Novel N,O-Cu(OAc)2 complex catalysed diastereo- and enantioselective 1,4-addition of glycine derivatives to alkylidene malonates†
Ming
Wang
a,
Yu-Hua
Shi
a,
Jun-Fei
Luo
a,
Wenting
Du
a,
Xiao-Xin
Shi
a,
John S.
Fossey
ab and
Wei-Ping
Deng
*a
aSchool of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: weiping_deng@ecust.edu.cn; Tel: +86 (21) 64252431
bSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, UK B15 2TT
First published on 4th February 2011
Abstract
Newly developed chiral N,O-Cu(OAc)2 complexes were found to catalyse the diastereo- and enantioselective 1,4-addition of glycine derivatives to alkylidene malonates to afford 3-aryl glutamic acids derivatives in good yields (82–98%) and high stereoselectivities (up to 83% for anti and 90% for syn adducts, respectively). High optical purity anti adducts (up to 99% ee) can be obtained after simple recrystallisation, and their conversion to free 3-aryl glutamic acids was demonstrated by a representative example, chlorpheg, via a one pot process in 72% yield.
Introduction
The synthesis of optically active (un)natural α-amino acid derivatives is of great importance in current organic synthesis, especially in the post-genomic era.1Glutamic acid and its β-substituted derivatives are biologically important α-amino acid derivatives because they work not only as essential components of peptides and proteins but also as signal mediators.2 For example, chlorpheg,33-p-chlorophenyl glutamic acid, is a selective L-homocysteic acid (HCA) uptake inhibitor, which selectively enhances the excitatory, depolarising activities of l-HCA on amphibian and mammalian central nervous system neurons. Among the most efficient synthetic methods of β-substituted glutamic acid derivatives are diastereoselective4 or catalytic51,4-additions of glycine derivatives to α,β-unsaturated esters or amides. Kobayashi and co-workers reported the first example of alkaline earth metal calcium-catalysed diastereo- and enantioselective 1,4-additions of glycine derivatives to β-alkyl-α,β-unsaturated esters,6 3-alkyl glutamic acid derivatives were prepared in excellent yields with up to 99% ee. It was shown that a tert-butylphenylmethylene glycine tert-butyl ester was the best substrate in terms of product distribution and selectivity. When less sterically hindered diphenylene glycine tert-butyl ester was employed a useful [3 + 2] cycloaddition reaction predominated to afford chiral multi-substituted pyrrolidines rather than 1,4-addition adducts. Furthermore, Kobayashi and co-workers have used this methodology to perform important transformations of nitroalkenes and enolate protonation.7 Nevertheless, the application of such a catalytic methodology to the synthesis of 3-aryl glutamic acid derivatives remains unreported.Results and discussion
To-date, a great challenge for organic synthesis is the preparation of biologically relevant 3-aryl glutamic acid derivativesviacatalytic, diastero- and enantioselective 1,4-addition protocols. On the other hand, alkylidene malonates serve as excellent Michael acceptors and have been successfully employed in a variety of 1,4-addition reactions.5e,8 To the best of our knowledge, catalytic enantioselective 1,4-addition of alkylidene malonates 1 to glycine ester derivatives 2 have not yet been reported.Consequently, we envisaged that such catalytic enantioselective 1,4-additions may provide a practical approach for the production of valuable optically pure 3-aryl glutamic acid derivatives. In the course of developing new catalytic systems for asymmetric reactions we designed and synthesised a series of new 1,3-dihydroimidazole based N,O-bidentate ligands 4–8,9 which we envisioned as new chiral ligands for asymmetric transition metal catalysis. Herein, catalytic asymmetric 1,4-addition reactions of glycine derivatives to various alkylidene malonates to afford 3-aryl glutamic acid derivatives, under control of new N,O ligands, in good to high diasteroselectivities and enantioselectivities is presented (Scheme 1).
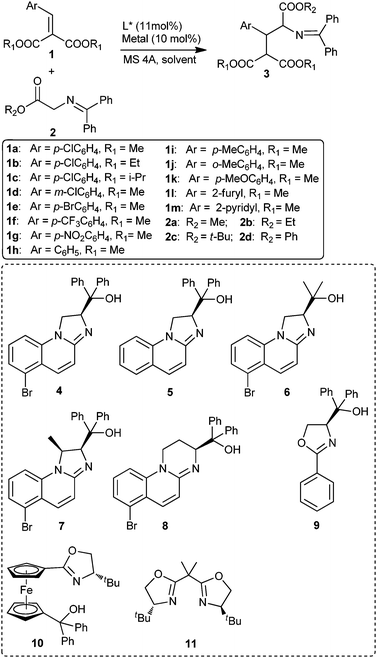 | ||
| Scheme 1 Catalytic asymmetric 1,4-addition of glycine Schiff bases 2 to alkylidene malonates 1. | ||
Initially, we examined N,O ligand 4 in the reaction of methyl cinnamate with glycine methyl ester 2a in the presence of 11 mol% of ligand and 10 mol% of Cu(OAc)2 in THF at room temperature, unfortunately no reaction was observed, perhaps due to the relatively low reactivity of methyl cinnamate. Considering the stronger electrophilicity of alkylidene malonates over methyl cinnamate, dimethyl p-chlorobenzylidene malonate 1a was next used in a reaction with glycine methyl ester 2a under the same conditions. Pleasingly, the desired 1,4-adduct 3aa was obtained in 33% yield with moderate diastereo- and enantioselectivity (Table 1, entry 1, anti/syn = 85/15, 50% ee for anti adduct). Moreover, the addition of a catalytic amount of KOtBu (10 mol%) was found to facilitate the reaction to afford 3aa in 85% yield with higher enantioselectivity (Table 1, entry 2, anti/syn = 91/9, 71% ee for anti adduct). At 0 °C the enantiomeric excess of 3aa could be further increased to 75% at the expense of yield under otherwise identical conditions (Table 1, entry 3). Screening solvents revealed THF to be optimal of those tried, in terms of both yield and enantioselectivity (Table 1, entries 4–8). Next, a series of Lewis acids were screened using 11 mol% of 4 and 10 mol% of KOtBu at room temperature in THF. Copper salts with stronger Lewis acidity, such as Cu(OTf)2 and CuCl2 were found to be inactive for this reaction (Table 1, entries 12–15). Other metal Lewis acids such as NiCl2·2H2O, Ni(OAc)2·4H2O, Zn(OAc)2·2H2O were completely inactive either (Table 1, entries 9–11). Silver acetate allowed the reaction to proceed smoothly to afford the desired product 3aa in 80% yield, albeit with low enantiomeric excess (Table 1, entry 16, 26% ee for anti adduct). Screening bases suggested that variation does not change the enantioselectivity but does affect the yield, as such KOtBu was selected as the optimal base. Since increasing the amount of KOtBu to 20 mol% only increased the yield of product slightly without changing the enantioselectivity of corresponding product (Table 1, entry 2 versus 22) 10 mol% of KOtBu was chosen as the optimal loading for further study.
| Entry | Metal salts | Bases | Solvent | Yield (%)a |
anti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :synb :synb |
eec/(anti:syn) |
|---|---|---|---|---|---|---|
| a Isolated yields, N.P. means no product observed (TLC analysis). b Ratio determined by HPLC. c Determined by HPLC. | ||||||
| 1 | Cu(OAc)2 | — | THF | 33 | 85:15 |
50/— |
| 2 | Cu(OAc)2 | KOtBu(10%) | THF | 87 | 91:9 |
71/26 |
| 3 | Cu(OAc)2 | KOtBu(10%) | THF(0 °C) | 65 | 91:9 |
75/— |
| 4 | Cu(OAc)2 | KOtBu(10%) | Toluene | 30 | 73:27 |
51/86 |
| 5 | Cu(OAc)2 | KOtBu(10%) | CH2Cl2 | 28 | 52:48 |
20/81 |
| 6 | Cu(OAc)2 | KOtBu(10%) | CH3CN | Trace | — | — |
| 7 | Cu(OAc)2 | KOtBu(10%) | Et2O | 57 | 89:11 |
59/10 |
| 8 | Cu(OAc)2 | KOtBu(10%) | Dioxane | 80 | 89:11 |
67/26 |
| 9 | NiCl2·6H2O | KOtBu(10%) | THF | NP | — | — |
| 10 | Ni(OAc)2·4H2O | KOtBu(10%) | THF | NP | — | — |
| 11 | Zn(OAc)2·2H2O | KOtBu(10%) | THF | NP | — | — |
| 12 | Cu(OTf)2 | KOtBu(10%) | THF | Trace | — | — |
| 13 | CuCl2 | KOtBu(10%) | THF | Trace | — | — |
| 14 | Cu(ClO4)2·6H2O | KOtBu(10%) | THF | Trace | — | — |
| 15 | Cu(acac)2·2H2O | KOtBu(10%) | THF | Trace | — | — |
| 16 | AgOAc | KOtBu(10%) | THF | 80 | 79:21 |
26/0 |
| 17 | Cu(OAc)2 | NaHMDS(10%) | THF | 91 | 89:11 |
63/19 |
| 18 | Cu(OAc)2 | PS (10%) | THF | 50 | 89:11 |
67/23 |
| 19 | Cu(OAc)2 | CsCO3(10%) | THF | 46 | 92:8 |
69/27 |
| 20 | Cu(OAc)2 | Et3N(10%) | THF | 56 | 90:10 |
68/— |
| 21 | Cu(OAc)2 | KOtBu(5%) | THF | 57 | 89:11 |
71/23 |
| 22 | Cu(OAc)2 | KOtBu(20%) | THF | 93 | 90:10 |
69/25 |
The effect of variation in the ester moieties (R1 and R2) was next probed, the results are summarised in Table 2. For both R1 and R2, the smallest group employed gave the optimal yield and enantioselectivity of corresponding 1,4-addition product (Table 2, entry 1, 71% ee, and anti:syn = 91:9). The diastereoselectivity was dramatically increased to 99:1 when the methyl ester group was replaced by a t-butyl ester, however yield and enantioselectivity were moderate (Table 2, entry 3). Substrate 1c, with an isopropyl ester, was found to be inactive for this catalytic system due to unconfirmed steric hindrance between isopropyl group and the catalytic complex.
| Entry | R1 | R2 | Yield (%) |
anti:syn |
ee (anti:syn) |
|---|---|---|---|---|---|
| a All reactions performed at room temperature, with THF as solvent, using 11 mol% of ligand 4, 10 mol% of Cu(OAc)2 and 10 mol% of KOtBu. | |||||
| 1 | Me | Me | 87 | 91:9 |
71/26 |
| 2 | Me | Et | 74 | 89:11 |
68/29 |
| 3 | Me | t-Bu | 27 | >99:1 |
55/— |
| 4 | Me | Ph | 65 | 89:11 |
51/7 |
| 5 | Et | Me | 68 | 91:9 |
67/27 |
| 6 | i-Pr | Me | Trace | — | — |
Next, a series of N,O-ligands 5–8 were examined under the optimal reaction condition, the results are summarised in Table 3. It was found that the enantioselectivity of the 1,4-addition adduct was increased to 81% ee and 87% ee for both the anti and syn isomer when N,O-ligand 7, bearing two stereogenic centres on imidazole ring, was employed. Two other known oxazoline based N,O-ligands 910 and 1011 also gave the 1,4-addition adduct, with lower diastereoselectivities and low enantioselectivities (Table 3, entries 6 and 7). Commercially available bisoxazoline ligand 11 delivered the desired product in moderate yields and diastereoselectivity, in close to racemic form (Table 3, entry 8).
| Entry | L | Yield (%) |
anti:syn |
ee (anti:syn) |
|---|---|---|---|---|
| a All reactions performed at room temperature, with THF as solvent, using 11 mol% of ligand 4, 10 mol% of Cu(OAc)2 and 10 mol% of KOtBu. | ||||
| 1 | 4 | 87 | 91:9 |
71/26 |
| 2 | 5 | 85 | 90:10 |
66/19 |
| 3 | 6 | 73 | 56:44 |
36/54 |
| 4 | 7 | 86 | 87:13 |
81/87 |
| 5 | 8 | 60 | 91:9 |
70/60 |
| 6 | 9 | 61 | 80:20 |
31/5 |
| 7 | 10 | 71 | 80:20 |
−19/−15 |
| 8 | 11 | 49 | 79:21 |
7/rac |
Having established the optimal catalytic system, the scope and generality of substrates with regard to the aryl funtionality of alkylidene malonates was examined. As shown in Table 4, a wide array of arylidene malonates 1a–k derived from various of aromatic aldehydes, bearing electron-rich, -neutral and -deficient groups, reacted smoothly with glycine methyl ester 2a to afford the corresponding 1,4-adducts (3aa–3ka) in high yields (82–98%) with moderate diastero- (4:1 to 9:1) and good enantioselectivities (79–83% ee for anti adducts and 83–90% ee for syn) (Table 4, entries 1–9). It is apparent that both the position and electronic properties of the substituents on the phenyl ring have little influence on the reaction activity and stereoselectivity. Notably, heteroaromatic substrates 1l and 1m were also tolerated and gave the corresponding adducts in good yields and similar stereoselectivities (Table 4, entries 10 and 11). The enantiomeric excess of the anti isomer of 3aa, 3ea and 3ha were improved to >98% after a simple recrystallisation.

| Entry | R1 | Yield (%) |
anti:syn |
ee (anti:syn) |
|---|---|---|---|---|
| a KOtBu is not need. b The reaction time was 24 h. c Data in parentheses were the ee of anti adduct after simple crystallisation. | ||||
| 1 | p-Cl-Ph (1a) | 86 | 87:13 |
81/87(98/—)c |
| 2 | m-Cl-Ph (1d) | 90 | 85:15 |
79/89 |
| 3 | p-Br-Ph (1e) | 98 | 87:13 |
81/88(98/—)c |
| 4 | p-CF3-Ph (1f) | 96 | 89:11 |
80/88 |
| 5a,b | p-NO2-Ph (1g) | 83 | 82:18 |
80/86 |
| 6 | Ph (1h) | 89 | 88:12 |
83/90(99/—)c |
| 7 | p-Me-Ph (1i) | 93 | 89:11 |
82/83 |
| 8 | o-Me-Ph (1j) | 82 | 82:18 |
80/90 |
| 9 | p-OMe-Ph (1k) | 82 | 87:13 |
81/86 |
| 10 | 2-furyl (1l) | 93 | 82:18 |
81/86 |
| 11 | 3-pyridyl (1m) | 88 | 80:20 |
78/80 |
Transformation of 3aa was also conducted to afford selective L-homocysteic acid (HCA) uptake inhibitor chlorpheg (12) in 72% yield via the hydrolysis of esters, subsequent decarboxylation and hydrolysis of imine in one pot (Scheme 2).
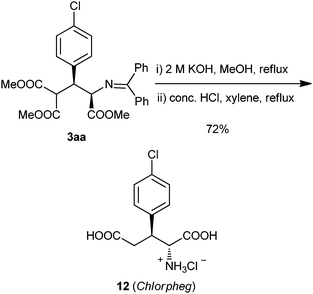 | ||
| Scheme 2 One pot imine and ester hydrolysis of 3aa to give Chlorpheg 12 in 72% yield. | ||
The relative configuration of major adducts of 3 were assigned as the anti isomer by X-ray diffraction analysis of 3ha (see ESI)† and comparison of other spectroscopic data. The absolute configuration of 3 was established as (2R,3S) by comparing the optical rotation of 12 to that of literature precedents.12
Conclusions
In summary, the first highly efficient catalytic diastereo- and enantioselective 1,4-addition of glycine derivatives to alkylidene malonates in the presence of chiral N,O-Cu(OAc)2 complexes to afford 3-aryl glutamic acids derivatives in good yields (82–98%) and high stereoselectivities (up to 83% for anti and 90% for syn adducts respectively) has been developed. High optical purity anti adducts were obtained after simple recrystallisation, and their conversion to free 3-aryl glutamic acids was demonstrated by a representative example, chlorpheg (12), via a one pot process in 72% yield. Further mechanistic studies and development of applications are now in progress.Acknowledgements
WPD thanks financial support from the New Century Excellent Talents in University, the Ministry of Education, China (no. NCET-07-0283), the Shanghai Committee of Science and Technology (grant no. 06PJ14023) and “111” Project (no. B07023). JSF thanks ECUST for visiting Professorship, ERDF AWMII and the University of Birmingham for support. WPD and JSF thank the CAtalysis and Sensing for our Environment (CASE) network, and the CASE09 workshop at ECUST for networking opportunities.Notes and references
- C. Najera and J. M. Sansano, Chem. Rev., 2007, 107, 4584–4671 CrossRef CAS.
- (a) M. Kotnik, J. Humljan, C. Contreras-Martel, M. Oblak, K. Kristan, M. Herve, D. Blanot, U. Urleb, S. Gobec, A. Dessen and T. Solmajer, J. Mol. Biol., 2007, 370, 107–115 CrossRef CAS; (b) H. Brauner-Osborne, J. Egebjerg, E. O. Nielsen, U. Madsen and P. Krogsgaard-Larsen, J. Med. Chem., 2000, 43, 2609–2645 CrossRef; (c) M. G. Moloney, Nat. Prod. Rep., 1998, 15, 205–219 RSC.
- (a) J. Davies, A. A. Francis, D. J. Oakes, M. J. Sheardown and J. C. Watkins, Neuropharmacology, 1985, 24, 177–180 CrossRef CAS; (b) M. L. Zeise, T. Knopfel and W. Zieglgansberger, Brain Res., 1988, 443, 373–376 CrossRef CAS; (c) S. Ito, L. Provini and E. Cherubini, Neurosci. Lett., 1991, 124, 157–161 CrossRef CAS.
- (a) V. A. Soloshonok, Curr. Org. Chem., 2002, 6, 341–364 CrossRef CAS; (b) T. K. Ellis, H. Ueki, R. Tiwari and V. A. Soloshonok, Tetrahedron: Asymmetry, 2009, 20, 2629–2634 CrossRef CAS; (c) M. Oba, T. Saegusa, N. Nishiyama and K. Nishiyama, Tetrahedron, 2009, 65, 128–133 CrossRef CAS; (d) V. A. Soloshonok, C. Z. Cai, T. Yamada, H. Ueki, Y. Ohfune and V. J. Hruby, J. Am. Chem. Soc., 2005, 127, 15296–15303 CrossRef CAS; (e) C. Herdeis and B. Kelm, Tetrahedron, 2003, 59, 217–229 CrossRef CAS; (f) V. A. Soloshonok, C. Z. Cai, V. J. Hruby, L. Van Meervelt and N. Mischenko, Tetrahedron, 1999, 55, 12031–12044 CrossRef CAS; (g) J. Ezquerra, C. Pedregal, I. Merino, J. Florez, J. Barluenga, S. Garcia-Granda and M. A. Llorca, J. Org. Chem., 1999, 64, 6554–6565 CrossRef CAS; (h) S. Kanemasa, A. Tatsukawa and E. Wada, J. Org. Chem., 1991, 56, 2875–2883 CrossRef CAS; (i) S. Kanemasa, A. Tatsukawa, E. Wada and O. Tsuge, Chem. Lett., 1989, 1301–1304 CAS; (j) A. Elachqar, M. Boumzebra, M. L. Roumestant and P. Viallefont, Tetrahedron, 1988, 44, 5319–5332 CrossRef; (k) A. B. Mauger, J. Org. Chem., 1981, 46, 1032–1035 CrossRef CAS.
- (a) S. Arai, F. Takahashi, R. Tsuji and A. Nishida, Heterocycles, 2006, 67, 495–+ CrossRef CAS; (b) B. Lygo, B. Allbutt and E. H. M. Kirton, Tetrahedron Lett., 2005, 46, 4461–4464 CrossRef CAS; (c) M. J. O'Donnell, Acc. Chem. Res., 2004, 37, 506–517 CrossRef CAS; (d) T. Ohshima, T. Shibuguchi, Y. Fukuta and M. Shibasaki, Tetrahedron, 2004, 60, 7743–7754 CrossRef CAS; (e) T. Akiyama, M. Hara, K. Fuchibe, S. Sakamoto and K. Yamaguchi, Chem. Commun., 2003, 1734–1735 RSC; (f) T. Shibuguchi, Y. Fukuta, Y. Akachi, A. Sekine, T. Ohshima and M. Shibasaki, Tetrahedron Lett., 2002, 43, 9539–9543 CrossRef CAS; (g) S. Arai, R. Tsuji and A. Nishida, Tetrahedron Lett., 2002, 43, 9535–9537 CrossRef CAS; (h) T. Ishikawa, Y. Araki, T. Kumamoto, H. Seki, K. Fukuda and T. Isobe, Chem. Commun., 2001, 245–246 RSC; (i) F. Y. Zhang and E. J. Corey, Org. Lett., 2000, 2, 1097–1100 CrossRef CAS; (j) E. J. Corey, M. C. Noe and F. Xu, Tetrahedron Lett., 1998, 39, 5347–5350 CrossRef CAS.
- (a) S. Saito, T. Tsubogo and S. Kobayashi, J. Am. Chem. Soc., 2007, 129, 5364–5365 CrossRef CAS; (b) S. Kobayashi, T. Tsubogo, S. Saito and Y. Yamashita, Org. Lett., 2008, 10, 807–809 CrossRef CAS; (c) T. Tsubogo, S. Saito, K. Seki, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 13321–13332 CrossRef CAS.
- (a) T. Poisson, T. Tsubogo, Y. Yamashita and S. Kobayashi, J. Org. Chem., 2010, 75, 963–965 CrossRef CAS; (b) T. Tsubogo, Y. Yamashita and S. Kobayashi, Angew. Chem., Int. Ed., 2009, 48, 9117–9120 CrossRef CAS.
- (a) Q. Li, C. H. Ding, X. L. Hou and L. X. Dai, Org. Lett., 2010, 12, 1080–1083 CrossRef CAS; (b) C. G. Chen, X. L. Hou and L. Pu, Org. Lett., 2009, 11, 2073–2075 CrossRef CAS; (c) Y. G. Wang, T. Kumano, T. Kano and K. Maruoka, Org. Lett., 2009, 11, 2027–2029 CrossRef CAS; (d) X. X. Yan, Q. Peng, Q. Li, K. Zhang, J. Yao, X. L. Hou and Y. D. Wu, J. Am. Chem. Soc., 2008, 130, 14362–14363 CrossRef CAS; (e) P. Elsner, L. Bernardi, G. Dela Salla, J. Overgaard and K. A. Jorgensen, J. Am. Chem. Soc., 2008, 130, 4897–4905 CrossRef CAS; (f) L. Bernardi, J. Lopez-Cantarero, B. Niess and K. A. Jorgensen, J. Am. Chem. Soc., 2007, 129, 5772–5778 CrossRef CAS; (g) T. Shibuguchi, H. Mihara, A. Kuramochi, S. Sakuraba, T. Ohshima and M. Shibasaki, Angew. Chem., Int. Ed., 2006, 45, 4635–4637 CrossRef CAS.
- Ligands 4–8 are now also being investigated in other catalytic enantioselective reactions.
- W. P. Deng, X. L. Hou and L. X. Dai, Tetrahedron: Asymmetry, 1999, 10, 4689–4693 CrossRef CAS.
- A. L. Braga, R. M. Rubim, H. S. Schrekker, L. A. Wessjohann, M. W. G. de Bolster, G. Zeni and J. A. Sehnem, Tetrahedron: Asymmetry, 2003, 14, 3291–3295 CrossRef CAS.
- D. E. Jane, D. J. Chalmers, J. A. K. Howard, I. C. Kilpatrick, D. C. Sunter, G. A. Thompson, P. M. Udvarhelyi, C. Wilson and J. C. Watkins, J. Med. Chem., 1996, 39, 4738–4743 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The synthetic procedures for N,O-ligands 4 and 7 along with corresponding spectral data; general experimental procedure, spectral data for all 1,4-addition adducts 3; X-ray analysis data of 3ha in CIF format is provided. CCDC reference number 795643. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0cy00001a |
| This journal is © The Royal Society of Chemistry 2011 |