Achieving maximum photo-oxidation reactivity of Cs0.68Ti1.83O4−xNx photocatalysts through valence band fine-tuning
Gang
Liu
a,
Ping
Niu
a,
Lianzhou
Wang
b,
Gao Qing (Max)
Lu
b and
Hui-Ming
Cheng
*a
aShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, 72 Wenhua Road, Shenyang, 110016, China. E-mail: cheng@imr.ac.cn; Fax: +86 24 23903126; Tel: +86 24 23971611
bARC Centre of Excellence for Functional Nanomaterials, School of Engineering and Australian Institute of Bioengineering and Nanotechnology, The University of Queensland, Qld., 4072, Australia
First published on 10th February 2011
Abstract
Does a wider absorption range of the photocatalyst always result in a higher photoreactivity? By investigating a set of Cs0.68Ti1.83O4−xNx (x = 0–0.31) photocatalysts with a continuously tuned absorption edge, we found that although the absorption edge of Cs0.68Ti1.83O4−xNx is gradually shifted to the low-energy region with the increase of the nitrogen dopant, the photoreactivity of the photocatalyst in terms of generating the ˙OH radical does not correspondingly increase under the irradiation of monochromatic light of 254 nm and continuous spectrum of >300 nm. The fundamental mechanism of the anion dopant enhanced photocatalytic activity through the interplay of three key factors—absorbance, oxidative potential and mobility of charge carriers—is proposed to explain the observed photocatalytic activity change.
1. Introduction
The process of photocatalysis with inorganic semiconductors as photocatalysts involves three mechanistic steps: the excitation, bulk diffusion and surface transfer of photoexcited charge carriers. The photocatalytic reactivity can be enhanced through the synergistic effects of absorbance, redox potential and mobility of charge carriers, which are determined by the bandgap, edge and width of the conduction/valence band of semiconductors, as well as the surface structure of a photocatalyst.1–7 Among them, the adequate absorption of available photons is a prerequisite to enable an efficient photocatalytic activity. However, most stable oxide photocatalysts such as TiO2 suffer from negligible low-energy photon absorption in the visible light range due to their large wide bandgaps. To overcome this serious shortcoming, doping particularly anion doping has been widely employed to modify the electronic structures of wide bandgap photocatalysts.1,8–34 One of the two major hypotheses put forward for this purpose is to elevate the valence band (VB) maximum through mixing of dopant states with the upper VB states of the bulk material at an electronic structure level,11 which can be imparted by homogeneous doping of dopants within the whole bulk particles.33,34 As a consequence, the bandgap can be narrowed leading to increased light absorbance.Two concomitant but rarely considered important properties as a result of such VB maximum elevation include the reduced oxidation potential and the increased mobility of holes in the modified VB. They have completely opposite roles in determining photo-oxidation reactivity of photocatalysts. Therefore, an optimal reactivity must arise from the balance of absorbance, oxidation potential of photoexcited holes and mobility of the holes in doped photocatalysts. Unfortunately, this fundamental issue has not been clarified yet.
In our previous work,34 it was found that a homogeneous nitrogen doping can be easily reached by calcining lepidocrocite-type layered caesium titanate (Cs0.68Ti1.83O4) with an average particle size of ca. 300 nm in a gaseous ammonia atmosphere, which can result in a maximum band-to-band redshift of the pristine absorption edge up to ca. 472 nm. The key discovery in that work is that the homogeneous distribution of the nitrogen dopant is the determining factor in realizing the upshift of VB maximum, which contributes to two favourable characteristics of high absorbance and high mobility of holes generated in the VB for photoreactivity. The superior photoreactivity of Cs0.68Ti1.83O4−xNx with an absorption edge of 472 nm to Cs0.68Ti1.83O4 was also ascribed to both characteristics. Unfortunately, the lowered oxidative potential of holes by the upshift of VB maximum was neglected due to the limited understanding of the time.
This work aims to shed light on the essential role of each factor (absorbance, oxidative potential and mobility of charge carriers) in affecting the photo-oxidation capability of photocatalysts so that a generic rule may be discovered for designing the possible best photocatalyst under different conditions. Through fine-tuning of the valence band of Cs0.68Ti1.83O4−xNx photocatalysts, significant insights into the fundamental mechanism of the anion dopant enhanced photocatalytic activity are thereby achieved.
2. Experimental methods
Sample preparation
The layered Cs0.68Ti1.83O4 was prepared according to a modified procedure.35 In detail, raw powder materials of Cs2CO3 and TiO2 were fully mixed in an agate mortar. The molar percentage of Cs to Ti in the mixture was higher by 3% than that in the stoichiometric Cs0.68Ti1.83O4. The powder was then calcined at 750 °C for 12 h in static air with a ramping rate of 3 °C min−1 to obtain the layered Cs0.68Ti1.83O4 powder. The resultant sample is fully crystalline and has an average primary particle size of ca. 300 nm. A series of Cs0.68Ti1.83O4−xNx (x = 0.02–0.31) was obtained through calcining the resultant Cs0.68Ti1.83O4 powder in a gaseous ammonia atmosphere in a tube furnace. The amount of nitrogen dopant in Cs0.68Ti1.83O4−xNx can be tuned simply by controlling calcination temperature (500–750 °C) and duration (30 min–2 h). Specifically, the detailed condition of calcination temperature and duration for each sample, x = 0.02, 0.09, 0.18 and 0.31, is 500 °C and 30 min, 600 °C and 30 min, 700 °C and 1 h, 750 °C and 2 h, respectively. The flux rate of gaseous ammonia is kept at 25 mL min−1 during calcination.Characterization
Chemical compositions of Cs0.68Ti1.83O4−xNx were analyzed using X-ray photoelectron spectroscopy (Thermo Escalab 250, a monochromatic Al KαX-ray source). All binding energies were referenced to the C 1s peak (284.6 eV) arising from adventitious carbon. The optical spectra of the samples were recorded in a UV-visible spectrophotometer (JACSCO-550). The fluorescence emission spectrum was recorded at room temperature excited by incident light of 320 nm with a fluorescence spectrophotometer (Hitachi, F-4500). Raman spectra were recorded by LabRam HR800.Photocatalysis reactivity measurements
˙OH radical reactions were performed as follows. 50 mg of the photocatalyst was suspended in 50 mL aqueous solution containing 0.01 M NaOH and 3 mM terephthalic acid. Before exposure to light irradiation, the suspension was stirred in dark for 30 min. Then 5 mL of the solution was taken out after 5 h under λ = 254 nm or 2.5 h under λ > 400 nm or 1 h under λ > 400 nm, and centrifuged for fluorescence spectrum measurements. During the photoreactions, no oxygen was bubbled into the suspension. A fluorescence spectrophotometer (Hitachi, F-4500) was used to measure the fluorescence signal of the 2-hydroxy terephthalic acid generated. The excitation light employed in recording fluorescence spectra was 320 nm.The continuous light source was a 300 W Xe lamp (Beijing Trusttech Co. Ltd, PLS-SXE-300UV). The wavelength of incident light in the photocatalytic reactions was satisfied by employing 400 nm long-pass glass filters. The monochromatic light was obtained by using a glass filter of 254 nm to cut light from a high-pressure mercury lamp (CHG-200).
3. Results and discussion
We prepared a series of Cs0.68Ti1.83O4−xNx by carefully controlling calcination temperature and the duration of treatment in a gaseous ammonia atmosphere. The value of x in Cs0.68Ti1.83O4−xNx prepared varies from 0.02 to 0.31. Evidenced by the binding energy of N 1s core electrons of Cs0.68Ti1.83O4−xNx at ca. 395 eV using X-ray photoelectron spectroscopy (see Fig. 1A), the chemical state of nitrogen species can be attributed to the substitutional nitrogen for lattice oxygen and thus formed Ti–N bonds in the Ti–O–Ti network.34 With the increase in the nitrogen dopant, the intrinsic absorption edge of Cs0.68Ti1.83O4−xNx can be continuously extended from ca. 356 nm to 472 nm as shown in Fig. 1B. The nearly parallel absorption edge of Cs0.68Ti1.83O4−xNx from each other indicates the nature of band-to-band photon excitation behavior, which is attributed to the homogeneous nitrogen doping in the bulk of Cs0.68Ti1.83O4−xNx.33,34 The bandgap of Cs0.68Ti1.83O4−xNx extrapolated from the plot of the Kubelka–Munk function versus the energy of the light decreases from 3.62 eV to 2.73 eV with increasing the nitrogen dopant. The narrowed bandgap is due to elevation of VB maximum by mixing N 2p states with O 2p states according to both experimental and theoretical results.34 This means that the VB width has correspondingly increased by a maximum 0.89 eV in Cs0.68Ti1.83O4−xNx (x = 0.31). The illumination of a modified band structure of Cs0.68Ti1.83O4−xNx is also given in Fig. 1C, where the VB maximum gradually shifts upwards and VB width gradually increases with the bandgap narrowing.34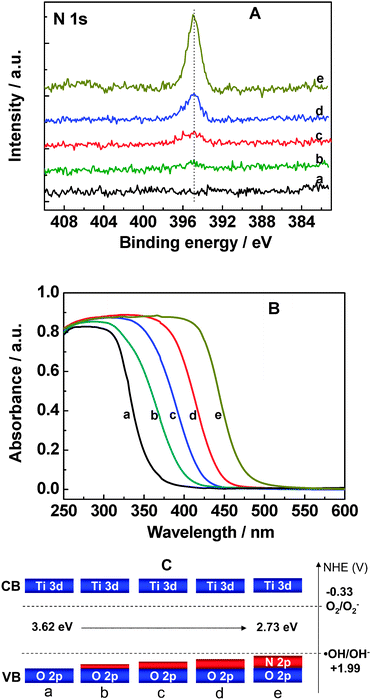 | ||
| Fig. 1 High resolution X-ray photoelectron spectra (A) of N 1s, UV-visible absorption spectra (B), and schematic (C) of revealed electronic structures of Cs0.68Ti1.83O4−xNx: a, x = 0; b, x = 0.02; c, x = 0.09; d, x = 0.18; e, x = 0.31. CB: conduction band; VB: valence band. | ||
In addition, it is known that the nitrogen dopant can cause oxygen vacancies in doped TiO2 in order to keep charge balance.31 As a result of a large amount of generated oxygen vacancies, a tail absorption band usually beyond 550 nm can be formed as observed in nitrogen doped TiO2 by Irie et al.31 In our case, no such band can be detected as shown in Fig. 1B, indicating that the amount of oxygen vacancies is formed in Cs0.68Ti1.83O4−xNx at a negligible level. In principle, however, the replacement of lattice O2− with N3− in Cs0.68Ti1.83O4 should also form some oxygen vacancies in order to keep charge balance, in particular at a high percentage of the nitrogen dopant. By analyzing the molar ratio of Cs+ to Ti4+ in Cs0.68Ti1.83O4−xNx, it is found that the ratio decreases by ca. 11.7% with the increase of x from zero to 0.31. It indicates a partial loss of Cs+ during the thermal treatment at an ammonia atmosphere, which can effectively compensate the charge imbalance caused by nitrogen substitution for lattice oxygen. This is the reason for the absence of oxygen vacancies in our case. Due to that the conduction band (CB) of Cs0.68Ti1.83O4 consists of major Ti 3d states and minor O 2p states, the partial loss of Cs+ will exert a negligible influence on the CB edges.34 The nearly identical redshift value of the absorption edge to that of VB maximum of Cs0.68Ti1.83O4−xNx (x = 0.31) in the previous work34 also validates this claim.
The fine-tuned VB structures in the above Cs0.68Ti1.83O4−xNx allow us to investigate the effects of absorbance, oxidation potential of holes and mobility of holes on the photo-oxidation reactivity of the photocatalyst. It is well established that photo-oxidation reaction mainly proceeds with active species ˙OH radicals besides the direct involvement of holes, which are generated from the reaction of holes with surface adsorbed water and hydroxyl groups.1,2 We estimated the photo-oxidation reactivity of Cs0.68Ti1.83O4−xNx by determining the amount of ˙OH radicals generated. It has been documented that the origin of these ˙OH radicals in basic medium is dominantly from the contribution of holes instead of electrons.34 As shown in Fig. 2, the dependence of ˙OH radicals generated on the amount of nitrogen dopant changes significantly upon various irradiation conditions. Under the irradiation of monochromatic light of 254 nm where no absorption factor is involved, the capability of generating ˙OH with Cs0.68Ti1.83O4−xNx drastically decreases before x = 0.09 and then slightly increases after x = 0.09 (see Fig. 2A). This indicates that the oxidation potential of holes is the key-controlling factor of the absolute reactivity—the stronger the oxidation power, the higher the reactivity; but the mobility of holes plays an obvious positive role in counteracting the negative role of the decreased oxidation power caused by doping: the higher the mobility, the better the counteracting effects. These results are also indicative that the combination of homogeneous doping to increase the VB width with other strategies to lower the VB edge might lead to drastically enhanced photocatalytic reactivity. Very recent results have validated this indication by combining quantum confinement effects with homogeneous sulfur doping in graphitic C3N4.36
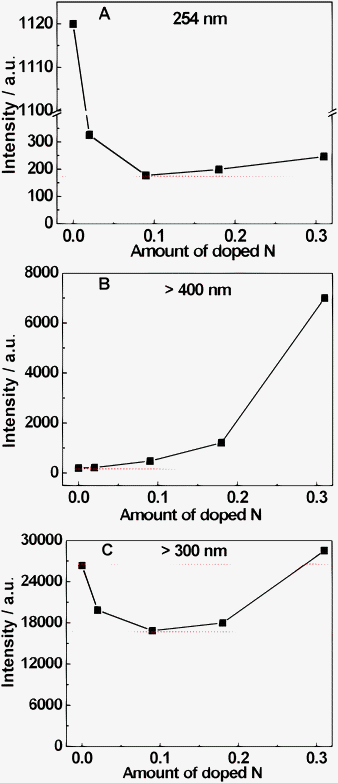 | ||
| Fig. 2 Dependence of the intensity of the fluorescence signal associated with 2-hydroxyterephthalic acid (TAOH) from the reaction of terephthalic acid (TA) with ˙OH radicals generated from the different Cs0.68Ti1.83O4−xNx on doped nitrogen under the irradiation of (A) 254 nm, (B) >400 nm and (C) >300 nm, respectively. | ||
We further examine the reactivity of Cs0.68Ti1.83O4−xNx under continuous spectrum irradiation (see Fig. 2B and C). Under λ > 400 nm, the amount of ˙OH radicals generated monotonously increases as nitrogen doping. Under λ > 300 nm, a similar trend to that under 254 nm is also observed. It is worthy of noting that the reactivity at x = 0.31 is much higher than that at x = 0.18 under both λ > 400 nm and λ > 300 nm. This can be rationalized as the synergistic effects of a wide absorption and wide VB. All these results clearly demonstrate that the optimal reactivity under continuous spectrum irradiation, which is also close to the practical application condition with solar light, can be acquired by maximizing the absorbance and VB width. Very interestingly, the above homogeneous nitrogen-concentration dependent photoreactivity is completely different from that of surface nitrogen doped TiO2−xNx (x ≤ 0.019),31 where both UV and visible light photocatalytic activity of TiO2−xNx drastically decayed with the increase of the nitrogen dopant. This clearly demonstrates the obvious advantage of homogeneous doping in developing efficient doped photocatalysts.
Finally but importantly, the possible influence of the surface structure change upon nitrogen doping on reactivity should be considered,32 though no obvious bulk structure change is observed.34Raman spectroscopy is sensitive to the surface structure.32 As shown in Fig. 3, obvious features can be derived from the Raman spectra of Cs0.68Ti1.83O4−xNx: a gradual intensity increase of the 289 cm−1 mode and gradual shift of the 435 cm−1 mode up to 445 cm−1 with the increase of the nitrogen dopant; while other Raman modes well retain. It is clear that the surface structure has been linearly changed upon introducing Ti–N bonds in the surface Ti–O–Ti network, which will exert its linear influence on the reactivity. Thus, the reactivity trend revealed in Fig. 2 is overwhelmingly determined by VB structures.
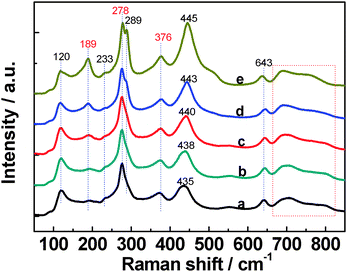 | ||
| Fig. 3 Raman spectra of Cs0.68Ti1.83O4−xNx: a, x = 0; b, x = 0.02; c, x = 0.09; d, x = 0.18; e, x = 0.31. | ||
4. Conclusions
This work provides new insights into the critical roles of rationally tailored VB related properties (absorbance, oxidative potential and mobility of holes) on acquiring the maximum photo-oxidation reactivity of Cs0.68Ti1.83O4−xNx by its VB under various irradiation conditions, which may offer some important implications in designing efficient photocatalysts.Acknowledgements
The financial support from Major Basic Research Program, Ministry of Science and Technology of China (No. 2009CB220001), NSFC (No. 50921004, 51002160, 21090343), Solar Energy Initiative of the Chinese Academy of Sciences is gratefully acknowledged. Gang Liu thanks the IMR SYNL-T.S. Kê Research Fellowship.References
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- A. L. Linsebigler, G. Q. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95, 735 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- H. Tada, T. Kiyonaga and S. Naya, Chem. Soc. Rev., 2009, 38, 1849 RSC.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782 CrossRef CAS.
- R. Abe, H. Takami, N. Murakami and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 7780 CrossRef CAS.
- X. L. Hu, G. S. Li and J. C. Yu, Langmuir, 2010, 26, 3031 CrossRef CAS.
- J. C. Yu, G. S. Li, X. C. Wang, X. L. Hu, C. W. Leung and Z. D. Zhang, Chem. Commun., 2006, 2717 RSC.
- J. Choi, H. Park and M. R. Hoffmann, J. Phys. Chem. C, 2010, 114, 783 CrossRef CAS.
- J. G. Yu, Q. J. Xiang and M. H. Zhou, Appl. Catal., B, 2009, 90, 595 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243 CrossRef CAS.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908 CrossRef CAS.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454 CrossRef CAS.
- Z. B. Lei, G. J. Ma, M. Y. Liu, W. S. You, H. J. Yan, G. P. Wu, T. Takata, M. Hara, K. Domen and C. Li, J. Catal., 2006, 237, 322 CrossRef CAS.
- C. Burda, Y. B. Lou, X. B. Chen, A. C. S. Samia, J. Stout and J. L. Gole, Nano Lett., 2003, 3, 1049 CrossRef CAS.
- X. F. Qiu, Y. X. Zhao and C. Burda, Adv. Mater., 2007, 19, 3995 CrossRef CAS.
- W. Balcerski, S. Y. Ryu and M. R. Hoffmann, J. Phys. Chem. C, 2007, 111, 15357 CrossRef CAS.
- K. Maeda, Y. Shimodaira, B. Lee, K. Teramura, D. Lu, H. Obayashi and K. Domen, J. Phys. Chem. C, 2007, 111, 18264 CrossRef CAS.
- H. X. Li, J. X. Li and Y. I. Huo, J. Phys. Chem. B, 2006, 110, 1559 CrossRef CAS.
- W. Zhao, H. Ma, C. C. Chen, J. C. Zhao and Z. G. Shuai, J. Am. Chem. Soc., 2004, 126, 4782 CrossRef CAS.
- S. In, A. Orlov, R. Berg, F. Garcia, S. Pedrosa-Jimenez, M. S. Tikhov, D. S. Wright and R. M. Lambert, J. Am. Chem. Soc., 2007, 129, 13790 CrossRef CAS.
- G. Liu, Y. N. Zhao, C. H. Sun, F. Li, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4516 CrossRef CAS.
- W. Ho, J. C. Yu and S. Lee, Chem. Commun., 2006, 1115 RSC.
- S. Livraghi, M. C. Paganini, E. Giamello, A. Selloni, C. Di Valentin and G. Pacchioni, J. Am. Chem. Soc., 2006, 128, 15666 CrossRef CAS.
- E. Martinez-Ferrero, Y. Sakatani, C. Boissiere, D. Grosso, A. Fuertes, J. Fraxedas and C. Sanchez, Adv. Funct. Mater., 2007, 17, 3348 CrossRef CAS.
- B. Chen and C. Burd, J. Am. Chem. Soc., 2008, 130, 5018 CrossRef CAS.
- J. Wang, D. N. Tafen, J. P. Lewis, Z. L. Hong, A. Manivannan, M. J. Zhi, M. Li and N. Q. Wu, J. Am. Chem. Soc., 2009, 131, 12290 CrossRef CAS.
- N. Serpone, J. Phys. Chem. B, 2006, 110, 24287 CrossRef CAS.
- X. K. Li, N. Kikugawa and J. H. Ye, Adv. Mater., 2008, 20, 3816 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483 CrossRef CAS.
- M. Batzill, E. H. Morales and U. Diebold, Phys. Rev. Lett., 2006, 96 Search PubMed.
- G. Liu, L. Z. Wang, H. G. Yang, H. M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831 RSC.
- G. Liu, L. Z. Wang, C. H. Sun, S. C. Smith, H. M. Cheng and G. Q. Lu, Chem. Mater., 2009, 21, 1266 CrossRef CAS.
- T. Sasaki, M. Watanabe, H. Hashizume, H. Yamada and H. Nakazawa, J. Am. Chem. Soc., 1996, 118, 8329 CrossRef CAS.
- G. Liu, P. Niu, C. H. Sun, S. Smith, Z. G. Chen, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |