Understanding the effect of thermal treatments on the structure of CuAu/SiO2 catalysts and their performance in propene oxidation†
Charlotte L.
Bracey
a,
Albert F.
Carley
a,
Jennifer K.
Edwards
a,
Peter R.
Ellis
b and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, UK CF10 3AT. E-mail: hutch@cardiff.ac.uk
bJohnson Matthey Technology Centre, Blount's Court, Sonning Common, Reading, UK RG4 9NH
First published on 31st January 2011
Abstract
The synthesis and catalytic application of supported CuAu is discussed. Different thermal treatments of a dried precursor of copper nitrate and tetrachloroauric acid on silica lead to catalysts with significantly different structures and properties. Direct calcination gives a catalyst which contains very large gold ensembles with minimal interaction with the copper present. Hydrogen reduction of the dried precursor leads to the formation of copper–gold alloy nanoparticles. Subsequent high temperature calcination de-alloys the copper from the gold to a significant extent. The presence of gold stabilises the formation of Cu+ on this catalyst. The activity and selectivity observed in the oxidation of propene with molecular oxygen, with or without co-fed hydrogen, depends on the pre-treatment, reaction conditions and the ratio of copper to gold in the catalyst. A number of different catalytic active sites are identified and discussed.
Introduction
The structure and properties of nanoalloy particles is a subject which has attracted much recent attention.1,2 Bimetallic copper–gold systems are especially interesting3 as a number of bulk alloy phases are known,4 but the structures of small particles can be complex5 and affected by temperature and interaction with a support or gas environment. The presence of even low levels of copper can significantly alter the structure of small gold particles.6Oxidation of propene can give rise to a number of products (Scheme 1) depending on the catalyst and reaction conditions used. Many of the products are valuable intermediates, such as propene oxide or acrolein. An alternative process to the chlorhydrin route for propene oxide production is desirable for environmental reasons,7 as the chlorhydrin process generates significant amounts of chlorinated waste.8 At the same time, considerable effort is being devoted to the sustainable production of acrolein from glycerol9 derived as a by-product of biodiesel manufacture. Acrolein is currently made by air oxidation of propene using bismuth molybdate catalysts;10 the replacement of these heavy metal-containing catalysts with a high selectivity alternative would be both economically and environmentally desirable. High selectivity to a single product is vital if the reaction is to find real industrial application. Propene oxidation is also relevant as part of the mechanism of propane conversion to valuable products.11
![Oxidation reactions of propene. Reactions indicated by [H] are hydrogenation reactions which could occur in a mixed H2–O2 feed.](/image/article/2011/CY/c0cy00003e/c0cy00003e-s1.gif) | ||
| Scheme 1 Oxidation reactions of propene. Reactions indicated by [H] are hydrogenation reactions which could occur in a mixed H2–O2 feed. | ||
Understanding the oxidation of propene over CuAu catalysts is an interesting challenge as both Cu12 and Au13catalysts are active for this reaction. Copper catalysts can be used in reduced or oxidised form. A pre-reduced Cu/SiO2 catalyst was found to give a mixture of CO2, acrolein and propene oxide when tested for propene oxidation with molecular oxygen between 200 and 400 °C. The selectivity changed significantly with temperature, propene oxide being observed at lower temperatures whilst acrolein production reached a maximum above 300 °C.12 A Cu/SBA-15 catalyst prepared using copper phthalocyanin14 as precursor also gave acrolein with moderate selectivity (20–40%) at high conversion (up to 90%). Other copper precursors also gave active catalysts, whilst catalysts not supported on silica (such as alumina, titania or zeolite) merely burnt the propene to give CO2. The copper in these catalysts was present as oxidised species. These two results suggest that there are differences in performance between oxidised and reduced copper catalysts.
Gold catalysts are also active for the oxidation of propene,15 the key breakthrough being the report by Haruta in 1998 that Au/TiO2 catalysts are selective for the formation of propene oxide at 30–80 °C16 when hydrogen was co-fed to the catalyst. The characteristic of these catalysts is a high selectivity to propene oxide at low propene conversion (typically 1–2%). A Cu/TiO2 catalyst tested by Haruta and co-workers was found to give only CO2. When the reaction temperature was increased, with titanosilicates as supports, propanal was formed over gold catalysts,17 possibly by sequential reaction of propene oxide. These initial observations have led to a large amount of work on Au catalysts for propene epoxidation, which has been reviewed.15,18 Variables such as the nature of the support19 and catalyst preparation method20 are important in determining catalytic performance. Co-feeding of hydrogen is necessary for propene oxide to form. This observation suggests that a hydroperoxide (OOH) intermediate is responsible for the selective oxidation observed. Gold catalysts possess high selectivity for hydrogen peroxide formation from hydrogen and oxygen,21 which supports this view.
CuAu catalysts have been previously examined for the oxidation of propene. Sinfelt and Barnett22 reported the production of acrolein with 70% selectivity at conversions between 10% and 40%. They used a catalyst with a novel reduction-calcination treatment which resulted in bimetallic nanoparticles which had a degree of core-shell structure.23 Exposure to air gave a copper-rich surface, whilst exposure to hydrogen gave a gold-rich surface. Llorca et al. used a CuAu/TiO2 catalyst prepared by supporting thiol-capped nanoparticles on anatase to oxidise propene using N2O as oxidant.24 They found the selectivity to propene oxide decreased with increasing calcination temperature, which they ascribed to increased amounts of copper oxide in the catalysts. The selectivity to carbon oxides and to acrolein increased with increasing calcination temperature. The bimetallic catalysts were also shown to be more active than the corresponding monometallic Au/TiO2 catalyst, and to exhibit higher selectivity to propene oxide. The catalysts made from pre-formed nanoparticles were significantly more active than those prepared by impregnation using copper chloride and tetrachloroauric acid followed by reduction in hydrogen at 400 °C.25 These impregnated catalysts were shown to contain CuAu alloy particles. CuAu catalysts are also active for the oxidation of CO26,27 and benzyl alcohol,28 and for the chlorination of alkenes.29
In this paper we examine the effects of the copper–gold ratio, calcination method and the co-feeding of hydrogen in the reaction on the structure and activity of AuCu/SiO2 catalysts in the oxidation of propene.
Experimental
Materials
Silica was supplied by Grace (reference number SP550-10022, BET surface area 340 m2 g−1). Copper nitrate hemipentahydrate was supplied by Alfa Aesar and tetrachloroauric acid solution (41% Au) by Johnson Matthey. All were used as received.Catalyst preparation
The catalyst precursor was prepared by co-impregnation of copper nitrate and tetrachloroauric acid into silica by the incipient wetness method. The water pore volume of the silica was measured as 1.5 mL g−1. A solution of copper nitrate and tetrachloroauric acid was prepared by dissolving the reagents in the appropriate amount of water to fill the pores of the silica. The silica was added to the solution, stirred well, then transferred to a glass beaker and dried at 105 °C for 3 h. This gave a catalyst precursor which was treated thermally in different ways.The catalyst precursor was calcined directly on a silica tray by heating in static air to 400 °C at 10 °C min−1, holding at 400 °C for 2 h then cooling to room temperature. Reduction of the catalyst precursor was performed in a tube furnace under flowing 5% H2/N2 gas mixture, heating to 315 °C at 10 °C min−1, holding for 2 h at 315 °C then cooling to room temperature. Subsequent calcination of the reduced material was by heating in flowing air to 675 °C at 10 °C min−1, holding at 675 °C for 16 h then cooling to room temperature.22
Characterisation
The metal contents of the catalysts were determined by Inductively Coupled Plasma (ICP-ES) following digestion in aqua regia. XRD patterns were recorded on a Bruker A-500 diffractometer, with crystallite sizes measured by Rietveld refinement. In situXRD analyses were performed using a Panalytical XPert powder X-ray diffractometer equipped with an in situ Anton Parr reaction chamber. Evolved Gas Analysis (EGA) was performed using a Netzsch TG-MS instrument. BET analyses were performed using a five point method on a Quantachrome Autosorb instrument. XPS measurements were made on a Kratos Axis Ultra DLD spectrometer using monochromatic Al-Kα radiation. Samples were mounted using double-sided adhesive tape, and binding energies referenced to the C(1s) binding energy of adventitious carbon contamination taken to be 284.7 eV. Visible spectra were obtained using a Datacolor International Spectraflash 600+ colorimeter. Scanning Electron Microscopy (SEM) was performed using a Carl Zeiss Evo-40 scanning electron microscope fitted with an Oxford Instruments energy dispersive X-ray elemental microanalysis. TEM images were acquired using a Tecnai F20. Powder samples were set in resin and sectioned to give thin slices for analysis, or the powder was dusted onto a holey carbon grid. Nickel TEM grids were used to avoid interference between Cu grids and the Cu in the samples. Visible microscope images were obtained using a Nikon SMZ-U microscope with a maximum magnification of around 50×.Catalyst testing
Propene oxidation was performed using a flow microreactor and the products were analysed using on-line gas chromatography. The catalyst (0.2 g) was heated in a flow of reactant gases with a total space velocity of 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 h−1. Initial experiments were performed without hydrogen addition under rich conditions to minimise over-oxidation. In this case the reactant gas mixture was C3H6:O2:He = 22:9:69. Later experiments were performed with hydrogen addition using the same gas mixture as Haruta's group16 (H2:C3H6:O2:He = 1:1:1:716).
500 h−1. Initial experiments were performed without hydrogen addition under rich conditions to minimise over-oxidation. In this case the reactant gas mixture was C3H6:O2:He = 22:9:69. Later experiments were performed with hydrogen addition using the same gas mixture as Haruta's group16 (H2:C3H6:O2:He = 1:1:1:716).
Results and discussion
Catalyst preparation and characterisation
The dried copper–gold on silica catalyst precursor was prepared by the incipient wetness method. This method is often favoured for its simplicity and reliability, but it does not give as high dispersion as, for example, deposition methods. The catalyst precursor was then treated thermally, either by direct calcination in air, by reduction using hydrogen, or by a sequential reduction-high temperature calcination method. The metal contents of the materials are shown in the supplementary information. The structures of the three resulting catalysts were investigated by a number of techniques.:Au = 3:1), is confirmed by XPS (Fig. 3) by the presence of a distinctive shake-up satellite structure. The degree of interaction between the copper and gold in the calcined catalysts was investigated by visible spectroscopy. Visible spectroscopy suggests that the interaction is minimal, as the position of the plasmon band is not shifted when the copper–gold ratio is changed (Fig. 4).
 | ||
| Fig. 1 XRD spectra of (i) Au/SiO2; (ii) Au3Cu/SiO2; (iii) AuCu/SiO2; (iv) AuCu3/SiO2 and (v) Cu/SiO2 after different thermal treatments: (a) calcined in air at 400 °C; (b) reduced in H2/N2 at 315 °C; (c) reduced in H2/N2 at 315 °C then calcined in air at 675 °C. The reflections for Au metal are labelled at the top of each figure, whilst the reflections for relevant Cu species are labelled at the bottom of the figures: (a) CuO; (b) Cu2O and (c) CuO. (d) shows an expansion of the XRD spectrum of AuCu3/SiO2 reduced at 315 °C in H2/N2. The location of reflections due to Cu and Au metal are indicated by bold lines, and discrete alloy phases are indicated by circles. | ||
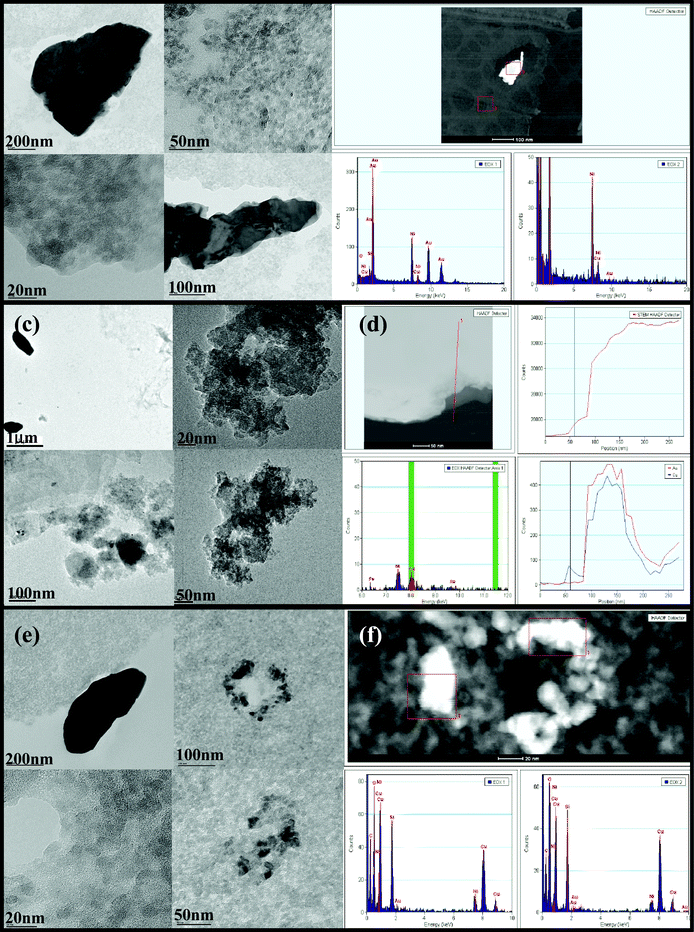 | ||
| Fig. 2 (a) TEM bright field images and (b) EDX analysis of CuAu/SiO2 calcined at 400 °C in air. (c) TEM bright field images and (d) EDX linescans of CuAu/SiO2 reduced at 315 °C in H2/N2. (e) TEM bright field images and (f) EDX analysis of CuAu/SiO2 reduced at 315 °C in H2/N2 and calcined at 675 °C in air. | ||
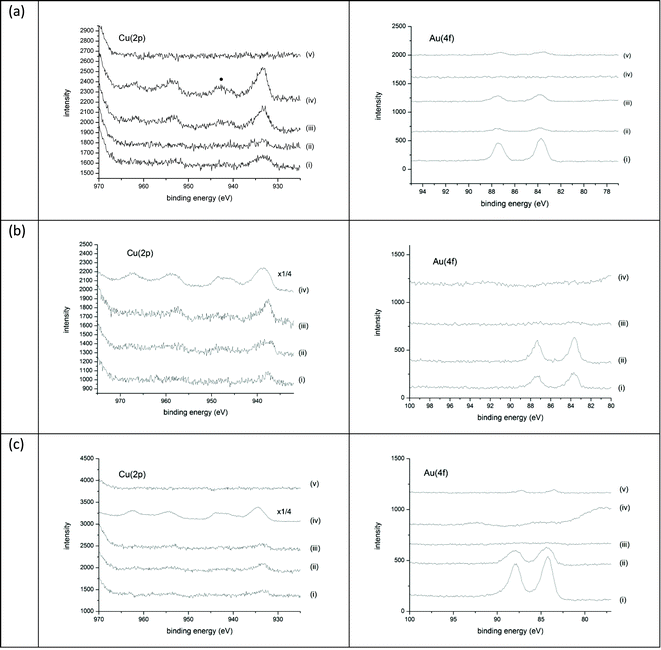 | ||
| Fig. 3 XPS analysis showing Cu 2p (left) and Au 4f (right) spectra for: (a) calcined catalysts, (b) reduced catalysts and (c) reduced and calcined catalysts: (i) CuAu3/SiO2, (ii) CuAu/SiO2, (iii) Cu3Au/SiO2, (iv) Cu/SiO2, (v) Au/SiO2. The position of the Cu2+ satellite peak in the Cu 2p spectra is marked with a dot (•) in curve (a) (iv). | ||
 | ||
| Fig. 4 Variation in visible spectrum peak maximum with copper gold ratio for calcined (circles), reduced (squares) and reduced-calcined (triangles) AuCu/SiO2 catalysts. | ||
The surface Cu:Au ratio measured by XPS (ESI, Table S4)† increases monotomically with the expected bulk ratio, but the catalysts exhibit a significant surface enrichment in Cu. Since the ICP results confirm a bulk composition close to the nominal value, this observation could arise for two reasons: (i) the particles are bimetallic and exhibit a core-shell structure with a gold core, or (ii) the copper is more highly dispersed on the support than the gold. The TEM and XRD results are consistent with the latter.
The Cu(2p) XP spectrum for the reduced Cu/SiO2 catalyst (Fig. 3b curve (d)) is remarkably intense compared with that for the calcined material (Fig. 3a curve (d)), which may reflect a much higher metal dispersion. This may be a direct result of the reduction step, since a similar increase is observed for the reduced and calcined catalyst. Not surprisingly, the Cu2+ satellites are either very weak or insignificant (Fig. 3b, curves (a)–(c)). The derived Cu:Au ratios indicate an enhancement in the surface Cu concentration, and this is particularly so for the sample with the highest nominal Cu content (ESI, Table S4);† this may reflect the presence of highly mobile metallic Cu during the reduction procedure.
As with the reduced materials, XPS does not observe a strong satellite peak for Cu2+ in the Cu 2p spectrum (Fig. 3c). However, the binding energy suggests an oxidised Cu species, not Cu metal. This is not without precedent; Au has previously been shown to stabilise the formation of Cu2O in alloy nanoparticles which have been prepared by oxidation of a CuAu alloy.30,31 In contrast to the calcined samples and the reduced catalysts, the XPS-derived surface Cu:Au ratios are close to the expected bulk ratios, except for the high Cu content (Cu:Au = 3:1) sample.
It is of interest to investigate the processes which lead to these different structures. In the direct calcination, copper nitrate decomposes to copper oxide at a lower temperature than that at which the chloride is removed from gold (ESI, Fig. S4a).† However, the gold and copper oxide do not interact, and so the gold forms large ensembles (in contrast to the highly dispersed CuO species) as it does not interact strongly with the silica either. When the decomposition occurs under hydrogen, again the copper is decomposed first, with loss of NO as the major product (ESI, Fig. S4b).† However, this leads to the formation of copper metal, which interacts strongly with metallic gold as it is formed to give an alloy. The copper–gold alloy phase is very stable, and needs calcination at high temperatures (e.g. the 675 °C used in this study) to decompose it to copper oxide and gold metal. TPO analysis of a bulk CuAu alloy26 found that the oxidation of the copper present proceeded between 650 °C and 800 °C, forming Au metal and CuO.
The synthesis of the reduced-calcined catalyst was investigated by in situXRD (Fig. 5). During the reduction step, the main reflections of gold metal are observed throughout the experiment. It is interesting to see that gold metal is already present at 150 °C, possibly due to HAuCl4 decomposition during the drying step. This could be the source of the unalloyed Au particles observed. Changes to the preparation to reduce the copper at lower temperatures could lead to greater degrees of alloying and so better catalysts. EGA (ESI, Fig. S4b)† shows that NO is produced at 100 °C on reduction, with HCl from the decomposition of the gold precursor observed above 200 °C. By XRD, however, broad reflections were observed at higher 2θ than the gold reflection starting from 275 °C (Fig. 5a). These are assigned to the formation of copper–gold alloy species. The reduced catalyst therefore contains a mixture of gold and copper–gold species. The intensity of the broad XRD reflection increases as the temperature and reaction time increase. It is possible that changes in the temperature and times of the reduction could be used to ‘tune’ the CuAu nanoparticles produced. On oxidation (Fig. 5b), the alloy reflections disappear, and the final material contains crystalline gold. The fate of the copper is not clear from the in situ analyses, although ex situXRD shows small reflections due to CuO, which may be related to particles not interacting with gold. XPS shows the presence of Cu+ on the surface of the catalyst (Fig. 3c). This has been observed before in Cu-based oxidation catalysis,32 where a copper–ceria catalyst was found to contain Cu+ after contact with CO for thirty minutes.
 | ||
| Fig. 5 (a) In situXRD spectra taken during reduction of the CuAu/SiO2 precursor. The spectra measured during heating from room temperature to 315 °C are shown in grey, and those measured during the hold at 315 °C are shown in black. (b) In situXRD spectra taken during calcination of the reduced CuAu/SiO2. The spectra measured during the ramp to 675 °C are shown in grey, and those measured at 675 °C are shown in black. In both figures, the positions of reflections due to Au metal and CuAu alloy phases are indicated. | ||
Propene epoxidation
The activity and selectivity of these catalysts in the oxidation of propene with or without hydrogen addition was very much affected by their composition and thermal pre-treatments (Tables 1 and 2 and supplementary information). In general, the catalysts exhibited a low conversion of propene (<2%) due to the high gas space velocity used in the experiments. Most did not make any C3 products at lower temperature (ca. 200 °C) but the yields of acrolein and/or propene oxide increased with increasing temperature (up to 300 °C). The conversion increased on co-feeding of hydrogen, and in some cases this changed the product distribution.| Heat treatment | Cu/Au molar | Propene conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|
| CO2 | Acrolein | Propene oxide | Ethanal | |||
| Calcination | 0/1 | 0.01 | 0 | 100 | 0 | 0 |
| 1/3 | 0.03 | 18 | 44 | 39 | 0 | |
| 1/1 | 0.03 | 23 | 42 | 33 | 0 | |
| 3/1 | 0.04 | 19 | 46 | 35 | 0 | |
| 1/0 | 0.23 | 19 | 81 | 0 | 0 | |
| Reduction | 0/1 | 0.03 | 18 | 35 | 46 | 0 |
| 1/3 | 0.13 | 21 | 22 | 70 | 0 | |
| 1/1 | 0.05 | 14 | 23 | 63 | 0 | |
| 3/1 | 0.03 | 19 | 81 | 0 | 0 | |
| 1/0 | 0.06 | 26 | 47 | 27 | 0 |
| Reduction and calcination | 0/1 | <0.01 | 100 | 0 | 0 | 0 |
| 1/3 | 0.05 | 18 | 59 | 23 | 0 | |
| 1/1 | 0.20 | 19 | 81 | 0 | 0 | |
| 3/1 | 0.03 | 16 | 46 | 38 | 0 | |
| 1/0 | <0.01 | 100 | 0 | 0 | 0 |
| Heat treatment | Cu/Au molar | Propene conversion (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|
| CO2 | Acrolein | Propene oxide | Ethanal | |||
| Calcination | 0/1 | 0.02 | 100 | 0 | 0 | 0 |
| 1/3 | 0.13 | 19 | 53 | 29 | 0 | |
| 1/1 | 0.06 | 11 | 59 | 30 | 0 | |
| 3/1 | 0.34 | 23 | 77 | 0 | 0 | |
| 1/0 | 0.19 | 19 | 35 | 25 | 21 | |
| Reduction | 0/1 | 0.29 | 16 | 84 | 0 | 0 |
| 1/3 | 0.41 | 6 | 69 | 25 | 0 | |
| 1/1 | 0.28 | 36 | 41 | 23 | 0 | |
| 3/1 | 0.63 | 12 | 81 | 7 | 0 | |
| 1/0 | 0.36 | 11 | 50 | 28 | 17 |
| Reduction and calcination | 0/1 | 0.06 | 100 | 0 | 0 | 0 |
| 1/3 | 0.27 | 17 | 83 | 0 | 0 | |
| 1/1 | 0.20 | 18 | 82 | 0 | 0 | |
| 3/1 | 0.07 | 33 | 67 | 0 | 0 | |
| 1/0 | 2.36 | 68 | 27 | 0 | 5 |
| Cu/Au | 0/1 | 1/3 | 1/1 | 3/1 | 1/0 |
|---|---|---|---|---|---|
| a Two CuAu phases identified, one very gold rich and the other more mixed. The gold rich phase has the larger crystallite size. b Crystallite sizes could not be determined due to the complex nature of the material (see Fig. 1d). | |||||
| Calcined | Au | Au | Au | Au | CuO |
| 53 nm | 64 nm | 54 nm | 45 nm | 21 nm | |
| Reduced | Au | CuAu a | CuAu a | CuAu b | Cu2O |
| 60 nm | 24 nm & 8.5 nm | 14 nm & 9.2 nm | 4.3 nm | ||
| Reduced and calcined | Au | Au | Au | Au | CuO |
| 60 nm | 59 nm | 54 nm | 49 nm | 9 nm |
| Cu/Au ratio | Calcined | Reduced | Reduced and calcined |
|---|---|---|---|
| Fresh catalysts | |||
| 1/3 | 0.068 | 0.079 | 0.054 |
| 1/1 | 0.071 | 0.063 | 0.061 |
| 3/1 | 0.110 | 0.045 | 0.082 |
Directly calcined monometallic catalysts were poorly active and tended to produce small amounts of CO2, acrolein and propene oxide.
:Au ratio after reaction, especially after reaction with added hydrogen (ESI, Table S4).† As with the directly calcined materials, propene oxidation led to carbon deposition (Table 4) which was greatest after reaction with added hydrogen.
Considering the major oxidation products, acrolein requires C–H bond cleavage whilst propene oxide forms viaactivation of the carbon–carbon double bond. This in turn suggests that the adsorption mode of the propene molecule onto the surface is different in each case. Significantly, Nijhuis et al. demonstrated that propene binds through the carbon–carbon double bond on gold nanoparticles.40 Monometallic gold catalysts are active for the production of propene oxide15 in the presence of hydrogen. Production of acrolein over copper(I) oxide, meanwhile, was found to proceed via an allyl alkoxide-type intermediate41 which was formed by abstraction of a hydrogen atom from propene. Changes in the product distribution, therefore, reflect changes in the nature of the active site or sites on the catalyst through different binding modes and therefore reaction pathways. The nature of the active sites is discussed in the next section.
Discussion
The catalytic test results suggest the existence of a number of different types of active site:• A copper(II) active site, which possesses low activity and selectivity to both propene oxide and acrolein. Co-feeding of hydrogen and promotion with gold increases the selectivity to acrolein. Surface science studies41 concluded that CuO favoured the combustion of propene.
• A copper(I) active site, observed in the reduced and calcined catalyst. This has high selectivity to acrolein, as reported for Cu(I) previously.41 Zhu and co-workers found that their K-promoted Cu/SiO2 catalyst contained Cu(I) and gave improved selectivity to propene oxide over acrolein at 250–300 °C.42 This was ascribed as Cu(I) being less active for the subsequent oxidation of propene oxide to acrolein and carbon oxides. The other feature of the reduced and calcined catalyst is its stability, with little hysteresis observed with temperature.
• A bimetallic CuAu active site, which is very active but tends to form many products. XRD shows that these catalysts contain many different CuAu species. There is also the possibility that the reduced catalyst contains some Cu+, which contributes to the mixed selectivity. The activity of this site is increased markedly by the co-feeding of hydrogen. The reason for this enhancement is not clear but may involve a change in reaction mechanism or oxidising species in the presence of hydrogen. Llorca et al.24 found a mixed selectivity for CuAu/TiO2 catalysts prepared using thiol-stabilised nanoparticles, and higher activity with increased copper content, both of which are in agreement with our results, even though they used N2O rather than O2 or H2–O2 as oxidant.
• A copper(0) active site which has low activity and a mixed selectivity. This is in agreement with the results of Vaughan, who produced acrolein, propene oxide and CO2 over a reduced Cu/SiO2 catalyst.12 Their characterisation data confirmed a Cu(0) active site; the differences between their results and those in this work are likely to be related to the higher dispersion in their catalyst resulting from the microemulsion preparation route.
• A gold metal active site, which tended to give more CO2 than the copper-containing sites, especially in the presence of co-fed hydrogen. This is unexpected in the light of Haruta's original results16 for the co-feeding of hydrogen to Au/TiO2 catalysts. This may highlight the role of the support in the Au/TiO2 system.43
Although the dispersion of these catalysts is hard to measure directly, XRD crystallite sizes can be used to give an indication of the average crystallite size. It should also be noted that some of these materials contain a significant amount of amorphous material. However, the catalysts which have the smallest crystallites (Table 3) are those prepared by direct reduction, and these give the highest propene conversion.
XPS spectra showed that carbon laydown occurred on the catalysts during propene oxidation reactions. Interestingly, more carbon was observed when the reaction was carried out in the presence of hydrogen. This may be related to the formation of surface hydrocarbon-like species through hydrogenation of tightly-bound intermediates. The exception to this is the reduced-only catalysts, where carbon levels are similar with or without hydrogen co-feeding. Unfortunately, XPS was not able to give any information on the carbon speciation.
Conclusions
This paper has described and discussed the preparation chemistry, structure and catalytic performance of bimetallic copper–gold/silica catalysts prepared using different thermal processes.• Direct calcination of the precursor comprising dried copper nitrate-tetrachloroauric acid on silica gives micron-sized copper–gold alloy particles containing very low levels of copper along with dispersed copper(II) oxide and copper(II) ions on the silica support. In this case, the copper nitrate precursor decomposes to copper oxide, which then does not interact with the large gold particles formed when tetrachloroauric acid is decomposed. These catalysts generally have low activity and give some selectivity to acrolein, but propene oxide and carbon dioxide are also observed.
• Direct reduction of the dried precursor gives a dispersed copper–gold alloy on the silica support. The difference in the preparation is that the copper forms metal by reduction immediately after decomposition, which is then able to react with the gold formed on reduction of the tetrachloroauric acid. The bimetallic copper–gold site is the most active of any observed in this work, but unfortunately has a modest selectivity.
• High temperature calcination of the reduced material leads to a significant level of de-alloying of the copper and gold. There is some evidence that gold is able to stabilise copper(I) against further oxidation. These catalysts have moderate activity, but good selectivity to acrolein, especially with co-feeding of hydrogen.
• Hydrogen co-feeding increases activity, presumably by increasing the surface coverage of active oxygen species. Different oxidising species may be responsible for the selectivities observed with different catalysts under different operating regimes.
Acknowledgements
The authors would like to thank Godson Nnorom-Junior for ICP analysis, Hoi Jobson and James McNaught for XRD analysis and Dr Gregory Goodlet and Dr Jingshan Dong for TEM analysis. CLB acknowledges Johnson Matthey for a PhD studentship. We also thank Drs Emma Schofield, David Thompsett and Gemma Moxham for useful discussions.References
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS.
- “Nanoalloys: From Theory to Application”, Faraday Discussions, 2008, 138.
- C. L. Bracey, P. R. Ellis and G. J. Hutchings, Chem. Soc. Rev., 2009, 38, 2231–43 RSC.
- M. Hansen and K. Anderko, Constitution of Binary Alloys, McGraw-Hill, 1958, pp. 198–203 Search PubMed.
- R. E. Schaak, A. K. Sra, B. M. Leonard, R. E. Cable, J. C. Bauer, Y.-F. Han, J. Means, W. Teizer, Y. Vasquez and E. S. Funck, J. Am. Chem. Soc., 2005, 127, 3506–15 CrossRef CAS.
- S. Darby, T. V. Mortimer-Jones, R. L. Johnston and C. Roberts, J. Chem. Phys., 2002, 116, 1536–1550 CrossRef CAS.
- T. A. Nijhuis, M. Makkee, J. A. Moulijn and B. M. Weckhuysen, Ind. Eng. Chem. Res., 2006, 45, 3447–59 CrossRef CAS.
- D. L. Trent, Propylene Oxide, in Kirk-Othmer Dictionary of Chemical Technology, ed. J. I. Kroschwitz and M. Howe-Grant, John Wiley & Sons, New York, 4th edn, 1996, vol. 1, pp. 232–51.20 and 271–301 Search PubMed.
- For example (a) S.-H. Chai, H.-P. Weng, Y. Liang and B.-Q. Xu, Appl. Catal., A, 2009, 353, 213–222 CrossRef CAS; (b) L. Ott, M. Bicker and H. Vogel, Green Chem., 2006, 8, 214–220 RSC.
- W. G. Etzkorn, J. J. Kurland and W. D. Neilsen, Acrolein and Derivatives, in Kirk-Othmer Dictionary of Chemical Technology, ed. J. I. Kroschwitz and M. Howe-Grant, John Wiley & Sons, New York, 4th edn, 1996, vol. 1, pp. 232–51 Search PubMed.
- M. Gasior, B. Grzybowska, K. Samson, M. Ruszel and J. Haber, Catal. Today, 2004, 91–92, 131–135 CrossRef CAS.
- O. P. H. Vaughan, G. Kyriakou, N. Macleod, M. Tikhov and R. M. Lambert, J. Catal., 2005, 236, 401–404 CrossRef CAS.
- T. A. Nijhuis, B. J. Huizinga, M. Makkee and J. A. Moulijn, Ind. Eng. Chem. Res., 1999, 38, 884–891 CrossRef CAS.
- H. Tüysüz, J. Llamas Galilea and F. Schüth, Catal. Lett., 2009, 131, 49–53 CrossRef.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–24 CrossRef CAS.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–75 CrossRef CAS.
- B. S. Uphade, S. Tsubota, T. Hayashi and M. Haruta, Chem. Lett., 1998, 1277–8 CrossRef CAS.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Top. Catal., 2004, 29, 95–102 CrossRef.
- N. Weiher, A. M. Beesley, N. Tsapatsaris, L. Delannoy, C. Louis, J. A. van Bokhoven and S. L. M. Schroeder, J. Am. Chem. Soc., 2007, 129, 2240–1 CrossRef CAS.
- T. V. Choudhary and D. W. Goodman, Res. Chem. Intermed., 2002, 21, 25–34 CAS.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058–9 RSC.
- J. Sinfelt and A. E. Barnett, US Patent 3989674, 1976.
- G. Meitzner, G. H. Via, F. W. Lytle and J. H. Sinfelt, J. Chem. Phys., 1985, 83, 4793–99 CrossRef CAS.
- J. Llorca, M. Dominguez, C. Ledesma, R. J. Chimentao, F. Medina, J. Sueiras, I. Angurell, M. Seco and O. Rossell, J. Catal., 2008, 258, 187–98 CrossRef CAS.
- R. J. Chimentao, F. Medina, J. L. G. Fierro, J. Llorca, J. E. Sueiras, Y. Cesteros and P. Salagre, J. Mol. Catal. A: Chem., 2007, 274, 159 CrossRef CAS.
- S. Kameoka and A. P. Tsai, Catal. Today, 2008, 132, 88–92 CrossRef CAS.
- X. Liu, A. Wang, X. Wang, C.-Y. Mou and T. Zhang, Chem. Commun., 2008, 3187–9 RSC.
- C. Della Pina, E. Falletta and M. Rossi, J. Catal., 2008, 260, 384–6 CrossRef CAS.
- (a) Dutch Patent Application 6411608, 1964; (b) French Patent 1509768, 1967; (c) S. Wang, B. Shen and Q. Song, Catal. Lett., 2009, 134, 102–9.
- G.-W. Zhou, J. A. Eastman, R. C. Birtcher, P. M. Baldo, J. E. Pearson, L. J. Thompson, L. Wang and J. C. Yang, J. Appl. Phys., 2007, 101, 033521 CrossRef.
- K. Koga and D. Zubia, J. Phys. Chem. C, 2008, 112, 2079–85 CrossRef CAS.
- D. Gamarra, G. Munuera, A. B. Hungria, M. Fernandez-Garcia, J. C. Conesa, P. A. Midgley, X. Q. Wang, J. C. Hanson, J. A. Rodriguez and A. Martinez-Arias, J. Phys. Chem. C, 2007, 111, 11026–38 CrossRef CAS.
- D. Gamarra, C. Belver, M. Fernandez-Garcia and A. Martinez-Arias, J. Am. Chem. Soc., 2007, 129, 12064–5 CrossRef CAS.
- A. M. Joshi, W. N. Delglass and K. T. Thomson, J. Phys. Chem. C, 2007, 111, 7384–95 CrossRef CAS.
- J. K. Edwards, B. Solsona, E. Ntainjua N, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–41 CrossRef CAS.
- J. A. Lopez-Sanchez and D. Lennon, Appl. Catal., A, 2005, 291, 230–7 CrossRef CAS.
- B. Bridier, N. Lopez and J. Perez-Ramirez, J. Catal., 2010, 269, 80–92 CrossRef CAS.
- S. M. Long, T. M. Bernhardt, R. N. Barnett, B. Yoon and U. Landman, J. Am. Chem. Soc., 2009, 131, 8939–51 CrossRef CAS.
- B. Jørgensen, S. E. Christiansen, M. L. D. Thomsen and C. H. Christensen, J. Catal., 2007, 251, 332–7 CrossRef CAS.
- T. A. Nijhuis, E. Sacaliuc, A. M. Beale, A. M. J. van der Eerden, J. C. Schouten and B. M. Weckhuysen, J. Catal., 2008, 258, 256–64 CrossRef CAS.
- J. B. Reitz and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 11467–8 CrossRef CAS.
- W. Zhu, Q. Zhong and Y. Wang, J. Phys. Chem. C, 2008, 112, 7731–4 CrossRef CAS.
- E. E. Stangland, B. Taylor, R. P. Andres and W. N. Delglass, J. Phys. Chem. B, 2005, 109, 2321–2330 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tabulated ICP and XPS data; visible spectroscopy; microscope images and SEM images; TGA-MS profiles and further catalyst test data. See DOI: 10.1039/c0cy00003e |
| This journal is © The Royal Society of Chemistry 2011 |