A water-soluble phosphite derived from sulfonated calix[4]arene. The remarkable stability of its rhodium complexes and two phase hydroformylation studies
Christopher J.
Cobley
and
Paul G.
Pringle
*
School of Chemistry, University of Bristol, Cantocks Close, Bristol, UK BS8 1TS. E-mail: paul.pringle@bristol.ac.uk
First published on 10th February 2011
Abstract
The reaction of tetrasulfonated calix[4]arene (trioctylammonium salt) with P(NMe2)3 followed by treatment with gaseous NMe2H gave a zwitterionic six-coordinate phosphorus(V) species 1 containing a P(H)(NHMe2) group which can be stored for months without decomposition. In water, 1 loses NHMe2 and rearranges to form the water soluble phosphite Lcc. Phosphite Lcc has a half-life in water of ca. 5 h decomposing to H3PO3 and free tetrasulfonated calix[4]arene. Compound 1 serves as a convenient precursor to Lcc and complexes of Lcc are formed by dissolving 1 in water in the presence of labile metal complexes. The products have been identified by comparison of their 31P NMR data with well established analogues of calix[4]arene derived phosphites. In this way the water soluble complexes [Rh(acac)(CO)(Lcc)2] (2c) [Rh2Cl2(CO)2(Lcc)2] (3c), [Pt2Cl4(Lcc)2] (4c) and [PtCl2(Lcc)2] have been tentatively identified. The rhodium complex 3c has a half-life in water of 4 months. Two-phase (water/toluene) hydroformylation of 2-methylpentenoate with 2c as a catalyst has been investigated and the results compared with the same reaction in toluene with lipophilic analogues of 2c. Under mild conditions, with 2ccatalyst, branched aldehydes are the only products of hydroformylation and one of the branched aldehydes is selectively hydrogenated to give the lactone derivative.
Introduction
Sulfonated arylphosphines have been extensively studied ever since the advent of triphenylphosphine trisulfonate (TPPTS) and its commercial application in two-phase hydroformylation catalysis.1,2 Triaryl phosphites are excellent ancillary ligands for Rh-catalysed hydroformylation3 but the kinetic instability of P–OR bonds to hydrolysis would appear to preclude making water-soluble sulfonated phosphites with any useful lifetime. Indeed Fell et al.4 showed that a mixture of sulfonated triphenylphosphites (TPOPS) (as tri-isooctylammonium salts) completely decompose in aqueous acetone after 24 h and Karakhanov et al.5 also reported that a polyether phosphite rapidly decomposed in the presence of water.
Phosphite ligands derived from calix[4]arene have been extensively studied6–8 and we reported7 that phosphites Laa and Lbb are stable to hydrolysis under conditions in which P(OPh)3 is rapidly hydrolysed. We now report the synthesis of phosphite Lcc derived from sulfonated calix[4]arene and show that its complexes are sufficiently stable in water to allow their application in two-phase catalysis.

Results and discussion
Ligand synthesis and stability
The hydrophilic phosphite Lcc was made according to the route shown in Scheme 1. Treatment of the rigorously dried trioctylammonium salt of the commercially available calix[4]arene tetrasulfonic acid with P(NMe2)3 in CH2Cl2 followed by bubbling gaseous NHMe2 through the solution gave a white precipitate which was assigned to the six-coordinate phosphorus species 1 (31P NMR in D2O, δ −124.5 1J(PH) 730 Hz) from the close similarity of its 31P NMR data to the analogous non-sulfonated species.6 Solid 1 could be stored for weeks without change but in water, 1 converted over a period of 1 hour to phosphite Lcc as shown by its characteristic 31P chemical shift of 113.5 ppm (cf. 112.9 ppm for Laa). Since Lcc is unstable in water (see below), compound 1 is a convenient precursor for the generation of Lccin situ. | ||
| Scheme 1 | ||
Phosphite Lcc decomposes in water over 24 h to give H3PO3 and free sulfonated calix[4]arene, as shown by 31P and 1H NMR spectroscopy. The kinetics of the decomposition was investigated by following the decline in the mole fraction Lcc (estimated from the 31P NMR integrals) as a function of time. A plot of ln[Lcc] against time was linear showing that the decomposition is a pseudo first order process with a t1/2 of 5 h. In view of our previous observations of the very high hydrolytic stability of Laa and Lbb and Fell's results which showed that sulfonation of triphenyl phosphite leads to enhanced hydrolytic stability, we had anticipated that Lcc would be more stable in water than is observed. The explanation for the lability of Lcc may lie in its conformation: assuming the conformation of Lcc is analogous to Laa and Lbb (i.e. the partial cone shown in Scheme 1), the SO3−group would be held in close proximity to the P and may therefore provide anchimeric assistance to the hydrolysis. Consistent with this hypothesis is that the rate of hydrolysis was independent of pH, being the same in buffers of pH 4.0, 7.0, and 9.0.
Coordination chemistry of Lcc
The coordination chemistry of Lcc with Rh, Pt, and Pd has been investigated by generating aqueous solutions of Lccin situ from 1 in the presence of a labile metal complex as described below. Attempts to isolate the product complexes from their aqueous solutions by removal of the water or by precipitation were unsuccessful, invariably giving intractable oily residues which contained partially decomposed complex and several by-products. The structural assignments of the complexes of Lcc are therefore tentative, being based on comparison of solution 31P NMR data with the data for the analogous well-characterised complexes of Laa or Lbb.We were interested in making a water-soluble analogue of the previously reported7hydroformylation catalysts 2a,b. Thus a solution of the precursor 1 in D2O was added to an acetone solution of [Rh(CO)2(acac)] and after 24 h the main rhodium–phosphite complex present (δ 93.2 ppm, J(RhP) 282 Hz) was assigned structure 2c on the basis of the similarity of the 31P NMR parameters to those for 2a (δ 109.8 ppm, J(RhP) 309 Hz) and 2b (δ 110.5 ppm, J(RhP) 311 Hz). Addition of precursor 1 to an aqueous solution of Na[RhCl2(CO)2] gave a rhodium complex whose 31P NMR parameters (δ 107 ppm, J(RhP) 329 Hz) were very similar to those for 3b (δ 103 ppm, J(RhP) 317 Hz) and therefore it was assigned the structure 3c. The stabilisation of the hydrophilic phosphite upon coordination to rhodium(I) is phenomenal: we have monitored the first order decomposition of 2c over six months in aqueous solution and thereby estimated its half-life in water to be 4 months at ambient temperatures.
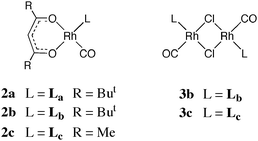
Treatment of Lcc (generated from 1) in D2O with an acetone solution of K[PtCl3(η-C2H4)] gave two platinum-containing complexes according to the 31P NMR spectrum. One species was assigned the binuclear structure 4c on account of its 31P NMR parameters (δ 7.2 ppm, J(PtP) 7320 Hz) being similar to those for the crystallographically characterised7 analogue 4b (in CDCl3, δ 2.2 ppm, J(PtP) 7348 Hz). The other product was assigned the mononuclear structure 5c on account of its 31P NMR parameters (δ 32.6 ppm, J(PtP) 6246 Hz) being similar to those for complex 5a (in (CD3)2SO, δ 37.9 ppm, J(PtP) 6629 Hz).

Like the rhodium(I) complexes 2c and 3c, the platinum species 4c and 5c were much more stable to hydrolysis than the free phosphite Lcc. However treatment of Na2[PdCl4] with Lcc in D2O gave initially a bright yellow solution but within 30 seconds a black precipitate had formed. The 31P NMR spectrum of the colourless supernatant showed the presence of a major peak at −22 ppm which is tentatively assigned to the phosphate Lcc(O) on the basis of the similarity of the δ(P) with that for the previously reported6phosphate Laa(O) of −22 ppm; this is consistent with oxidation of Lcc by Pd(II) and the formation of metallic palladium.

Hydroformylation catalysis
The high stability of aqueous solutions of rhodium(I) complexes of Lcc prompted us to investigate two-phase (aqueous/organic) hydroformylation catalysis with this ligand. The hydroformylation of 2-methylpentenoate (2-MP) was studied, first with 2a and 2b as catalysts in a single toluene phase and then with hydrophilic complex 2c as a catalyst in an aqueous/toluene mixture. The matrix of products that are formed is shown in Scheme 2. The aldehydes A2 and A3 are derived from hydroformylation of the internal alkene 2-MP and hydrogenation of these aldehydes followed by lactonisation with loss of MeOH would give the corresponding lactones Lt2 and Lt3. Isomerisation of 2-MP to 1-MP followed by hydroformylation would give aldehydes A1 and A2. This last reaction is of particular commercial interest9 because 2-MP is the product of methoxycarbonylation of butadiene and linear aldehyde A1 is a precursor to adipic acid. | ||
| Scheme 2 Products formed in the hydroformylation of 2-methylpentenoate (2-MP). | ||
The results from the catalysis are collected in Table 1 and the details of the conditions are given in the Experimental section. Pre-prepared complexes 2a–c were used and for each run, the conversion of 2-MP was >75%. The results with 2a–c (entries 2–7) can be compared with the control run using [Rh(CO)2(acac)] (entry 1). The main effect of the catalysts derived from the lipophilic phosphites Laa and Lbb (entries 2–5) is to reduce the amount of A1 produced as a proportion of the total aldehyde yield from 33% with the control to ca. 5% (entries 4 and 5).
| Run | Ligand | Conversion | Aldehyde | Linearityb | A1 | A2 | A3 | Lt2 | Lt3 |
|---|---|---|---|---|---|---|---|---|---|
a Unless otherwise stated, products obtained after reaction for 3 h in toluene at 160 °C and 60 atm H2/CO with Rh catalyst of 6.8 mM, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 ratio of L∶Rh, and ca. 150∶1 ratio of 2-methylpentenoate∶Rh. Product amounts are in % as determined by GC using toluene or cumene as internal standard; the balance (1–4%) is 2-methylpentenoate. See Experimental under single phase catalysis for full details.
b Mol of A1/total mol of aldehyde.
c 10∶1 ratio of L∶Rh.
d Two phase catalysis, at 160 °C for Run 6 and 60 °C for Run 7. See Experimental for details. ∶1 ratio of L∶Rh, and ca. 150∶1 ratio of 2-methylpentenoate∶Rh. Product amounts are in % as determined by GC using toluene or cumene as internal standard; the balance (1–4%) is 2-methylpentenoate. See Experimental under single phase catalysis for full details.
b Mol of A1/total mol of aldehyde.
c 10∶1 ratio of L∶Rh.
d Two phase catalysis, at 160 °C for Run 6 and 60 °C for Run 7. See Experimental for details.
|
|||||||||
| 1 | None | 87 | 46 | 33 | 15 | 14 | 17 | 23 | 31 |
| 2 | Laa | 79 | 40 | 22 | 9 | 17 | 14 | 29 | 30 |
| 3 | Lbb | 78 | 31 | 16 | 5 | 16 | 10 | 28 | 40 |
| 4c | Laa | 87 | 23 | 5 | 1 | 18 | 4 | 34 | 42 |
| 5c | Lbb | 88 | 24 | 5 | 1 | 20 | 3 | 28 | 46 |
| 6d | Lcc | 83 | 13 | 0 | 0 | 12 | 1 | 38 | 45 |
| 7d | Lcc | 94 | 46 | 0 | 0 | 46 | 0 | 4 | 48 |
The results of the two-phase catalysis with 2c carried out at 160 °C (entry 6) are suspect because at the end of the run, substantial amounts of rhodium metal precipitate were observed in the autoclave. However at 60 °C (entry 7), the catalysis proceeded with no Rh metal deposited and the highest conversion (94%) of all the runs was obtained. Elemental analysis of the aqueous and organic layers at the end of the run showed that ca. 60% of the rhodium resided in the organic layer showing that considerable leaching had occurred. Over 90% of the product was accounted for by A2 and Lt3 in approximately equal amounts. This suggests that the branched aldehydes A2 and A3 are the exclusive products of hydroformylation and subsequently A3 is selectively hydrogenated to the alcohol which, after lactonisation, gives Lt3. The selective hydrogenation of A3 may imply a selective binding of the A3 to the Rh; inspection of the proposed intermediates (Scheme 3) might suggest that the smaller chelate formed by A3 would be favoured over the one formed by A2.
 | ||
| Scheme 3 Proposed chelates formed prior to hydrogenation by (a) A3 and (b) A2. | ||
It is clear that more work is required to further characterise the catalysis but we have shown that water-soluble phosphite complexes are stable in water for long periods and therefore could be used for two-phase catalysis.
Experimental section
General procedures
Unless otherwise stated, all reactions were carried out under a dry nitrogen atmosphere using standard Schlenk line techniques. Commercial reagents were used as supplied unless otherwise stated. The phosphites Laa and Lbb were made as previously described6,7 and the starting materials [Rh2Cl2(CO)4]10 and K[PtCl3(C2H4)]11 were prepared by literature methods. NMR spectra were measured on a Jeol EX 90 or Jeol GX 400. 31P{1H}, 13C{1H}, and 1H NMR spectra were recorded at ambient temperature of the probe using a deuterated solvent to provide the field/frequency lock.Synthesis and stability of water soluble phosphite precursor 1
Trioctylamine (9.15 cm3, 21 mmol) was added to a suspension of p-(tetrasulfonic acid)calix[4] arene (3.00 g, 3.52 mmol) in CH2Cl2 (30 cm3) and after 1 min a clear yellow solution was obtained. Toluene (50 cm3) was added and then any water was azeotropically removed by concentration of the solution to 10 cm3 by distillation of the solvent. The remaining volatiles were removed under reduced pressure to give a tan oil. The oil was dissolved in CH2Cl2 (30 cm3) and P(NMe2)3 (0.88 cm3, 0.79 g, 4.84 mmol) added. A white precipitate began to form after 30 s and the mixture was stirred for 25 min after which time gaseous NHMe2 was bubbled through the mixture at a rate of 3 bubbles per second for 5 min. The white solid product was then filtered off and washed with CH2Cl2 (10 cm3). 31P NMR (D2O, 36.4 MHz): δ −124.5, 1J(PH) 730 Hz. Elemental analysis was consistent with the formulation 1·N(C8H17)3 (calc. for C62H107N6O16PS4 in parentheses): C 53.57 (55.09); H 8.01 (7.98); S 9.08 (9.49); P 2.29 (2.29); N 6.13 (6.22). A sample of compound 1 (65 mg) was dissolved in D2O (0.6 cm3) and the 31P NMR spectrum of the solution was recorded every 10 min for the first 1 h and then hourly for the next 12 h and then finally after 24 h (see Results and Discussion).Generation of aqueous solutions of rhodium complexes of Lcc
(1) [Rh(CO)2(acac)] (17 mg, 0.070 mmol) and precursor 1 (65 mg, 0.070 mmol) were added together to a mixture of D2O (0.5 cm3) and (CD3)2CO (0.5 cm3) in an NMR tube. A bright red precipitate forms immediately but then dissolves upon shaking the tube. After 24 h, the 31P NMR spectrum showed the presence of one main (75%) Rh-phosphite species in solution (δ 93.2 ppm, J(RhP) 282 Hz) assigned the structure 2c (see Results and Discussion). Attempts to isolate the product by evaporation of the water under reduced pressure led to a viscous brown oil that contained a mixture of several unidentified Rh-containing species and ligand decomposition products.(2) [Rh2Cl2(CO)4] (12 mg, 0.030 mmol) was dissolved in a solution of NaCl (40 mg, 0.68 mmol) in D2O (1.0 cm3) in an NMR tube and then precursor 1 (60 mg, 0.065 mmol) was added. After 1 h the 31P NMR spectrum showed the presence of a single Rh-phosphite species in solution (δ 107.0 ppm, J(RhP) 329 Hz) assigned the structure 3c. The decomposition of 3c in D2O to several P-containing species was monitored by 31P NMR spectroscopy over six months. Attempts to isolate the product by evaporation of the water under reduced pressure or by precipitation of the product by addition of [NBu4]Cl led to viscous red-brown oils that contained a mixture of products including unidentified Rh-containing species and ligand decomposition products.
Generation of aqueous solutions of platinum complexes of Lcc
A solution of precursor 1 (30 mg, 0.032 mmol) in D2O (0.5 cm3) was added to a solution of K[PtCl3(C2H4)] (11 mg, 0.032 mmol) in (CD3)2CO (0.5 cm3) in an NMR tube. After 3 h the 31P NMR spectrum showed the presence of two Pt-phosphite species in solution in the ratio 10∶1, the major species with δ 7.2 ppm, J(PtP) 7320 Hz, assigned structure 4c and the minor species with δ 32.6 ppm, J(PtP) 6246 Hz assigned structure 5c (see Results and Discussion). Attempts to isolate the product by evaporation of the water under reduced pressure or by precipitation of the product by addition of [NBu4]Cl led to viscous yellow-orange oils that contained a mixture of products including unidentified Pt-containing species and ligand decomposition products.
Hydroformylation of 2-methylpentenoate
∶1 mixture of H2 and CO. The pressure was then increased to 30 atm with H2/CO and heated to 160 °C. Then the pressure was increased to 60 atm and the reaction mixture stirred for 3 h at 800 rpm. The autoclave was then cooled to ambient temperature and the pressure slowly released and the product analysed by GC.
We would like to thank Rhône-Poulenc for a studentship (for CJC) and Johnson-Matthey for a loan of rhodium compounds.
References
- E. G. Kuntz, Fr. Patent, 2314910, 1975, (to Rhone-Poulenc) Search PubMed.
- Aqueous Phase Organometallic Catalysis: Concepts and Applications, ed. B. Cornils and W. A. Herrmann, Wiley, 2004 Search PubMed.
- Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer Academics Publishers, Dordrecht, 2000 Search PubMed.
- B. Fell, G. Papadogianakis, W. Konkol, J. Weber and H. Bahrmann, J. Prakt. Chem., 1993, 335, 75 CAS.
- E. A. Karakhanov, Y. S. Karasheva, E. A. Runova and V. A. Semernina, J. Mol. Catal. A: Chem., 1999, 142, 339 CrossRef CAS.
- (a) D. V. Khasnis, J. M. Burton, M. Lattman and H. Zhang, J. Chem. Soc., Chem. Commun., 1991, 562 RSC; (b) D. V. Khasnis, J. M. Burton, J. D. McNeil, C. J. Santini, H. Zhang and M. Lattman, Inorg. Chem., 1994, 33, 2657 CrossRef CAS.
- (a) C. J. Cobley, D. D. Ellis, A. G. Orpen and P. G. Pringle, J. Chem. Soc., Dalton Trans., 2000, 1101 RSC; (b) C. J. Cobley, D. D. Ellis, A. G. Orpen and P. G. Pringle, J. Chem. Soc., Dalton Trans., 2000, 1109 RSC.
- (a) F. J. Parlevliet, C. Keiner, J. Fraanje, K. Goubitz, M. Lutz, A. L. Spek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 2000, 1113 RSC; (b) C. Dieleman, S. Steyer, C. Jeunesse and D. Matt, J. Chem. Soc., Dalton Trans., 2001, 2508 RSC; (c) S. Steyer, C. Jeunesse, J. Harrowfield and D. Matt, Dalton Trans., 2005, 1301 RSC; (d) S. Steyer, C. Jeunesse, J. Harrowfield, D. Matt, R. Welter and M. Wesolek, J. Chem. Soc., Dalton Trans., 2002, 4264 RSC; (e) C. Kunze, D. Selent, I. Neda, M. Freytag, P. G. Jones, R. Schmutzler, W. Baumann and A. Borner, Z. Anorg. Allg. Chem., 2002, 628, 779 CrossRef CAS; (f) P. Maji, S. S. Krishnamurthy and M. Nethaji, Polyhedron, 2008, 27, 3519 CrossRef CAS; (g) A. Sarkar, M. Nethaji and S. S. Krishnamurthy, J. Organomet. Chem., 2008, 693, 2097 CrossRef CAS; (h) D. Semeril, D. Matt and L. Toupet, Chem.–Eur. J., 2008, 14, 7144 CrossRef CAS; (i) L. Monnereau, D. Semeril, D. Matt and L. Toupet, Adv. Synth. Catal., 2009, 351, 1629 CrossRef CAS; (j) L. Monnereau, D. Semeril and D. Matt, Eur. J. Org. Chem., 2010, 3068 CrossRef CAS; (k) L. Monnereau, D. Semeril and D. Matt, Green Chem., 2010, 12, 1670 RSC; (l) S. Liu and C. A. Sandoval, J. Mol. Catal. A: Chem., 2010, 325, 65 CrossRef CAS.
- (a) E. Drent and W. W. Jager, Eur. Patent, EP 577205, 1992, (to Shell) Search PubMed; (b) M. Beller, A. Krotz and W. Baumann, Adv. Synth. Catal., 2002, 344, 517 CrossRef CAS.
- R. Cramer, Inorg. Synth., 1974, 19, 218.
- J. Chatt and M. L. Searle, Inorg. Chem., 1950, 5, 210.
| This journal is © The Royal Society of Chemistry 2011 |