Hierarchised luminescent organic architectures: design, synthesis, self-assembly, self-organisation and functions
Laura
Maggini
a and
Davide
Bonifazi
*ab
aDepartment of Chemistry, University of Namur (FUNDP), B-5000 Namur, Belgium. E-mail: davide.bonifazi@fundp.ac.be
bDipartimento di Scienze Chimiche e Farmaceutiche and INSTM UdR Trieste, Università degli Studi di Trieste, 34127 Trieste, Italy
First published on 12th July 2011
Abstract
This critical review aims at highlighting the prevailing supramolecular approaches employed nowadays in the preparation of luminescent hierarchised materials. Specifically, it has the ambition to illustrate how progresses in the control of the supramolecular interaction toolbox ultimately led to the development of spectacular luminescent nano- and micro-architectures, through a combination of molecular self-assembly and self-organisation processes involving organic π-conjugated molecules. The reader will be guided through a systematic exploration of the most common avenues to prepare and characterise luminescent self-assembled/self-organised materials embedded into one-, two- or three-dimensional networks, accompanied by a critical discussion of their main advantages and limitations. Key representative examples of this research field will be thoroughly described, with a particular focus on those systems displaying potential on the device application scene. Particular attention will be devoted to the design and synthetic approaches aimed at the preparation of the primary π-conjugated molecular modules, the chemical, structural and electronic properties of which dramatically influence the fate and the features of the self-assembled/self-organised material (215 references).
 Laura Maggini and Davide Bonifazi | Laura Maggini was born in La Spezia, Italy, in 1984. She received her Master degree in Chemistry from the University “La Sapienza” in 2008. Then, she moved to the University of Namur as a Marie-Curie fellow in the frame of the European FINELUMEN network under the supervision of Prof. Davide Bonifazi, to accomplish a PhD degree working on the preparation of novel luminescent hybrid materials based on the functionalisation of carbon nanotubes with lanthanide complexes and π-conjugated molecular modules. |
Davide Bonifazi was born in Guastalla (Italy) in 1975. After obtaining the “Laurea” from the University of Parma (1994–1999), and working with Prof. Enrico Dalcanale, he joined the group of Prof. François Diederich at the Swiss Federal Institute of Technology, Zürich (2000–2004). He was awarded the Silver Medallion of the ETH for his doctoral dissertation (2005). After a one-year postdoctoral fellowship with Prof. Maurizio Prato at the University of Trieste, he joined the Department of Pharmaceutical Science at the same University as a research associate first and then as permanent researcher. Since September 2006, he has also joined the Department of Chemistry at the Facultés Universitaires Notre-Dame de la Paix in Namur (FUNDP) as junior professor in organic chemistry. In 2010, he was awarded the “Ciamician Medal” from the Organic Division of the Italian Chemical Society. His research interests focus on the self-assembly and self-organisation of electronically and optically active molecules at interfaces, porphyrin chemistry and functionalisation of carbon nanostructures. |
1. Introduction
In the dialogue “Politéia” (the Republic),1Plato reasons on human beings. In his view the soul of men is divided into different parts: Temperance, Wisdom, and Courage, and only as a result of their harmonious assembly arises Justice. However, since man alone is not endowed to satisfy his needs, he inevitably started to “self-organise” into communities. According to Plato's vision, in these living entities every single human has to do his part in interacting and self-adapting to his neighbour avoiding destructive interferences, thereby allowing the achievement of synergetic outcomes dramatically enhanced with respect to those obtained by single individuals.These concepts are as true in the human societies as they are at the molecular scale. Structurally-ordered nano- and micro-sized architectures based on the controlled assembly/organisation of fundamental molecular modules2–8 can act as seeds for the growth of novel structured materials, in which the molecular properties result largely modified or completely different from those of the constituting molecular modules.9–13 Perceiving the value of structural- and order-dependent properties emerging from such architectures, chemists have then focused their attention on the construction of novel materials trying to gain fundamental understanding on the assembly process and on its repercussions on the features of the final architecture. The knowledge obtained through these pioneering investigations disclosed the possibility of easily tuning the properties of such molecularly ordered materials by simply modifying the structure of their composing units. Because of their effective versatility, molecularly organised supramolecular materials hold great promises for a general change in different everyday-use devices, such as organic semiconductors,8,14 solar cells,15 chemical16 and bio-sensors.17–19
A large number of recent reviews summarising the research on nanostructured materials, focus their attention on the structural properties of the resulting architectures, obtained via both covalent and non-covalent approaches.20 The present review will instead focus on the different strategies to prepare functional fluorescent multidimensional architectures exclusively via a combination of the self-assembly and self-organisation methodologies. The different self-assembly and self-organisation approaches towards the construction of these structured materials will be described along with all the resulting properties and consequent promising applications (OLED, FET, NLO, sensors and organic conductors). A general overview will also be devoted to the design and synthetic approaches aimed at the preparation of the primary π-conjugated molecular modules, the chemical, structural and electronic properties of which dramatically influence the fate of the self-assembly/self-organisation processes and consequently the luminescent properties of the architecture.
1.1 Self-assembly vs. self-organisation: two sides of the same medal
In order to obtain predefined engineering of structured functional materials, pivotal is the understanding and control of both the self-assembly and self-organisation mechanisms, which can give rise to a collective molecular-scale order via weak non-covalent interactions. Despite the intrinsic differences between the two mechanisms,21,22 these terms are often used with little care in the contemporary chemical literature, leading to perplexity and imprecise meanings, thus urging the scientific community to recall their exact consistent definitions.23 Although both these phenomena occur through non-covalent interactions (e.g., hydrogen bonds, halogen bonding, ionic interactions, metal chelation, dipolar and van der Waals interactions, etc.), they are profoundly different on the thermodynamic basis. Indeed, whilst self-assembly implies spontaneous processes tending towards equilibrium products, self-organisation is intended for non-equilibrium processes.As clearly observed in biology, the self-organisation of an undefined number of molecular components is the result of a set of dynamic dissipative non-linear non-equilibrium processes, which ultimately lead to a structural order at the nano and microscopic levels, driven by a delicate interplay between internal (coordination interaction, hydrogen bonds and dipole–dipole to name a few) and external (concentration, temperature, stoichiometries, solvent and time) factors. With respect to self-assembled systems, the self-organisation process can lead to non-thermodynamically stable products, each one favoured by a combination of the above-mentioned external and internal factors, and thus displaying peculiar assembly-related properties (both morphological and electronic). Exploiting this dependence, more complex hierarchised self-organised structures could be envisaged through a sequential stepwise building-up approach.
In contrast, the self-assembly process depends on equilibrium conditions and the fundamental molecular modules interact specifically among themselves through molecular recognition relationships encoded within the module structure, leading to the controlled formation of a pre-defined architecture. Thus, an important difference between self-assembly and self-organisation lies in the initial structural encoding of the molecular components, which is not required for the latter. If generalised, it is clear that due to the general reversible nature of the non-covalent interactions between the self-assembling components, any self-assembled system has the intrinsic capability for self-correcting. Indeed self-assembled architectures reflect those encoded molecular information, determining the structural and functional properties of the final material at the equilibrium (i.e., the most thermodynamically favourable structure).
The fine control and prediction of such an ensemble of dissipative and non-dissipative, equilibrium and non-equilibrium processes constitute the major challenges towards the design and engineering of superior and reproducible complex matters displaying exploitable properties for applications in material sciences. A predictive molecular programming of the self-assembling and self-organising molecular modules strictly depends on the driving structural and electronic information as encoded in the fundamental molecular components and how these parameters are responding and sensitive towards the internal and external factors.
1.2 Methodological approaches towards structured materials
Understanding the self-assembly and self-organisation mechanisms that have a significant impact on the structural features of the aggregates,24 together with their direct visualisation by scanning probe (STM and AFM), and electronic microscopies (SEM and TEM), is important in order to engineer, in a controllable fashion and hence optimise, the photonic properties of self-assembled/self-organised systems. Several approaches to arrange the chromophoric modules into functional architectures have been developed with time exploiting colloidal,5,25 sol–gel polycondensation,26 templated27,28 and non-covalent10,29,30 methods. Among all these possibilities the supramolecular approach31 is still the one which produces the broadest variety of easily accessible and interesting molecular architectures,6,32–39 thanks to the versatility of the non-covalent interactions toolbox, which includes hydrogen bonding, π–π and electrostatic interactions together with solvophobic effects.As shown in Scheme 1, the discussion will first take into account the design and the synthesis of the chromophores themselves. This review wants to acknowledge also the synthetic efforts aimed to obtain the building molecular modules (the “bricks”) of these luminescent nano-architectures, the peculiar features of which characterise both the assembly process and the properties of the final material. Consequently, both templated and non-templated self-organisation of chromophoric modules yielding molecularly organised materials will be approached. In the first case, ranging from the linear polymeric to the fibrous and nanotubular arrays, and from micellar to toroidal and vesicular spherical ones, we will reason on the way the chromophores assemble, and on the efficiency of each approach regarding the production of luminescent material.
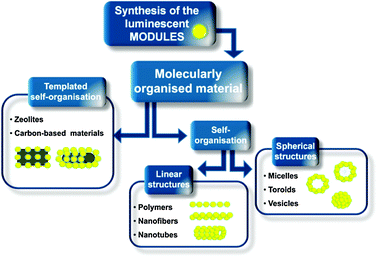 | ||
| Scheme 1 Schematic overview of the approaches undertaken for the preparation of luminescent hierarchised multidimensional nanoarchitectures. | ||
We will finally focus our discussion on the supramolecular functionalisation of two specific classes of scaffolds, namely zeolites40–42 and carbonaceous materials (nano-diamonds,43,44graphene45–47 and carbon-nanotubes),48,49 taking into account not only their templating action, but also the effects of their presence on the final properties of the resulting hybrid materials.
The different synthetic protocols along with the non-covalent-based preparative methodologies towards the construction of luminescent nanoarchitectures will be thoroughly described, taking especially into account their potential applications currently holding great promises in materials sciences.
2. Self-organised fluorescent nanostructures
2.1 Linear assemblies
Scheme 2 illustrates the synthetic route undertaken for the preparation of rigid monomer 1. Nucleophilic attack of the ortho-lithiated biphenyl derivative on methoxy-truxenone, followed by demethylation by BBr3 and alkylation with n-dodecylbromide under basic conditions afforded the first rigid intermediate. Subsequent Pd(0)-catalysed Suzuki coupling reaction56,57 with 4-(methoxycarbonyl) phenylboronic acid, followed by saponification reaction with LiOH yielded the desired carboxylic hexaacid 1. When mixed in THF, hexaacid 1 undergoes self-assembly and successively self-organisation leading to nanofibre-like structures. Scanning electron microscopy (SEM) images (Fig. 1) demonstrated that uniform and well-dispersed nanowires with diameters ranging from 100 to 200 nm and length achieving several hundred micrometres were formed. Comparison of the photoluminescence (PL) spectra of 1 and that of the ester precursor, unable to self-assemble into polymeric structures, showed comparable molecular absorption features (peak at 324 nm) due to the similar molecular effective conjugation length in the ground state (Fig. 1).58 However, the assembly-event caused a 12 nm blue shift for the 1-centred emission peak (411 nm), in comparison to that of the precursor ester module (423 nm). Despite the identical absorption maximum, in thin solid films the ester displayed a 15-nm blue-shift (408 nm) emission with respect to the value from solution while a 13 nm red-shift was observed for the nano-wires of molecule 1. This is caused by the presence of the H-bonds and a different conformation adopted by the phenyl rings in the solid state. The absolute PL efficiency of the supramolecular nano-rod at the solid state resulted to be 22% (compared to 32% in solution), a relatively high value among organic nanofibres.59,60
 | ||
| Scheme 2 Synthetic pathway towards hexacarboxylic monomer 1. | ||
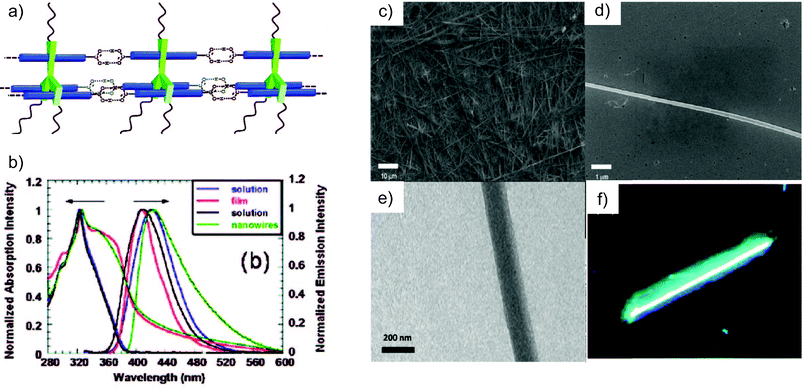 | ||
| Fig. 1 (a) Schematic representation of the self-assembled structure of 1 at the molecular level. (b) UV-Vis and PL spectra for 1 and for the ester precursor in THF solution and solid state; (c–e) SEM and TEM images for the nanowires materials; (f) fluorescence micrograph of the supramolecular nanowires (400 magnitude). (Adapted with permission fromref. 50. Copyright 2009, American Chemical Society). | ||
Another example of a supramolecular-induced enhancement of the molecular luminescent properties has been reported by Meijer and Schenning and co-workers.61 Three π-conjugated monomers (a blue, a green and a red-emitting one) symmetrically functionalised with the self-complementary H-bonding ureido-pyrimidinone recognition unit (UPy)62,63 were prepared. These monomers resulted to self-assemble into mixed supramolecular polymers bearing the three chromophores, which at a certain mixing ratio led to white luminescence. The same white emission could not be achieved using mixtures of the bare unfunctionalised chromophores, thus demonstrating the key role of the organisation on the output properties through a fine tuning of the energy transfer pathways between the chromophoric species.
In Schemes 3 and 4 the synthetic pathway towards the preparation of the three chromophores is depicted. The green-emitting oligo(p-phenylenevinylene) 2 was prepared by stepwise Wittig couplings64,65 of benzyl triphenylphosphonium salts with aromatic aldehydes.66 Subsequent reduction of the peripheral cyano groups to benzyl amines, and final coupling with an activated UPy derivative67 yielded 2. The red-emitting 3 was instead synthesised by reaction of a diaryloxy substituted perylene anhydride68 with an excess of 1,4-diamino-2,6-di-tert-butylbenzene, followed by coupling with the activated UPy.67 Finally, the blue-emitting oligofluorene 4 was obtained by stepwise Pd(0) catalysed Suzuki coupling of two mono-amino fluorenes to a symmetrically bis-functionalised fluorene core.69 Attachment of activated UPys to bis-NH2-terminated oligofluorenes afforded the desired chromophore. As displayed in the inset of Fig. 2, all chromophoric monomers self-assemble into supramolecular polymers through strong (Ka = 6 × 107 M−1 in CDCl3) quadruple self-complementary H-bonding interactions established between the UPy termination units. Fluorescence titrations demonstrated that the blue emitter 4 (at 407 nm) can act as an efficient energy donor when coupled with appropriate energy acceptors such as the green and red emissive chromophores 2 (at 436 nm) and 3 (at 515 and 553 nm).69 Simultaneous addition of aliquots of 2 and 3 into a solution of 4 led to the proposed supramolecular copolymer, which at the ratio 2/3/4 = 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:59, afforded white photoluminescence of comparable intensity throughout the whole visible spectrum. In addition, when the supramolecular polymer was spin coated from a concentrated o-dicholorobenzene solution (4.5–9 × 10−3 M), a white photoluminescent film (ratio of 2/3/4 = 10:6:84) was also formed, showing higher effectiveness of the energy transfer process with respect to the solution. At appropriate mixing ratios, prototypical electroluminescent OLED devices emitting blue, green, red, or white light have been also engineered greatly displaying the real technological promises of these materials.
:8:59, afforded white photoluminescence of comparable intensity throughout the whole visible spectrum. In addition, when the supramolecular polymer was spin coated from a concentrated o-dicholorobenzene solution (4.5–9 × 10−3 M), a white photoluminescent film (ratio of 2/3/4 = 10:6:84) was also formed, showing higher effectiveness of the energy transfer process with respect to the solution. At appropriate mixing ratios, prototypical electroluminescent OLED devices emitting blue, green, red, or white light have been also engineered greatly displaying the real technological promises of these materials.
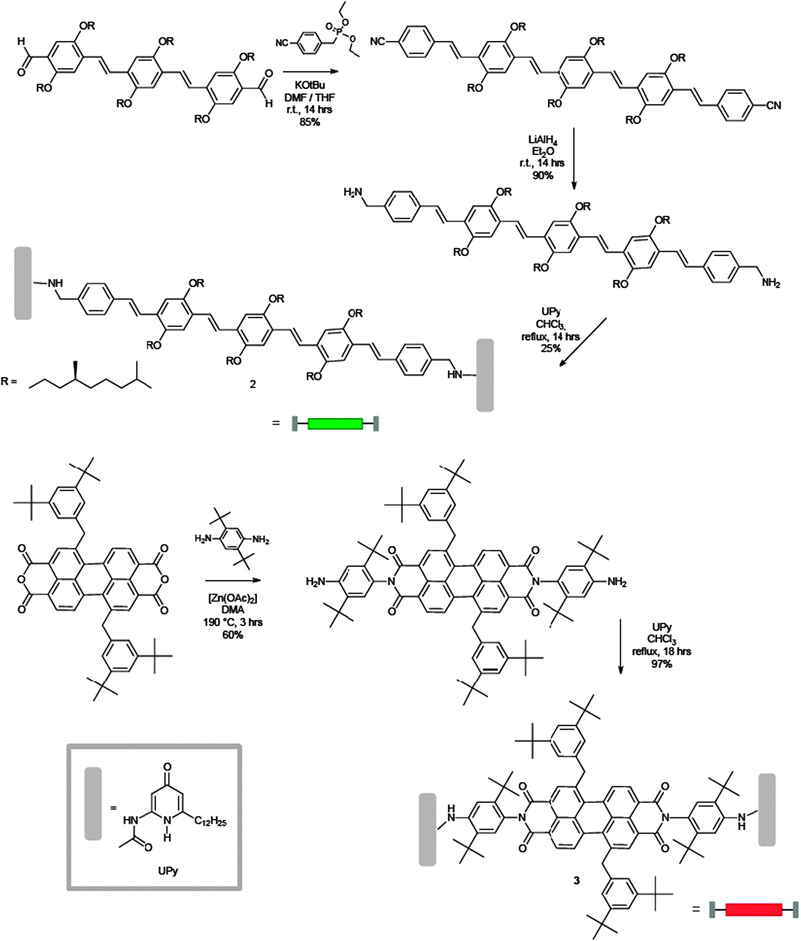 | ||
| Scheme 3 Synthetic pathway towards monomers 2 (green) and 3 (red). Inset: the structure of the recognition unit UPy. | ||
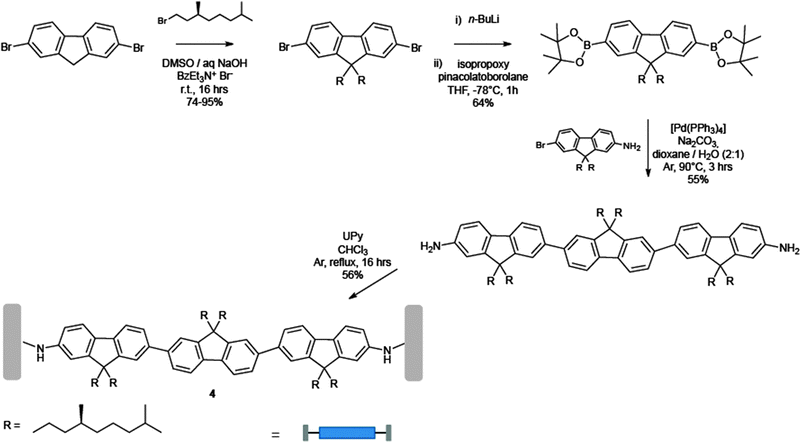 | ||
| Scheme 4 Synthetic pathway towards the blue monomer 4. | ||
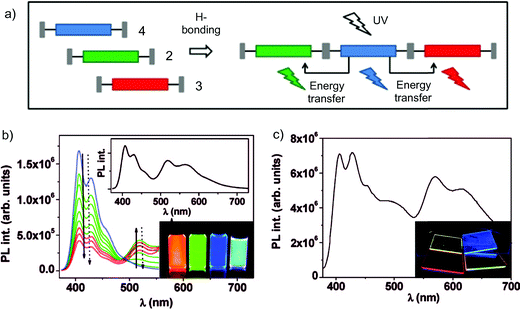 | ||
| Fig. 2 (a) Schematic representation of the assembly between emitters 2, 3 and 4; (b) fluorescence titration experiment in CHCl3 solution (blue: pure 4; green: upon stepwise addition of 2; red: upon final addition of 3). Inset: spectrum displaying the white light luminescence originated from a (33:8:59) mixture; (c) simultaneous emission of molecules 2–4 (ratio 10:6:84) in a thin spin-coated film upon UV excitation (λexc = 365 nm). (Adapted with permission fromref. 61. Copyright 2009, American Chemical Society.) | ||
As shown in Scheme 5, these symmetric p-quaterphenylenes derivatives were prepared by applying a two-fold Pd(0)-catalysed Suzuki cross-coupling reaction from commercially available p-substituted phenylboronic acids and 4,4′-dibromobiphenyl. Deposition of the differently substituted p-quaterphenylenes performed in high vacuum (pressure 5 × 10−8 mbar) viasublimation from a Knudsen cell resulted in the well-organised formation of light-emitting nano-fibers whose differing optical properties were strictly dependent on the functional group present on the molecular building block.
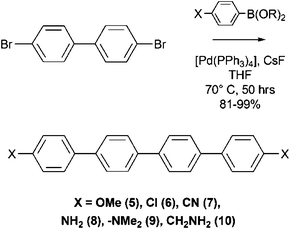 | ||
| Scheme 5 Synthetic route to the symmetric quaterphenylene derivatives 5–7. | ||
Fluorescence spectra recorded upon continuous UV irradiation (λexc = 325 nm) of a 1 mm2 sample area of aggregates on a mica substrate highlighted a shift of the peak emission frequency of the symmetric nanoaggregates from 383 nm (5, X = OMe) to 452 nm (9, X = NMe2), probably due to inductive and mesomeric effects of the functional groups on the conjugated π-electron system.74
Fluorescence microscopy images of self-organized 5, 6 and 7 (excitation wavelength λexc = 365 nm) show brightly fluorescent and well-defined nanostructures with different morphologies (Fig. 3). Deposition of 5 on muscovite led to mutually aligned, almost parallel fibers with mean widths and heights of several hundreds and several tens of nanometres, respectively, and lengths up to several hundreds of micrometres. The nanofibers from 6 are significantly shorter with a length of up to 30 mm, but exhibit widths and heights comparable to the fibers from 5. Nanofibers grown from 7 show diverse structures within a single domain. Besides mutually aligned fiber-like structures, aggregates with increased heights are visible with a shape that reminds one of swallow wings. The fibers have a mean length of about 7 mm as well as a typical width of several 100 nm and a typical height of several 10 nm, respectively.
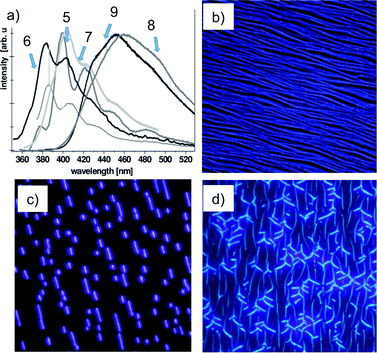 | ||
| Fig. 3 (a) Fluorescence spectra of the symmetrically functionalized p-quaterphenylenes 5–9; (b–d) fluorescence microscopy images of nanoaggregates on muscovite mica (λexc = 365 nm) from 5 (a, 200 × 200 μm2, Ts = 340 K), 6 (b, 95 × 95 μm2, Ts = 380 K) and 7 (c, 83 × 83 μm2, Ts = 300 K). (Adapted with permission fromref. 72. Copyright 2008, Royal Society of Chemistry.) | ||
Another interesting feature regarding these fibers may be noticed: an increased nonlinear optical response arises from the molecular building block upon introduction of two different functional groups in the 1,4′′′-para positions of quaterphenylenes. Upon irradiation with femtosecond laser pulses the asymmetrically substituted nanofibers emitted a strong second-harmonic signal.75,76 Such a property is of obvious importance for future integrated optical circuits, but is also a powerful technique for understanding the correlation between morphology and optoelectronic response of nano-aggregates.
More specific supramolecular interactions were then employed in the preparation of luminescent nanofibers from Ajayaghosh and coworkers. Cholesterol moieties were for the first time used to obtain aligned oligo(p-phenylenevinylene) (OPV) chromophoric supramolecular nano-architectures, allowing also the control over nanoscopic properties of the resultant hierarchical self-assembly (Scheme 6).77
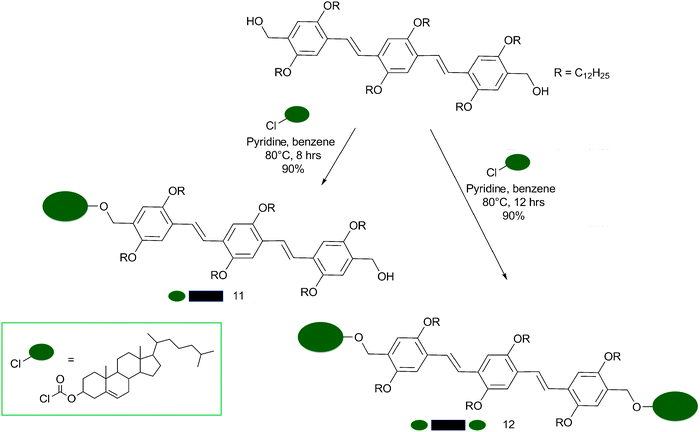 | ||
| Scheme 6 Synthetic pathway for the production of the cholesterol-OPV derivatives 11 (mono-substituted) and 12 (di-substituted). | ||
The mono- and di-substituted cholesterol-appended OPVs 11 and 12 were prepared by treatment of the corresponding bis(hydroxymethyl)-OPVs78 with cholesteryl chloroformate. Solutions of the mono- and di-substituted OPVs in chloroform (3 × 10−4 M) showed identical absorption and emission spectra with an absorption maximum at λ = 408 nm (π–π* transition) and emission maxima at λ = 466 and 494 nm (quantum yields of fluorescence (Φf) were measured to be 0.76 and 0.82 for 11 and 12 respectively, using quinine sulfate as standard). In decane, at room temperature, 11 exhibited a broad absorption (λmax = 402 nm) with a red-shifted shoulder around 470 nm.78–80 In contrast, 12 showed a structured absorption with a blue-shifted maximum (Δλmax = 10 nm) and two new bands at λ = 414 and 444 nm.81,82 Differences between the two chromophores were also found in their emission spectra in decane at 20 °C. A broad structureless emission band between 500–700 nm with a maximum around 560 nm was detected for 11, whereas 12 displayed a structured emission profile with two emission maxima at 494 and 528 nm. These differences in the absorption and emission spectra between 11 and 12 in a decane solution, together with the observation of a more intense emission spectrum for 12 instead of 11, could not be rationalised by simply invoking H-type81–83 and J-type79 aggregation. Indeed, if the blue-shifted absorption spectrum of 12 was to be assigned to the presence of H aggregates, it should have been less intense relatively to the spectrum of the J aggregates for 11. For these reasons, the two aggregates were addressed as “pseudo-H” and “pseudo-J” with twisted and tilted chromophore stacks for 12 and 11, respectively (Fig. 4), indicating the formation of two aggregates, whose electronic properties are different from each other.
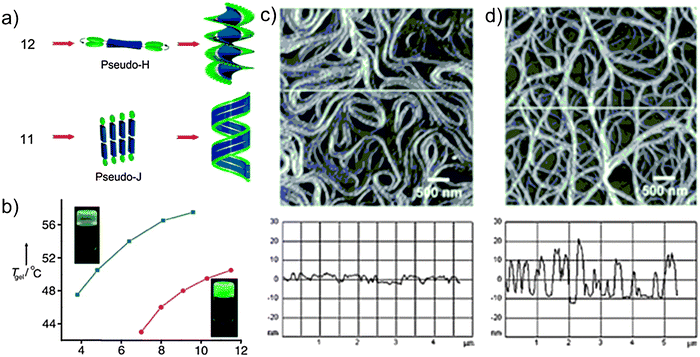 | ||
| Fig. 4 (a) Probable self-assembly process for OPV derivatives 11 and 12; (b) melting temperature profiles for the gel (Tgel) vs. the concentration of the gelators 11 (blue line) and 12 (red line) in decane, showing an enhanced stability for 11-based gel. Insets: photographs of the corresponding gels; (c and d) AFM images of the assemblies of 11 (a) and 12 (b) with the corresponding height profiles shown. (Adapted with permission fromref. 77. Copyright 2006, Wiley-VCH.) | ||
These differences also reflected in a disparity in the gelation behaviour between the two chromophores. Indeed, although both compounds induce gelation of a variety of hydrocarbon solvents, gels with molecule 12 were relatively weaker than those with 11 (critical gelator concentration, CGC, in decane resulted to be 7.0 and 3.4 mm for 12 and 11 respectively). Furthermore, the gel with oligomer 12 displayed a relatively strong green emission, whilst 11 showed a weak yellow emission, revealing stronger π interaction and hence better electronic conjugation between the chromophores relative to those of the latter (Fig. 4).
Atomic force microscopy (AFM) images of 11 and 12 obtained from solutions in decane showed significant differences in the morphology of the gels, although both assemblies adopt right-handed helical structures (Fig. 4). AFM analysis revealed flat ribbon-like structures for 11, aligned sideways to form coiled superstructures. The width of the individual ribbons varies from 30 to 70 nm, with an average height of 4–8 nm, corresponding to two individual tapes lying one over the other. Interestingly, molecule 12 showed a morphology comprised of entangled helical fibers of different sizes. The width of the smallest fiber resulted to be 40 nm, with a height of 4 nm and several micrometres in length. Section analysis along the axis revealed that assemblies of 11 have an irregular helical pitch of 40–80 nm characteristic of a flexible and flattened coiled tape-like morphology, whereas those of 12 are almost uniform with zigzag patterns and have a pitch length of 46 nm indicating the presence of intertwined twisted helical fibrillar assemblies.
Intrigued by the possibility of exploiting the moderate self-assembly and partial energy transfer occurring for 12 to obtain white light emission materials (WLE) in the presence of an appropriate acceptor, Ajayaghosh et al. performed the gelation of 12 in the presence of the red-light-emitting conjugated oligomer 13 (Fig. 5).84
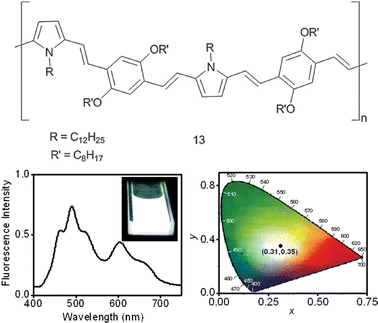 | ||
| Fig. 5 Chemical structure of molecule 13. Fluorescence emission of a decane gel of 1 (3.2 × 10−4 M) in the presence of 2.1 mol% of 13 (λ = 380 nm), and the corresponding CIE coordinate diagram (x = 0.31, y = 0.35). Inset shows the white-light emission of the gel upon irradiation at 365 nm. (Adapted with permission fromref. 84. Copyright 2009, Wiley-VCH.) | ||
Energy transfer studies were carried out by adding appropriate aliquots of a solution of 13 in decane (from 0 to 2 mol%) to a decane solution of the donor (3 × 10−4 M). The mixture was then heated to 80 °C to form a homogeneous solution and slowly cooled to room temperature to obtain co-assembled gels.
The emission of a decane gel of 12 upon excitation at 380 nm in the absence of 13 showed two maxima, at 492 and 528 nm, with shoulders on either side. The shoulder at 464 nm (blue-light-emission colour) corresponds to the residual monomer emission of the donor. Upon excitation at 380 nm in the presence of small amounts of 13, the emission signal is quenched with the concomitant formation of a new peak at 607 nm, which corresponds to the acceptor emission, without affecting the blue light-emitting shoulder at 464 nm. It must be noted that a further increase in the amount of 13 while maintaining the same donor concentration dissociates the donor gel scaffold, leading to a significant decrease in the energy-transfer efficiency.
Surprisingly, when the concentration of the acceptor reached around 2.0 mol%, the decane gel of 12 exhibited a near-WLE (Commission Internationale de L'Eclairage, CIE, coordinates of (0.28,0.34)). The partial quenching of the emission of the donor-based self-assembly results in a broad emission spectrum in the range of 400–750 nm, which contains red, green, and blue colours that yield a white-light emission. From this observation, it is clear that the blue emission of the donor monomers, green emission of the donor aggregates, and red emission from the acceptor as a result of partial energy transfer occur simultaneously. The purity of the white-light emission could be improved by optimising the concentration of 12 (3.2 × 10−4 M) and 13 (2.1 mol%). At this stage, the emission spectrum covers the visible light range (Fig. 7) with a better balance of red, green, and blue, which results in a pure WLE-gel.
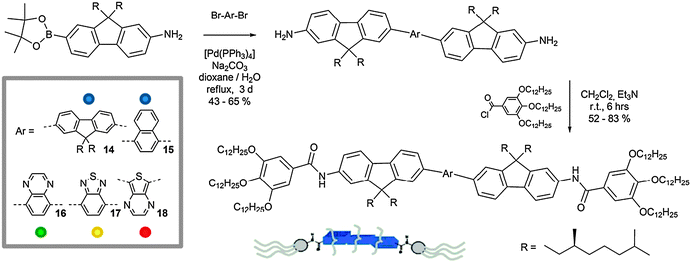
Schenning and co-workers85 described the formation of self-organised fibrils in apolar solvent utilising a series of pentameric fluorine oligomers. These chromophores (14–18) consist of two chiral dialkylfluorene units linked by a central aromatic moiety (fluorene, naphthalene, quinoxaline, benzothiadiazole or thienopyrazine) that are able to emit light in the vi range, from blue (14, 15) to green (16), yellow (17) and red (18).
In Scheme 7 the synthetic strategy towards the preparation of 14–18 is shown. The bis(aminofluorene) oligomers were prepared through a Suzuki coupling reaction between the dibromide aromatic cores86–89 and an aminofluorene boronic ester.61 Subsequent amidation capping of the di-amino chromophores with tris(dodecyloxy)benzoic acid chloride prepared in situ from the corresponding acid90 led to the final emitters. The fibril self-organisation process revealed to be dependent on the solution concentration. In fact, it emerged that, whilst at sufficiently low concentrations (≤10 × 10−6 M, in methylcyclohexane, MCH)91,92 all compounds are molecularly dissolved, upon stepwise increase of the concentration of 14–18, a progressive precipitation was observed for compounds 15–17. In contrast, molecules 14 and 18 organise into elongated aligned aggregates with fibrillar morphology as shown by AFM imaging (Fig. 6). At concentrations higher than 2.5 × 10−6 M, molecular emitters 14 and 18 formed stable blue and red fluorescent organogels. The gelation process was confirmed by variable concentration (from 0.25 to 5 × 10−6 M) luminescence studies, which showed the appearance of a low-energy shoulder in the UV-Vis absorption profile and a red shift of ∼5 nm in the PL spectrum of fluorenyl derivative 14.
 | ||
| Scheme 7 Synthetic route to the preparation of compounds 14–18. | ||
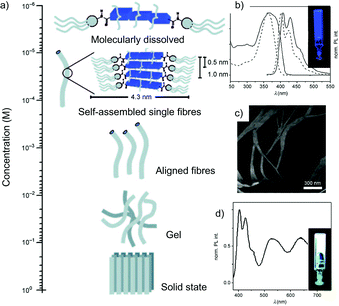 | ||
| Fig. 6 (a) Schematic representation of the self-organisation of molecules 14–18, (b) normalised UV-Vis and PL spectra of 14 in the gel state (solid line) and in MCH dilute solutions (0.7 × 10−6 M; dashed line) at rt; (c) AFM topographic images of 14-based morphologies as obtained from a drop-cast solution of 14 in MCH; (d) PL spectrum of a gel containing chromophores 14 (4.5 × 10−3 M) and 16–18 (λexc = 375 nm; molar ratio 14/16/17/18 = 4.5:0.7:0.2:0.1). Inset: PL lifetime-decay (λexc = 400 nm) monitored at 415 nm of a gel containing only 14 and another gel containing also the chromophores 14, 16, 17 and 18. (Adapted with permission fromref. 85. Copyright 2009, Wiley-VCH.) | ||
Crucial for the formation of luminescent gels resulted the ability of the aromatic parts to form stable stacks via π–π stacking interactions. In fact, from molecular modelling (MM) simulations it has been possible to attribute the differences in the π-stacking behaviour to the chromophoric conformational properties. The structural differences in the molecular shapes (quasi linear for 14 and 18 and slightly curved for 15, 16 and 17), and in the torsion angles between the neighbouring units (less than 40° for 15 and 18 and greater than or equal to 40° for 15, 16 and 17), may explain the different self-organisation behaviours for each one of the described emitters. Upon addition of 16 (green), 17 (yellow) and 18 (red) into a gel of 14 in a molar ratio of 4.5:0.7:0.2:0.1 (14/16/17/18), white-light emitting organogels were obtained (Fig. 6). Proof of the energy transfer phenomena is the decrease in the fluorescence lifetimes (τf) of the bi-exponential decay of the excited singlet state of 14 in MCH (from τ = 0.93 (94.5%) and 1.81 ns (5.5%) to τ = 0.28 (95.4%) and 1.10 ns (5.6%) for the mixed system).
Another example of linear fibrillar morphologies has been reported by Park and co-workers who described the preparation of self-organised nano-fibers with 1-cyano-trans-1,2-bis-(3′,5′-bis-trifluoromethyl-biphenyl)ethylene (19) exhibiting aggregation-induced enhanced emission (AIEE).93 Pivotal for this peculiar behaviour is the simultaneous occurrence of several non-covalent interactions during the organisation process, influencing all the functional moieties of 19: the cyano group (CN), the trifluoromethyl (CF3) groups and the aromatic backbone (Fig. 7). In fact, the CN group increases the polarisation of 19 giving rise to an anisotropic electrostatic distribution, causing an electrostatic attraction between neighbouring molecules. As the molecules come closer, the phenyl rings begin to planarise, but because of the repulsion induced by the peripheral CF3 groups, the approaching molecules tend to slide away along the molecular axis. This off-set disposition is further stabilised by intermolecular C–F–H–C H-bond interactions.94–96 Similar studies conducted on analogues of molecule 19 lacking CN or CF3 functional groups showed no formation of nanofibres and/or organogels, thus highlighting the essential role of all the functional groups of 19.
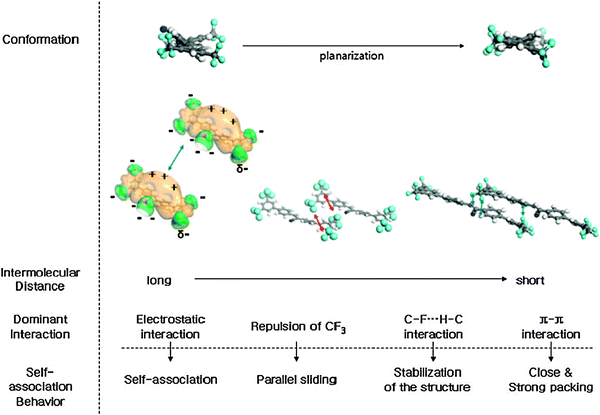 | ||
| Fig. 7 Schematic representation of the conformational changes, dominant interactions, and the self-association behaviour of molecules 19 with varying the intermolecular distances. (Adapted with permission fromref. 93. Copyright 2004, American Chemical Society.) | ||
As shown in Scheme 8, module 19 was prepared by connecting the two bis-CF3-phenyl extremities by a Knovenagel reaction, in the presence of Et4NOH.97,98 The two precursors were first prepared by Pd(0)-catalysed Suzuki couplings between the brominated and the boronic acid starting materials. Because of the conformational changes induced by the supramolecular architecture in the solid state, the non-emissive molecule 19 becomes highly fluorescent in the aggregated state, showing more than a 100-fold enhancement in its fluorescence intensity (Fig. 8). To gain insight into the organogel morphology of 19, a dried sample was studied by SEM. All images suggest that molecule 19 undergoes three-dimensional entanglement forming luminescent spaghetti-like networks of fibrous bundles (Fig. 8).
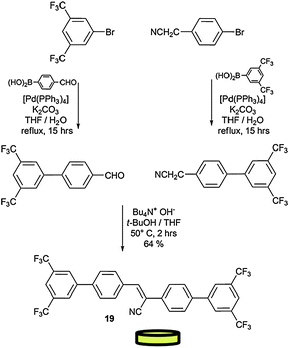 | ||
| Scheme 8 Synthetic pathway for the preparation of module 19. | ||
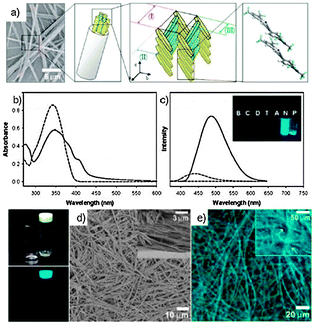 | ||
| Fig. 8 (a) Schematic representation of the assembly of 19; (b and c) UV-Vis and PL spectra of 19 (2 × 10−5 M in THF; dashed line) and its nanoparticle suspension (THF/H2O = 1:4; plain line); (d) SEM images of a dried gel of 19 in 1,2-dichloroethane and (e) fluorescence microscopy images of a 19 organogel. (Adapted with permission fromref. 93. Copyright 2004, American Chemical Society.) | ||
On the basis of these results, Park and co-workers prepared a series of structurally equivalent compounds (11–13) characterised by AIEE fluorescent emissions covering the whole visible range,99 namely green (λmax = 543 nm), yellow (λmax = 564 nm), and red (λmax = 598 nm) respectively (Fig. 9). These colour-tuned highly fluorescent nanofibres easily form mechanically-stable nano-fabrics-like arrays when the dye concentration in the solution exceeds 2.0 wt%, showing fairly high PL quantum yields (PLQY) in the solid state (19–22 show 93.9% (91.7%), 22.5%, 15.3%, and 9.5% PLQY respectively). Furthermore, these fluorescent NFs could be shaped into various desired forms with active areas ranging from ∼0.3 cm2 to ∼1.2 cm2 by moulding or stamping techniques (Fig. 9).
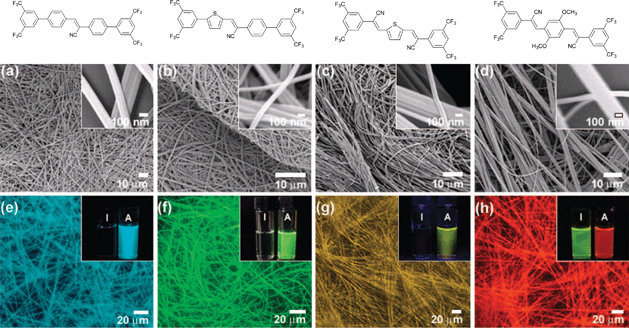 | ||
| Fig. 9 (a–d) SEM micrographs and (e–h) fluorescence microscopy images of the nanostructures composed of 19–22 generated by solution drop-casting. Inset photos show the fluorescence colours for the THF solution (I) and the nanostructured fibres as obtained from solution of THF/H2O (1:4 v/v; A). (Adapted with permission fromref. 93. Copyright 2009, American Chemical Society.) | ||
Another similar system explored by Park et al. regards a cyano distyrylbenzene (DSB) derivative, (2Z,2′Z)-2,2′-(1,4-phenylene)bis(3-(4-butoxyphenyl)) acrylonitrile (23, Fig. 10), which besides a very high solid-state fluorescence quantum yield (62%) due to the characteristic AIEE process, was also observed to exhibit reversible piezochromism behaviour.100 This luminescent module possesses the ability to assemble into fluorescent single crystals composed of several “molecular sheets”, thanks to the contemporaneous occurrence of multiple C–H⋯N and C–H⋯O hydrogen bonds. On the basis of several structural, optical, photophysical and computational characterisations,100 two different phases were identified for the crystal of molecule 23: the metastable green-emitting G-phase (λmax = 533 nm) and the thermodynamically stable blue-emitting B-phase (λmax = 458 nm).
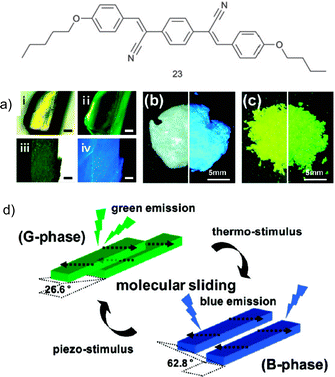 | ||
| Fig. 10 Graphical representation of 23; (a) photo of a single crystal of 23: before annealing, under room light (i), and UV light (ii), and after annealing, under room light (iii), and UV light (iv) (scale bar = 0.2 mm); (b) photo of the pristine powder under room (left) and UV light (right); (c) photo of the ground powder under room light (left) and UV light (right); (d) Illustration of the two different modes of slip-stacking in 23 molecular sheets, dictated by different ways of antiparallel/head-to-tail coupling of local dipoles (Adapted with permission fromref. 100. Copyright 2009, American Chemical Society.) | ||
This structural dichotomy can be attributed to the effect of the substituents present on the molecule. Due to the presence of the butoxy-substituents, the outer phenyl rings of 23 are richer in electrons when compared to the central phenyl ring which at the contrary suffers an electron-density reduction because of the effect of the cyano groups. As a result of these opposite actions, 23 becomes polarised and presents two local dipoles that together lead to a zero net dipole moment.101–104 Interactions between the different sheets of the crystals are hence governed by these dipoles, namely by the way dipoles belonging to two different molecules interact with one another; in the G-phase occurrence of anti-parallel coupling between dipoles belonging to the molecules present in two different molecular sheets positioned one on top of the other gives rise to a metastable crystalline structure. Upon annealing of the G-phase crystal, a smooth slip of the molecular sheets occurs leading to the more stable B-phase, characterised by a head-to-tail parallel arrangement of the local dipoles (Fig. 10).
Interestingly, these aggregates resulted to be able to undergo such internal reorganisation upon subjection to external stimuli, namely temperature and pressure (Fig. 10). Indeed, as grown from ethyl acetate, crystals of 23 are in the G-phase; heating the sample at 125 °C for 1 hour immediately brings the crystals to the B-phase. Grinding the blue powder in a mortar or rubbing it with a spatula also triggered an immediate colour change to the green-emitting G-phase. This piezochromic transition thus represents the reversal of the annealing process.
Such piezochromic transition105–112 is the basis for the preparation of a rewritable fluorescent optical recording media that showed fast-responding multi-stimuli luminescence switching (Fig. 11). A green-emitting (G-phase) composite film of 23 in PMMA (20 wt%) was prepared by spin coating on glass; upon thermal stimulus (125 °C for 10 s with hot letter stamp), the green fluorescence changed to blue in accordance with the G- to B-phase transition. The thermally annealed B-phase film showed high pressure-sensitivity, changing the emission colour immediately to green even with a very small shear force, thus allowing for sensitive piezo-writing. Upon exposing the whole films to organic vapors (CH2Cl2 for 30 s), the piezo-writing was erased changing the entire emission colour to green. Under all these conditions, a reproducible and fully reversible two-color fluorescence writing/erasing process was achieved and both emission colours of blue and green were found to be persistent over one year of observation under ambient conditions.
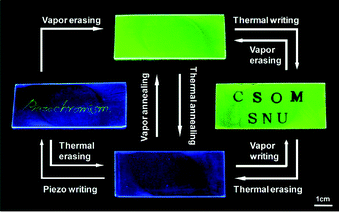 | ||
| Fig. 11 Photos of the luminescence writing/erasing cycle on a 23-PMMA film. (Adapted with permission fromref. 100. Copyright 2009, American Chemical Society.) | ||
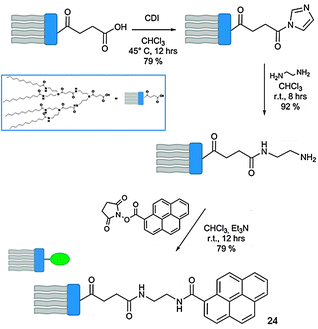
Carboxylic terminated dendron, synthesised according to the convergent procedures reported in the literature,114–116 was initially treated with 1,1-carbonyldiimidazole (CDI), activating the starting material towards an amidation reaction with ethylenediamine (Scheme 9). This intermediate was hence reacted with the N-hydroxysuccinimmide (NHS) ester of 1-pyrene carboxylic acid, yielding target dendron 24. Dendron 24 alone self-organises into vesicles in aqueous solutions. Upon addition of CDs into the vesicular solution, CD–pyrene complexation occurs through the inclusion of the hydrophobic luminescent pyrenyl unit into the cavity of the CD, leading to the formation of the NT-like architecture.113PL studies revealed major differences between the emission of the vesicular solution of 24 and a solution of self-assembled NTs. In the first case, a broad excimer at 420–550 nm deriving from the pyrenyl–pyrenyl interactions between different dendrons was detected. This signal resulted drastically reduced upon addition of the CDs, which led to a progressive increase of the emission signal of isolated pyrenyl moieties (370–410 nm).
The dramatic transformation of the self-organised structures from the vesicular to NT architecture upon addition of CDs to the solution of dendron 24 was confirmed by using TEM and SEM techniques (Fig. 12). From the TEM image a clear hollow tubular structure is observed, with outer and inner diameters of ∼45 and 22 nm, respectively. The wall thickness is 11.5 nm, corresponding to a unilamellar bilayer of dendron 24 (extended length = 53.5 Å) with the focal pyrenyl moiety included into the CDs' cavity. As the functional groups at the C6 position of the CD macrocycles are externally exposed, the self-organised NTs could be efficiently functionalised at the outer surface through the use of differently C6-functionalised CDs. A library of externally-derivatised functional NTs was thus constructed exploiting the hierarchical self-assembly/self-organisation of dendron 24 with differently functionalised CDs bearing at the C6 position a iodo, amine, carboxyl or biotin functional group.117 Depending on the functional moiety employed, different uses could be envisaged. As an example, amine functionalised CDs-based NTs could be externally decorated with anionic gold nanoparticles (Au-NPs) through columbic interactions (Fig. 13), and the golden decoration was detected by PL measurements. In particular, the fluorescence spectra of the hybrid luminescent material show a quenching of the pyrene emission (at 370–600 nm) in the presence of the Au-NPs with an approximate efficiency of 95%.118,119
 | ||
| Fig. 12 (a) Schematic representation of the assembly of 24; (b–d) TEM images of the NT formed by 24 in the presence of CDs; (e) SEM image of the same sample. (Adapted with permission fromref. 113. Copyright 2006, National Academy of Sciences.) | ||
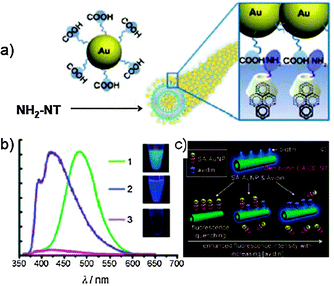 | ||
| Fig. 13 (a) Schematic representation of the assembled hybrid between Au-NPs and NH2-functionalised NTs; (b) PL spectra of vesicles of dendron 24 (green line), NH2-NTs (blue line) and Au-NPs-NTs (pink line); (c) schematic representation of the inhibition assay on biotin-NTs. (Adapted with permission fromref. 117. Copyright 2008, Wiley-VCH.) | ||
Biotin-covered NTs were also prepared and used as a biosensing platform to detect streptavidin receptor-bearing analytes. Upon addition of the streptavidin–AuNP conjugate (SA–AuNP) to a solution of biotin-NTs, the fluorescence of the NTs resulted progressively quenched because of the proximity of the pyrenyl moieties to the SA–AuNP, demonstrating the effectiveness of biotin-NTs as a potential biosensor vehicle.
2.2 Spherical assemblies
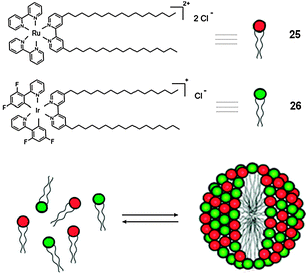 | ||
| Scheme 10 Structure and schematic representation of the self-organisation process towards the formation of the MSs containing surfactants 25 and 26. (Adapted with permission fromref. 122. Copyright 2008, American Chemical Society.) | ||
The absorption and emission spectra of surfactants 25 and 26 are shown in Fig. 14. 25 in H2O presents a maximum at 455 nm, due to the metal-to-ligand charge transfer (MLCT) transition. Under excitation and below the critical micelle concentration (CMC) (0.05 × 10−3 M), compound 25 displays a broad and rather intense emission (λmax = 635 nm).12826 (CMC = 3 × 10−6 M) presents two intense absorption bands between 250 and 300 nm, which can be assigned to ligand-centred transitions, whilst the emission spectrum at rt exhibits a maximum at 544 nm, resulting from the mixing of metal-to-ligand and ligand-centred excited states.129,130 Upon excitation at 350 nm of a 1:1 mixture of 25 (2.5 × 10−5 M) and 26 (2.5 × 10−5 M), these MSs intimately self-assemble into micellar structures, and because of the favourable 26 → 25 ET process (ΔGET# = −0.5 eV), the characteristic 16-centred emission is quenched and only the 25-centred emission at 645 nm is present.131 Yet, because of the low value of the CMC of the mixed system, the tunability of the ET process resulted difficult. In order to overcome this drawback, the conventional optically-inactive surfactant cetyltrimethylammonium bromide (CTAB) displaying a higher CMC value (0.9 × 10−3 M) was employed132 to prepare inert micelles, inside which the MSs could be encapsulated in a closed preserved environment, allowing effective ET. Indeed, the maximum distance possible inside the CTAB micelle, corresponding to the micellar dimensions (34 Å sphere for CTAB at 3 × 10−3 M),132 allows an efficient Förster-type ET process.
Fig. 14 shows the emission spectra of a 1:1 mixture (2 × 10−6 M) of 25 and 26 recorded at different concentrations of CTAB. As a consequence of the increasing concentration of CTAB's in the system, the intensity of Ru(II)-centred emission at 635 nm also increases, whilst the emission of Ir(III)-centred gradually decreases. This behaviour suggests that with an increase in the amount of CTAB above its CMC, the concentration of complexes 25 and 26 dissolved within the conventional micelles increases, allowing an effective triplet–triplet ET between the donor and acceptor MSs. This work clearly depicts the double role of micellar structures, which, as previously shown, not only can be passively involved in the control properties of the luminescent material but can also be actively involved in its construction.
![(a) Schematic representation of the electron transfer process; (b) UV-Vis and PL spectra of 25 (dotted line), 26 (dashed line), and a mixture of 25 (0.025 mM) and 26 (0.025 mM) (plain line) at rt (λexc = 350 nm). The emission spectra are indicated by asterisks; (c) intensity ratio emission, I/I0 (I0 is the intensity in the absence of CTAB) of 25 (■), 26 (●), and a 1 : 1 (2 mM) mixture of 25 (□) and 26 (○) at different concentrations of CTAB (the dashed line shows the CMC of CTAB; λexc = 350 nm); (d) emission spectra of a 1 : 1 mixture at different concentrations of CTAB ([CTAB] = 10−3 M): (a) 0.0, (b) 1.0, (c) 1.5, (d) 2.0, (e) 3.0; λexc = 350 nm). (Adapted with permission fromref. 122. Copyright 2008, American Chemical Society.)](/image/article/2012/CS/c1cs15031f/c1cs15031f-f14.gif) | ||
| Fig. 14 (a) Schematic representation of the electron transfer process; (b) UV-Vis and PL spectra of 25 (dotted line), 26 (dashed line), and a mixture of 25 (0.025 mM) and 26 (0.025 mM) (plain line) at rt (λexc = 350 nm). The emission spectra are indicated by asterisks; (c) intensity ratio emission, I/I0 (I0 is the intensity in the absence of CTAB) of 25 (■), 26 (●), and a 1:1 (2 mM) mixture of 25 (□) and 26 (○) at different concentrations of CTAB (the dashed line shows the CMC of CTAB; λexc = 350 nm); (d) emission spectra of a 1:1 mixture at different concentrations of CTAB ([CTAB] = 10−3 M): (a) 0.0, (b) 1.0, (c) 1.5, (d) 2.0, (e) 3.0; λexc = 350 nm). (Adapted with permission fromref. 122. Copyright 2008, American Chemical Society.) | ||
In this context, Yao and co-workers recently reported the supramolecular synthesis of triblock microrods of 1,3-diphenyl-2-pyroline (27, a blue-emissive molecule) doped with an orange dye, 4-(dicyanomethylene)-2-methyl-6-(p-dimethyl-aminostyryl)-4H-pyran (28), selectively at both ends, induced by CTAB micelles.133 Although these triblock microrods show a certain degree of heterogeneity, they exhibit macroscopic high-quality white light emission (WLE) in both the colloidal suspension and in the solid state with a PL efficiency of 36 ± 5%.
Scheme 11 illustrates the formation mechanism of triblock micro-rods. In a typical preparation, an aliquot of a stock solution of 27 (5 × 10−3 M) and 28 (0.01 × 10−3 M) in ethanol (27/28 = 2:1000) was rapidly injected into H2O in the presence of CTAB (0.9 × 10−3 M) under vigorous stirring, leading to a colloidal suspension within several minutes. Under the present conditions, some of the 27 and 28 molecules would first dissolve into the hydrophobic core of spherical CTAB micelles due to a process called solubilisation. 27 molecules which dissolve into spherical micelles can induce the formation of rod-like micelles,134 which subsequently act as templates directing the primary rod-like growth of monodimensional cylindrical nanoparticles 27. Finally, the micelle melting process caused by the micellar sphere-to-rod transition leads to co-deposition of 27 and 28 molecules at both ends of the as-formed primary 1D particles, generating the triblock microrods.
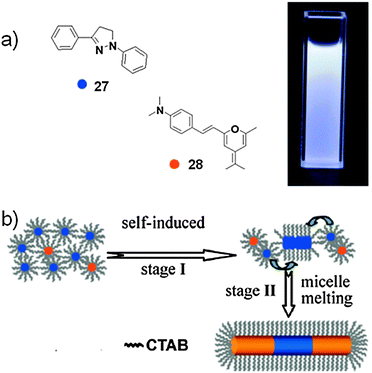 | ||
| Scheme 11 (a) Molecular structures of 27 and 28. Inset: optical photograph of a colloidal suspension of the micro-rods under UV illumination (λ = 365 nm; 17/18 = 1000:4); (b) schematic representation of the formation of the micro-rods. (Adapted with permission fromref. 133. Copyright 2010, American Chemical Society.) | ||
The absorption spectrum of the colloidal suspension of triblock microrods is similar to that of 27nanowires,134 including n–π* (at 320 nm), π–π* (at 375 nm), and J-aggregate (at 420 nm) bands, respectively. The absorption of 28 in the region 450–550 nm is hardly observed as a result of the 27/28 ratio (2:1000). Therefore, the orange emission from the end stripes of the triblock microrods might be a result of Förster energy transfer (FET) from excited 27 to 28 molecules.15,135 To selectively excite 28 components, a switch of the excitation source to blue light (440–480 nm) was actuated. Interestingly, the blue-emissive middle stripes of the triblock microrods disappear, while the yellow-emissive end stripes remain detectable. This suggests that the middle stripe of the triblock microrod is mainly composed of component 27, while molecule 28 distributes at both ends. The colloidal suspension exhibits macroscopic high-quality WLE under a UV lamp (365 nm). The steady-state excitation spectrum recorded by monitoring the 28-centred emission at 560 nm reveals energy levels including not only 28 but also 27. This is clear evidence for the efficient FET from the excited molecule 27 to 28. Therefore, the efficient FET process suggests that derivative 28 is uniformly embedded in a 27 crystalline matrix at both end stripes of the triblock microrods.15SEM and TEM microscopy (Fig. 15) images show rod-like structures with a smooth outer surface. Each individual rod has a quite uniform diameter (∼1 μm) throughout the entire length (20–25 μm). Fluorescence microscopy images of microrods placed on a quartz slide by excitation with unfocused UV light (330–380 nm) highlight the triblock heterostructures with striping patterns: the middle stripe exhibits blue photoluminescence, while the two ends are orange.
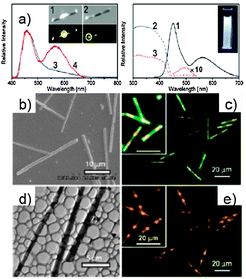 | ||
| Fig. 15 (a) Spatially resolved spectra recorded by local excitation at the middle (blue line) and end (red line) stripes of a single micro-rod, PL (black line) and excitation (blue and red dotted lines) spectra of a colloidal suspension of triblock micro-rods (λexc = 450, 560 nm); (b) SEM and (d) TEM images of the micro-rods; (c and e) fluorescence microscopy images excited with unfocused UV (λ = 330–380 nm) and blue light (λ = 460–490 nm). (Adapted with permission fromref. 133. Copyright 2010, American Chemical Society.) | ||
The synthesis of amphiphilic dumbbell-shaped molecules consisting of a rod segment connected by hydrophilic oligoether dendrons at one end and hydrophobic branched alkyl chains at the other end is outlined in Scheme 12. Dendritic oligoether dendrons and branched alkyl chains are prepared according to the procedures described in the literature.140–142 The meta-linked dihydroxy triphenylene derivative was involved in an etherification reaction with a tosylated oligoether dendron. Suzuki coupling reaction with 4-(t-butyldimethylsilyloxy)-biphenyl-4′-boronic acid in the presence of a Pd(0)-based catalyst allowed the linkage of the hydrophilic moiety.141–143 The Y-shaped hydrophobic alkyl dendrons have been synthesised using a similar procedure. Final coupling of the two moieties to 4,4′-bis(bromomethyl)-biphenyl by a Williamson-like reaction in the presence of K2CO3 yielded the final dumbbell-shaped amphiphilic rods 29–32.
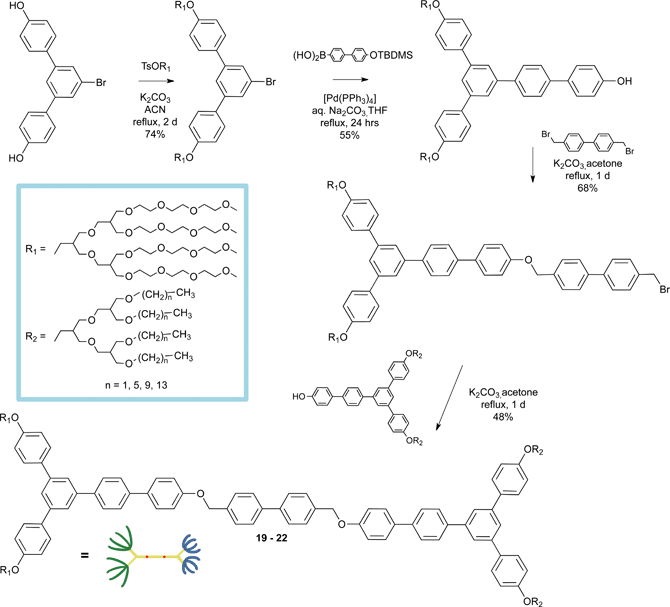 | ||
| Scheme 12 Synthetic pathway for the synthesis of the dumbbell-shaped rods 29–32. | ||
Molecular amphiphiles 29–32 when dissolved in a solvent suitable for the oligoether segments can self-assemble into aggregation structures.144 Direct visualisation of the aggregates by a cryogenic-TEM technique was performed in aqueous solution.145Amphiphile 29 based on an ethyl group self-assembles into discrete spherical objects with an average diameter of 11 nm (Fig. 16). This diameter corresponds to approximately twice the extended hydrophobic segments (∼5.6 nm by CPK modeling), implying that aggregates of molecule 29 are micellar in nature. Increasing the alkyl chain from ethyl to hexyl gives rise to a significant structural change, namely from spheres to toroids. TEM images clearly show dark toroidal objects with a uniform diameter of ∼12 nm, dimension in reasonable agreement with twice the length of the hydrophobic segments, including the rod and alkyl chains. A further increase in the length of the alkyl chain to a decyl group drives the system to form 2-D sheets. The magnification of the 2-D objects by cryo-TEM showed that the sheets consist of in-plane nanopores, most probably implying that the toroidal structure directly transforms into planar nets without passing through an intermediate structure such as extended cylindrical micelles.
 | ||
| Fig. 16 (a) Schematic representation of the aggregation behaviour of the rod amphiphiles in aqueous solution; (b) TEM image of a cast film and (c) cryo-TEM image exhibiting a spherical structure of 29; (d) cryo-TEM image showing a toroidal structure of 30 (e) cryo-TEM image of a planar network structure formed from 31; (d) cryo-TEM image of vesicles formed from 32. (Adapted with permission fromref. 136. Copyright 2007, American Chemical Society.). | ||
TEM experiments showed that branched oligophenylene 32 undergoes a hollow vesicular structure with diameters ranging from 500 nm to a few micrometres and a uniform bilayer thickness of 13 nm. This result indicates that an increase in the hydrophobic alkyl length decreases the interfacial curvature to relieve the energetic loss associated with the loosely-packed edge parts.146 Consequently, the nanoporous 2-D nets transform into hollow vesicular structures to avoid energetically unfavourable edges. The increment in the extent of parallel arrangement at the expense of radial arrangement is reflected in the systematic shift of the absorption edge towards higher wavelengths in UV-Vis absorption spectra, indicating that π-stacking interactions between adjacent rod segments increase with increasing alkyl length.147,148 As a result, the strong propensity of the elongated rod segments to align in a parallel fashion, together with the repulsive interactions between bulky oligoether dendrons, seem to be the primary driving forces in this structural progression.
Ajayaghosh and co-workers recently reported the hierarchised assembly behaviour of the melamine-functionalised oligo(p-phenyleneethynylene) (OPE) 33, which in the presence of a barbituric acid derivative (CA) in aliphatic solvents self-assembles into luminescent toroidal-like objects of nanometre dimension (Scheme 13).149
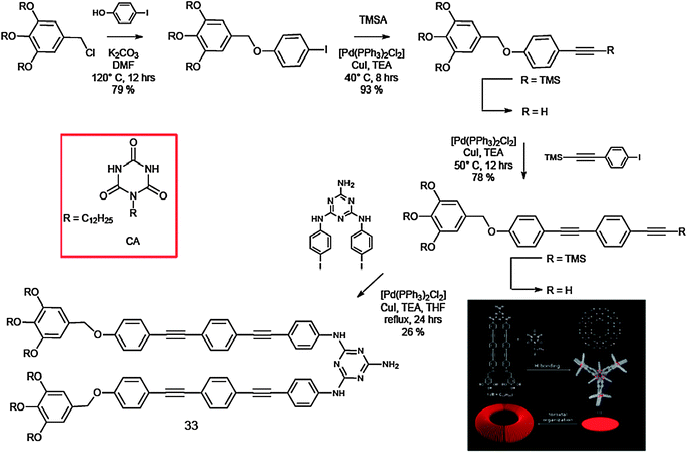 | ||
| Scheme 13 Synthetic pathway for the preparation of molecule 33 and representation of its self-assembly with CA. | ||
The melamine-linked OPE derivative 33 was prepared by a Sonogashira coupling150 of N,N′-di(p-iodophenyl)triaminotriazine with 3,4,5-tridodecyloxybenzyloxy-substituted ethynyltolan. Co-assembly of 33 with CA resulted in discrete rosettes.151 A unique stacking property might be evolved for the present rosette, for which 33 π-electronic segments play an important role. As shown by the absorption measurements, a major driving force for the association of rosettes is H-type aggregation between 33 segments. However, molecular modelling shows that 33 π planes are rotated out of the hydrogen-bonded plane by 45°. Extended association of rosettes in such a tilted stacking fashion could lead to the formation of columns with a curvature, and eventually to the formation of toroidal architectures in solution. The absorption spectrum of the equimolar mixture of 33 and CA in CHCl3 (1 × 10−2 M) showed a maximum at λ = 334 nm, which is slightly blue-shifted from that of 33 alone in CHCl3 (λmax = 339 nm), indicating H-type aggregation of OPE segments upon conformational fixation of 33 to form a H-bonded rosette assembly with CA.152,153 The emission of the OPE co-assembly showed a red shift of ca. 30 nm compared to that of the monomer (λem-max = 378 to 407 nm), which is in good agreement with the H-type excitonic coupling. An insight into the morphology of the hierarchical structures formed from the rosette in decane is obtained from tapping-mode AFM images, which displayed a large number of toroidal nanoobjects (Fig. 17).
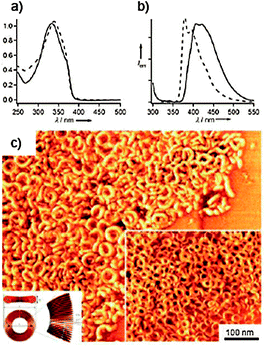 | ||
| Fig. 17 (a) Absorption and (b) fluorescence spectra of 33 in the presence of 1 equivalent of CA (solid curves, C = 1 × 10−2 M) and 33 alone (dashed curves, C = 1 × 10−5 M) in CHCl3 (λexc = 300 nm); (c) AFM phase image of an equimolar mixture of 33 and CA spin-coated from decane solution on HOPG. (Adapted with permission fromref. 149. Copyright 2008, Wiley-VCH.) | ||
Getting deeper into the matter, Ajayaghosh and Kitamura et al. demonstrated that toroidal structures can transform into fibrous assemblies upon increasing the concentration. In fact, the melamine–oligo(p-phenylenevinylene) conjugate (OPV) 34, upon complexation with 1/3 equivalents of CA, provides well-defined, ring-shaped nanostructures at micromolar concentrations, which open to form fibrous assemblies at submillimolar concentrations and organogels in the millimolar concentration range.154 Apparently, the enhanced aggregation ability of 34 by CA is a consequence of columnar organisation of the resulting discotic complex [343·CA].
In Scheme 14 the preparation of the desired OPV 34 is described. Subsequent nucleophilic attack on 2,4,6-trichloro-1,3,5-triazine, first by the precursor amino-OPV,155 and subsequently by dodecylamine and by dioctylamine afforded the target 34. The UV-Vis and fluorescence spectra of dilute solutions of 34 (1 × 10−5 M) in CHCl3 and methyl-cyclohexane (MCH) were measured to compare their self-aggregating behaviour (Fig. 18). 34 in MCH showed the π–π* transition at λmax = 376 nm, which is identical to that observed in CHCl3 (λmax = 377 nm), indicating that 34 exists in the molecularly dissolved state (or more precisely, free from π–π stacking interactions) at this concentration.155 The PL spectrum of the solution in MCH presented no noticeable change in the peak positions, if compared to the spectra of the solution in CHCl3, confirming the absence of aggregation. Self-aggregation of 34 starts to occur at around 5 × 10−4 M, shifting the π–π* transition to λmax = 365 nm.156 However, the fluorescence spectrum at this concentration only shows a slight increase in the relative fluorescence intensity of the peak at 439 nm compared with that of the 1 × 10−5 M solution. This observation indicates that the 365 nm absorbing species are weakly interacting molecular assemblies, which undergo rapid exchange with molecularly dissolved species. These spectral changes are characteristic of the π–π stacking of OPV chromophores.155,157–160 The AFM images confirmed the formation of the toroidal nanostructures. Remarkably, the sample prepared from the mixtures of 34 and CA showed agglomerated and isolated ring-shaped nanostructures (Fig. 18).
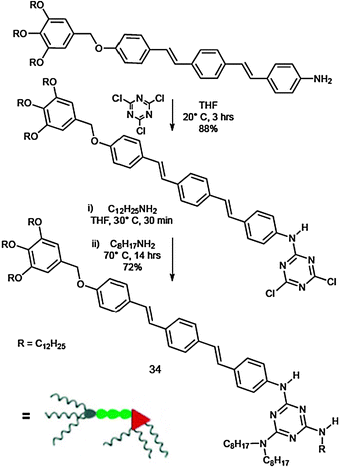 | ||
| Scheme 14 Synthetic pathway for the preparation of 34. | ||
![(a) Schematic representation of the assembly of 34 in the presence of CA; (b and c) UV-Vis and PL spectra of 34 (1 × 10–5 M, λexc = 349 nm) in CHCl3 (red curve) and MCH (blue curve) at 20 °C; (d–f) AFM images of [343·CA] spin-coated from a solution in MCH (2 × 10−4 M); (g) AFM height image of [343·CA] spin-cast from a solution in MCH (5 × 10−4 M). (Adapted with permission fromref. 154. Copyright 2010, Wiley-VCH.)](/image/article/2012/CS/c1cs15031f/c1cs15031f-f18.gif) | ||
| Fig. 18 (a) Schematic representation of the assembly of 34 in the presence of CA; (b and c) UV-Vis and PL spectra of 34 (1 × 10–5 M, λexc = 349 nm) in CHCl3 (red curve) and MCH (blue curve) at 20 °C; (d–f) AFM images of [343·CA] spin-coated from a solution in MCH (2 × 10−4 M); (g) AFM height image of [343·CA] spin-cast from a solution in MCH (5 × 10−4 M). (Adapted with permission fromref. 154. Copyright 2010, Wiley-VCH.) | ||
When a homogeneous solution of the 3:1 mixture of 34 (5 × 10−3 M) and CA in MCH was kept at rt, a transparent gel was formed after one day. The AFM observation of a dried gel did not show any fibrous structures typically observed for low-molecular-weight gels. Subsequently, the gel was diluted to 1 × 10−4 M. AFM images of the spin-cast solution exhibited short wormlike structures (Fig. 18), which are considered to be intermediate species between the gel-forming extended columnar structures and the toroids formed in solutions at lower concentration. These structures were <300 nm in length and 10–30 nm in width.
Molecule 35 was synthesised by using multistep synthetic strategies involving Pd(0)-catalysed Sonogashira coupling reactions, followed by reduction in the presence of sodium boron hydride (Scheme 15).164 The UV-Vis spectrum of 35 in CHCl3 (1 × 10−5 M) at 25 °C showed a strong π–π* absorption band at 377 nm. In decane, the absorption maximum is red-shifted to 385 nm and the formation of an additional band at 419 nm was observed. The emission maximum in CHCl3 was at 414 nm, which was significantly red-shifted to 443 nm in decane. These observations are attributed to the J-type aggregation of 35 in decane.
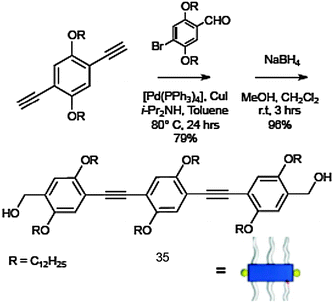 | ||
| Scheme 15 Synthetic route to OPE 35. | ||
AFM images of the self-assemblies of 1 × 10−6 M and 1 × 10−5 M solutions of OPE 35 in decane at different magnifications are shown in Fig. 19. These images reveal the concentration-dependent self-assembly of 35 to individually dispersed particles of different sizes, with an average diameter of 94 ± 2 nm and a height of 4 ± 1 nm at 1 × 10−6 M. In contrast, the analysis of the particles formed at 1 × 10−5 M revealed an average diameter of 535 ± 10 nm and a height of 20 ± 5 nm. The wall thickness of the vesicle is approximately 10 nm and has a bilayer structure, each layer having a thickness of about 4 nm. Further increase of the concentration (1 × 10−4 M) resulted in the formation of fluorescent microspheres with diameters as large as 10 mm. These microspheres were agglomerated and fused in many places, in contrast to the individual nanospheres obtained at low concentrations. A self-supporting gel with strong blue fluorescence (λmax = 443 nm, Fig. 19) is obtained at 3.4 × 10−3 M. SEM image of a dried 35 gel obtained from decane (3.4 × 10−3 M) reveals the formation of superstructures of micrometre size. The morphological features indicate that these extended structures could be formed from fused microspheres at higher concentrations.
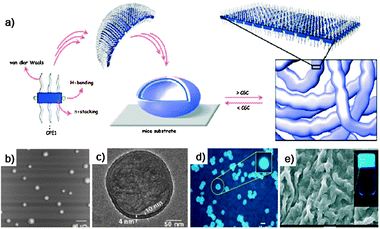 | ||
| Fig. 19 (a) Schematic representation of the self-assembly process of 35; (b) AFM tapping mode height images of 35 in decane; (c) HR-TEM image of a sphere of 35 from decane; (d) optical microscopic picture of 35 (1 × 10−4 M in decane) under UV illumination (λ = 365 nm); (e) SEM image of 35 gel (3.4 × 10−3 M) in decane. Inset: photograph of the gel in decane under UV illumination (λexc = 365 nm). (Adapted with permission fromref. 161. Copyright 2006, Wiley-VCH.) | ||
In a collaborative work with the group of Armaroli, our team has recently demonstrated that under temperature and solvent polarity control, supramolecular adducts between two π-conjugated molecules bearing complementary H-bonding end groups can be formed in solution and further self-organise evolving into hollow spherical nanoparticles with a vesicular structure (Scheme 16).165 Most notably, the nanoparticle size distribution and shape could be tuned by changing the stoichiometric ratio of the two molecular components, as a consequence of the specific solvophobic/solvophilic interactions established within the supramolecular adducts.
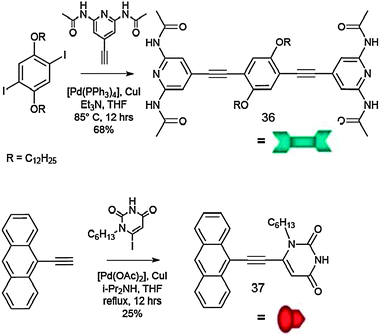 | ||
| Scheme 16 Synthetic pathway towards complementary molecules 36 and 37. | ||
Molecule 36 consists of a p-disubstituted central benzene ring surrounded at both sides by 2,6-di(acetylamino)-pyridyl terminal groups; it was prepared through a Pd(0)-catalysed Sonogashira coupling reaction between the bis-iodo derivative of the alkylated hydroquinone166 and 2,6-di(acetylamino)-4-ethynylpyridine.167 Module 37 possesses only one H-bonding active site, as it bears only one uracyl unit on the anthracenyl moiety, to which it was coupled through a Pd(0)-catalysed Sonogashira reaction.167 These molecular units are known to undergo self-assembly through triple H-bonding with very high association constants in apolar solvents, i.e. of the order of 104–105 M−1.168
Molecules 36 and 37 exhibit intense absorption bands in the UV region and are strong luminophores (ΦF of 9 and 63%, respectively) in cyclohexane (CHX) solution (Fig. 20). The absorption spectrum of the ternary complex [36·(37)2] presented a red shift over the low energy absorption band of 37 (not observed for 37 alone), indicating the presence of π–π stacking interactions, most likely promoted by the preliminary formation of the H-bond system between the complementary uracil and 2,6-di(acetylamino)pyridil moieties.167
![(a) Schematic representation of the assembly of 36 and 37 into vesicular adducts; (b) experimental and calculated absorption spectra of 36, 37 and [36·(37)2] (left), and variable-T absorption spectra of 36 in cyclohexane (right). TEM (c and d) and AFM (e and f) images of the self-assembled structures of 36 (c and e) and [36·(37)2] (d and f). (Adapted with permission fromref. 165. Copyright 2009, Royal Society of Chemistry.)](/image/article/2012/CS/c1cs15031f/c1cs15031f-f20.gif) | ||
| Fig. 20 (a) Schematic representation of the assembly of 36 and 37 into vesicular adducts; (b) experimental and calculated absorption spectra of 36, 37 and [36·(37)2] (left), and variable-T absorption spectra of 36 in cyclohexane (right). TEM (c and d) and AFM (e and f) images of the self-assembled structures of 36 (c and e) and [36·(37)2] (d and f). (Adapted with permission fromref. 165. Copyright 2009, Royal Society of Chemistry.) | ||
The morphology of the self-assembled structures was characterised using TEM and AFM microscopy techniques (Fig. 20). The obtained images highlighted the ability of 36 to self-assemble in CHX into both spherical micro- and nanoparticles, with sizes ranging from 10 nm to over 1 mm. Both TEM and AFM images of a drop-cast solution of 36 and 37 (1:2) in cyclohexane instead showed the presence of luminescent vesicles of highly uniform size distribution (diameter range of 80–180 nm, Fig. 20). These results demonstrated that the presence of 37 increases the solute–solvent interaction as a consequence of the change of the solvophobic 2,6-di(acetylamino)pyridil into the solvophilic anthracenyl moiety, thus favouring the formation of the thermodynamically stable nanostructures in which the unfavourable interactions of the polar di(acetylamino)pyridyl units with the apolar solvent are minimised. In the light of these results and aimed at the understanding of the influence of the molecular geometrical properties on the final microstructured morphology, a regioisomeric library of chromophoric acetylenic scaffolds equipped with 2,6-di(acetylamino)pyridil or uracil moieties has also been constructed . Depending on the regioisomers, several hierarchised nanostructured architectures, namely nanoparticles, nanovesicles, nanofibers and nanorods (Fig. 21), could be obtained through a fine tuning of the self-assembling conditions.169
![(a) Structure of the different molecular modules bearing complementary H-bonding units; schematic representation of the different possible assemblies; tapping mode-AFM (TM-AFM) images of assemblies [36·38] (b), [36·39]n (c) and [36·40] (d) on the mica surface. (Adapted with permission fromref. 169. Copyright 2011, Wiley-VCH.)](/image/article/2012/CS/c1cs15031f/c1cs15031f-f21.gif) | ||
| Fig. 21 (a) Structure of the different molecular modules bearing complementary H-bonding units; schematic representation of the different possible assemblies; tapping mode-AFM (TM-AFM) images of assemblies [36·38] (b), [36·39]n (c) and [36·40] (d) on the mica surface. (Adapted with permission fromref. 169. Copyright 2011, Wiley-VCH.) | ||
As previously discussed, module 36 alone self-assembles into fluorescent spherical particles, whilst when mixed with 37 in a 2:1 ratio, it affords hollow vesicular spheres. Upon mixing molecules 36 and 38 in toluene (1:1 molar ratio), the formation of a supramolecular polymer aggregate occurred. AFM images indicate the presence of nano-wires of length above 400 nm and width ∼30 nm (Fig. 21). To control the length of the polymer [36·38]n, addition of molecular stopper 37 was envisaged, in the presence of which a large portion of nanorods (length and width between 80–200 nm and 40–60 nm, respectively) along with some amounts of spherical aggregates were detected. For assembly [36·39]n, the AFM images indicate the formation of nano-fibres, exhibiting humps and lumps suggesting the formation of helicoidally-organised nanostructures (Fig. 21). The combination of molecules 36 and 40 led to the formation of a toroid-like structure (Fig. 21), with average dimensions ranging from 680 to 860 nm and 290 nm to 390 nm in outer and inner diameters, respectively, and from 1 to 4 nm in height. As a consequence of the two-dimensional molecular character of module 40, assembly [36·40]n could give birth to 2D nanostructures that, depending on the concentration of the single modules, could cover the entire surface. At higher concentration (1.2 × 10−3 M), these nano-structured objects cover the entire mica surface, still conserving their circular structures. These examples suggest how a univocal correlation between the molecular structure of the organic module and the architecture of the nano-objects is difficult to achieve and a controlled design is still far from being predicted.
In an another recent report,170 we have also demonstrated that it is possible to thermally trigger171 the self-assembly/self-organisation processes to induce the formation of objects on a mica surface that exhibit crater-like morphologies and a very homogeneous size distribution (Fig. 22). Specifically, a tetra-substituted pyrenyl bearing four N-tert-butoxycarbonylamino (N-BOC) protected uracilic moieties (Py-U4) could be selectively and thermally deprotected to induce supramolecular polymerisationin situ in the presence of complementary linear module 38, and consequently the formation of the above-described morphologies. Confirmation of the presence of the hydrogen-bonding-driven self-assembly/self-organisation process was obtained by variable-temperature (VT) steady-state UV-Vis absorption and emission measurements in o-dichlorobenzene (o-DCB). Interestingly, addition of an equimolar solution of linear module 38 to a preheated (150 °C for different time intervals and then cooled to 20 °C) 17 μM solution of N-BOC protected tetratopic pyrenyl derivative resulted in a progressive decrease in the absorption feature at 480 nm and the concomitant appearance of a new band centered at 505 nm. The emission spectra recorded under identical conditions exhibited a decrease in fluorescence intensity.
![Schematic representation of the nanostructuration process between molecule 36 and Py-U4 triggered by the T-dependent removal of the BOC protecting group; (a and b) TM-AFM topography of the crater-like self-organised morphologies as obtained from the assembly of molecules [Py-U4·36]n from a solution deposited on mica surfaces at 145 °C and (d) their 3D representation; (e) cross-sectional profiles of three different nanocraters marked in (b). (Adapted with permission fromref. 170. Copyright 2011, American Chemical Society.)](/image/article/2012/CS/c1cs15031f/c1cs15031f-f22.gif) | ||
| Fig. 22 Schematic representation of the nanostructuration process between molecule 36 and Py-U4 triggered by the T-dependent removal of the BOC protecting group; (a and b) TM-AFM topography of the crater-like self-organised morphologies as obtained from the assembly of molecules [Py-U4·36]n from a solution deposited on mica surfaces at 145 °C and (d) their 3D representation; (e) cross-sectional profiles of three different nanocraters marked in (b). (Adapted with permission fromref. 170. Copyright 2011, American Chemical Society.) | ||
These observations, along with the formation of a new red-shifted absorption band and the quenching of the pyrene-centered fluorescence, are attributed to the formation of H-bonded supramolecular assemblies initiated by the thermal cleavage of the BOC protecting group.165,167 This was further proven by the addition of 0.2 mL of dimethylsulfoxide (DMSO), in which a complete restoration of absorption and emission spectral profiles was observed. The morphologic and geometric characterisation of nanostructures [Py-U4·38]n, as obtained by drop casting a solution of the functionalised pyrene and module 38 (1:1) onto mica surfaces heated to different temperatures, showed the formation of very regular nano-objects. As clearly seen in Fig. 22, the AFM imaging showed the formation of discrete nanostructures, the morphology of which resembles that of a crater. The variation of the geometrical and spatial features of the morphologies as monitored by AFM imaging at different temperatures allowed the first elaboration of a thermodynamic theoretical model, showing that the self-organisation process is transport-driven (i.e., depending on the solute diffusivity D0 and solvent kinematic viscosity ν) and thermal-dependent, as only crater-like structures have been observed at the temperature cleavage of the BOC functions thus definitely showing to be depending on the H-bonding recognition.
3. Templated self-assembly and self-organisation processes
3.1 Zeolites
Exploiting the peculiar orthogonal functionalisation methodologies of zeolites, Calzaferri and De Cola's groups recently reported a multi-chromophoric loaded zeolite, in which the green-emitting Atto-425 (molecule 41, λabs = 425 nm, λem = 468 nm) and the red-emitting Atto-610 chromophore (molecule 42, λabs = 614 nm, λem = 630 nm) were orthogonally positioned on the coat and at the channel entrances, respectively.172 Notably, due to the spatial separation between the two dyes exceeding the critical distance, at which electronic energy transfer might occur, no evidences of parasite energy transfer processes between the two chromophores have been observed.Molecular emitter 42 was placed at the channels' entrances of the zeolites both by covalent and non-covalent strategies. In the first case, the zeolite crystals were reacted with a molar amount of 9-fluorenylmethyl carbamate-3-aminopropyl-methoxydimethylsilane (FMOC–APMS), known to selectively react at the channel entrances of zeolite crystals.173 The FMOC protecting group was then removed in the presence of piperidine, affording zeolite exposing –NH2groups exclusively localised at channel entrances, which were subsequently coupled with the N-hydroxysuccinimide (NHS) ester of the red 42dye, obtaining an amidic linked system (AL). Because of its cationic nature and linear aromatic geometry, the cationic molecular fragment of 42 can also enter into the zeolites' channels.174 Thus, functionalisation of the dye with a terminating tail group composed of a bulky 2,2-diphenyl ethylamine (DPA) group, displaying a ring-to-ring distance of 9.0 Å, does not allow penetration of the chromophore inside the channel due to its superior diameter with respect to the zeolite channel entrances (7.1 Å).175 By this means, chromophore 42 remains stuck at the entrances, behaving as a “stop-cocked” system. Both the covalently and non-covalently base-functionalised zeolites were subsequently reacted with 3-aminopropyl-triethoxysilane (APTES) in order to functionalise their coats with free NH2 groups, which were subsequently reacted with the NHS ester of 41.173Epifluorescence microscopy of both samples clearly shows red fluorescence at the bases and a uniform green fluorescence covering the surface of the crystals. Interestingly, a region of yellow emission is seen at the interface between the two different emitting zones, resulting from mixed red and green emissions (Fig. 23). For comparison purposes, a mixed model system (MM) comprising an equimolar mixture of 41 and 42 bonded to a zeolite crystal with 100% amine coverage was prepared. Epifluorescence microscopy showed the crystals to have a homogenous red-orange colour (Fig. 23), distinctively different from the emission of the orthogonal systems. Employing a similar procedure, the same group prepared novel phototherapeutic tools based on the functionalised zeolite L capable of targeting and eliminating antibiotic-resistant bacteria, such as Gram-negative. These bacteria are known to be very resistant towards photoinactivation owing to the presence of an additional outer lipo-polysaccharidic membrane.176 Since amino-functionalised zeolites have been demonstrated to be able to bind E. colicells,177 these systems provide an effective anchor and vehicle to target the bacterial surface owing to their capacity to interact through hydrogen bonding and electrostatic interactions. For these purposes, a trifunctional zeolite-based nanomaterial composed of a bio-labelling dye, a photosynthesiser moiety such as phthalocyanine (PC) for the in situ photoproduction of therapeutic 1O2, and an amino coating for the specific binding of bacteria has recently been developed by De Cola's group (Fig. 24).178
 | ||
| Fig. 23 (a) Schematic representation of the chromophores 41 and 42, and the different functionalisation techniques employed; (b) epifluorescence micrograph of the mixed sample containing both 41 and 42 (MM); (c) epifluorescence micrograph image of the “orthogonal” stop-cocked sample containing 41 and 42 (SC); (d) confocal micrographs of multiple and single amide-linked orthogonal systems (AL). (Adapted with permission fromref. 172. Copyright 2008, Wiley-VCH.) | ||
 | ||
| Fig. 24 (a) Schematic representation of the multifunctional nanomaterial used to target, label and photoinactivate antibiotic-resistant bacteria. The zeolite L nanocrystal is loaded with 43, and its surface is functionalised with a PC through covalent linkage with the amino groups present on the surface of the zeolite; (b) bright-field microscopy, (c) fluorescence microscopy (λexc = 470–490 nm) and (d) SEM images of the bacteria treated with the functionalised zeolite. (Adapted with permission fromref. 178. Copyright 2009, Wiley-VCH.) | ||
Green-luminescent dye, N,N′-bis(2,6-dimethylphenyl)perylene-3,4,9,10-tetracarbodiimide (43), was inserted into the channels of zeolite L nanocrystals by gas-phase insertion.175 The loading of 43 was kept below 1% to avoid the formation of J aggregates inside the zeolite L channels, which are known to lower the photoluminescence quantum yield and to cause a bathochromic shift of the emitted wavelength.179 Such low concentrations give rise to uniform green luminescence. The 43-loaded zeolite L nanocrystals were further functionalised with tetra-t-butyl-substituted Si(IV) PC by axial attachment of the macrocycle's central silicon atom to OH groups of the zeolite L surface. Finally, the surface of the multifunctional zeolites was coated with amino groups to promote the adhesion to bacteria.177 Distinctive excitation and emission spectra of PC180,181 demonstrated that both the zeolite L surface and the presence of the tert-butyl substituents on the PC hindered the formation of aggregates thus ultimately preserving its photoactivity.176,182,183 Upon excitation of PC at 670 nm an intense emission with maximum at 1275 nm is obtained, which matches the characteristic phosphorescence of 1O2.180,181 The use of the 1O2 quencher 1,4-diazabicyclo[2.2.2]octane (DABCO) completely suppressed the emission at 1275 nm confirming the effectiveness in the production of 1O2. Moreover no electronic energy transfer has been observed occurring from perylene derivative 43 to PC, which is a consequence of the electronic decoupling between the chromophores localised inside the zeolite L channels and those attached to the outer surface.172,175
Photodynamic treatments have been carried out by suspending a batch of E. colicells with the trifunctional hybrid zeolite (Fig. 25) and irradiating the sample for 2.5 h in the wavelength range in which only the PC is excited to produce 1O2 (570–900 nm). Whilst no significant cell death was detected in the absence of PC or of irradiation, the photodynamic inactivation of a batch of E. coli in the presence of the photoactive hybrid material (i.e., with PC) was complete (95%) after 2 h, corresponding to an unprecedented low light dose of 27 J cm−2, considerably smaller than other values described in the literature with other materials.184
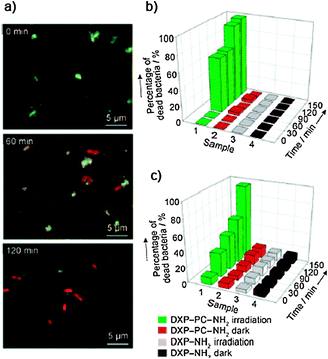 | ||
| Fig. 25 (a) Time-dependent fluorescence microscopy images of E. colicells during photodynamic treatment (λexc = 470–490 nm). The green emission comes from the 43-filled hybrid nanomaterial, while the red one arises from the PI–DNA complex, which is only formed inside the dead bacteria; (b) percentage of inactivated E. coli and (c) N. gonorrhoeaecells as a function of time and experimental conditions. (Adapted with permission fromref. 178. Copyright 2009, Wiley-VCH.) | ||
3.2 Carbonaceous material
Alternatively, covalently functionalised fluorescent NDs have been thoroughly reported. The group of Mochalin and Gogotsi recently developed the synthesis of hydrophobic blue fluorescent material (ND-44) by covalent linking of octadecylamine (44) to 5 nm ND particles.186
Functionalisation of NDs with 44 through amide bond formation occurs with a procedure similar to that used for SWCNTs (Scheme 17).187 Oxidized NDs188 were first refluxed with SOCl2 and the acyl-chloride derivative was then reacted with terminal amino functions. While ND completely precipitates within 1 h, ND-44 without any sonication or other means forms a clear, stable pale yellowish colloidal solution in toluene displaying high stability. With respect to known red and green fluorescent NDs, ND-44 displays a bright-blue fluorescence signal under UV light excitation (emission at 450 nm), whilst neither pristine ND nor neat 44 show any fluorescence.189,190
 | ||
| Scheme 17 Synthetic pathway for the preparation of ND-44. Excitation (violet line, λem = 450 nm) and emission (blue line, λexc = 410 nm) spectra of ND-44 dispersion in CH2Cl2. Inset: photograph of ND-44 dispersion in CH2Cl2 under UV irradiation (λ = 365 nm). (Adapted with permission fromref. 186. Copyright 2009, American Chemical Society.) | ||
![(a) Synthetic procedure towards the exfoliation of graphite producing monolayer graphene·45 hybrids; (b) images of the aqueous solutions of 45, [EG·45] and [HG·45] and (c) corresponding images under UV illumination (λ = 365 nm); (d) schematic representation of the exfoliation of few-layer graphene with 45 to yield monolayer graphene·45 hybrids. (Adapted with permission fromref. 191. Copyright 2010, Wiley-VCH.)](/image/article/2012/CS/c1cs15031f/c1cs15031f-f26.gif) | ||
| Fig. 26 (a) Synthetic procedure towards the exfoliation of graphite producing monolayer graphene·45 hybrids; (b) images of the aqueous solutions of 45, [EG·45] and [HG·45] and (c) corresponding images under UV illumination (λ = 365 nm); (d) schematic representation of the exfoliation of few-layer graphene with 45 to yield monolayer graphene·45 hybrids. (Adapted with permission fromref. 191. Copyright 2010, Wiley-VCH.) | ||
Graphene samples were prepared by two different methods, namely thermal exfoliation of graphite oxide (EG) or arc evaporation of graphite under a H2 atmosphere (HG).194–196 Aqueous solutions of graphene were prepared by the addition of coronene derivative 45 to a dispersion of EG or HG in distilled H2O and heating the mixture at 100 °C for 24 h and subsequently sonicated at 70 °C for 2 h to ensure maximum solubilisation. The ultrasonication step is expected to overcome the van der Waals inter-plane graphene interactions helping the intercalation of polar coronene derivative 45. Characterisation of hybrid graphene 45 in solution was carried out by absorption and fluorescence spectroscopies. The spectrum of 45 in H2O exhibits strong absorption at 310 nm and weak bands between 400 and 450 nm, consistent with the spectra of coronene-tetraester derivatives.192 Interestingly, solutions of [EG·45] and [HG·45] showed absorption bands that were significantly red-shifted, appearing at 470 and 505 nm, thus proving the strong interaction between perylene 45 and the graphene layers. Fluorescence measurements showed significant quenching of the coronene-centred emission in all hybrid samples (reduced by a factor of 27 and 20 for the solutions containing [EG·45] and [HG·45], respectively). The quenching of fluorescence in all hybrids is accompanied by the appearance of a weak red-shifted emission around 600 nm, suggesting the effectiveness of the interaction between the two molecular counterparts. Solid thin films of the hybrids deposited on glass show similar optical changes to those observed in solution. Furthermore, emission bands at 435 and 462 nm, corresponding to loosely-bound molecules, are quenched in the film confirming efficient charge-transfer processes in the solid state, and thus the potential use in organic electronics.
Attachment of the fluorophores to PVP was accomplished via the tyramide-horseradish peroxidase reaction,198 in which horseradish peroxidase is used to activate tyramide-fluorophore conjugates generating active tyramide radicals. Addition of PVP to this reaction mixture results in fluorescent labelling of PVP. Fluorescent coating of SWCNTs was accomplished by mixing the suspension of SWCNTs stabilized in 1% SDS with various amounts of fluorescent PVP (PVP-46, 47, 48). Electrophoresis was then employed as a method to strip the excess of the PVP-46, 47, 48 from the hybrid in a controlled fashion. Direct examination of the fluorescently-labeled SWCNTs using a fluorescent microscope allowed the detection of multiple fluorescent single nanotubes, demonstrating that the prevention of fluorescence quenching had been achieved (Fig. 27). TEM images of SWCNT/PVP-47 showed individual nanotubes and small bundles associated with a monolayer of polymer. Highly ordered packing of the polymer around the SWCNT was observed, similar to that of a wire in an induction coil, characterized by areas with different thicknesses (∼2.5 nm and ∼0.7 nm thick areas) which alternated periodically. Being driven by strong thermodynamic forces,199 the tense polymer coiling requires very high molecular flexibility, creating strong internal stress, which has to be relaxed by zones of less tight packaging. These results pointed out that SWCNTs could be fluorescently labelled via non-covalent coating with a fluorescent polymer. Such nanotubes have a potential to serve as unique fluorescent probes because multiple fluorophores attached to the surface of a single nanotube make it a convenient composite applicable for detection of single molecule events.
 | ||
| Fig. 27 (a) TEM images of PVP-47·NT; (b) magnification of the area and in the inset representation of the stacking of the polymer coils (scale bar = 10 nm); (c) combined light and fluorescent microscopy of nanotube bundles (scale bar = 7 μm). Individual fluorescent CNTs are only seen using fluorescence detection. (Adapted with permission fromref. 197. Copyright 2005, American Chemical Society.) | ||
Ajayaghosh and Praveen investigated the influence of CNTs on gel-forming oligo(p-phenylene vinylene)s (OPVs),8 demonstrating that the self-organisation of the OPV molecules results accelerated through the interaction with CNTs in hydrocarbon solvents.200 The individually dispersed CNTs are significantly aligned within the OPV gel, thereby reinforcing the OPV supramolecular tapes, acting as physical cross-linkers between the single tapes, thus enhancing the stability of the gel. As the CNTs are not chemically modified, these hybrid π-conjugated gels retain the physical and electronic properties of the CNTs (Fig. 28).
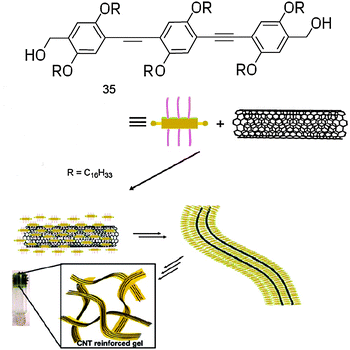 | ||
| Fig. 28 Chemical structures of OPV 35 and schematic representation of hybrid gel 35·CNT. (Adapted with permission fromref. 200. Copyright 2008, Wiley-VCH.) | ||
OPV 35 is known to form organogels above a critical concentration in nonpolar solvents.8 However, in relatively polar solvents, such as toluene, higher concentrations of molecule 35 lead to the formation of a gel mechanically unstable. Interestingly, addition of small amounts of SWCNTs (HiPco) to this solution with sonication and subsequent standing at rt resulted in a soft dense solid, thus indicating that the gelation process had occurred. During the process, SWCNTs become uniformly dispersed in the gel matrix. The absorption spectrum of the composite exhibited a shoulder at 464 nm corresponding to the tape-like thermo-reversible aggregates of 35 (Fig. 29). The characteristic electronic transitions corresponding to the metallic (400–600 nm) and the semiconducting (600–900 nm and 1200–1500 nm, respectively) SWCNTs have also been clearly detected, in agreement with the reported spectra of dispersed HiPco SWCNTs.4,201–203 Addition of SWCNTs to a solution of molecule 35 (1 × 10−4 M) in toluene resulted in a decrease of the band emission centred at 464, 500, and 525 nm and the appearance of a broad emission between 500 and 650 nm, the latter being characteristic of self-assembled OPV aggregates. Upon heating to 55 °C the gel turned into a solution and the emission bands at 464 and 500 nm intensified, indicating a reversible self-assembly process.
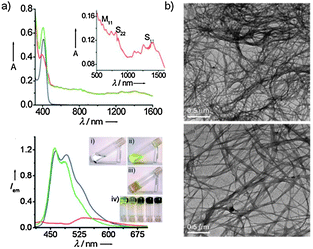 | ||
| Fig. 29 (a) UV-Vis spectra of 35 at 20 °C (blue line) and 35·SWCNT in toluene at 55 °C (green line) and 20 °C (red line); PL spectra of 35 (blue line) and 35·SWCNT in toluene at 55 °C (green line) and 20 °C (red line, λexc = 390 nm). Inset photographs of SWCNTs in toluene (i), 35 (3 × 10−4 M, ii), gel of 35 in toluene with SWNTs (iii) and 35 gels in toluene (5 × 10−4 M) with 0, 0.22, 0.45, 0.9, and 1.3 wt% of MWNTs with respect to toluene (iv); (b) TEM images of pristine SWNT and 35·SWNT nano-hybrid. (Adapted with permission fromref. 200. Copyright 2008, Wiley-VCH.) | ||
It is clear that the gel stability increased significantly with the increase in the proportion of CNTs present. Only 8 wt% of the SWCNTs is required to form a stable gel of 35 in toluene at a concentration of 3 × 10−4 M. A TEM image of SWCNTs shows the presence of bundled nanotubes. A TEM image of the self-assembled OPV exhibited the characteristic nano-tape morphology with a width of 10–200 nm and length of several micrometres. Unbundled nanotubes are seen for the [35·SWCNT] hybrid and these are aligned in a side-wise fashion within the self-assembled OPV. This design of a hybrid gel consisting of a total π-system, CNTs, OPVs, and an aromatic solvent (toluene) is expected to lead to further studies on the potential use of CNT–OPV nano-hybrid gels as optoelectronic materials.
Chen and co-workers developed a facile in situ fabrication method for the preparation of H2O-soluble gold nanoparticles (Au-NPs)–MWCNT hybrids with high density and homogeneous distribution of the Au-NPs.204 This luminescent and homogeneous hybrid was obtained thanks to the presence of N,N-bis-(2-mercaptoethyl)-perylene-3,4,9,10-tetracarboxylic diimide (49), which had the double role to first act as an interlinker between MWCNTs and Au-NPs and then as a stabiliser for the control of the nucleation and growth of the Au-NPs on the MWNTs (Scheme 18).
![Synthetic procedure for emitter 49 and schematic representation of the preparative protocol of [AuNPs·49·MWCNTs]. MWCNTs were first added to deionized H2O consisting of 0.05 wt% sodium dodecyl benzene sulfonate (SDBS) and sonicated for 1 h. Perylene derivative 49 was then added into the above MWCNT suspension, as a solution in DMF, followed by a HAuCl4 aqueous solution. The mixture was stirred for another 12 h at rt, affording a H2O-soluble and well-organised nanohybrid [AuNPs·49·MWCNTs].](/image/article/2012/CS/c1cs15031f/c1cs15031f-s18.gif) | ||
| Scheme 18 Synthetic procedure for emitter 49 and schematic representation of the preparative protocol of [AuNPs·49·MWCNTs]. MWCNTs were first added to deionized H2O consisting of 0.05 wt% sodium dodecyl benzene sulfonate (SDBS) and sonicated for 1 h. Perylene derivative 49 was then added into the above MWCNT suspension, as a solution in DMF, followed by a HAuCl4 aqueous solution. The mixture was stirred for another 12 h at rt, affording a H2O-soluble and well-organised nanohybrid [AuNPs·49·MWCNTs]. | ||
Scheme 18 depicts the synthetic pathway for the preparation of 49. The UV-Vis absorption spectra of the hybrid exhibited absorption around 310 nm, while the PL spectrum exhibited a strong blue emission at 378 nm. The loss of characteristic surface plasmon resonance (SPR) absorption around 520 nm for Au-NPs with a size smaller than 3 nm205,206 is caused either by a strong ground-state interaction between the plasmon electrons of Au-NPs and the π-electron cloud of MWCNTs or by the masking effect of the black MWCNTs material which absorbing throughout the visible range ultimately covers the characteristic Vis-absorption of AuNPs.207TEM images (Fig. 30) show the isolated AuNPs (mean diameter = 2.73 ± 1.8 nm) uniformly ornamenting the external walls of the MWCNTs.
![(a) PL spectra of AuNPs·49·MWNTs in H2O (λexc = 314 nm); (b) TEM and HR-TEM images of [AuNPs·49·MWNTs]. MWCNTS: multi-walled carbon nanotubes. (Adapted with permission fromref. 204. Copyright 2007, IOP Publishing.)](/image/article/2012/CS/c1cs15031f/c1cs15031f-f30.gif) | ||
| Fig. 30 (a) PL spectra of AuNPs·49·MWNTs in H2O (λexc = 314 nm); (b) TEM and HR-TEM images of [AuNPs·49·MWNTs]. MWCNTS: multi-walled carbon nanotubes. (Adapted with permission fromref. 204. Copyright 2007, IOP Publishing.) | ||
Recently, in a collaborative joint effort, our group has reported a simple method to prepare luminescent SWCNTs by adsorbing in situ an Eu(III) complex, [Eu(DBM)3(Phen)] (50) onto the surfaces of oxidised SWCNTs (ox-SWCNT) via hydrophobic interactions (Fig. 31).208
![(a) Schematic representation of the hybrid luminescent material [50·SWCNTs]; (b) absorption spectra of 50 (full line) and ox-SWCNT (dotted line) in the polystyrene (PS) matrix at rt. Excitation profile of 50 (dashed line, kem = 614 nm) is also depicted. Inset: emission spectra and lifetime decay of 50 (OD = 0.4, kexc = 350 nm) in the polystyrene matrix at rt; (c) HR-TEM images of the hybrid taken under Scherzer defocus; (d) Z-contrast ADF-STEM images of a 50 deposit. (Adapted with permission fromref. 208. Copyright 2007, Wiley-VCH.)](/image/article/2012/CS/c1cs15031f/c1cs15031f-f31.gif) | ||
| Fig. 31 (a) Schematic representation of the hybrid luminescent material [50·SWCNTs]; (b) absorption spectra of 50 (full line) and ox-SWCNT (dotted line) in the polystyrene (PS) matrix at rt. Excitation profile of 50 (dashed line, kem = 614 nm) is also depicted. Inset: emission spectra and lifetime decay of 50 (OD = 0.4, kexc = 350 nm) in the polystyrene matrix at rt; (c) HR-TEM images of the hybrid taken under Scherzer defocus; (d) Z-contrast ADF-STEM images of a 50 deposit. (Adapted with permission fromref. 208. Copyright 2007, Wiley-VCH.) | ||
The oxidised SWCNTs209,210 were suspended in a MeOH solution of 50. Sonication of the suspension for 1 h and subsequent stirring at rt afforded the final hybrid material 50·SWCNTs. PL investigations conducted in solution and in PS revealed that the presence of the SWCNT frameworks does not affect the Eu(III)-centred luminescence, therefore yielding emitting [50·SWCNTs] materials that maintain the structural and electronic properties of ox-SWCNTs. Z-contrast images show that the Eu(III)-containing complex adheres to the SWCNTs surface, which exhibits red emission in the VIS range as measured via light emission measurements. Despite the very easy and straightforward preparation, the desorption of 50 and the difficulty to control the loading of the complex turned out to be the main limitations towards the technological exploitation of these hybrids. Inspired by the columbic-directed layer-by-layer methodology,58,211 we have recently described the assembly of negatively-charged Eu(III) complexes onto cationic MWCNTs, previously obtained by the adsorption of an imidazolium-based ionic liquid.212 Such ionic liquids (ILs) are known from literature to be able to enter the coordination sphere of the Eu(III) ion through an anion exchange reaction.213–215
Pristine MWCNTs (p-MWCNTs) were first dispersed in a solution of an imidazolium-based IL in mesitylene by ultrasonication. The mixture was then heated at 170 °C for 1 h and the positively charged scaffold was promptly obtained. In order to perform the anion exchange reaction, tetrakis(2-naphtoyltrifluoro-acetonato)Eu(III) complex (51) was added to a dispersion of IL-bearing a carbon structure in toluene and stirred for 14 h at rt. PL studies showed that the ensuing hybrid [51·IL·MWCNTs] resulted a bright red luminescent material (upon UV excitation, λ = 365 nm). In the solid state (KBr matrix), hybrid [51·IL·MWCNTs] showed an intense and sharp emission peak at 615 nm with the satellite weaker features at 593, 654 and 702 nm, characteristic of Eu(III) ions (Scheme 19). The solid state luminescence quantum yield and excited state lifetime of 51·IL·MWCNT were measured as 0.44 and 0.29 ms, respectively, in accordance with the data of the pristine complex in KBr (Φf = 0.44 and τf = 0.28 ms), which suggests high loading of the luminophore on the surface of MWCNTs through the non-covalent columbic interactions. Moreover, preliminary measurements in polymeric matrices (Scheme 19) showed that such hybrid luminescent CNTs could also be easily embedded in composites providing exploitable materials easy to be implemented in practical devices opening the way to original approaches for the design of novel high performing electroluminescent devices (OLED, LEC) where the presence of the carbon material could provide substantial improvement of the mechanical and charge conductivity properties of the active layers within the real devices.
![(a) Synthetic pathway to the obtainment of the luminescent hybrid [51·IL·MWCNTs]; (b) schematic representation of the luminescent material and [51·IL·MWCNTs] embedded into PMMA polymeric matrices, under UV light irradiation (λ = 365 nm). (Adapted with permission fromref. 212. Copyright 2011, Royal Society of Chemistry.)](/image/article/2012/CS/c1cs15031f/c1cs15031f-s19.gif) | ||
| Scheme 19 (a) Synthetic pathway to the obtainment of the luminescent hybrid [51·IL·MWCNTs]; (b) schematic representation of the luminescent material and [51·IL·MWCNTs] embedded into PMMA polymeric matrices, under UV light irradiation (λ = 365 nm). (Adapted with permission fromref. 212. Copyright 2011, Royal Society of Chemistry.) | ||
4. Conclusions
In this critical review, an exhaustive overview on the different supramolecular routes towards the achievement of organised luminescent organic materials has been given through a selection of key examples in the field. By acknowledging a detailed description of templated and non-templated self-assembly and self-organisation protocols undertaken to supramolecularly organise chromophoric modules, this article discusses the main strategies towards the engineering of one-, two- or three-dimensional nanostructured luminescent materials. Therefore, the reader has been guided through a systematic exploration of the most common avenues to prepare and characterise luminescent organised materials arranged in polymeric, fibrous, nanotubular, micellar, toroidal, vesicular or spherical nanostructures, accompanied by a critical discussion of their main advantages/limitations and potential applications.The self-assembly and self-organisation process of ruling the control of the final emission and morphological properties has been particularly highlighted, driving the focus on the developed synthetic and preparative protocols. Since such molecular materials feature distinct properties as a consequence of both molecular modules and their supramolecular arrangement, a critical discussion of the predictive tools aiming at encoding (i.e., molecular programming) the fundamental molecular components with selected structural and electronic information to engineer in controllable fashion reproducible self-assembled/self-organised functional materials has been given. Clearly, it appeared that the fine control and prediction of the self-assembly and self-organisation mechanisms pass through an ensemble of dissipative and non-dissipative, equilibrium and non-equilibrium processes, the control of which constitutes the major challenge towards the design and engineering of reproducible complex sufficiently stable materials, displaying exploitable fluorescent properties. As it transpires from the above-discussed examples, the predictive character of the molecular programming of the molecular modules strictly depends on how the structural and electronic information, as encoded in the fundamental molecular components, are responding and sensitive towards internal and external factors. Thus, further experimental and computational studies aiming at the discovery of new paradigms for establishing the necessary experimental conditions for the programmable and controllable engineering of organic soft materials are necessary to rationally link macroscopic properties to the hierarchisation mechanisms at the nano- and microscale levels.
Although prototypes of various functional luminescent materials and preparative approaches have been proposed, the examples reported in this article represent the initial steps of an increasing effort aimed at the design and preparation of further soft materials which possess all the necessary characteristics (such as structural stability under thermal and electrical stresses) for technological applications in real devices. Although the research on such nanomaterials based on small organic compounds is still in its infancy, architectures will continue to be developed, as more types of luminescent molecular derivatives will be manufactured to high degrees of perfection and become commercially interesting.
Acknowledgements
This work was supported by the European Union through the Marie-Curie Initial Training Network “FINELUMEN” (grant agreement PITN-GA-2008-215399), INSTM, the Belgian National Research Foundation (through the FRFC contracts no. 2.4.625.08, 2.4.550.09 and 2.4.617.07.F and the MIS no. F.4.505.10.F), the “Loterie Nationale”, the Région Wallonne through the “SOLWATT” program (contract no. 850551), the FSR, the ‘TINTIN’ ARC project from the Belgian French Community (contract no. 09/14-023) and the University of Namur. We would like to gratefully acknowledge Silvia Maggini, Silvia Tasini and Francesco Forgnone for the artworks.References
- I. A. Richards, Plato's Republic, CUP Archive, Cambridge, 1966 Search PubMed.
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS.
- P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502–518 CrossRef CAS.
- D. M. Guldi, G. M. A. Rahman, F. Zerbetto and M. Prato, Acc. Chem. Res., 2005, 38, 871–878 CrossRef CAS.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS.
- J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS.
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS.
- A. Ajayaghosh, R. Varghese, S. Mahesh and V. K. Praveen, Angew. Chem., Int. Ed., 2006, 45, 7729–7732 CrossRef CAS.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- Y. Gao, X. Zhang, C. Ma, X. Li and J. Jiang, J. Am. Chem. Soc., 2008, 130, 17044–17052 CrossRef CAS.
- A. Muñoz-Bonilla, E. Ibarboure, E. Papon and J. Rodriguez-Hernandez, Langmuir, 2009, 25, 6493–6499 CrossRef.
- S. Cui, H. Liu, L. Gan, Y. Li and D. Zhu, Adv. Mater., 2008, 20, 2918–2925 CrossRef CAS.
- E. Gomar-Nadal, J. Puigmarti-Luis and D. B. Amabilino, Chem. Soc. Rev., 2008, 37, 490–504 RSC.
- Y. S. Zhao, H. B. Fu, F. Q. Hu, A. D. Peng, W. S. Yang and J. N. Yao, Adv. Mater., 2008, 20, 79–83 CrossRef CAS.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS.
- L. Chen, D. W. McBranch, H.-L. Wang, R. Helgeson, F. Wudl and D. G. Whitten, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12287–12292 CrossRef CAS.
- R. M. Jones, L. Lu, R. Helgeson, T. S. Bergstedt, D. W. McBranch and D. G. Whitten, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14769–14772 CrossRef CAS.
- Y. Kim and T. M. Swager, Macromolecules, 2006, 39, 5177–5179 CrossRef CAS.
- E. Schwartz, S. Le Gac, J. J. L. M. Cornelissen, R. J. M. Nolte and A. E. Rowan, Chem. Soc. Rev., 2010, 39, 1576–1599 RSC.
- I. Prigogine, Self-Organisation in Nonequilibrium Systems, Wiley-VCH, New York, 1977 Search PubMed.
- J.-M. Lehn, Supramolecular Chemistry, Wiley-VCH, Weinheim, New York, 1995 Search PubMed.
- J. D. Halley and D. A. Winkler, Complexity, 2008, 14, 10–17 CrossRef.
- Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564 RSC.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- N. Mizoshita, Y. Goto, Y. Maegawa, T. Tani and S. Inagaki, Chem. Mater., 2010, 22, 2548–2554 CrossRef CAS.
- X.-L. Fang, S.-L. Deng, J. Wang, X.-F. Wang, C. Chen, Y. Li, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, Chem. Mater., 2009, 21, 5763–5771 CrossRef CAS.
- C. M. Veziri, M. Palomino, G. N. Karanikolos, A. Corma, N. K. Kanellopoulos and M. Tsapatsis, Chem. Mater., 2010, 22, 1492–1502 CrossRef CAS.
- Z. Wang, L. Wang, X. Zhang, J. Shen, S. Denzinger and H. Ringsdorf, Macromol. Chem. Phys., 1997, 198, 573–579 CrossRef CAS.
- B.-S. Kim, D.-J. Hong, J. Bae and M. Lee, J. Am. Chem. Soc., 2005, 127, 16333–16337 CrossRef CAS.
- J.-M. Lehn, Angew. Chem., Int. Ed., 1988, 27, 89–112 CrossRef.
- J. Alper, Science, 2002, 295, 2396–2397 CrossRef CAS.
- M. W. Hosseini, Chem. Commun., 2005, 5825–5829 RSC.
- M. Fujita, Chem. Soc. Rev., 1998, 27, 417–425 RSC.
- Special issue on “Supramolecular Chemistry Anniversary”. Chem. Soc. Rev. 2007, 36, 125–126.
- P. Samorì, F. Cacialli, H. L. Anderson and A. E. Rowan, Adv. Mater., 2006, 18, 1235–1238 CrossRef.
- A. Ajayaghosh, V. K. Praveen, C. Vijayakumar and S. J. George, Angew. Chem., Int. Ed., 2007, 46, 6260–6265 CrossRef CAS.
- F. Gao, Q. Liao, Z.-Z. Xu, Y.-H. Yue, Q. Wang, H.-L. Zhang and H.-B. Fu, Angew. Chem., Int. Ed., 2010, 49, 732–735 CAS.
- H. Nakanotani, M. Saito, H. Nakamura and C. Adachi, Adv. Funct. Mater., 2010, 20, 1610–1615 CrossRef CAS.
- A. Corma, M. J. Diaz-Cabanas, J. L. Jorda, C. Martinez and M. Moliner, Nature, 2006, 443, 842–845 CrossRef CAS.
- M. Ganschow, C. Hellriegel, E. Kneuper, M. Wark, C. Thiel, G. Schulz-Ekloff, C. Bräuchle and D. Wöhrle, Adv. Funct. Mater., 2004, 14, 269–276 CrossRef CAS.
- G. S. Lee, Y.-J. Lee and K. B. Yoon, J. Am. Chem. Soc., 2001, 123, 9769–9779 CrossRef CAS.
- A. M. Schrand, S. A. C. Hens and O. A. Shenderova, Crit. Rev. Solid State Mat. Sci., 2009, 34, 18 CrossRef CAS.
- H. Schwertfeger, A. A. Fokin and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 1022–1036 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS.
- C. N. R. Rao, B. C. Satishkumar, A. Govindaraj and M. Nath, ChemPhysChem, 2001, 2, 78–105 CrossRef CAS.
- P. Singh, S. Campidelli, S. Giordani, D. Bonifazi, A. Bianco and M. Prato, Chem. Soc. Rev., 2009, 38, 2214–2230 RSC.
- J. Luo, T. Lei, L. Wang, Y. Ma, Y. Cao, J. Wang and J. Pei, J. Am. Chem. Soc., 2009, 131, 2076–2077 CrossRef CAS.
- A. Arduini, L. Domiano, L. Ogliosi, A. Pochini, A. Secchi and R. Ungaro, J. Org. Chem., 1997, 62, 7866–7868 CrossRef.
- S. C. Zimmerman, F. W. Zeng, D. E. C. Reichert and S. V. Kolotuchin, Science, 1996, 271, 1095–1098 CAS.
- T. Yamaguchi, T. Kimura, H. Matsuda and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 6350–6355 CrossRef CAS.
- A. Tsuda, M. A. Alam, T. Harada, T. Yamaguchi, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 8198–8202 CrossRef CAS.
- J. D. Badjic, A. Nelson, S. J. Cantrill, W. B. Turnbull and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 723–732 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- J. Luo, T. Lei, X. Xu, F.-M. Li, Y. Ma, K. Wu and J. Pei, Chem.–Eur. J., 2008, 14, 3860–3865 CrossRef CAS.
- S. S. Babu, V. K. Praveen, S. Prasanthkumar and A. Ajayaghosh, Chem.–Eur. J., 2008, 14, 9577–9584 CrossRef CAS.
- T. Naddo, Y. Che, W. Zhang, K. Balakrishnan, X. Yang, M. Yen, J. Zhao, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2007, 129, 6978–6979 CrossRef CAS.
- R. Abbel, C. Grenier, M. J. Pouderoijen, J. W. Stouwdam, P. E. L. G. Leclere, R. P. Sijbesma, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 833–843 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS.
- G. Wittig and U. Schöllkopf, Chem. Ber., 1954, 87, 1318–1330 CrossRef.
- B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 CrossRef CAS.
- B. Wang and M. R. Wasielewski, J. Am. Chem. Soc., 1997, 119, 12–21 CrossRef CAS.
- A. T. ten Cate, P. Y. W. Dankers, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2003, 125, 6860–6861 CrossRef CAS.
- T. Van der Boom, R. T. Hayes, Y. Zhao, P. J. Bushard, E. A. Weiss and M. R. Wasielewski, J. Am. Chem. Soc., 2002, 124, 9582–9590 CrossRef CAS.
- S. P. Dudek, M. Pouderoijen, R. Abbel, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 11763–11768 CrossRef CAS.
- A. C. Simonsen and H. G. Rubahn, Nano Lett., 2002, 2, 1379–1382 CrossRef CAS.
- J. Brewer, H. H. Henrichsen, F. Balzer, L. Bagatolli, A. C. Simonsen and H. G. Rubahn, Proc. SPIE–Int. Soc. Opt. Eng., 2005, 5931, 250–257 Search PubMed.
- M. Schiek, F. Balzer, K. Al-Shamery, A. Lützen and H. G. Rubahn, Soft Matter, 2008, 4, 277–285 RSC.
- M. Schiek, K. Al-Shamery and A. Lützen, Synthesis, 2007, 4, 613–621 Search PubMed.
- B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, 2001 Search PubMed.
- J. Brewer, M. Schiek, A. Lützen, K. Al-Shamery and H. G. Rubahn, Nano Lett., 2006, 6, 2656–2659 CrossRef CAS.
- M. Schiek, J. Brewer, F. Balzer, A. Lützen, K. Al-Shamery and H.-G. Rubahn, Proc. SPIE–Int. Soc. Opt. Eng., 2007, 6475, 647517 Search PubMed.
- A. Ajayaghosh, C. Vijayakumar, R. Varghese and S. J. George, Angew. Chem., Int. Ed., 2006, 45, 456–460 CrossRef CAS.
- A. Ajayaghosh and S. J. George, J. Am. Chem. Soc., 2001, 123, 5148–5149 CrossRef CAS.
- A. P. H. J. Schenning, P. Jonkheijm, E. Peeters and E. W. Meijer, J. Am. Chem. Soc., 2001, 123, 409–416 CrossRef CAS.
- S. J. George and A. Ajayaghosh, Chem.–Eur. J., 2005, 11, 3217–3227 CrossRef CAS.
- P. A. van Hal, M. M. Wienk, J. M. Kroon and R. A. J. Janssen, J. Mater. Chem., 2003, 13, 1054–1057 RSC.
- T. Mori, T. Watanabe, K. Minagawa and M. Tanaka, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1569–1578 CrossRef CAS.
- S.-I. Kawano, N. Fujita and S. Shinkai, Chem.–Eur. J., 2005, 11, 4735–4742 CrossRef CAS.
- C. Vijayakumar, V. K. Praveen and A. Ajayaghosh, Adv. Mater., 2009, 21, 2059–2063 CrossRef CAS.
- R. Abbel, R. van der Weegen, W. Pisula, M. Surin, P. Leclère, R. Lazzaroni, E. W. Meijer and A. P. H. J. Schenning, Chem.–Eur. J., 2009, 15, 9737–9746 CrossRef CAS.
- R. Abbel, A. P. H. J. Schenning and E. W. Meijer, Macromolecules, 2008, 41, 7497–7504 CrossRef CAS.
- M. J. Edelmann, J.-M. Raimundo, N. F. Utesch, F. Diederich, C. Boudon, J.-P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 2002, 85, 2195–2213 CrossRef CAS.
- B. A. DaSilveira Neto, A. S. A. Lopes, G. Ebeling, R. S. Gonçalves, V. E. U. Costa, F. H. Quina and J. Dupont, Tetrahedron, 2005, 61, 10975–10982 CrossRef CAS.
- D. D. Kenning, K. A. Mitchell, T. R. Calhoun, M. R. Funfar, D. J. Sattler and S. C. Rasmussen, J. Org. Chem., 2002, 67, 9073–9076 CrossRef CAS.
- L. Brunsveld, H. Zhang, M. Glasbeek, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 6175–6182 CrossRef CAS.
- M. Knaapila, V. M. Garamus, F. B. Dias, L. Almásy, F. Galbrecht, A. Charas, J. Morgado, H. D. Burrows, U. Scherf and A. P. Monkman, Macromolecules, 2006, 39, 6505–6512 CrossRef CAS.
- M. Knaapila, F. B. Dias, V. M. Garamus, L. Almásy, M. Torkkeli, K. Leppänen, F. Galbrecht, E. Preis, H. D. Burrows, U. Scherf and A. P. Monkman, Macromolecules, 2007, 40, 9398–9405 CrossRef CAS.
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS.
- L. Shimoni, H. L. Carrell, J. P. Glusker and M. M. Coombs, J. Am. Chem. Soc., 1994, 116, 8162–8168 CrossRef CAS.
- F. Emmerling, I. Orgzall, B. Dietzel, B. W. Schulz, G. Reck and B. Schulz, J. Mol. Struct., 2007, 832, 124–131 CrossRef CAS.
- H. I. Kim, T. Koini, T. R. Lee and S. S. Perry, Langmuir, 1997, 13, 7192–7196 CrossRef CAS.
- E. Knoevenagel, Ber. Dtsch. Chem. Ges., 1898, 31, 2596–2619 CrossRef CAS.
- S. R. Mahalle, P. D. Netankar, S. P. Bondge and R. A. Mane, Green Chem. Lett. Rev., 2008, 1, 103 CrossRef CAS.
- B.-K. An, S. H. Gihm, J. W. Chung, C. R. Park, S.-K. Kwon and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 3950–3957 CrossRef CAS.
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS.
- I. Cacelli, A. Ferretti, M. Girlanda and M. Macucci, Chem. Phys., 2006, 320, 84–94 CrossRef CAS.
- T. Haino, M. Tanaka and Y. Fukazawa, Chem. Commun., 2008, 468 RSC.
- K. Kishikawa, S. Furusawa, T. Yamaki, S. Kohmoto, M. Yamamoto and K. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 1597–1605 CrossRef CAS.
- T. Yasuda, T. Imase, Y. Nakamura and T. Yamamoto, Macromolecules, 2005, 38, 4687–4697 CrossRef CAS.
- Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS.
- J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119–122 CrossRef CAS.
- V. N. Kozhevnikov, B. Donnio and D. W. Bruce, Angew. Chem., Int. Ed., 2008, 47, 6286–6289 CrossRef CAS.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS.
- S. Mizukami, H. Houjou, K. Sugaya, E. Koyama, H. Tokuhisa, T. Sasaki and M. Kanesato, Chem. Mater., 2005, 17, 50–56 CrossRef CAS.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 3367–3371 CrossRef CAS.
- J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 8163–8172 CrossRef CAS.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS.
- C. Park, I. H. Lee, S. Lee, Y. Song, M. Rhue and C. Kim, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1199–1203 CrossRef CAS.
- C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef CAS.
- S. P. Rannard and N. J. Davis, Org. Lett., 1999, 1, 933–936 CrossRef CAS.
- S. P. Rannard and N. J. Davis, Org. Lett., 2000, 2, 2117–2120 CrossRef CAS.
- C. Park, M. S. Im, S. Lee, J. Lim and C. Kim, Angew. Chem., Int. Ed., 2008, 47, 9922–9926 CrossRef CAS.
- B. I. Ipe, K. G. Thomas, S. Barazzouk, S. Hotchandani and P. V. Kamat, J. Phys. Chem. B, 2002, 106, 18–21 CrossRef CAS.
- C. Fan, S. Wang, J. W. Hong, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297–6301 CrossRef CAS.
- G. B. Behera, B. K. Mishra, P. K. Behera and M. Panda, Adv. Colloid Interface Sci., 1999, 82, 1–42 CrossRef CAS.
- I. Capek, Adv. Colloid Interface Sci., 2002, 97, 91–147 CrossRef CAS.
- A. Guerrero-Martínez, Y. Vida, D. Domínguez-Gutiérrez, R. Q. Albuquerque and L. De Cola, Inorg. Chem., 2008, 47, 9131–9133 CrossRef.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- M. R. Arkin, E. D. A. Stemp, C. Turro, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1996, 118, 2267–2274 CrossRef CAS.
- R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS.
- S. Anderson, E. C. Constable, K. R. Seddon, J. E. Turp, J. E. Baggott and M. J. Pilling, J. Chem. Soc., Dalton Trans., 1985, 2247–2261 RSC.
- S. Sprouse, K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1984, 106, 6647–6653 CrossRef CAS.
- D. Dominguez-Gutierrez, G. De Paoli, A. Guerrero-Martinez, G. Ginocchietti, D. Ebeling, E. Eiser, L. De Cola and C. J. Elsevier, J. Mater. Chem., 2008, 18, 2762–2768 RSC.
- M. G. Colombo, A. Hauser and H. U. Guedel, Inorg. Chem., 1993, 32, 3088–3092 CrossRef CAS.
- F. Lafolet, S. Welter, Z. Popovic and L. D. Cola, J. Mater. Chem., 2005, 15, 2820–2828 RSC.
- S. Welter, F. Lafolet, E. Cecchetto, F. Vergeer and L. De Cola, ChemPhysChem, 2005, 6, 2417–2427 CrossRef CAS.
- A. B. Mandal and B. U. Nair, J. Phys. Chem., 1991, 95, 9008–9013 CrossRef CAS.
- Y. Lei, Q. Liao, H. Fu and J. Yao, J. Am. Chem. Soc., 2010, 132, 1742–1743 CrossRef CAS.
- H. Fu, D. Xiao, J. Yao and G. Yang, Angew. Chem., Int. Ed., 2003, 42, 2883–2886 CrossRef CAS.
- A. D. Peng, D. B. Xiao, Y. Ma, W. S. Yang and J. N. Yao, Adv. Mater., 2005, 17, 2070–2073 CrossRef CAS.
- E. Lee, Y.-H. Jeong, J.-K. Kim and M. Lee, Macromolecules, 2007, 40, 8355–8360 CrossRef CAS.
- D. R. M. Williams and G. H. Fredrickson, Macromolecules, 1992, 25, 3561–3568 CrossRef CAS.
- M. A. Horsch, Z. Zhang and S. C. Glotzer, Phys. Rev. Lett., 2005, 95, 056105 CrossRef.
- A. Halperin, Macromolecules, 1990, 23, 2724–2731 CrossRef CAS.
- M. Jayaraman and J. M. J. Fréchet, J. Am. Chem. Soc., 1998, 120, 12996–12997 CrossRef CAS.
- J.-H. Ryu, N.-K. Oh and M. Lee, Chem. Commun., 2005, 1770–1772 RSC.
- C.-J. Jang, J.-H. Ryu, J.-D. Lee, D. Sohn and M. Lee, Chem. Mater., 2004, 16, 4226–4231 CrossRef CAS.
- J.-H. Ryu, J. Bae and M. Lee, Macromolecules, 2005, 38, 2050–2052 CrossRef CAS.
- S. Förster and T. Plantenberg, Angew. Chem., Int. Ed., 2002, 41, 688–714 CrossRef.
- G. Battaglia and A. J. Ryan, J. Am. Chem. Soc., 2005, 127, 8757–8764 CrossRef CAS.
- Z. Li, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2006, 6, 1245–1249 CrossRef CAS.
- P. Jonkheijm, F. J. M. Hoeben, R. Kleppinger, J. van Herrikhuyzen, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2003, 125, 15941–15949 CrossRef CAS.
- M. Lee, Y.-S. Jeong, B.-K. Cho, N.-K. Oh and W.-C. Zin, Chem.–Eur. J., 2002, 8, 876–883 CrossRef CAS.
- S. Yagai, S. Mahesh, Y. Kikkawa, K. Unoike, T. Karatsu, A. Kitamura and A. Ajayaghosh, Angew. Chem., Int. Ed., 2008, 47, 4691–4694 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- F. J. M. Hoeben, M. J. Pouderoijen, A. P. H. J. Schenning and E. W. Meijer, Org. Biomol. Chem., 2006, 4, 4460–4462 CAS.
- P. Samorí, V. Francke, K. Müllen and J. P. Rabe, Chem.–Eur. J., 1999, 5, 2312–2317 CrossRef.
- U. H. F. Bunz, Acc. Chem. Res., 2001, 34, 998–1010 CrossRef CAS; G. De Luca, A. Liscio, P. Maccagnani, F. Nolde, V. Palermo, K. Mullen and P. Samori, Adv. Funct. Mater., 2007, 17, 379–3798 CrossRef.
- S. Yagai, H. Aonuma, Y. Kikkawa, S. Kubota, T. Karatsu, A. Kitamura, S. Mahesh and A. Ajayaghosh, Chem.–Eur. J., 2010, 16, 8652–8661 CrossRef CAS.
- S. Yagai, S. Kubota, T. Iwashima, K. Kishikawa, T. Nakanishi, T. Karatsu and A. Kitamura, Chem.–Eur. J., 2008, 14, 5246–5257 CrossRef CAS.
- S. Yagai, S. Kubota, K. Unoike, T. Karatsu and A. Kitamura, Chem. Commun., 2008, 4466–4468 RSC.
- R. Matmour, I. De Cat, S. J. George, W. Adriaens, P. Leclere, P. H. H. Bomans, N. A. J. M. Sommerdijk, J. C. Gielen, P. C. M. Christianen, J. T. Heldens, J. C. M. van Hest, D. W. P. M. Lowik, S. De Feyter, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2008, 130, 14576–14583 CrossRef CAS.
- J.-F. Eckert, J.-F. Nicoud, J.-F. Nierengarten, S.-G. Liu, L. Echegoyen, F. Barigelletti, N. Armaroli, L. Ouali, V. Krasnikov and G. Hadziioannou, J. Am. Chem. Soc., 2000, 122, 7467–7479 CrossRef CAS.
- S. J. George and A. Ajayaghosh, Chem.–Eur. J., 2005, 11, 3217–3227 CrossRef CAS.
- B. W. Messmore, J. F. Hulvat, E. D. Sone and S. I. Stupp, J. Am. Chem. Soc., 2004, 126, 14452–14458 CrossRef CAS.
- A. Ajayaghosh, R. Varghese, V. K. Praveen and S. Mahesh, Angew. Chem., Int. Ed., 2006, 45, 3261–3264 CrossRef CAS.
- A. Ajayaghosh, S. J. George and V. K. Praveen, Angew. Chem., Int. Ed., 2003, 42, 332–335 CrossRef CAS.
- A. Ajayaghosh and S. J. George, J. Am. Chem. Soc., 2001, 123, 5148–5149 CrossRef CAS.
- J.-F. Nierengarten, T. Gu, G. Hadziioannou, D. Tsamouras and V. Krasnikov, Helv. Chim. Acta, 2004, 87, 2948–2966 CrossRef CAS.
- K. Yoosaf, A. Belbakra, N. Armaroli, A. Llanes-Pallas and D. Bonifazi, Chem. Commun., 2009, 2830–2832 RSC.
- Q. Zhou, P. J. Carroll and T. M. Swager, J. Org. Chem., 1994, 59, 1294–1301 CrossRef CAS.
- A. Llanes-Pallas, C.-A. Palma, L. Piot, A. Belbakra, A. Listorti, M. Prato, P. Samorì, N. Armaroli and D. Bonifazi, J. Am. Chem. Soc., 2009, 131, 509–520 CrossRef CAS.
- F. Würthner, C. Thalacker, A. Sautter, W. Schärtl, W. Ibach and O. Hollricher, Chem.–Eur. J., 2000, 6, 3853–3886 Search PubMed.
- K. Yoosaf, A. Llanes-Pallas, T. Marangoni, A. Belbakra, R. Marega, E. Botek, B. Champagne, D. Bonifazi and N. Armaroli, Chem.–Eur. J., 2011, 17, 3262–3273 CrossRef CAS.
- T. Marangoni, S. A. Mezzasalma, A. Llanes-Pallas, K. Yoosaf, N. Armaroli and D. Bonifazi, Langmuir, 2011, 27, 1513–1523 CrossRef CAS.
- V. H. Rawal and M. P. Cava, Tetrahedron Lett., 1985, 26, 6141–6142 CrossRef CAS.
- M. Busby, H. Kerschbaumer, G. Calzaferri and L. De Cola, Adv. Mater., 2008, 20, 1614–1618 CrossRef CAS.
- S. Huber and G. Calzaferri, Angew. Chem., Int. Ed., 2004, 43, 6738–6742 CrossRef CAS.
- A. Zabala Ruiz, H. Li and G. Calzaferri, Angew. Chem., Int. Ed., 2006, 45, 5282–5287 CrossRef.
- G. Calzaferri, S. Huber, H. Maas and C. Minkowski, Angew. Chem., Int. Ed., 2003, 42, 3732–3758 CrossRef CAS.
- M. R. Hamblin and T. Hasan, Photochem. Photobiol. Sci., 2004, 3, 436–450 CAS.
- Z. Popović, M. Otter, G. Calzaferri and L. DeCola, Angew. Chem., Int. Ed., 2007, 46, 6188–6191 CrossRef.
- C. A. Strassert, M. Otter, R. Q. Albuquerque, A. Höne, Y. Vida, B. Maier and L. DeCola, Angew. Chem., Int. Ed., 2009, 48, 7928–7931 CrossRef CAS.
- M. Busby, C. Blum, M. Tibben, S. Fibikar, G. Calzaferri, V. Subramaniam and L. De Cola, J. Am. Chem. Soc., 2008, 130, 10970–10976 CrossRef CAS.
- K. Ishii, Y. Kikukawa, M. Shiine, N. Kobayashi, T. Tsuru, Y. Sakai and A. Sakoda, Eur. J. Inorg. Chem., 2008, 2975–2981 CrossRef CAS.
- K. Ishii, M. Shiine, Y. Kikukawa, N. Kobayashi, T. Shiragami, J. Matsumoto, M. Yasuda, H. Suzuki and H. Yokoi, Chem. Phys. Lett., 2007, 448, 264–267 CrossRef CAS.
- H. T. J. B. W. Dougherty, C. J. Gomer, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef.
- A. Juzeniene, Q. Peng and J. Moan, Photochem. Photobiol. Sci., 2007, 6, 1234–1245 CAS.
- V. Mantareva, V. Kussovski, I. Angelov, E. Borisova, L. Avramov, G. Schnurpfeil and D. Wöhrle, Bioorg. Med. Chem., 2007, 15, 4829–4835 CrossRef CAS.
- N. M. Gibson, T. J. M. Luo, O. Shenderova, Y. J. Choi, Z. Fitzgerald and D. W. Brenner, Diamond Relat. Mater., 2010, 19, 234–237 CrossRef CAS.
- V. N. Mochalin and Y. Gogotsi, J. Am. Chem. Soc., 2009, 131, 4594–4595 CrossRef CAS.
- J. Chen, M. A. Hamon, H. Hu, Y. Chen, A. M. Rao, P. C. Eklund and R. C. Haddon, Science, 1998, 282, 95–98 CrossRef CAS.
- S. Osswald, G. Yushin, V. Mochalin, S. O. Kucheyev and Y. Gogotsi, J. Am. Chem. Soc., 2006, 128, 11635–11642 CrossRef CAS.
- S.-J. Yu, M.-W. Kang, H.-C. Chang, K.-M. Chen and Y.-C. Yu, J. Am. Chem. Soc., 2005, 127, 17604–17605 CrossRef CAS.
- C.-C. Fu, H.-Y. Lee, K. Chen, T.-S. Lim, H.-Y. Wu, P.-K. Lin, P.-K. Wei, P.-H. Tsao, H.-C. Chang and W. Fann, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 727–732 CrossRef CAS.
- A. Ghosh, K. V. Rao, S. J. George and C. N. R. Rao, Chem.–Eur. J., 2010, 16, 2700–2704 CrossRef CAS.
- U. Rohr, P. Schlichting, A. Böhm, M. Gross, K. Meerholz, C. Bräuchle and K. Müllen, Angew. Chem., Int. Ed., 1998, 37, 1434–1437 CrossRef CAS.
- S. Alibert-Fouet, I. Seguy, J.-F. Bobo, P. Destruel and H. Bock, Chem.–Eur. J., 2007, 13, 1746–1753 CrossRef CAS.
- K. S. Subrahmanyam, S. R. C. Vivekchand, A. Govindaraj and C. N. R. Rao, J. Mater. Chem., 2008, 18, 1517–1523 RSC.
- H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- K. S. Subrahmanyam, L. S. Panchakarla, A. Govindaraj and C. N. R. Rao, J. Phys. Chem. C, 2009, 113, 4257–4259 CAS.
- V. V. Didenko, V. C. Moore, D. S. Baskin and R. E. Smalley, Nano Lett., 2005, 5, 1563–1567 CrossRef CAS.
- A. J. Gross and I. W. Sizer, J. Biol. Chem., 1959, 234, 1611–1614 CAS.
- M. J. O'Connell, P. Boul, L. M. Ericson, C. Huffman, Y. Wang, E. Haroz, C. Kuper, J. Tour, K. D. Ausman and R. E. Smalley, Chem. Phys. Lett., 2001, 342, 265–271 CrossRef CAS.
- S. Srinivasan, S. S. Babu, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2008, 47, 5746–5749 CrossRef CAS.
- T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072–2074 CrossRef CAS.
- K. Keren, R. S. Berman, E. Buchstab, U. Sivan and E. Braun, Science, 2003, 302, 1380–1382 CrossRef CAS.
- J. Chen, H. Liu, W. A. Weimer, M. D. Halls, D. H. Waldeck and G. C. Walker, J. Am. Chem. Soc., 2002, 124, 9034–9035 CrossRef CAS.
- R. J. Zhou, M. M. Shi, X. Q. Chen, M. Wang, Y. Yang, X. B. Zhang and H. Z. Chen, Nanotechnology, 2007, 18, 485603–485610 CrossRef.
- R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- Y.-Y. Ou and M. H. Huang, J. Phys. Chem. B, 2006, 110, 2031–2036 CrossRef CAS.
- G. Accorsi, N. Armaroli, A. Parisini, M. Meneghetti, R. Marega, M. Prato and D. Bonifazi, Adv. Funct. Mater., 2007, 17, 2975–2982 CrossRef CAS.
- K. J. Ziegler, Z. Gu, H. Q. Peng, E. L. Flor, R. H. Hauge and R. E. Smalley, J. Am. Chem. Soc., 2005, 127, 1541–1547 CrossRef CAS.
- D. Bonifazi, C. Nacci, R. Marega, S. Campidelli, G. Ceballos, S. Modesti, M. Meneghetti and M. Prato, Nano Lett., 2006, 6, 1408–1414 CrossRef CAS.
- D. M. Guldi, F. Pellarini, M. Prato, C. Granito and L. Troisi, Nano Lett., 2002, 2, 965–968 CrossRef CAS.
- L. Maggini, H. Traboulsi, K. Yoosaf, J. Mohanraj, J. Wouters, O. Pietraszkiewicz, M. Pietraszkiewicz, N. Armaroli and D. Bonifazi, Chem. Commun., 2011, 47, 1625–1627 RSC.
- P. Nockemann, E. Beurer, K. Driesen, R. Van Deun, K. Van Hecke, L. Van Meervelt and K. Binnemans, Chem. Commun., 2005, 4354–4356 RSC.
- K. Lunstroot, K. Driesen, P. Nockemann, C. Görller-Walrand, K. Binnemans, S. Bellayer, J. Le Bideau and A. Vioux, Chem. Mater., 2006, 18, 5711–5715 CrossRef CAS.
- S. M. Bruno, R. A. S. Ferreira, F. A. Almeida Paz, L. D. Carlos, M. Pillinger, P. Ribeiro-Claro and I. S. Gonçalves, Inorg. Chem., 2009, 48, 4882–4895 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |