromatography (n-hexane–ethyl acetate 1:5). Rf 0.74. 1H NMR (CDCl3, 200 MHz) δ = 7.31–7.37 (m, 10 H), 3.65 (s, 4 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 138.0, 128.9, 128.4, 126.9, 35.5 ppm; MS (ESI) m/z = 214 (59, M+), 123 (31), 91 (100), 65 (17).
tert-Butyl methylsulfide (13a)
1H NMR (CDCl3, 200 MHz) δ = 3.18 (s, 3 H, CH3), 1.76 (s, 9 H, tBu) ppm; 13C NMR (CDCl3, 50 MHz) δ = 47.1, 29.9, 11.0 ppm; MS (ESI) m/z = 104 (36, M+), 57 (82), 41 (100).tert-Dibutylsulfide (14a)
1H NMR (CDCl3, 200 MHz) δ = 1.42 (s, 12 H, CH3) ppm; 13C NMR (CDCl3, 50 MHz) δ = 45.3, 33.2 ppm; MS (ESI) m/z = 146 (12, M+), 57 (100).Dibutylsulfide (15a)17
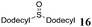
1H NMR (CDCl3, 200 MHz) δ = 2.28 (t, 4 H, J = 7.20 Hz, S(CH2CH2CH2CH3)2), 1.14–1.49 (m, 8 H, S(CH2CH2CH2CH3)2), 0.75 (t, 6 H, J = 7.20 Hz, CH3) ppm; 13C NMR (C6D6, 50 MHz) δ = 32.1, 32.0, 22.3, 13.8 ppm; MS (ESI) m/z = 146 (40, M+), 103 (15), 90 (31), 61 (97), 56 (100).Didodecylsulfide (16a)17
1H NMR (CDCl3, 200 MHz) δ = 0.47–2.80 (m, 52 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 38.0, 31.8, 29.5, 29.4, 29.3, 29.1, 28.7, 22.6, 22.4, 14.0 6.7, 6.3 ppm. MS (ESI) m/z = 370 (M+, not detected), 216 (58), 201 (100), 111 (20), 103 (23), 97 (38), 83 (48), 75 (11), 69 (60), 61 (58), 55 (60).Tetrahydrothiophene (17a)17
1H NMR (CDCl3, 200 MHz) δ = 2.42–2.55 (m, 4 H), 1.36–1.48 (m, 4 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 32.1, 32.0 ppm; MS (ESI) m/z = 87 (13, M+), 59(49), 43 (100).2,4-Dithiapentane (18a)17
1H NMR (CDCl3, 200 MHz) δ = 3.18 (s, 2 H), 1.76 (s, 6 H) ppm; 13C NMR (CDCl3, 50 MHz) δ = 40.2, 14.0 ppm; MS (ESI) m/z = 110 (14, M+), 108 (99), 61 (100).Procedure for the deoxygenation of sulfoxides in the presence of TEMPO
A pressure tube was charged with Zn(OTf)2 (0.036 mmol, 5.0 mol%), di(p-methylphenyl)sulfoxide (0.72 mmol), PhSiH3 (1.1 mmol) and TEMPO (0.72 mmol) in an atmosphere of nitrogen. After addition of dry toluene (2.0 mL) the reaction mixture was stirred in a preheated oil bath at 100 °C for 24 h. The mixture was cooled on an ice bath and dodecane (internal standard) was added. The solution was diluted with dichloromethane and an aliquot was taken for GC-analysis (30 m Rxi-5ms column, 40–300 °C).Single-crystal X-ray structure determination
A NMR tube was charged with Zn(OTf)2 (0.036 mmol, 5.0 mol%) and phenyl methylsulfoxide (0.72 mmol) or diphenylsulfoxide (0.72 mmol). After addition of benzene-d6 (0.6 mL) the mixture was heated at 60 °C for 1 h. Slow cooling to room temperature yielded colourless crystals suitable for single-crystal X-ray diffraction analysis. Crystals were each mounted on a glass capillary in perfluorinated oil and measured under a flow of nitrogen. The data was collected on an Oxford Diffraction Xcalibur S Sapphire at 150 K (Mo-Kα radiation, λ = 0.71073 Å). The structures were solved by direct methods and refined on F2 with the SHELX-9718 software package. The positions of the H atoms were calculated and considered isotropically according to a riding model. 29: crystal system: trigonal; space group: R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ; unit cell dimensions a = 12.5877(4) Å α = 90°, b = 12.5877(4) Å, β = 90°, c = 29.1803(11) Å, γ = 120°; volume: 4004.2(2) Å3; Z = 3; Dcalcd: 1.499 Mg m−3; μ: 0.850 mm−1; F(000): 1860; theta range for data collection: 3.24 to 24.99°; reflections collected: 5363; independent reflections: 1575 [Rint = 0.0315]; completeness to theta = 24.99°: 99.7%; refinement method: full-matrix least-squares on F2; goodness-of-fit on F2: 1.001; final R indices [I > 2σ(I)] R1 = 0.0301, wR2 = 0.0716; R indices (all data): R1 = 0.0423, wR2 = 0.0746; largest diffraction peak and hole 0.306 and −0.241 e Å−3. 30: crystal system: trigonal; space group: P
; unit cell dimensions a = 12.5877(4) Å α = 90°, b = 12.5877(4) Å, β = 90°, c = 29.1803(11) Å, γ = 120°; volume: 4004.2(2) Å3; Z = 3; Dcalcd: 1.499 Mg m−3; μ: 0.850 mm−1; F(000): 1860; theta range for data collection: 3.24 to 24.99°; reflections collected: 5363; independent reflections: 1575 [Rint = 0.0315]; completeness to theta = 24.99°: 99.7%; refinement method: full-matrix least-squares on F2; goodness-of-fit on F2: 1.001; final R indices [I > 2σ(I)] R1 = 0.0301, wR2 = 0.0716; R indices (all data): R1 = 0.0423, wR2 = 0.0746; largest diffraction peak and hole 0.306 and −0.241 e Å−3. 30: crystal system: trigonal; space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; unit cell dimensions a = 10.5817(4) Å; α = 83.554(3)°, b = 13.8939(5) Å, β = 88.085(3)°, c = 19.8576(3) Å, γ = 73.177(3)°; volume: 2775.99(18) Å3; Z = 2; Dcalcd: 1.496 Mg m−3; μ: 0.746 mm−1; F(000): 1284; theta range for data collection: 3.31 to 25.00°; reflections collected: 21815; independent reflections: 9791 [Rint = 0.0768]; completeness to theta = 25.00°: 99.7%; refinement method: full-matrix least-squares on F2; goodness-of-fit on F2: 0.826; final R indices [I > 2σ(I)] R1 = 0.0561, R2 = 0.1010; R indices (all data): R1 = 0.1276, wR2 = 0.0930; largest diffraction peak and hole 0.559 and −0.372 e Å−3.
; unit cell dimensions a = 10.5817(4) Å; α = 83.554(3)°, b = 13.8939(5) Å, β = 88.085(3)°, c = 19.8576(3) Å, γ = 73.177(3)°; volume: 2775.99(18) Å3; Z = 2; Dcalcd: 1.496 Mg m−3; μ: 0.746 mm−1; F(000): 1284; theta range for data collection: 3.31 to 25.00°; reflections collected: 21815; independent reflections: 9791 [Rint = 0.0768]; completeness to theta = 25.00°: 99.7%; refinement method: full-matrix least-squares on F2; goodness-of-fit on F2: 0.826; final R indices [I > 2σ(I)] R1 = 0.0561, R2 = 0.1010; R indices (all data): R1 = 0.1276, wR2 = 0.0930; largest diffraction peak and hole 0.559 and −0.372 e Å−3.
Acknowledgements
Financial support from the Cluster of Excellence “Unifying Concepts in Catalysis” (funded by the Deutsche Forschungsgemeinschaft and administered by the Technische Universität Berlin) is gratefully acknowledged. The author thanks Sebastian Krackl for technical support.Notes and references
- (a) P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS; (b) J. L. Tucker, Org. Process Res. Dev., 2010, 14, 328–331 Search PubMed.
- (a) M. Beller and C. Bolm, ed. Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed; (b) B. Cornils and W. A. Herrmann, Applied Homogeneous Catalysis with Organometallic Compounds, Wiley-VCH, Weinheim, 1996 Search PubMed; (c) A. Behr, Angewandte homogene Katalyse, Wiley-VCH, Weinheim, 2008 Search PubMed; (d) B. Cornils, W. A. Herrmann and I. T. Horvath, Multiphase Homogenous Catalysis, Wiley-VCH, Weinheim, 2005 Search PubMed.
- (a) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS; (b) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS; (c) B. Plietker, ed. Iron catalysis in organic chemistry, Wiley-VCH, Weinheim, 2008 Search PubMed; (d) A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC.
- (a) H. Mimoun, J. Y. De Saint Laumer, L. Giannini, R. Scopelliti and C. Floriani, J. Am. Chem. Soc., 1999, 121, 6158–6166 CrossRef CAS; (b) H. Mimoun, J. Org. Chem., 1999, 64, 2582–2589 CrossRef CAS; (c) V. Bette, A. Mortreux, C. W. Lehmann and J.-F. Carpentier, Chem. Commun., 2003, 332–333 RSC; (d) V. Bette, A. Mortreux, F. Ferioli, G. Martelli, D. Savoia and J.-F. Carpentier, Eur. J. Org. Chem., 2004, 3040–3045 CrossRef CAS; (e) V. Bette, A. Mortreux, D. Savoia and J.-F. Carpentier, Tetrahedron, 2004, 60, 2837–2842 CrossRef CAS; (f) V. M. Mastranzo, L. Quinterno, C. Anaya de Parrodi, E. Juaristi and P. J. Walsh, Tetrahedron, 2004, 60, 1781–1789 CrossRef CAS; (g) H. Ushio and K. Mikami, Tetrahedron Lett., 2005, 46, 2903–2906 CrossRef CAS; (h) V. Bette, A. Mortreux, D. Savoia and J.-F. Carpentier, Adv. Synth. Catal., 2005, 347, 289–302 CrossRef CAS; (i) S. Gérard, Y. Pressel and O. Riant, Tetrahedron: Asymmetry, 2005, 16, 1889–1891 CrossRef CAS; (j) B.-M. Park, S. Mun and J. Yun, Adv. Synth. Catal., 2006, 348, 1029–1032 CrossRef CAS; (k) M. Bandini, M. Melucci, F. Piccinelli, R. Sinisi, S. Tommasi and A. Umani-Ronchi, Chem. Commun., 2007, 4519–4521 RSC; (l) T. Inagaki, Y. Yamada, L. T. Phong, A. Furuta, J.-i. Ito and H. Nishiyama, Synlett, 2009, 253–256 CAS; (m) J. Gajewy, M. Kwit and J. Gawroński, Adv. Synth. Catal., 2009, 351, 1055–1063 CrossRef CAS; (n) S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770–1771 CrossRef CAS; (o) N. A. Marinos, S. Enthaler and M. Driess, ChemCatChem, 2010, 2, 846–853 Search PubMed; (p) S. Enthaler, B. Eckhardt, S. Inoue, E. Irran and M. Driess, Chem.–Asian J., 2010, 5, 2027–2035 CrossRef CAS; (q) S. Enthaler, K. Schröder, S. Inoue, B. Eckhardt, K. Junge, M. Beller and M. Driess, Eur. J. Org. Chem., 2010, 4893–4901 CrossRef CAS; (r) S. Enthaler, Catal. Lett., 2010 DOI:10.1007/s10562-010-0463-4.
- Selection for reductions of sulfoxides, see also references therein: (a) A. C. Fernandes, J. A. Fernandes, C. C. Romão, L. F. Veiros and M. J. Calhorda, Organometallics, 2010 DOI:10.1021/om100450a; (b) P. M. Reis, P. J. Costa, C. C. Romão, J. A. Fernandes, M. J. Calhorda and B. Royo, Dalton Trans., 2008, 1727–1733 RSC; (c) K. Bahrami, M. M. Khodaei and A. Karimi, Synthesis, 2008, 2543–2546 CrossRef CAS; (d) J. M. Khurana, V. S. Sharma and A. Chacko, Tetrahedron, 2007, 63, 966–969 CrossRef CAS; (e) B. W. Yoo, M. C. Park and M. S. Song, Synth. Commun., 2007, 37, 4079–4083 CrossRef CAS; (f) B. W. Yoo, M. S. Song and M. C. Park, Bull. Korean Chem. Soc., 2007, 28, 171–172 CrossRef CAS; (g) B. W. Yoo, M. S. Song and M. C. Park, Synth. Commun., 2007, 37, 3089–3093 CrossRef CAS; (h) L. K. Pandey, U. Pathak and A. N. Rao, Synth. Commun., 2007, 37, 4105–4109 CrossRef CAS; (i) K. Bahrami, M. M. Khodaei and M. Khedri, Chem. Lett., 2007, 36, 1324–1325 CrossRef CAS; (j) A. C. Fernandes and C. C. Romão, Tetrahedron Lett., 2007, 48, 9176–9179 CrossRef CAS; (k) A. C. Fernandes and C. C. Romão, Tetrahedron, 2006, 62, 9650–9654 CrossRef CAS; (l) C. D. Roy and H. C. Brown, Journal of Chemical Research, 2006, 2006, 642–644 Search PubMed; (m) B. R. Raju, G. Devi, Y. S. Nongpluh and A. K. Saikia, Synlett, 2005, 358–360 CAS; (n) J. H. Espenson, Coord. Chem. Rev., 2005, 249, 329–341 CrossRef CAS; (o) R. Sanz, J. Escribano, Y. Fernández, R. Aguado, M. R. Pedrosa and F. J. Arnáiz, Synthesis, 2004, 1629–1632 CrossRef CAS; (p) D. J. Harrison, N. C. Tam, C. M. Vogels, R. F. Langler, R. T. Baker, A. Decken and S. A. Westcott, Tetrahedron Lett., 2004, 45, 8493–8496 CrossRef CAS; (q) N. Koshino and J. H. Espenson, Inorg. Chem., 2003, 42, 5735–5742 CrossRef CAS; (r) B. W. Yoo, K. H. Choi, D. Y. Kim, K. I. Choi and J. H. Kim, Synth. Commun., 2003, 33, 53–57 CrossRef CAS; (s) J. Arias, C. R. Newlands and M. M. Abu-Omar, Inorg. Chem., 2001, 40, 2185–2192 CrossRef CAS; (t) K. C. Nicolaou, A. E. Koumbis, S. A. Snyder and K. B. Simonsen, Angew. Chem., Int. Ed., 2000, 39, 2529–2533 CrossRef CAS; (u) M. M. Abu-Omar and S. I. Khan, Inorg. Chem., 1998, 37, 4979–4985 CrossRef CAS; (v) J. B. Arterburn and M. C. Perry, Tetrahedron Lett., 1996, 37, 7941–7944 CrossRef CAS; (w) M. M. Abu-Omar, E. H. Appelman and J. H. Espenson, Inorg. Chem., 1996, 35, 7751–7757 CrossRef CAS; (x) V. Y. Kukuskin, Coord. Chem. Rev., 1995, 139, 375–407 CrossRef CAS; (y) Z. Zhu and J. H. Espenson, J. Mol. Catal. A: Chem., 1995, 103, 87–94 CrossRef CAS.
- Selection for reductions of sulfoxides, see also references therein: (a) V. Y. Kukuskin, Russ. Chem. Rev., 1990, 59, 844–852 Search PubMed; (b) M. Madesclaire, Tetrahedron, 1988, 44, 6537–6551 CrossRef CAS; (c) J. C. Bryan, R. E. Stenkamp, T. H. Tulip and J. M. Mayer, Inorg. Chem., 1987, 26, 2283–2288 CrossRef CAS; (d) J. S. Cha, J. E. Kim and J. D. Kim, Tetrahedron Lett., 1985, 26, 6453–6456 CrossRef CAS; (e) H. C. Brown and N. Ravindran, Synthesis, 1973, 42–43 CrossRef CAS; (f) Y. Guidon, J. G. Atkinson and H. E. Morton, J. Org. Chem., 1984, 49, 4538–4540 CrossRef; (g) S. C. A. Sousa and A. C. Fernandes, Tetrahedron Lett., 2009, 50, 6872–6876 CrossRef CAS; (h) M. G. Voronkov, N. N. Vlasova, S. A. Bol'shakova, N. K. Gusarova and G. G. Efremova, Zh. Obshchei Khim., 1985, 55, 1034–1035 Search PubMed.
- (a) R. Hille, J. Reètey, U. Bartlewski-Hof, W. Reichenbecher and B. Schink, FEMS Microbiol. Rev., 1998, 22, 489–501 CrossRef; (b) R. Hille, Chem. Rev., 1996, 96, 2757–2816 CrossRef CAS; (c) C. Kisker, H. Schindelin and D. C. Rees, Annu. Rev. Biochem., 1997, 66, 233–267 CrossRef CAS; (d) J. H. Enemark, J. J. A. Cooney, J.-J. Wang and R. H. Holm, Chem. Rev., 2004, 104, 1175–1200 CrossRef CAS.
- (a) E. M. McGarrigle, E. L. Myers, O. Illa, M. A. Shaw, S. L. Riches and V. K. Aggarwal, Chem. Rev., 2007, 107, 5841–5883 CrossRef CAS; (b) D. Rickard and G. W. Luther III, Chem. Rev., 2007, 107, 514–562 CrossRef CAS; (c) E. Nicolas, M. Vilaseca and E. Giralt, Tetrahedron, 1995, 51, 5701–5710 CrossRef CAS.
- For a radical based reduction of sulfoxides see: J. W. Cubbage, T. A. Tetzlaff, H. Groundwater, R. D. McCulla, M. Nag and W. S. Jenks, J. Org. Chem., 2001, 66, 8621–8628 Search PubMed.
- i Pr3SiOH was applied as mimic for Et3SiOH.
- THF was chosen because of better solubility of Zn(OTf)2.
- E. V. Amel'chenkova, T. O. Denisova and S. E. Nefedov, Russ. J. Inorg. Chem. (Transl. of Zh. Neorg. Khim.), 2006, 51, 1218–1263 CrossRef.
- A similar motif was reported for Zn(dmso)6(ClO4)2 I. Persson, Acta Chem. Scand., Ser. A, 1982, 36a, 7–13 Search PubMed.
- See for instance: ref. 5a and references therein.
- J. Drabowicz and M. Mikolajczyk, Synthesis, 1976, 527–528 CrossRef CAS.
- Hydrogen was not detectable during the 1H NMR studies.
- (a) T. Itoh and T. Mase, Org. Lett., 2004, 6, 4587–4590 CrossRef CAS; (b) V. Dichiarante, M. Fagnoni and A. Albini, Chem. Commun., 2006, 3001–3003 RSC; (c) G. H. Penner and R. E. Wasylishen, Can. J. Chem., 1989, 67, 525–534 CrossRef CAS; (d) R. Sanz, Y. Fernandez, M. P. Castroviejo, A. Perez and F. J. Fananas, J. Org. Chem., 2006, 71, 6291–6294 CrossRef CAS; (e) D. Guijarro, B. Mancheno and M. Yus, Tetrahedron, 1992, 48, 4593–4600 CrossRef CAS; (f) J. C. Dyer, S. A. Evans and Slayton Jr., Magn. Reson. Chem., 1991, 29, 286–288 CrossRef CAS; (g) K. Ajiki, M. Hirano and K. Tanaka, Org. Lett., 2005, 7, 4193–4195 CrossRef CAS; (h) G. Barbarella, P. Dembech, A. Garbesi and A. Fava, Org. Magn. Reson., 1976, 8, 108–114 CrossRef CAS; T. Gandhi and B. R. Jagirdar, Inorg. Chem., 2005, 44, 1118–1124 CrossRef CAS.
- G. M. Sheldrick, SHELXL93, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † CCDC reference numbers 782191 and 796905. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cy00039f |
| This journal is © The Royal Society of Chemistry 2011 |