DOI:
10.1039/C1CS15009J
(Critical Review)
Chem. Soc. Rev., 2012,
41, 192-210
In situ solid-state NMR for heterogeneous catalysis: a joint experimental and theoretical approach
Received
10th January 2011
First published on 11th July 2011
Abstract
In situ solid-state NMR is a well-established tool for investigations of the structures of the adsorbed reactants, intermediates and products on the surface of solid catalysts. The techniques allow identifications of both the active sites such as acidic sites and reaction processes after introduction of adsorbates and reactants inside an NMR rotor under magic angle spinning (MAS). The in situ solid-state NMR studies of the reactions can be achieved in two ways, i.e. under batch-like or continuous-flow conditions. The former technique is low cost and accessible to the commercial instrument while the latter one is close to the real catalytic reactions on the solids. This critical review describes the research progress on the in situ solid-state NMR techniques and the applications in heterogeneous catalysis under batch-like and continuous-flow conditions in recent years. Some typical probe molecules are summarized here to detect the Brønsted and Lewis acidic sites by MAS NMR. The catalytic reactions discussed in this review include methane aromatization, olefin selective oxidation and olefin metathesis on the metal oxide-containing zeolites. With combining the in situ MAS NMR spectroscopy and the density functional theoretical (DFT) calculations, the intermediates on the catalyst can be identified, and the reaction mechanism is revealed. Reaction kinetic analysis in the nanospace instead of in the bulk state can also be performed by employing laser-enhanced MAS NMR techniques in the in situ flow mode (163 references).

Weiping Zhang
| Weiping Zhang obtained his PhD in physical chemistry at Dalian Institute of Chemical Physics, Chinese Academy of Sciences in 2000. From 2000 until 2004 he was a postdoctoral fellow at Swiss Federal Institute of Technology (ETH, Zurich) and National Research Council of Canada in Ottawa. Then he returned to DICP where he became an associate professor in 2005 and professor in 2006. In 2011 he moved to Dalian University of Technology as a full professor. His research interests are the development of in situ NMR techniques and their applications in material science and catalysis, especially the synthesis and reaction mechanisms as revealed by in situ 1D and 2D NMR spectroscopy and theoretical calculations. |

Shutao Xu
| Shutao Xu was born in 1982 in Anhui, China. He received his BSc in chemistry from Fudan University, China in 2004. He received his PhD in physical chemistry from Dalian Institute of Chemical Physics, Chinese Academy of Sciences in 2010. He is currently a research assistant at DICP, and his research interest is focusing on the heterogeneous catalytic mechanism by in situ solid-state NMR spectroscopy. |

Xiuwen Han
| Xiuwen Han received her BSc in chemistry at Beijing University of Technology in 1964. Since then she has been working at Dalian Institute of Chemical Physics. She was appointed as a full professor in 1994 at DICP. From 1987 till 1988 she was a visiting scientist at Bruker-Spectrospin Instruments Ltd in Switzerland and Germany. From 1995 till 2007 she was a visiting professor at many universities such as Chinese University of Hong Kong in 1995, University of Connecticut in 1998, SUNY at Stony Brook in 2005, Northeastern University, USA in 2006, etc. Her research interests are structural analysis of functional saponins, polypeptides and polysaccharides by 1D and 2D NMR, catalytic mechanism by in situ MAS NMR. |

Xinhe Bao
| Xinhe Bao received his PhD in physical chemistry from Fudan University, China in 1987. He moved to Germany in 1989 and worked as an Alexander von Humboldt Fellow at Fritz-Haber Institute in Berlin. In 1995 he came back to China and joined Dalian Institute of Chemical Physics as a full professor. He is a member of Chinese Academy of Sciences, and a Fellow of the Royal Society of Chemistry, UK. His research activities focus on the fundamental understanding of catalysis by nanotechnologies and in situ analytical techniques including surface probes and NMR spectroscopy, and their applications to the development of new nanocatalysts and novel catalytic processes related to energy conversion, in particular clean coal and natural gas utilization. |
1. Introduction
Heterogeneous catalysis plays an important role in chemical industry due to the advantages of easy separation, good persistence and recyclability.1,2 In general, the heterogeneous catalytic processes take place in an opaque medium.3,4 The entrance and exit of the system can be transparently examined during the course of the reaction. Although there is some understanding of what happens inside a solid catalyst with the existing techniques, non-invasive measurements of the active sites, reaction mechanism and kinetics under the real working conditions are still a challenge. Many spectroscopic methods have been developed for in situ study of the working catalysts, among which in situ magic-angle-spinning (MAS) NMR spectroscopy is of great potential value for investigating the active species and catalytic reactions at high temperatures.5 The MAS NMR rotor with catalyst particles is used as a microreactor to simulate the reaction process under batch-like or continuous-flow conditions. The former technique is low cost and accessible to the commercial instrument while the latter one is close to the real catalytic process in chemical industry. During past decades in situ MAS NMR has been mostly performed with samples fused in a glass ampoule or put into a gas-tight MAS NMR rotor,6–9 which is comparable to the batch reaction. In order to investigate the heterogeneous catalysis in large-scale industrial processes, many attempts have been made to develop in situ solid-state NMR under continuous-flow conditions.10–14 The most successful and practical design is that reported by Hunger et al. directly utilizing the NMR rotor as a fix-bed reactor with continuous flowing of reactants under magic-angle spinning.11 In some cases, continuous-flow MAS NMR spectroscopy is combined with other analytical techniques such as gas chromatography,15,16 UV/Vis spectroscopy17 or laser hyperpolarized xenon18 to give additional information on the reaction intermediates, products and kinetics. In this review, firstly the experimental setups for in situ MAS NMR are briefly described, then more recent research progress on the applications of in situ MAS NMR in the acidity characterizations, reaction mechanisms and kinetics of some important organic reactions mainly catalyzed by zeolites are summarized. The aim of this paper is to emphasize the importance of a joint experimental–theoretical approach in understanding the nature of the acid sites and the reaction intermediates. Additionally, the catalytic kinetics performed by laser-enhanced in situ MAS NMR in the nanospace instead of in the bulk state has also been highlighted in the present review. Only a few selected reactions will be covered here since the field of in situ NMR is very broad, and it has already been demonstrated to be a powerful tool in studying many aspects of heterogeneous catalysis. For further information, the reader could refer to previous reviews.19–28
In situ studies of catalytic reactions on solids require controlling the atmosphere and sealing the NMR tube or rotor to keep the reaction conditions similar to real reactors and to exclude air or moisture. On the other hand, in order to eliminate the line broadening effect of solid-state NMR spectra, the rotor with catalysts needs magic-angle spinning. Generally, there are two types of NMR experimental approaches for in situ studies in which the conditions of a catalytic reactor are simulated in the NMR rotor under batch-like conditions or continuous-flow conditions.
2.1 Batch-like conditions
Although the batch-like mode is an ex situ method which needs the samples activated (or dehydrated) beforehand and adsorbed with adsorbates outside of the NMR probe, it is very suitable for low temperature studies of active adsorbates on solid acids.29 Moreover, an important advantage of it is that most of the materials and equipment are commercially available.25 Furthermore, if the catalyst–adsorbate samples are air or moisture sensitive and require preparation on a vacuum line or the adsorbate is only weakly adsorbed, the static batch-like conditions are desirable to suit the mentioned requirements. Two general methods are widely employed to reconcile the awkward requirements of maintaining sample integrity while loading it into a MAS rotor. The first method makes use of glass ampoules prepared by flame sealing on the vacuum line.6,30,31 In the glass ampoules, solid catalyst is activated in vacuum and at temperatures up to 723 K. After activation and adsorption of reactants, the glass ampoules are fused at the waist while the glass ampoule needs to be cooled by liquid nitrogen to prevent heating of the catalyst.32 Obviously, the glass ampoules must be highly symmetrical in order to obtain high rotation speed. In addition, the re-addition of reagents is impossible once the sample is sealed. In order to balance the rotor containing the glass ampoule, Miyoshi et al.33 packed powder glass or KBr into a MAS rotor with the glass ampoule. The endurable pressure of the glass ampoule depends on the tensile strength and the ratio of the outer diameter to the inner diameter. Recently, a design of a high pressure up to 10 MPa insert made from polyether ether ketones (PEEK) polymer was used to fit inside a standard commercial 7 mm MAS rotor spun at the maximum frequency of 5 kHz.34
The second method performed by Haw and co-workers8,35,36 applies a special rotor design, abbreviated as CAVERN (Cryogenic Adsorption Vessel Enabling Rotor Nestling). This approach makes use of rotor caps with 10 to 15 deformable ridges that create an air-tight seal when the cap is driven into the rotor. The most significant advantage of it is that there is no need to handle the activated catalyst in a glove box in order to transfer it into the MAS rotor. Thus, sample preparation is greatly simplified and speeded up. Any possibility of contamination from the glove box is precluded by use of this device. In order to recreate catalytic reaction conditions in an environment suitable for NMR spectroscopy, we designed and manufactured a special device as shown in Fig. 1 for on-line treatment of powder samples.9 By this system, the solid sample can be heated up to about 1000 K in vacuum for dehydration and exposed to several different gases parallelly or separately, depending on the requirements for adsorption and reaction. After the treatment, the sample can be placed in situ into an NMR rotor, sealed and transferred to the spectrometer without exposure to air.9 This equipment is suitable for ex situ or in situ NMR studies of solid-catalyzed reactions under batch-like conditions.
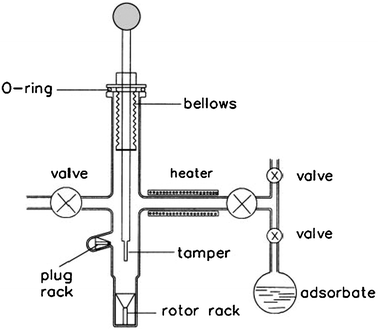 |
| | Fig. 1 Diagram of the evacuating, loading and sealing apparatus for solid samples for MAS NMR study. (Adapted from ref. 9). | |
The contact time of reactants and catalysts is relatively longer by the methods of catalyst and adsorbate sealed in glass ampoules or MAS rotors compared to the industrial catalytic processes. Haw et al.37 developed a pulse-quench catalytic reactor to apply NMR studies to catalysts which had been prepared under real reaction conditions. The organic species on the catalyst bed were characterized by NMR at room temperature following a rapid (∼200 ms) quench from the reaction temperature while the volatile products produced from the reactor at high temperature were analyzed by gas chromatography (GC) or mass spectrometry (MS). This method has been extensively used in the methanol-to-olefin (MTO) reaction, and the combination of in situ NMR and DFT calculations provided evidence in support of a carbon-pool mechanism for observation of the cyclopentenyl cation and pentamethylbenzenium cations.37–40
All the above mentioned methods under batch-like conditions suffer from the disadvantage that they could not provide information about adsorption or reaction at the earliest stages. Recently, Xu et al.41 proposed a method to trace the adsorption process at the earliest stages in porous materials under batch-like conditions. The rotor containing the porous material was spun for a short period of time to create a hollow space at the center of it. Adsorbate was sealed in a glass capillary and inserted into the rotor containing the sample. The “magic-angle spinning” (MAS) in the NMR experiments played a role as a “trigger” to start up the adsorption process. 1H MAS NMR spectra recorded the evolution of the adsorption process for acetone or water on MCM-41, in particular revealing the details of adsorbates redistributing among different sites before reaching the final equilibrium.41,42
2.2 Continuous-flow conditions
In situ NMR studies of catalysis should be performed under “ideal” NMR experimental conditions in which the temperature and flow rate are close to those used in conventional microreactor studies, and GC or MS should be appended for analysis of the product stream.43 Since most industrial processes based on heterogeneous catalysis are often flow, it has long been recognized that a worthy technical goal would be to develop an in situ flow reactor.44,45 Achieving MAS line narrowing in a flow reactor has been extremely difficult because of the need to spin the reactor at speeds of at least a few kHz, while simultaneously flowing a gas stream through the sample and maintaining the environmental integrity of the sample. In spite of these technical difficulties, a few laboratories have developed flow MAS probes that have yielded promising results.12–14,46–48
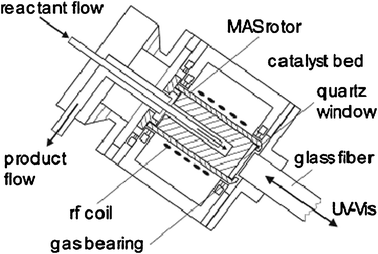
Among these protocols, the most widely used one was designed by Hunger and Horvath47 who first reported a MAS flow probe allowing reactant gas to flow over a catalyst. This equipment consists of a MAS rotor with an axially placed tube inserted into the rotor via a hole in the rotor cap. Fig. 2 shows a MAS NMR rotor modified for in situ MAS NMR investigations under continuous-flow conditions.16 With a specially constructed tool, the catalyst is pressed into a hollow cylinder. And the carrier gas loaded with vapors of the reactants is injected into the spinning MAS rotor via the injection tube. The exhaust gas leaves the rotor via an annular gap in the rotor cap. At an early stage, this design allows the heating of samples to 423 K under ambient flow pressures and MAS rates of 3 kHz. In a subsequent study, the authors coupled this continuous injection technique with on-line gas chromatography16 or UV/Vis spectroscopy17 (Fig. 3). It allows simultaneously in situ MAS NMR and UV/Vis spectroscopy studies of adsorbate complexes formed under flow conditions and gas chromatographical analysis of the reaction products. Moreover, the above-mentioned injection system appended on a Doty probe can allow temperatures for the NMR investigations up to 873 K with spinning rates of up to 3.5 kHz.49 This in situ technique is extensively used for studies of the MTO reaction mechanism which is involved with the formation of “carbon-pool species” such as polyenic and aromatic compounds and carbenium ions on the surface of solid catalysts.17,50,51 Hunger26 also suggested four different protocols for in situ flow MAS NMR experiments, i.e. continuous-flow, switched-flow, stopped-flow and pulsed-flow mode. Among them, stopped-flow NMR was a valuable approach to isolate and identify the intermediates with longer lifetimes on solid catalysts.52
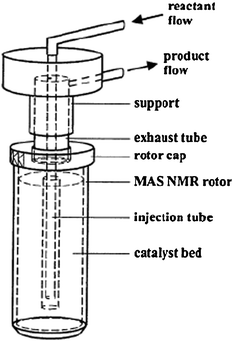 |
| | Fig. 2 Schematic representation of an MAS rotor reactor for MAS NMR study under continuous-flow conditions. (Adapted with permission from ref. 16. Copyright 1999 Springer). | |
In general, the catalytic reaction must be on the time scale of NMR spectroscopy. In the case of 13C NMR spectroscopy, the acquisition time is relatively long due to the spin lattice relaxation time of 1–30 s and a much higher number of scans. Application of the in situ continuous-flow MAS technique in laser-hyperpolarized 129Xe NMR experiments can be utilized to enhance the sensitivity of NMR spectroscopy. Laser-hyperpolarized xenon is achieved by spin-exchange optical pumping to tremendously enhance the signal intensity by 4 to 5 orders of magnitude compared to conventional 129Xe NMR.53–55 It has been shown that the laser-hyperpolarized 129Xe NMR technique is powerful for studying the porosity of porous materials,56–61 or even diffusion and combustion process.62,63 As shown in Fig. 4, we reported a design of laser-hyperpolarized xenon premixed with a reactant outside the probe head which then entered the high-field coil region with an MAS NMR rotor to investigate the catalytic kinetics in nanocages by in situ continuous-flow MAS NMR.18 The adsorption and reaction processes at the earliest stage can be monitored by 129Xe NMR spectroscopy with much higher sensitivity and shorter acquisition time (∼10 s per spectrum) under real working conditions.
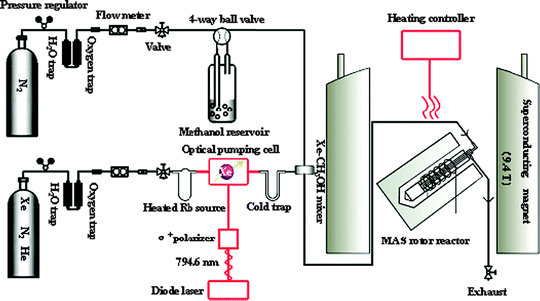 |
| | Fig. 4 Sketch of the experimental setup for in situ continuous-flow MAS NMR coupled with hyperpolarized 129Xe. (Adapted with permission from ref. 18. Copyright 2009 American Chemical Society). | |
3. Acidity detection on the solid surface
3.1 Direct observation of Brønsted acid sites by 1H MAS NMR after adsorption of probe molecules
Acidity characterization is a very important topic in heterogeneous catalysis since many reactions are involved in acidic sites on the solid surface. High-resolution 1H MAS NMR, a useful and direct method, has been employed routinely to characterize the acidic sites in zeolites. Compared with IR, it can provide quantitative information on the interaction between the supported metal ions and the hydroxyl species on the catalyst surface without the difficulties associated with extinction coefficients. 1H NMR shifts of bridging hydroxyl groups, i.e. Brønsted acidic sites, are in the range of 3.6–7.0 ppm. Such measurements have to be performed on carefully dehydrated samples, sealed in symmetric glass ampoules or placed in gastight NMR rotors as mentioned in section 2.1. The NMR detection of acidity on solid samples has been covered extensively in literature reviews,20,32,64 and only the more recent examples are described.
3.1.1 Perfluorotributyl amine to distinguish internal and external Brønsted sites.
Perfluorotributyl amine [(n-C4F9)3N] is a weak base with a diameter of 0.94 nm larger than the pore size of microporous zeolites such as ZSM-5 or Y, which can only be adsorbed on the external surface of most zeolites. We firstly introduced it to quantitatively distinguish the internal and external acidic sites, silanols and some non-framework Al species in zeolites by 1H MAS NMR spectroscopy.9,65 The bridging hydroxyl groups located on the external surface of the zeolites interacted with the perfluorotributyl amine to form perfluorotributylaminium ions whose resonance signal appeared at about 6.0 ppm. The concentration of Brønsted acidic sites on the external surface of the zeolites could be obtained by comparing the integral area of the peak at 3.9 ppm after and before adsorption of perfluorotributyl amine.9,65 With this probe molecule, it was found that nanosized HZSM-5 zeolites had a distinctly greater amount of external Brønsted acidic sites than microsized HZSM-5 zeolites.65 After loading molybdenum onto the HZSM-5 support, 1H MAS NMR indicated that impregnated Mo species remained predominantly on the external surface of nanosized HZSM-5 zeolites.66 For MCM-22 zeolites, nearly all the silanol species were located on the external surface and in the form of so-called terminal silanol groups.67 It seems that perfluorotributyl amine could be used quantitatively as an NMR probe molecule to distinguish the internal or external hydroxyls of zeolite based catalysts.
3.1.2 Deuterated pyridine to probe the Brønsted acid strength.
Many molecules such as deuterated acetonitrile,68–70 pyridine,32,71 2-13C-enriched acetone20,32,64etc. can be employed to probe the Brønsted acid strength on solids using MAS NMR spectroscopy. Deuterated pyridine (pyridine-d5) is one of the widely used NMR probe molecules.72–74 The formation of a hydrogen bond between pyridine and non-acidic hydroxyl groups (such as Si–OH) shifts the 1H MAS NMR signal position from 2 to about 10 ppm. In the case of acidic OH groups (Brønsted acidic sites), the adsorption of pyridine results in 1H NMR signals in the range of 12–20 ppm.72–74 The large downfield shift of the 1H NMR signal results from a proton transfer from the acidic OH group to the probe molecule to form a pyridinium ion. Besides the traditional zeolites, the acidity of mesoporous materials such as AlMCM-41, MoOx/ZrO2 WOx/ZrO2 has also been characterized by 1H MAS NMR using pyridine-d5 as a probe molecule.75,76 The correlation between the 1H chemical shift of adsorbed pyridine-d5 and the Brønsted acid strength of solid acids has been investigated by DFT calculations. Zheng et al.77 used an 8T zeolite model with different Si–H bond lengths to represent the Brønsted acidic sites with different strengths (from weak, strong, to superacid), and to predict the pyridine adsorption structure as well as the 1H chemical shift (Fig. 5). They found a linear correlation between the pyridine-d51H chemical shift and the proton affinity (PA) of the Brønsted acidic site.77 The theoretical calculations suggest that a smaller 1H chemical shift of the pyridinium ions on the solid acids indicates a stronger acid strength.
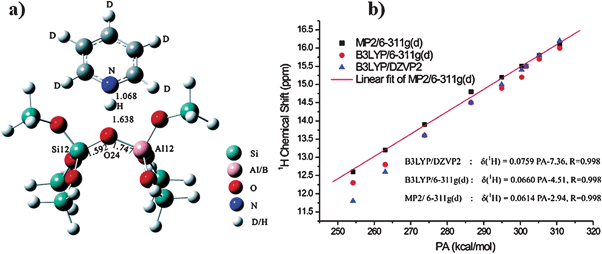 |
| | Fig. 5 (a) The optimized configuration of pyridine-d5 adsorbed on the 8T model for Al-ZSM-5. (b) Correlation of calculated 1H-NMR chemical shift of adsorbed pyridine-d5 and proton affinity (PA) of solid acids. (Adapted with permission from ref. 77. Copyright 2007 American Chemical Society). | |
3.2 Detection of Lewis and Brønsted acidic sites by probe molecules
The Lewis acidic sites can be detected only by the adsorption of probe molecules on solid catalysts. Feasible molecules for the Lewis acidity study are classified as trimethylphosphine and trimethylphosphine oxide by 31P MAS NMR spectroscopy, 15N-enriched pyridine by 15N MAS NMR spectroscopy, and 2-13C-enriched acetone by 13C MAS NMR spectroscopy. Meanwhile, the Brønsted acidic sites can also be detected using these probe molecules.
3.2.1 Trimethylphosphine.
Trimethylphosphine (TMP) is suitable for 31P MAS NMR studies as it has high natural abundance (100%), a nuclear spin of 1/2, and a large gyromagnetic ratio. Quantitative acidity measurements are possible with TMP adsorption since no cross-polarization (CP) is needed for NMR acquisition. Lunsford et al.78 first introduced it into the characterization of acid sites in zeolite Y by 31P MAS NMR. After that, it has been extensively used in acidity investigations of zeolites79–87 and mesoporous materials.88,89 The interaction of TMP with bridging hydroxyl groups in zeolites also leads to protonation of the probe molecules. 31P MAS NMR investigations show that the protonated TMP occurs at about −3 to −5 ppm corresponding to the Brønsted acidic sites while the chemical shift of Lewis-bound TMP is in the range from about −30 to −60 ppm.81,86 The oxidation of TMP as an in situ probe reaction was also performed to monitor the oxidation ability of different Ti species in TS-1 zeolite by 31P MAS NMR spectra, which gave direct experimental evidence on the oxidation center of TS-1 zeolite.8731P MAS NMR of TMP adsorption on TS-1 or HBeta zeolites demonstrated that there were various Brønsted and Lewis acidic sites on the surface, and DFT theoretical calculations further provided the specific assignments of these sites.90,91 Kao and Grey79,80,83 used 27Al–31P double resonance NMR to determine the structure of TMP bound to the Brønsted/Lewis acidic sites in HY zeolite. The Al–P distances for the acid site-TMP complex could be measured, demonstrating the use of double-resonance NMR methods for study of the binding of molecules on surfaces and in zeolites. In their following acidity study on the same zeolite, they proposed a diphosphine molecule to detect the distance between two Brønsted sites by 1D and 2D double quantum (DQ) 31P MAS NMR spectroscopy.92
3.2.2 Trimethylphosphine oxide.
Trimethylphosphine is a highly flammable and air-sensitive liquid at room temperature, and requires great care in its handling. Studies on amorphous silica-alumina indicate that the use of trialkylphosphine oxides offers several advantages for the study of acidic binding sites.93 These oxides are solids at room temperature, they are not susceptible to oxidation, and titration of the molecules onto acidic solids can be accomplished using solution-state chemistry.94 Trimethylphosphine oxide (TMPO) was used by Mueller's group to direct and simultaneous quantification of the Brønsted and Lewis-acid site content in USY zeolite via31P MAS NMR spectroscopy.94,95 Zhao et al. reported an approach to qualitatively and quantitatively characterize the internal and external acidic sites in zeolites using different adsorbed probe molecules namely TMP oxide (TMPO) and tributylphosphine oxide (TBPO) by the combination of MAS NMR spectroscopy and elemental analysis.96,97 Although there are many factors that influence the 31P chemical shift in these complexes, one primary source is due to electronic changes in the probe molecule leading to a change in the local magnetic field around the nucleus. In terms of the concept of molecular orbitals, Osegovic and Drago98 pointed out that the coordination of the phosphoryl oxygen of TMPO creates an overlap between the lowest unoccupied molecular orbital (LUMO) on the Brønsted or Lewis acid site and the highest occupied molecular orbital (HOMO) on the TMPO oxygen atom. The resulting adduct bond causes a loss of electron density from the probe that can be observed by a change in the 31P NMR shift. Assuming this to be the case, it may be possible to relate the 31P chemical shift of the TMPO/Brønsted complex with the strength of the acidic site.99 However, the effect of the alkyl-group on the 31P NMR shifts of the probe molecules adsorbed on acidic sites with varied acidic strengths is still not well understood. This, in turn, makes spectral assignments ambiguous especially in determining the types (Brønsted vs. Lewis acidity), strengths and concentrations, and locations (internal vs. external acidity) of acidic sites based on variations in 31P resonances probed by different R3PO molecules. In this case, the chemical shifts of adsorbed trialkylphosphine oxides (TAPO) and the configurations of the corresponding TAPOH+ complexes on Brønsted acidic sites with varying acidic strengths in modeled zeolites have been predicted theoretically by means of DFT calculations.100,101
3.2.3
15N-enriched pyridine.
The 15N chemical shifts of adsorbed 15N-pyridine have a wide range and it can be used to distinguish different acidic sites on the solids.102 Haw et al.103 used it to explore the effects of sulfated zirconia composition and treatment on the relative number of Brønsted sites. Quantitative studies revealed that the number of Brønsted sites capable of protonating pyridine corresponded to only ∼7% of the sulfur atoms on the catalyst. Additional Brønsted sites can be recreated on this catalyst with addition of water, a reaction not observed for sulfur-free zirconia. But this approach has been little used because the probe can undergo fast chemical exchange on some catalysts at room temperature and the sensitivity of 15N is low even with complete isotopic enrichment. These problems maybe made out by working out the experimental conditions for 15N CP/MAS spectra of pyridine.104 However, the chemical exchange of pyridine molecules at different adsorption sites with different efficiencies of cross polarization makes quantitative evaluation difficult.
3.2.4
13C-enriched acetone.
Adsorption of 2-13C-acetone can be used in conjunction with the 13C MAS NMR chemical shift as a measure of relative acid strength of various solid acids.20,64 The isotropic shift of 2-13C-acetone changes upon complexation with either Brønsted105,106 or Lewis acids.107 The formation of a hydrogen bond between the acidic proton and the carbonyl oxygen of adsorbed acetone will cause a downfield shift of the carbonyl carbon. The stronger the Brønsted acidity, the stronger the hydrogen bond between the carbonyl carbon and the acidic proton, and consequently the more downfield the 13C isotropic chemical shift. Therefore, 2-13C-acetone is a useful NMR probe molecule to determine the relative acid strength of solid acids. Biaglow et al.106 proposed an acidity scale based on their measurements of the 13C NMR chemical shift of the carbonyl carbon of acetone molecules adsorbed on different solid acid catalysts relative to the resonance of the carbonyl carbon in pure acetone. Haw et al.19 demonstrated that this scale reasonably reflects the acid strength of solid acids. They found the threshold acid strength of a solid superacid, which corresponds to a 100% H2SO4 solution, gives rise to an isotropic 13C NMR chemical shift of 245 ppm of adsorbed 2-13C-acetone.19 Solid-state NMR experiments and quantum chemical DFT calculations of acetone adsorption were used by the Deng group to study the location of protons in anhydrous 12-tungstophosphoric acid (HPW), and the acid strength of the anhydrous hydroxyl groups.10813C MAS NMR of acetone adsorption indicated there were two types of isolated protons with different acid strengths in the anhydrous Keggin HPW. Rotational Echo Double Resonance (REDOR) NMR experiments combined with quantum chemical DFT calculations demonstrated that acidic protons in anhydrous HPW are localized on both bridging (Oc) and terminal (Od) atoms of the Keggin unit. Using the same joint experimental–theoretical approach, they found the Brønsted/Lewis acid synergy in dealuminated HY zeolite.10913C NMR spectroscopy of adsorbed 2-13C-acetone demonstrated a remarkable increase in acid strength of dealuminated HY zeolite. With both NMR experiments and DFT calculations, the detailed structure of non-framework Al species and the mechanism of Brønsted/Lewis acid synergy were identified.109
4. Investigations of the reaction mechanisms and kinetics by in situ MAS NMR spectroscopy
4.1 Catalyst structures and reaction mechanisms on metal oxide-containing zeolites
4.1.1 Methane dehydroaromatization on MoO3/zeolites in the absence of oxygen.
The efficient utilization, particularly direct use of methane, remains a great challenge to catalysis science from both practical and theoretical points of view. Since our laboratory110 first reported that methane can be transformed into aromatics and H2 on the MoO3/HZSM-5 catalyst in the absence of oxygen in 1993, the methane dehydroaromatization (MDA) reaction has been extensively investigated.111–113 Our work on the applications of solid-state NMR in this reaction can be summarized as follows: (a) interaction between Mo species and zeolite support, (b) direct observation of the active site by ultrahigh field 95Mo NMR, (c) investigation of the catalytic process.
4.1.1.1 Interaction between Mo species and zeolite support.
The MoO3/HZSM-5 supported catalyst, normally abbreviated as Mo/HZSM-5, is the best among the vast number of tested catalysts. Multinuclear MAS NMR revealed that Mo species strongly interacted with the HZSM-5 support, which led to the new Al2(MoO4)3 crystallites and partial consumption of Brønsted acidic sites.66 Using perfluorotributyl amine as the NMR probe, 1H MAS NMR demonstrated that the Mo species might migrate into the lattice channels of HZSM-5 zeolite during the impregnation and subsequent treatment. The migration of some Mo species into the zeolite channels could be beneficial for the MDA reaction.66 To get better catalytic performance, zeolite HMCM-22 was also used as the support. It seemed that the Mo/HMCM-22 catalyst had a higher benzene yield and a longer lifetime, which was attributed to the unique two independent pore systems.114 Multinuclear MAS NMR characterizations indicated that there was still interaction between the Mo species and the HMCM-22 support.67 Using 1H MAS and 1H/27Al TRAPDOR NMR techniques, we found there was a condensation/reaction between Mo species and the hydroxyl species of HMCM-22. Among the hydroxyl species, shrinking of Brønsted acidic sites or extra-lattice Al–OH of the HMCM-22 after loading Mo was obvious.67 Most of the Brønsted acidic sites were located at the internal channels of HMCM-22 (>75%), while most of the silanol groups stayed at the external surface.115 With Brønsted sites serving as a powerful trap, molybdenum may migrate into the internal channels of the zeolite and reacted preferentially with bridging hydroxyl groups to anchor Mo species to the framework aluminium through an oxygen bridge.67 After adsorption of xenon at different pressures, 129Xe NMR found that Mo species were preferably located in the supercages of HMCM-22 at low coverage (Mo loading <6 wt%).97 Therefore, most Mo species were distributed in the internal zeolitic channels of HMCM-22. Two dimensional 27Al multiple-quantum (MQ) MAS NMR experiments were also used to further investigate interaction between the Mo species and HMCM-22.116 Combined DFT calculations and MQ MAS NMR measurements demonstrated that Mo species were directly bonded to the zeolite framework Al species with an isotropic chemical shift of 54.6 ppm and quadrupolar coupling constant of 6.2 MHz, which were responsible for the initial transformation of reactant in MDA. The other framework Al atoms that were inert to adsorption of Mo and still bound to protons retained their Brønsted nature and participated in succeeding catalytic reactions.116
4.1.1.2 Direct observation of the active site by ultrahigh-field 95Mo NMR.
It has been hypothesized that molybdenum may migrate into zeolitic channels and anchor on Brønsted acid sites during the synthesis.66,115–117 However, it is unclear whether the active molybdenum species are small crystallites or isolated exchanged species. Zheng et al. employed ultrahigh-field 95Mo NMR spectroscopy at 21.1 T to recognize different types of molybdenum species on the Mo/HZSM-5 catalyst.118,119
The left side of Fig. 6 shows the static 95Mo NMR spectra of MoO3 and the activated catalyst. Compared with MoO3, the spectrum of 2Mo/HZSM-5 has a much broader peak centered at about −250 ppm, spanning from about −639 to 168 ppm (Fig. 6e). This peak has a quite different line shape and a significant upfield shift relative to the spectrum from MoO3. Additionally, the decrease of 0.9 Brønsted acid sites per unit cell in the 2Mo/HZSM-5 sample matches the amount of the new type of molybdenum species (0.92/unit cell) detected by 95Mo NMR spectroscopy and elemental analysis. It is concluded that the dominant Mo species in the 2Mo/HZSM-5 sample are a new type of molybdenum species, which may be attributed to the exchanged molybdenum species previously suggested based on 1H MAS NMR and 27Al MQ MAS NMR results.116,117 The spectral line shapes of the samples with higher molybdenum loadings have features that resemble those of MoO3, but broader shoulder peaks, centered at about −250 ppm, can also be observed. The 95Mo NMR spectra of working catalysts after different reaction times to form molybdenum carbide are also obtained, and compared with the fresh catalysts. The right side of Fig. 6 shows a good correlation between the amount of the exchanged molybdenum species and aromatics formation rate. This demonstrates that the carburized molybdenum species originating from the exchanged Mo species are the active centers for the MDA reaction.
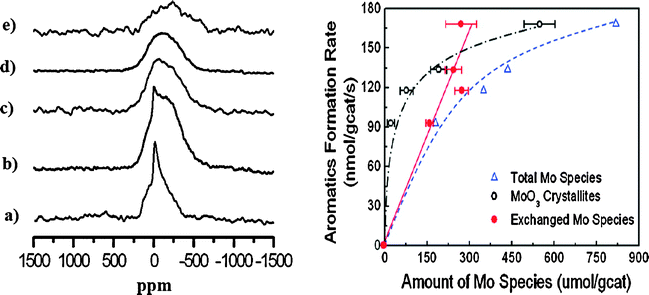 |
| | Fig. 6 (left) Static 95Mo NMR spectra of (a) MoO3, (b) 10Mo/HZSM-5, (c) 6Mo/HZSM-5, (d) 4Mo/HZSM-5 and (e) 2Mo/HZSM-5. (right) Correlating the aromatics formation rate with different molybdenum species. (Adapted with permission from ref. 118. Copyright 2008 American Chemical Society). | |
As shown in Fig. 7, the Brønsted acid signal at 4.1 ppm decreases dramatically, and two new resonances at 6.8 and 7.9 ppm emerge clearly, when the reaction time is prolonged. The signal at 6.8 ppm can be assigned to the water molecules formed and adsorbed during the reaction:
| | | MoO3 + CH4 → MoC2 + CO2 + CO + H2O | (1) |
The signal at 7.9 ppm is assigned to the hydrogen atoms of the aromatic species. At the same time, the intensity of the Brønsted acid signal decreases further, indicating that Brønsted acidic sites take part in the aromatization or hydrogen transfer process of the reaction.117 The reaction process is stable up to 1 h (Fig. 7d). After a further 2 h reaction time a new signal with similar chemical shift to the signal at 7.9 ppm but with a much broader form appears, while the amount of adsorbed water at 6.8 ppm is greatly reduced (Fig. 7e). A clearer 1H MAS NMR profile of Fig. 7e can be found in Fig. 7g in which an enlarged spectrum and the simulated lines are presented. The broad signal can be attributed to the aromatic carbonaceous deposits on the catalyst surface. The broad line width reveals its mixed composition and complicated nature. For the first time the variation of the amount of Brønsted acidic sites during a real high-temperature reaction at 973 K, as well as the formation of water, benzene, and aromatic carbonaceous deposits in the reaction process have been directly observed by in situ1H MAS NMR spectroscopy. This provides strong support for an acid-induced aromatization mechanism. Solid-state 13C MAS NMR can also be used to study the catalytic process of the MDA reaction under batch-like conditions.121 After reaction at 873 K for 1 h (Fig. 8b), the intensity of methane signal decreases dramatically and a strong CO2 signal at 126 ppm appears. When increasing the temperature to 973 K for 30 min, the color of the catalyst changed from blue-white to black, which is the typical color of molybdenum carbide with a 13C NMR chemical shift at 274 ppm (Fig. 8c), indicating the onset of the reaction. The resonance of molybdenum carbide (Fig. 8c) is actually an overlap of a broad peak and a narrow one, as a result of the two different kinds of molybdenum carbide formed during the reaction, which are assigned to molybdenum carbide dispersed on the external surface and internal channel of zeolite, respectively.121 For the deactivation process of the Mo/HZSM-5 catalyst for MDA, 13C MAS NMR measurements confirm the carbonaceous deposits on the Brønsted acidic sites, and thereafter the blockage of the channels as well as the deep dehydrogenation of live carbonaceous species to the formation of graphitic coke in the final stage.122
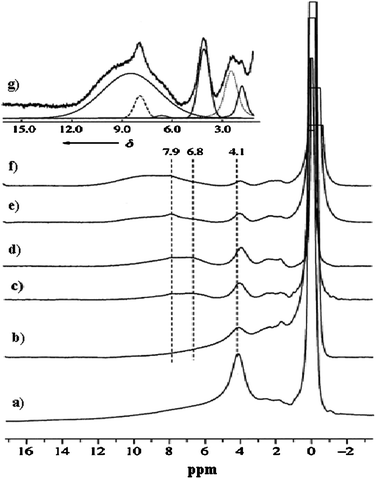 |
| | Fig. 7
In situ
1H MAS NMR spectra of 6 wt%Mo/HZSM-5 catalyst treated with (a) methane flow at 873 K for 1 h. (b)–(f) after the MDA reaction (973 K, 1500 mL g−1 h−1, 1 atm); reaction times: (b) 10 min, (c) 30 min, (d) 1 h, (e) 3 h, (f) 6 h. Spectrum (g) is an expansion of spectrum (e), with six lines simulating the original spectrum. (Adapted with permission from ref. 120. Copyright 2000 Wiley-VCH). | |
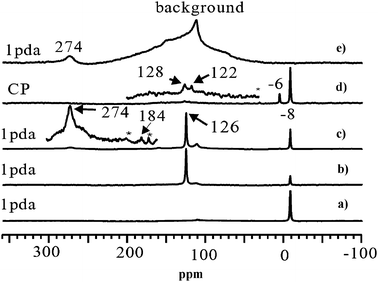 |
| | Fig. 8
13C MAS NMR spectra of methane (13C, 99%) reaction on 6 wt%Mo/HZSM-5 at (a) room temperature, 1 pulse with 1H decoupling (1 pda); (b) 873 K for 1 h, 1 pda; (c) 973 K for 30 min, 1 pda; (d) 973 K for 30 min, CP. For comparison, the 13C MAS NMR spectrum of molybdenum carbide powder is shown in (e). The spectra were recorded at room temperature. Asterisks denote spinning sidebands. The signal at 112 ppm is due to background of the spinning module. (Adapted from ref. 121). | |
4.1.2 Olefin metathesis on MoO3/HBeta-Al2O3 catalyst.
Olefin metathesis is one of the most important C–C bond formation reactions in organic chemistry. Chauvin, Grubbs and Schrock won the Nobel Prize in chemistry in 2005 for their development of olefin metathesis in homogeneous systems.123–125 Heterogeneous catalysts for metathesis reactions would be of further interest due to the ease of separation, good persistence, and recyclability.126 Recently, we reported that Mo/HBeta-Al2O3 was a highly active catalyst for metathesis of ethene and 2-butene to propene.127–129 This provides a promising alternative way for the production of propene, which is in high demand in the world-wide chemical industry. In contrast, Mo supported on HBeta zeolites performed poorly due to the strong interaction between Mo species and HBeta zeolites.130 So, the composition and structure of the support played an important role in the heterogeneous olefin metathesis reaction. For further application in industry the deactivation of the catalyst needs to be understood in order to find ways to improve the catalyst stability. We performed detailed investigations on the catalyst structures and deactivation mechanism during olefin metathesis using multinuclear solid-state NMR techniques.
4.1.2.1 The role of alumina in the composite support.
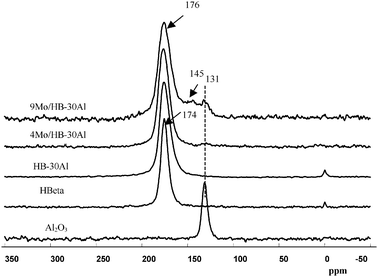
Among the tested catalysts, the optimized 4.0 wt% Mo supported on a composite support of 70% HBeta zeolite and 30% γ-alumina (HB-30Al) showed much higher performance for the metathesis of ethene and 2-butene to propene compared with the catalyst without alumina in the support.128 Quantitative one-dimensional 27Al MAS NMR measurements indicate that the framework Al content of HBeta zeolites decreases from 592 to 400 μmol g−1 after introduction of Mo species, which means the dealumination of the HBeta framework. However, the concentration of framework Al in HBeta zeolites increases to 486 μmol g−1 with addition of 30% alumina into the support. This reveals that addition of alumina into the support may protect the HBeta framework from being destroyed. Two-dimensional 27Al MQ MAS NMR spectra are employed to remove the anisotropic line broadening which allows species with similar isotropic chemical shifts but different quadrupolar coupling constants to be identified.128 As demonstrated in Fig. 9a, the overlapped peaks observed in the 27Al MAS spectra at 54 ppm are clearly resolved in the MQ MAS spectra for HBeta zeolites. Two distinct framework Al species designated as Al(IV)a and Al(IV)b are presented in the tetrahedral region, and a third one at ca. 0 ppm (Al(VI)a) in the octahedral environment is also observed. When HBeta was impregnated with Mo species, the 27Al MQ MAS spectra of Mo/HBeta reveals an additional species Al(IV)c with an isotropic chemical shift (δiso) at 58.8 ppm besides the line at 14 ppm assigned to a hydrated species of aluminium molybdate (Al2(MoO4)3). The signal of Al(IV)c appears in the resonance region of the tetrahedral aluminium framework, and experiences a large quadrupolar interaction with the second order quadrupolar effect (SOQE) of 4.0 MHz. This large anisotropic quadrupolar broadening makes it difficult to identify in the usual 27Al MAS NMR spectra, however, in the isotropic F1 projection, it is clearly noticeable. At the same time one could note that the intensity of the Al(IV)a signal is decreased readily from the HBeta framework after impregnating Mo species. The ratio of Al(IV)a/Al(IV)b in the framework aluminium content decreases from 0.6 to 0.06. This implies that introduction of Mo species preferentially removes aluminium atoms from crystallographic positions represented by Al(IV)a. In the meantime two new peaks of Al(IV)c and Al(VI)b were observed in the spectra. So, it is the introduction of Mo species that leads to the formation of a third framework Al(IV)c species, and subsequently the appearance of non-framework Al2(MoO4)3. This suggests that the Al atoms corresponding to peak Al(IV)c could be assigned to the distorted extra-framework tetrahedral aluminium similar to that observed by van Bokhoven et al. in the hydrothermal treatment of HBeta zeolites.131,132 For the 4Mo/HB-30Al catalyst besides the tetrahedral Al(IV)d in alumina a part of Al(IV)a can still be observed in the HBeta framework, and the ratio of Al(IV)a/Al(IV)b increases from 0.06 to 0.23. At the same time the peak belonging to the distorted tetrahedral Al(IV)c is not so evident as that in the 4Mo/HB catalyst. It experiences a smaller quadrupolar interaction (δiso = 58.3 ppm, SOQE = 3.2 MHz). So, alumina can protect the specific T-sites in the framework of HBeta zeolites when impregnating Mo species. The framework aluminium in HBeta zeolites can remain at the same crystallographic sites after addition of alumina into the composite support.128 As shown in Fig. 10, hyperpolarized 129Xe NMR can be used to detect the location of Mo species in the HBeta and alumina composite support.128 The peak at 174 ppm is from xenon adsorbed in the 12-membered ring channels of HBeta zeolite. The signal of xenon adsorbed in alumina pores appears at 131 ppm. For the HBeta-Al2O3 composite support only the peak corresponding to xenon in the HBeta pores is detectable. This may be due to the preferential adsorption of xenon in the micropores rather than in the mesopores. After introduction of Mo species a small downfield shift of the xenon signal in HBeta zeolites is observed. This indicates a very small portion of the Mo species is deposited into the pores of HBeta, which narrows the pore size of HBeta slightly. At the same time a small peak at ca. 131 ppm emerges upon Mo loading, which is consistent with the signal from the empty alumina. With the high Mo loading of 9 wt%, there is also an additional peak centered at ca. 145 ppm. This peak can be ascribed to the xenon adsorbed in the alumina with Mo species because the deposition of Mo species into the pores of alumina may decrease the average free path of xenon in alumina, which leads to the low-field shift of the xenon signal. From the hyperpolarized 129Xe NMR measurements it was clear that Mo species were deposited more in the pores of alumina than in the pores of HBeta zeolites at higher Mo loadings.128 Such a distribution may preserve the aluminium atoms at specific T-positions in HBeta zeolites as evidenced by the above 27Al MQ MAS NMR, and resulted in the moderate distribution of Mo species in the composite support. As demonstrated in section 3.1, high-resolution 1H MAS NMR is a powerful and direct method to characterize the acidic sites in solid catalysts. Fig. 11 shows the 1H MAS NMR spectra of composite support HB-30Al with different Mo loadings. During the catalyst preparation the Mo species first reacted with the basic Al hydroxyls at −0.2 ppm on alumina and silanols at 1.7 ppm on the surface of HBeta zeolite.129 When the Mo loading was as high as 4.0 wt%, the peak intensity of the Brønsted acidic sites at 3.9 ppm on HBeta zeolite was clearly reduced. So, there was moderate Brønsted acidity on the catalysts with a Mo content of 4.0–6.0 wt%, which was beneficial for the metathesis reaction.129 In order to give a comprehensive understanding of the mechanism, DFT calculations were performed on the Mo/HBeta catalyst with different acidities.133 The Si–H distance of the cluster model was set between 1.30 and 2.25 Å to represent the supports with different Brønsted acid strengths.77Fig. 12 shows the cluster models of the Brønsted acidic sites in HBeta and the Mo-carbene active site. The proton affinity (PA) values decreased from 350.4 to 326.4 kcal mol−1 when the Si–H distance increased from 1.30 to 2.25 Å, indicating a more acidic zeolite cluster. A detailed DFT study on the cross-metathesis of ethene and 2-butene to propene using the above models demonstrated that the acidity of the support would affect the transition-state structures as well as the activation barriers.133 The DFT calculations revealed that olefin metathesis would occur more easily over more acidic heterogeneous catalysts, which is consistent with the experimental results from our group and others.129,134–136 However, too much Brønsted acidity may also cause side reactions such as olefin isomerization and oligomerization. So, proper acidity would be advantageous for the olefin metathesis reaction on a practical heterogeneous catalyst.128,129
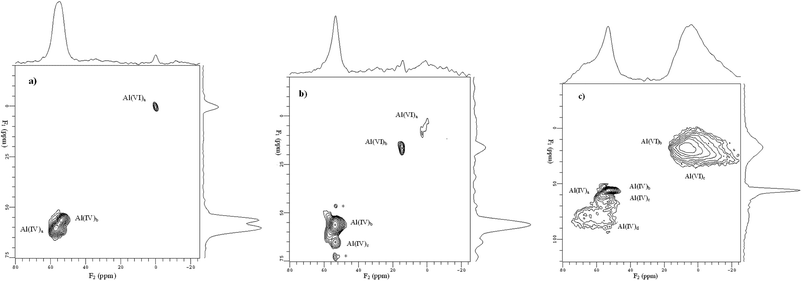 |
| | Fig. 9 Two-dimensional (2D) 27Al MQ MAS NMR spectra of samples: (a) HBeta, (b) 4Mo/HB, (c) 4Mo/HB-30Al. The corresponding 27Al MAS NMR spectrum is given on the top of the MQ MAS plot. The F1 projection is the pure isotropic spectrum. Asterisk denotes the sidebands. (Adapted with permission from ref. 128. Copyright 2007 Elsevier). | |
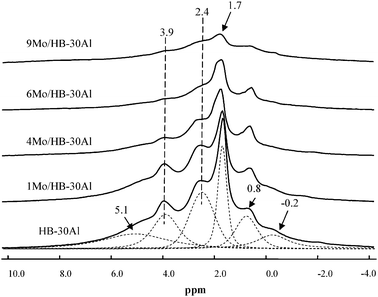 |
| | Fig. 11
1H MAS NMR spectra of HBeta-alumina composite supports with different Mo loadings. The spinning rate was 10 kHz, and 200 single-pulse scans were accumulated. (Adapted with permission from ref. 129. Copyright 2008 American Chemical Society). | |
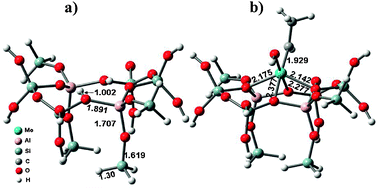 |
| | Fig. 12 The cluster models of (a) the Brønsted acidic site in HBeta and (b) the Mo-carbene active site. (Adapted with permission from ref. 133. Copyright 2010 Springer). | |
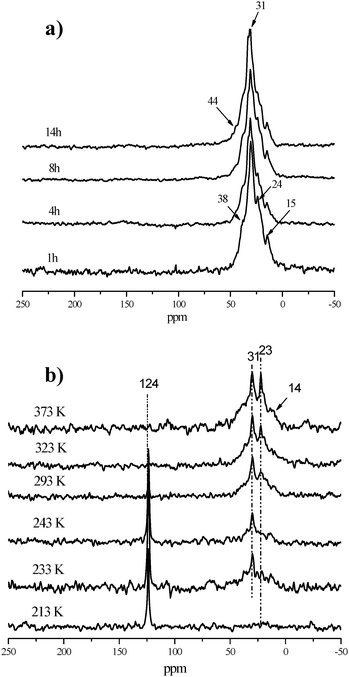
In order to get more information about the origin of the carbonaceous deposits on the Mo/HBeta-Al2O3 catalyst during the metathesis reaction, in situ13C CP/MAS NMR experiments of ethene adsorbed on the catalyst were performed from 213 to 373 K. As shown Fig. 13b, only gas signals from ethene at 124 ppm could be observed at 213 K in the spectrum indicating no reaction happens. With increasing temperature, some of the ethene begins to oligomerize, which leads to the appearance of a resonance peak at 31 ppm which is in accordance with the reports by Gutsze et al.140 At the same time, the peak at 23 ppm gradually appears when the temperature is above 293 K. The increase in signal intensity at 23 ppm upon temperature further implies that the oligomerization reaction happens. When the temperature is higher than 323 K, the gas ethene signal at 124 ppm disappears, and the lines at 31 and 23 ppm are more intense. This indicates the appearance of branched paraffins at higher temperatures. The 13C CP/MAS NMR spectra of 2-butene adsorbed on 4Mo/HBeta-Al2O3 catalyst have similar results (not shown here). So, in situ13C CP/MAS NMR spectra of the reaction of ethene or 2-butene on 4Mo/HBeta-Al2O3 catalyst demonstrate that the carbonaceous species on the used metathesis catalyst should mainly come from the olefin oligomerization.137129Xe NMR experiments clearly revealed that the deposited carbon was located at the positions of the channel intersections in HBeta zeolites, which may cause the diffusion problem in the catalyst.141 XPS measurements indicated the reduction of Mo6+ to Mo4+ after long reaction times.137 By combining the characterization results of fresh, used, and regenerated catalysts with their metathesis activity, it could be deduced that active sites poisoned by carbonaceous deposits from olefin oligomerization and deep reduction of the active site Mo species may be responsible for the deactivation of the olefin metathesis catalyst, which is consistent with the DFT calculation results.142 These findings may be beneficial for its further application in industry.
4.1.3 Styrene oxidation mechanism on titanium silicalite (TS-1) zeolites.
TS-1 zeolites have shown great potential in industry for their excellent catalytic properties in the selective oxidation of organic compounds. Particularly, oxidation of olefins to epoxides on TS-1 with aqueous H2O2 as the oxidant is highly efficient and environmentally benign.143,144 However, when the reaction was extended to styrene, the main product was phenylacetaldehyde (PADH), and only a small amount of epoxide was obtained.145–147 There was no direct evidence of how PADH and styrene epoxide were formed. We carried out a combination of in situ13C MAS NMR and DFT calculations to reveal the catalytic mechanism of styrene oxidation over TS-1 zeolites with urea-hydroperoxide (UHP).148 Two kinds of TS-1 were used in the study: one without Brønsted acidity (sample A) and the other with Brønsted acidity (sample B). The acidity was confirmed by 31P MAS NMR with trimethylphosphine as the probe molecule (see section 3.2.1). When β-13C enriched styrene was adsorbed on the sample without Brønsted acidity (sample A), one main peak corresponding to its β carbon atom is observed (Fig. 14a). When styrene was adsorbed on premixed UHP and sample A, two additional peaks centered at 50.2 and 102.2 ppm appear (Fig. 14b). The former is a typical signal for the β carbon atom of styrene epoxide, which is formed by the epoxidation of styrene with UHP.149 The latter signal is assigned to a hemiacetal species bound to framework Ti species of the zeolite, whose structure and 13C chemical shift were optimized by DFT calculations (Fig. 15a).148 The calculated chemical shift of this species was 102.7 ppm in agreement with the experimental value of 102.2 ppm. In addition, a broad peak at 64.0–68.0 ppm appeared, which can be assigned to a glycol species bound to the framework Ti species (Fig. 15b). This assumption was confirmed by calculation of the chemical shift at 64.7 ppm for the corresponding glycol structure.148 When the sample was heated at 313 K after adsorption, more and more hemiacetal species appeared with the decrease in styrene (Fig. 14c–d). However, no signal at 200 ppm corresponding to PADH appeared. Therefore, without Brønsted acidic sites, styrene could not be converted into PADH. When styrene was adsorbed on TS-1 zeolites with Brønsted acidity, in situ13C MAS NMR experiments indicated a different picture. After styrene reacted with a premixture of UHP and sample B (Fig. 16c–e) at 313 K, a new peak corresponding to PADH appeared at about 202.0 ppm. On the other hand, the intensity of the signal for the hemiacetal species at 102.2 ppm increased at first then decreased with the depletion of styrene and the formation of PADH. This strongly suggested that the hemiacetal species are intermediates for PADH production. It ruled out the previous assumption that PADH results from the rearrangement of styrene epoxide.149,150 The intermediate role of the hemiacetal species in the production of PADH was in agreement with the well-documented equilibrium between hemiacetal and acetal, that was, the hemiacetal can be changed to the acetal under acidic conditions.151 So, it can be concluded that the Brønsted acidic sites on TS-1 zeolites catalyze the transformation of the hemiacetal species to PADH. Additionally, protic solvents such as water and methanol can also provide acidic centers to convert the intermediates into PADH.152 Assisted by in situ EPR and DFT calculations, it was found that the Ti-superoxo radical is responsible for the formation of the hemiacetal species. Based on the facts from the joint experimental and theoretical investigations, a reasonable mechanism for the oxidation of styrene was proposed in Scheme 1.148
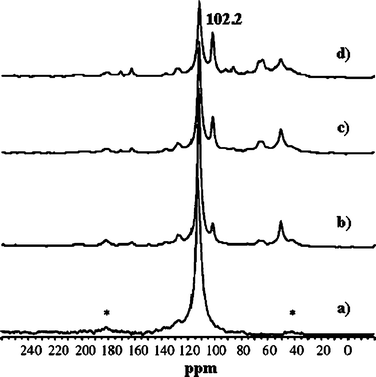 |
| | Fig. 14
In situ
13C MAS NMR spectra of styrene adsorbed on sample A (a), and for the reaction of styrene with a mixture of sample A and UHP at 313 K after 0 min (b), 15 min (c), and 45 min (d). The asterisks denote spinning side bands. (Adapted with permission from ref. 148. Copyright 2004 Wiley-VCH). | |
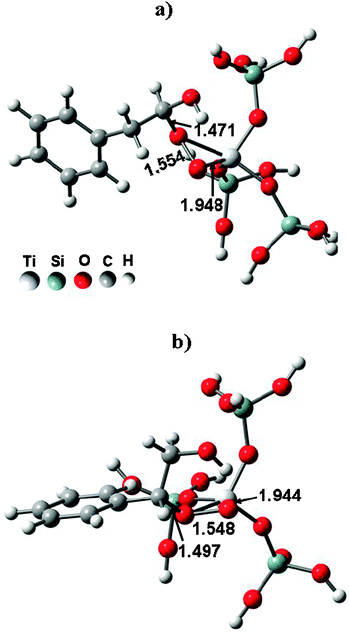 |
| | Fig. 15 The optimized structures of hemiacetal (a) and glycol (b) species bound to the framework of TS-1 zeolites. (Adapted with permission from ref. 148. Copyright 2004 Wiley-VCH). | |
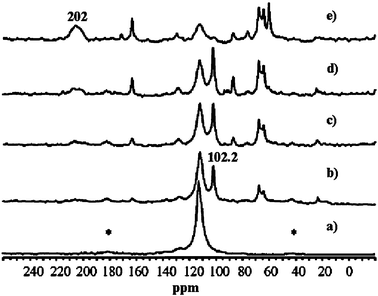 |
| | Fig. 16
In situ
13C MAS NMR spectra of styrene adsorbed on sample B (a), and for the reaction of styrene with a mixture of sample B and UHP at 313 K after 0 min (b), 15 min (c), 45 min (d), and 60 min (e). The asterisks denote spinning side bands. (Adapted with permission from ref. 148. Copyright 2004 Wiley-VCH). | |
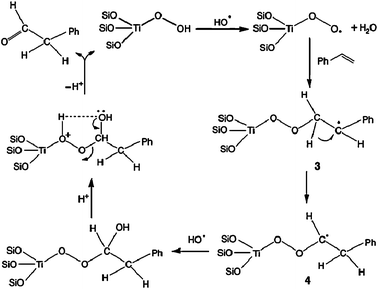 |
| | Scheme 1 The proposed mechanism for the radical reaction leading to the hemiacetal species and PADH. (Adapted with permission from ref. 148. Copyright 2004 Wiley-VCH). | |
4.2 Reaction kinetics in heterogeneous catalysis
In situ MAS NMR spectroscopy can provide not only information on reaction mechanism but also the kinetic process of heterogeneous catalysis. Normally, 1H and/or 13C NMR signal plots are continuously recorded versus reaction time for obtaining the kinetic parameters involving apparent rate constants and activation energies. Reaction kinetics from conventional 1H and 13C NMR are obtained by concentration variations of reactant or product in the bulk state,153 however, the kinetics in a confined space such as a nanocage or nanochannel can not be achieved easily. It has been shown that the laser-hyperpolarized (HP) 129Xe NMR technique is powerful for the studying of porous materials.56–63 The resultant 129Xe NMR spectrum is from xenon atoms adsorbed in pores with dimensions down to the nanoscale, which could make it possible for investigation of the reaction kinetics in a restricted geometry. The kinetic parameters obtained from the NMR spectra could serve as the basis for the verification of quantum chemical calculations for molecule activation and conversion pathways.154 Here the applications of in situ MAS NMR for reaction kinetics in the bulk state and in the nanoconfined space are described using conventional 1H, 13C NMR154–163 and our recently developed hyperpolarized 129Xe NMR approaches, respectively.18
4.2.1 Reaction kinetics in the bulk state by 1H and 13C MAS NMR.
Normally, the reaction kinetics are investigated by 1H and 13C MAS NMR to record the reactant or product concentration variations under batch-like conditions. The Stepanov, He and Hunger groups154–163 carried out detailed kinetic studies on alkane conversion and H/D exchange reactions on acidic catalysts. For instance, Stepanov et al.155–157 investigated n-butane conversion on sulfated zirconia by in situ monitoring of the kinetic processes of the reactant with a selectively 13C-labeled methyl group and product under batch reaction conditions. Firstly, they found that carbon scrambling in n-butane accompanied isomerization. Then, they demonstrated that scrambling in n-butane was a monomolecular process, and the isomerization process was a purely bimolecular process. Furthermore, a kinetic analysis of n-butane isomerization at 291–323 K showed that the observed rate constants of monomolecular carbon scrambling were twice as large as the rate constants of biomolecular isomerization, and the apparent activation energies of these reactions were similar (71 and 63 kJ mol−1, respectively).
The Hunger group used in situ1H and 13C MAS NMR to investigate the H/D exchange reactions of acetone on HZSM-5,161 and alkylaromatic hydrocarbons on HY and HZSM-5 zeolites.162,163 By in situ pulse-flow 1H MAS NMR coupled with UV/Vis spectroscopy, they observed regioselective H/D exchange at the side-chain methyl group of ethylbenzene adsorbed on dealuminated HY zeolites in the temperature range of 443–463 K (Fig. 17).163 A reaction mechanism involving both Lewis and Brønsted acidic sites in the H/D exchange reaction was also suggested. Furthermore, the activation energy of the regioselective H/D exchange was obtained by recording and analyzing the stack plots of in situ1H MAS NMR spectra at three different temperatures.163
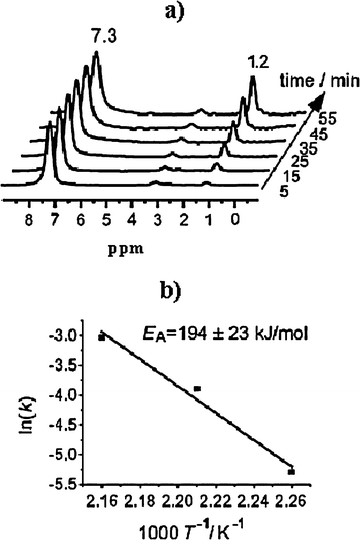 |
| | Fig. 17 (a) Stack plot of in situ1H MAS NMR spectra recorded during the H/D exchange of ethyl-d5-benzene on dehydrated zeolite HY at 453 K. (b) Arrhenius plot of the H/D exchange rates of the methyl group in ethylbenzene adsorbed on zeolite HY at temperatures of 443, 453, and 463 K. (Adapted with permission from ref. 163. Copyright 2008 Wiley-VCH). | |
4.2.2 Reaction kinetics in the nanospace by hyperpolarized 129Xe MAS NMR.
Recently, our group developed the new approach of in situ continuous-flow laser-hyperpolarized (HP) 129Xe MAS NMR together with 13C MAS NMR to study the adsorption and reaction kinetics in the nanospace.18 The advantages of hyperpolarized 129Xe with much higher sensitivity and shorter acquisition time allow the kinetics to be probed in a confined geometry under the continuous-flow conditions close to the real heterogeneous catalysis. The dynamic and kinetic processes of methanol adsorption and reaction to dimethyl ether (DME) in chabazite (CHA) nanocages were monitored by one- and two-dimensional hyperpolarized 129Xe MAS NMR. The kinetic curves and apparent activation energy of the nanocages involving the active site were also obtained quantitatively. The adsorption of methanol in CHA nanocages can be described as follows:| | | CH3OH(g) + [Cage]empty → CH3OH@[Cage] | (2) |
The rate function of eqn (2) can be expressed by r = k1[CH3OH]gm[Cage]nempty. [CH3OH]g remains constant due to the continuous injection of methanol vapour into the rotor by N2, thus r = ka[Cage]nempty, where ka = k1[CH3OH]gm. For quantitatively obtaining the kinetic information on methanol adsorption in the CHA cages, a series of 129Xe MAS NMR spectra were recorded at 298, 333 and 373 K, respectively, with increasing adsorption times. Thus, the adsorption rate function at different temperatures can be obtained as r = ka[Cage]1.57empty after fitting the kinetic curves. The activation energy of methanol adsorption in the CHA nanocages could be estimated as ca. 5.2 kJ mol−1 according to the Arrhenius equation. It is typically physical adsorption.
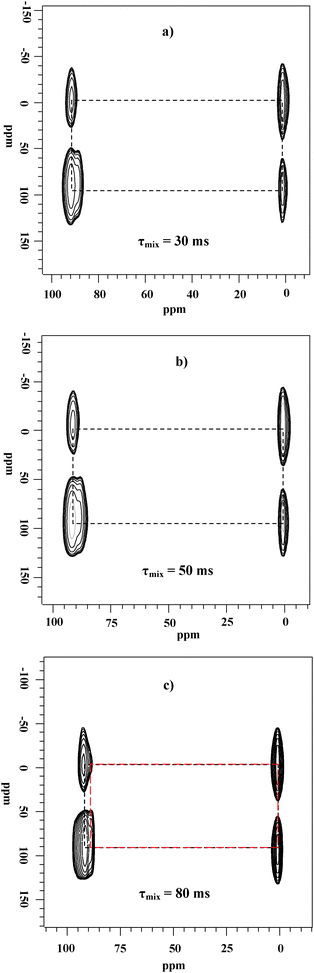
After saturation adsorption of methanol, the temperature is gradually increased from 298 to 453 K while still injecting the gas mixture of 13CH3OH and HP 129Xe. Interestingly, besides xenon in the empty cage at 84 ppm in the HP 129Xe NMR spectra the signal at lower field splits into two peaks with increasing the reaction time (Fig. 18). The main peak A is from Xe co-adsorbed with methanol in CHA cages. The shoulder peak B may be due to the occurrence of the methanol conversion because in the corresponding in situ13C CP/MAS NMR spectra dimethyl ether (DME) is detected at temperatures higher than 393 K.18 In order to further confirm the assignment of peak B, in situ two-dimensional HP 129Xe exchange spectra (2D-EXSY) were acquired during methanol conversion under the steady state (Fig. 19). The technique is extensively employed to investigate the dynamic processes of xenon in different domains in the porous materials.59,60,63 The cross-peaks indicate an exchange of xenon atoms between the corresponding environments on the diagonal within the period of mixing time. At the reaction temperature of 453 K, the presence of cross-peaks in Fig. 19a suggests that the exchange of Xe between the gas phase and peak A exists on a time scale of 30 ms. Another cross-peak emerging in Fig. 19c indicates that the exchange of Xe between the gas phase and peak B occurs when increasing the mixing time to 80 ms. Therefore, xenon exchange took place faster between the gas phase and peak A than between the gas phase and peak B. This could be caused by the desorption of products, which restricted the xenon atoms motion between the gas phase and the CHA cages. So, it further confirms that the new peak B can be assigned to xenon in the methanol reaction cage, in which methanol molecules are converted to DME and water on the Brønsted acid site. The overall reaction of methanol in CHA nanocages can be described as follows:
| | | CH3OH(g) + [Cage]empty → DME&H2O@[Cage] | (3) |
The reaction rate function of
eqn (3) can be expressed as
r =
k2[CH
3OH]
gm[Cage]
nempty, where
k2 denotes the apparent reaction rate constant. The HP
129Xe NMR spectra were also recorded at other reaction temperatures,
e.g. 413 and 433 K. Thus, the reaction rate function with different temperatures can also be obtained as
r =
kr[Cage]
1.27empty after fitting the kinetic curves, and the apparent
activation energy of the active site in the
methanol reaction could be calculated as
ca. 57 kJ mol
−1 according to the Arrhenius equation (
Fig. 20). In our study, the empty CHA
nanocage represents the active site and is described in the reaction rate function from the molecular level that is different from the other conventional kinetic studies which only concern the concentration of reactants or products in the bulk space. Our approach demonstrates the reaction kinetics in the nanospace under continuous-flow conditions.
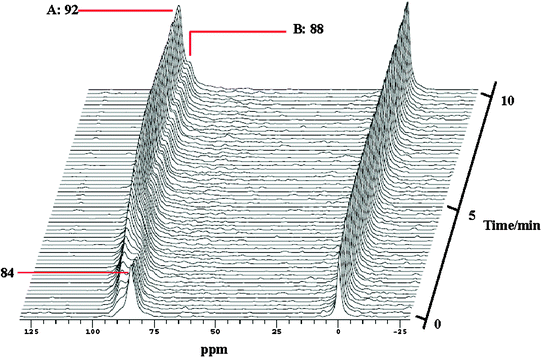 |
| | Fig. 18
In situ hyperpolarized 129Xe MAS NMR spectra recorded with a time resolution of 10 s per spectrum as a function of time during the methanol reaction in CHA nanocages at 453 K. (Adapted with permission from ref. 18. Copyright 2009 American Chemical Society). | |
 |
| | Fig. 19
In situ two-dimensional (2D) hyperpolarized 129Xe 2D-EXSY MAS NMR spectra recorded during the methanol reaction in CHA nanocages at 180 °C with different mixing times. The black dashed lines denote Xe exchange between the gas phase and peak A, and the red ones denote Xe exchange between the gas phase and peak B. (Adapted with permission from ref. 18. Copyright 2009 American Chemical Society). | |
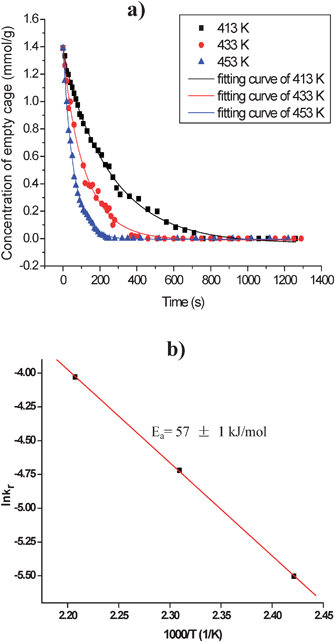 |
| | Fig. 20 (a) Kinetic curves of the methanol reaction in CHA nanocages at various temperatures. (b) Arrhenius plot of rate constants kr at different reaction temperatures, Ea is the apparent reaction activation energy. (Adapted with permission from ref. 18. Copyright 2009 American Chemical Society). | |
5. Concluding remarks
It is demonstrated in this review that in situ MAS NMR spectroscopy under batch-like or continuous-flow conditions is a powerful tool for investigating the nature and properties of acidic sites on solid surfaces, the reaction mechanisms and kinetics in heterogeneous catalysis. In the past decade, great achievements have made in the development of in situ MAS NMR techniques with the combination of other analytical methods such as gas chromatography, UV/Vis spectroscopy under the flow mode. This gives us a deeper understanding of the reaction mechanism on real working catalysts. As demonstrated, the introduction of hyperpolarized xenon into in situ MAS NMR spectroscopy enhances the sensitivity significantly, which makes kinetic study in a confined space possible. In the future, we can expect that (i) these approaches have the potential for further improvements and applications, for example increase in the sensitivity of in situ MAS NMR spectroscopy by employing the dynamic nuclear polarization (DNP) technique etc.; (ii) since many active sites in/on heterogeneous catalysts are low-γ nuclei, such as 95Mo, 107Ag, 47,49Ti, their direct NMR measurements under reaction conditions should go to the ultra-high field magnet using the Carr–Purcell–Meiboom–Gill (CPMG) sequence and/or two-dimensional MQ MAS NMR technique etc. to increase the detection resolution; (iii) joint in situ MAS NMR experiments and quantum chemical calculations and simulations using DFT, Monte Carlo etc. will be much more helpful in determination of the specific intermediate structures and reaction pathways, i.e. reaction mechanisms and kinetics on solid catalysts.
Acknowledgements
We are grateful for the financial support of the National Natural Science Foundation of China (Grant No. 20403017, 20573106, 20673111, 20873140), the Ministry of Science and Technology of China through the National Key Project of Fundamental Research (Grant No. G1999022400, 2005CB221400, 2003CB615806, 2009CB623507). W. Z. is also grateful to the Chinese Academy of Sciences for the financial support through the “Hundred-Talent” Program. W. Z., X. H. and X. B. thank our co-workers: Ding Ma, Xianchun Liu, Xiumei Liu and former students: Jianqin Zhuang, Zhimin Yan, Gang Yang, Heng Zheng, Yong Liu, Xiujie Li, Zhenchao Zhao, Xin Li and Renshun Xu for their contributions to this work. We also thank Dr Jian Zhi Hu, Dr Yong Wang and Dr Charles H. F. Peden of Pacific Northwest National Laboratory (PNNL), USA for helpful discussions.
References
- G. A. Somorjai, Top. Catal., 2008, 48, 1–7 CrossRef CAS.
- A. Corma and H. Garcia, Top. Catal., 2008, 48, 8–31 CrossRef CAS.
- K. Tamaru, Appl. Catal., A, 1997, 151, 167–177 CrossRef CAS.
- H. Topsoe, J. Catal., 2003, 216, 155–164 CrossRef CAS.
-
J. F. Haw, NMR Techniques in Catalysis, ed. A. Bell and A. Pines, Marcel Dekker, New York, 1994, p. 139 Search PubMed.
- I. D. Gay, J. Magn. Reson., 1984, 58, 413–420 CAS.
- M. W. Anderson and J. Klinowski, Nature, 1989, 339, 200–203 CrossRef CAS.
- J. F. Haw, B. R. Richardson, I. S. Oshiro, N. D. Lazo and J. A. Speed, J. Am. Chem. Soc., 1989, 111, 2052–2058 CrossRef CAS.
- W. P. Zhang, D. Ma, X. C. Liu, X. M. Liu and X. H. Bao, Chem. Commun., 1999, 1091–1092 RSC.
- G. W. Haddix, J. A. Reimer and A. T. Bell, J. Catal., 1987, 106, 111–115 CrossRef CAS.
- M. Hunger and T. Horvath, J. Chem. Soc., Chem. Commun., 1995, 1423–1424 RSC.
- P. Goguen and J. F. Haw, J. Catal., 1996, 161, 870–872 CrossRef CAS.
- E. MacNamara and D. Raftery, J. Catal., 1998, 175, 135–137 CrossRef CAS.
- P. K. Isbester, A. Zalusky, D. H. Lewis, M. C. Douskey, M. J. Pomije, K. R. Mann and E. J. Munson, Catal. Today, 1999, 49, 363–375 CrossRef CAS.
- M. Hunger and T. Horvath, J. Catal., 1997, 167, 187–197 CrossRef CAS.
- M. Hunger, M. Seiler and T. Horvath, Catal. Lett., 1999, 57, 199–204 CrossRef CAS.
- M. Hunger and W. Wang, Chem. Commun., 2004, 584–585 RSC.
- S. T. Xu, W. P. Zhang, X. C. Liu, X. W. Han and X. H. Bao, J. Am. Chem. Soc., 2009, 131, 13722–13727 CrossRef CAS.
- J. F. Haw, J. B. Nicholas, T. Xu, L. W. Beck and D. B. Ferguson, Acc. Chem. Res., 1996, 29, 259–267 CrossRef CAS.
- J. F. Haw and T. Xu, Adv. Catal., 1998, 42, 115–180 CrossRef CAS.
- E. G. Derouane, H. Y. He, S. B. Derouane-Abd Hamid and I. Ivanova, Catal. Lett., 1999, 58, 1–19 CrossRef CAS.
- X. W. Han, Z. M. Yan, W. P. Zhang and X. H. Bao, Curr. Org. Chem., 2001, 5, 1017–1037 CrossRef CAS.
- B. M. Weckhuysen, Chem. Commun., 2002, 97–110 RSC.
- M. Hunger, Catal. Today, 2004, 97, 3–12 CrossRef CAS.
- M. Hunger and W. Wang, Adv. Catal., 2006, 50, 149–225 CrossRef CAS.
- M. Hunger, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 53, 105–127 CrossRef CAS.
- T. Blasco, Chem. Soc. Rev., 2010, 39, 4685–4702 RSC.
- I. I. Ivanova and Y. G. Kolyagin, Chem. Soc. Rev., 2010, 39, 5018–5050 RSC.
- T. Xu and J. F. Haw, Top. Catal., 1997, 4, 109–118 CrossRef CAS.
- T. A. Carpenter, J. Klinowski, D. T. B. Tennakoon, C. J. Smith and D. C. Edwards, J. Magn. Reson., 1986, 68, 561–563 CAS.
- P. J. Giammatteo, W. W. Hellmuth, F. G. Ticehurst and P. W. Cope, J. Magn. Reson., 1987, 71, 147–150 Search PubMed.
- M. Hunger, Catal. Rev. - Sci. Eng., 1997, 39, 345–393 CAS.
- T. Miyoshi, K. Takegoshi and T. Terao, J. Magn. Reson., 1997, 125, 383–384 CrossRef CAS.
- T. Deuchande, O. Breton, J. Haedelt and E. Hughes, J. Magn. Reson., 2006, 183, 178–182 CrossRef CAS.
- E. J. Munson, D. B. Ferguson, A. A. Kheir and J. F. Haw, J. Catal., 1992, 136, 504–509 CrossRef CAS.
- E. J. Munson, D. K. Murray and J. F. Haw, J. Catal., 1993, 141, 733–736 CrossRef CAS.
- J. F. Haw, P. W. Goguen, T. Xu, T. W. Skloss, W. G. Song and Z. K. Wang, Angew. Chem., Int. Ed., 1998, 37, 948–949 CrossRef CAS.
- T. Xu, D. H. Barich, P. W. Goguen, W. G. Song, Z. K. Wang, J. B. Nicholas and J. F. Haw, J. Am. Chem. Soc., 1998, 120, 4025–4026 CrossRef CAS.
- J. F. Haw, J. B. Nicholas, W. G. Song, F. Deng, Z. K. Wang, T. Xu and C. S. Heneghan, J. Am. Chem. Soc., 2000, 122, 4763–4775 CrossRef CAS.
- D. M. McCann, D. Lesthaeghe, P. W. Kletnieks, D. R. Guenther, M. J. Hayman, V. Van Speybroeck, M. Waroquier and J. F. Haw, Angew. Chem., Int. Ed., 2008, 47, 5179–5182 CrossRef CAS.
- M. C. Xu, K. D. M. Harris, J. M. Thomas and D. E. W. Vaughan, ChemPhysChem, 2007, 8, 1311–1313 CrossRef CAS.
- M. C. Xu, K. D. M. Harris and J. M. Thomas, J. Am. Chem. Soc., 2008, 130, 5880–5882 CrossRef CAS.
- J. F. Haw, Top. Catal., 1999, 8, 81–86 CrossRef CAS.
- G. W. Haddix, A. T. Bell and J. A. Reimer, J. Phys. Chem., 1989, 93, 5859–5865 CrossRef CAS.
- M. S. Went and J. A. Reimer, J. Am. Chem. Soc., 1992, 114, 5768–5775 CrossRef CAS.
- A. Bendada, G. Chinchen, N. Clayden, B. T. Heaton, J. A. Iggo and C. S. Smith, Catal. Today, 1991, 9, 129–136 CrossRef CAS.
- M. Hunger and T. Horvath, J. Chem. Soc., Chem. Commun., 1995, 1423–1424 RSC.
- C. Keeler, J. C. Xiong, H. Lock, S. Dec, T. Tao and G. E. Maciel, Catal. Today, 1999, 49, 377–383 CrossRef CAS.
- M. Seiler, U. Schenk and M. Hunger, Catal. Lett., 1999, 62, 139–145 CrossRef CAS.
- W. Wang, Y. J. Jiang and M. Hunger, Catal. Today, 2006, 113, 102–114 CrossRef CAS.
- Y. J. Jiang, J. Huang, V. R. R. Marthala, Y. S. Ooi, J. Weitkamp and M. Hunger, Microporous Mesoporous Mater., 2007, 105, 132–139 CrossRef CAS.
- W. Wang, M. Seiler, I. I. Ivanova, U. Sternberg, J. Weitkamp and M. Hunger, J. Am. Chem. Soc., 2002, 124, 7548–7554 CrossRef CAS.
- W. Happer, E. Miron, S. Schaefer, D. Schreiber, W. A. Vanwijngaarden and X. Zeng, Phys. Rev. A, 1984, 29, 3092–3110 CrossRef CAS.
- D. Raftery, H. Long, T. Meersmann, P. J. Grandinetti, L. Reven and A. Pines, Phys. Rev. Lett., 1991, 66, 584–587 CrossRef CAS.
- D. Raftery, E. MacNamara, G. Fisher, C. V. Rice and J. Smith, J. Am. Chem. Soc., 1997, 119, 8746–8747 CrossRef CAS.
- I. L. Mourdrakovski, V. V. Terskikh, C. I. Ratcliffe, J. A. Ripmeester, L. Q. Wang, Y. Shin and G. J. Exarhos, J. Phys. Chem. B, 2002, 106, 5938–5946 CrossRef.
- A. Nossov, F. Guenneau, M. A. Springuel-Huet, E. Haddad, V. Montouillout, B. Knott, F. Engelke, C. Fernandez and A. Gedeon, Phys. Chem. Chem. Phys., 2003, 5, 4479–4483 RSC.
- W. P. Zhang, C. I. Ratcliffe, I. L. Moudrakovski, C. Y. Mou and J. A. Ripmeester, Anal. Chem., 2005, 77, 3379–3382 CrossRef CAS.
- Y. Liu, W. P. Zhang, S. J. Xie, L. Xu, X. W. Han and X. H. Bao, J. Phys. Chem. B, 2008, 112, 1226–1231 CrossRef CAS.
- Y. Liu, W. P. Zhang, Z. C. Liu, S. T. Xu, Y. D. Wang, Z. K. Xie, X. W. Han and X. H. Bao, J. Phys. Chem. C, 2008, 112, 15375–15381 CAS.
- L. Itani, Y. Liu, W. P. Zhang, K. N. Bozhilov, L. Delmotte and V. Valtchev, J. Am. Chem. Soc., 2009, 131, 10127–10139 CrossRef CAS.
- T. Meersmann, J. W. Logan, R. Simonutti, S. Caldarelli, A. Comotti, P. Sozzani, L. G. Kaiser and A. Pines, J. Phys. Chem. A, 2000, 104, 11665–11670 CrossRef CAS.
- S. Anala, G. E. Pavlovskaya, P. Pichumani, T. J. Dieken, M. D. Olsen and T. Meersmann, J. Am. Chem. Soc., 2003, 125, 13298–13302 CrossRef CAS.
- J. F. Haw, Phys. Chem. Chem. Phys., 2002, 4, 5431–5441 RSC.
- W. P. Zhang, X. H. Bao, X. W. Guo and X. S. Wang, Catal. Lett., 1999, 60, 89–94 CrossRef CAS.
- W. P. Zhang, D. Ma, X. W. Han, X. M. Liu, X. H. Bao, X. W. Guo and X. S. Wang, J. Catal., 1999, 188, 393–402 CrossRef CAS.
- D. Ma, Y. Y. Shu, X. W. Han, X. M. Liu, Y. D. Xu and X. H. Bao, J. Phys. Chem. B, 2001, 105, 1786–1793 CrossRef CAS.
- J. F. Haw, M. B. Hall, A. E. Alvarado-Swaisgood, E. J. Munson, Z. Y. Lin, L. W. Beck and T. Howard, J. Am. Chem. Soc., 1994, 116, 7308–7318 CrossRef CAS.
- C. Paze, A. Zecchina, S. Spera, G. Spano and F. Rivetti, Phys. Chem. Chem. Phys., 2000, 2, 5756–5760 RSC.
- A. Simperler, R. G. Bell and M. W. Anderson, J. Phys. Chem. B, 2004, 108, 7142–7151 CrossRef CAS.
- M. Hunger, Solid State Nucl. Magn. Reson., 1996, 6, 1–29 CrossRef CAS.
- D. Freude, M. Hunger and H. Pfeifer, Chem. Phys. Lett., 1982, 91, 307–310 CrossRef CAS.
- V. R. R. Marthala, W. Wang, J. Jiao, Y. J. Jiang, J. Huang and M. Hunger, Microporous Mesoporous Mater., 2007, 99, 91–97 CrossRef CAS.
- J. Huang, Y. Jiang, V. R. R. Marthala, B. Thomas, E. Romanova and M. Hunger, J. Phys. Chem. C, 2008, 112, 3811–3818 CAS.
- M. C. Xu, W. Wang, M. Seiler, A. Buchholz and M. Hunger, J. Phys. Chem. B, 2002, 106, 3202–3208 CrossRef CAS.
- J. Xu, A. M. Zheng, J. Yang, Y. C. Su, J. Q. Wang, D. L. Zeng, M. J. Zhang, C. H. Ye and F. Deng, J. Phys. Chem. B, 2006, 110, 10662–10671 CrossRef CAS.
- A. M. Zheng, H. L. Zhang, L. Chen, Y. Yue, C. H. Ye and F. Deng, J. Phys. Chem. B, 2007, 111, 3085–3089 CrossRef CAS.
- J. H. Lunsford, W. P. Rothwell and W. Shen, J. Am. Chem. Soc., 1985, 107, 1540–1547 CrossRef CAS.
- H. M. Kao and C. P. Grey, Chem. Phys. Lett., 1996, 259, 459–464 CrossRef CAS.
- H. M. Kao and C. P. Grey, J. Am. Chem. Soc., 1997, 119, 627–628 CrossRef CAS.
- J. H. Lunsford, Top. Catal., 1997, 4, 91–98 CrossRef CAS.
- B. Y. Zhao, H. J. Pan and J. H. Lunsford, Langmuir, 1999, 15, 2761–2765 CrossRef CAS.
- H. M. Kao, H. M. Liu, J. C. Jiang, S. H. Lin and C. P. Grey, J. Phys. Chem. B, 2000, 104, 4923–4933 CrossRef CAS.
- D. Ma, X. W. Han, S. J. Xie, X. H. Bao, H. B. Hu and S. C. F. Au-Yeung, Chem.–Eur. J., 2002, 8, 162–170 CrossRef CAS.
- J. Q. Zhuang, Z. M. Yan, X. M. Liu, X. C. Liu, X. W. Han, X. H. Bao and U. Mueller, Catal. Lett., 2002, 83, 87–91 CrossRef CAS.
- W. P. Zhang, X. W. Han, X. M. Liu and X. H. Bao, J. Mol. Catal. A: Chem., 2003, 194, 107–113 CrossRef CAS.
- J. Q. Zhuang, D. Ma, Z. M. Yan, F. Deng, X. M. Liu, X. W. Han, X. H. Bao, X. W. Liu, X. W. Guo and X. S. Wang, J. Catal., 2004, 221, 670–673 CrossRef CAS.
- W. Hu, Q. Luo, Y. C. Su, L. Chen, Y. Yue, C. H. Ye and F. Deng, Microporous Mesoporous Mater., 2006, 92, 22–30 CrossRef CAS.
- Q. Luo, F. Deng, Z. Y. Yuan, J. Yang, M. J. Zhang, Y. Yue and C. H. Ye, J. Phys. Chem. B, 2003, 107, 2435–2442 CrossRef CAS.
- G. Yang, J. Q. Zhuang, D. Ma, X. J. Lan, L. J. Zhou, X. C. Liu, X. W. Han and X. H. Bao, J. Mol. Struct., 2008, 882, 24–29 CrossRef CAS.
- J. Guan, X. J. Li, G. Yang, W. P. Zhang, X. C. Liu, X. W. Han and X. H. Bao, J. Mol. Catal. A: Chem., 2009, 310, 113–120 CrossRef CAS.
- L. M. Peng, P. J. Chupas and C. P. Grey, J. Am. Chem. Soc., 2004, 126, 12254–12255 CrossRef CAS.
- L. Baltusis, J. S. Frye and G. E. Maciel, J. Am. Chem. Soc., 1986, 108, 7119–7120 CrossRef CAS.
- E. F. Rakiewicz, A. W. Peters, F. Wormsbecher, K. J. Sutovich and K. T. Mueller, J. Phys. Chem. B, 1998, 102, 2890–2896 CrossRef CAS.
- K. J. Sutovich, A. W. Peters, E. F. Rakiewicz, R. F. Wormsbecher, S. M. Mattingly and K. T. Mueller, J. Catal., 1999, 183, 155–158 CrossRef CAS.
- Q. Zhao, W. H. Chen, S. J. Huang, Y. C. Wu, H. K. Lee and S. B. Liu, J. Phys. Chem. B, 2002, 106, 4462–4469 CrossRef CAS.
- S. J. Huang, Q. Zhao, W. H. Chen, X. W. Han, X. H. Bao, P. S. Lo, H. K. Lee and S. B. Liu, Catal. Today, 2004, 97, 25–34 CrossRef CAS.
- J. P. Osegovic and R. S. Drago, J. Phys. Chem. B, 2000, 104, 147–154 CrossRef CAS.
- H. M. Kao, C. Y. Yu and M. C. Yeh, Microporous Mesoporous Mater., 2002, 53, 1–12 CAS.
- A. M. Zheng, S. J. Huang, W. H. Chen, P. H. Wu, H. L. Zhang, H. K. Lee, L. C. de Menorval, F. Deng and S. B. Liu, J. Phys. Chem. A, 2008, 112, 7349–7356 CrossRef CAS.
- A. M. Zheng, H. L. Zhang, X. Lu, S. B. Liu and F. Deng, J. Phys. Chem. B, 2008, 112, 4496–4505 CrossRef CAS.
- G. E. Maciel, J. F. Haw, I. S. Chuang, B. L. Hawkins, T. A. Early, D. R. McKay and L. Petrakis, J. Am. Chem. Soc., 1983, 105, 5529–5535 CrossRef CAS.
- J. F. Haw, J. H. Zhang, K. Shimizu, T. N. Venkatraman, D. P. Luigi, W. G. Song, D. H. Barich and J. B. Nicholas, J. Am. Chem. Soc., 2000, 122, 12561–12570 CrossRef CAS.
- J. A. Ripmeester, J. Am. Chem. Soc., 1983, 105, 2925–2927 CrossRef CAS.
- T. Xu, E. J. Munson and J. F. Haw, J. Am. Chem. Soc., 1994, 116, 1962–1972 CrossRef CAS.
- A. I. Biaglow, R. J. Gorte, G. T. Kokotailo and D. White, J. Catal., 1994, 148, 779–786 CrossRef CAS.
- T. Xu, P. D. Torres, L. W. Beck and J. F. Haw, J. Am. Chem. Soc., 1995, 117, 8027–8028 CrossRef CAS.
- J. Yang, M. J. Janik, D. Ma, A. M. Zheng, M. J. Zhang, M. Neurock, R. J. Davis, C. H. Ye and F. Deng, J. Am. Chem. Soc., 2005, 127, 18274–18280 CrossRef CAS.
- S. H. Li, A. M. Zheng, Y. C. Su, H. L. Zhang, L. Chen, J. Yang, C. H. Ye and F. Deng, J. Am. Chem. Soc., 2007, 129, 11161–11171 CrossRef CAS.
- L. S. Wang, L. X. Tao, M. S. Xie, G. F. Xu, J. S. Huang and Y. D. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- Y. D. Xu and L. W. Lin, Appl. Catal., A, 1999, 188, 53–67 CrossRef CAS.
- Y. D. Xu, X. H. Bao and L. W. Lin, J. Catal., 2003, 216, 386–395 CrossRef CAS.
- Z. R. Ismagilov, E. V. Matus and L. T. Tsikoza, Energy Environ. Sci., 2008, 1, 526–541 CAS.
- Y. Y. Shu, D. Ma, L. Y. Xu, Y. D. Xu and X. H. Bao, Catal. Lett., 2000, 70, 67–73 CrossRef CAS.
- D. Ma, F. Deng, R. Q. Fu, X. W. Dan and X. H. Bao, J. Phys. Chem. B, 2001, 105, 1770–1779 CrossRef CAS.
- D. Ma, X. W. Han, D. H. Zhou, Z. M. Yan, R. Q. Fu, Y. Xu, X. H. Bao, H. B. Hu and S. C. F. Au-Yeung, Chem.–Eur. J., 2002, 8, 4557–4561 CrossRef CAS.
- D. Ma, W. P. Zhang, Y. Y. Shu, X. M. Liu, Y. D. Xu and X. H. Bao, Catal. Lett., 2000, 66, 155–160 CrossRef CAS.
- H. Zheng, D. Ma, X. H. Bao, J. Z. Hu, J. H. Kwak, Y. Wang and C. H. F. Peden, J. Am. Chem. Soc., 2008, 130, 3722–3723 CrossRef CAS.
- J. Z. Hu, J. H. Kwak, Y. Wang, C. H. F. Peden, H. Zheng, D. Ma and X. Bao, J. Phys. Chem. C, 2009, 113, 2936–2942 CAS.
- D. Ma, Y. Y. Shu, W. P. Zhang, X. W. Han, Y. D. Xu and X. H. Bao, Angew. Chem., Int. Ed., 2000, 39, 2928–2931 CrossRef CAS.
- J. Yang, D. Ma, F. Deng, Q. Luo, M. J. Zhang, X. H. Bao and C. H. Ye, Chem. Commun., 2002, 3046–3047 RSC.
- H. Zheng, D. Ma, X. M. Liu, W. P. Zhang, X. W. Han, Y. D. Xu and X. H. Bao, Catal. Lett., 2006, 111, 111–114 CrossRef CAS.
- Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef.
- R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef CAS.
- R. H. Grubbs, S. J. Miller and G. C. Fu, Acc. Chem. Res., 1995, 28, 446–452 CrossRef CAS.
- J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS.
- S. L. Liu, S. J. Huang, W. J. Xin, J. Bai, S. J. Xie and L. Y. Xu, Catal. Today, 2004, 93–95, 471–476 CrossRef CAS.
- X. J. Li, W. P. Zhang, S. L. Liu, L. Y. Xu, X. W. Han and X. H. Bao, J. Catal., 2007, 250, 55–66 CrossRef CAS.
- X. J. Li, W. P. Zhang, S. L. Liu, L. Y. Xu, X. W. Han and X. H. Bao, J. Phys. Chem. C, 2008, 112, 5955–5960 CAS.
- X. J. Li, W. P. Zhang, S. L. Liu, X. W. Han, L. Y. Xu and X. H. Bao, J. Mol. Catal. A: Chem., 2006, 250, 94–99 CrossRef CAS.
- A. Omegna, M. Vasic, J. A. van Bokhoven, G. Pirngruber and R. Prins, Phys. Chem. Chem. Phys., 2004, 6, 447–452 RSC.
- A. Abraham, S. H. Lee, C. H. Shin, S. B. Hong, R. Prins and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2004, 6, 3031–3036 RSC.
- X. Li, A. M. Zheng, J. Guan, X. W. Han, W. P. Zhang and X. H. Bao, Catal. Lett., 2010, 138, 116–123 CrossRef CAS.
- J. Handzlik, J. Ogonowski, J. Stoch and M. Mikolajczyk, Catal. Lett., 2005, 101, 65–69 CrossRef CAS.
- H. Aritani, O. Fukuda, T. Yamamoto, T. Tanaka and S. Imamura, Chem. Lett., 2000, 66–67 CrossRef CAS.
- J. C. Mol, Catal. Today, 1999, 51, 289–299 CrossRef CAS.
- X. J. Li, W. P. Zhang, X. Li, S. L. Liu, H. J. Huang, X. W. Han, L. Y. Xu and X. H. Bao, J. Phys. Chem. C, 2009, 113, 8228–8233 CAS.
- J. Weitkamp and S. Maixner, Zeolites, 1987, 7, 6–8 CrossRef CAS.
- J. L. White, N. D. Lazo, B. R. Richardson and J. F. Haw, J. Catal., 1990, 125, 260–263 CrossRef CAS.
- A. Gutsze, J. P. Lange, H. G. Karge and J. Allgeier, J. Catal., 1988, 113, 525–528 CrossRef CAS.
- H. J. Huang, X. C. Liu, S. L. Liu, X. M. Liu, L. Y. Xu, X. W. Han, W. P. Zhang and X. H. Bao, Chin. J. Catal., 2010, 31, 186–190 CAS.
- X. Li, J. Guan, A. M. Zheng, D. H. Zhou, X. W. Han, W. P. Zhang and X. H. Bao, J. Mol. Catal. A: Chem., 2010, 330, 99–106 CrossRef CAS.
- G. N. Vayssilov, Catal. Rev. - Sci. Eng., 1997, 39, 209–251 CAS.
- S. Bordiga, F. Bonino, A. Damin and C. Lamberti, Phys. Chem. Chem. Phys., 2007, 9, 4854–4878 RSC.
- S. B. Kumar, S. P. Mirajkar, G. C. G. Pais, P. Kumar and R. Kumar, J. Catal., 1995, 156, 163–166 CrossRef CAS.
- M. A. Uguina, D. P. Serrano, R. Sanz, J. L. G. Fierro, M. Lopez-Granados and R. Mariscal, Catal. Today, 2000, 61, 263–270 CrossRef CAS.
- J. Q. Zhuang, D. Ma, Z. M. Yan, X. M. Liu, X. W. Han, X. H. Bao, Y. H. Zhang, X. W. Guo and X. S. Wang, Appl. Catal., A, 2004, 258, 1–6 CrossRef CAS.
- J. Q. Zhuang, G. Yang, D. Ma, X. J. Lan, X. M. Liu, X. W. Han, X. H. Bao and U. Mueller, Angew. Chem., Int. Ed., 2004, 43, 6377–6381 CrossRef CAS.
- S. C. Laha and R. Kumar, J. Catal., 2001, 204, 64–70 CrossRef CAS.
- G. Bellussi, A. Carati, M. G. Clerici, G. Maddinelli and R. Millini, J. Catal., 1992, 133, 220–230 CrossRef CAS.
-
J. March, Advanced Organic Chemistry: Reaction, Mechanisms and Structure, McGraw-Hill, New York, 1977, p. 804 Search PubMed.
- J. Q. Zhuang, X. W. Han, X. H. Bao and U. Mueller, J. Catal., 2009, 267, 177–180 CrossRef CAS.
- M. V. Luzgin, A. G. Stepanov, S. S. Arzumanov, V. A. Rogov, V. N. Parmon, W. Wang, M. Hunger and D. Freude, Chem.–Eur. J., 2006, 12, 457–465 CrossRef.
- A. G. Stepanov, V. N. Parmon and D. Freude, Kinet. Catal., 2007, 48, 521–534 CrossRef CAS.
- A. G. Stepanov, M. V. Luzgin, S. S. Arzumanov, H. Ernst and D. Freude, J. Catal., 2002, 211, 165–172 CAS.
- A. G. Stepanov, M. V. Luzgin and V. N. Sidelnikov, Catal. Lett., 2002, 78, 153–156 CrossRef CAS.
- M. V. Luzgin, S. S. Arzumanov, V. P. Shmachkova, N. S. Kotsarenko, V. A. Rogov and A. G. Stepanov, J. Catal., 2003, 220, 233–239 CrossRef CAS.
- A. G. Stepanov, S. S. Arzurnanov, M. V. Luzgin, H. Ernst, D. Freude and V. N. Parmon, J. Catal., 2005, 235, 221–228 CrossRef CAS.
- M. V. Luzgin, V. A. Rogov, S. S. Arzumanov, A. V. Toktarev, A. G. Stepanov and V. N. Parmon, Angew. Chem., Int. Ed., 2008, 47, 4559–4562 CrossRef CAS.
- Z. N. Ma, Y. Zou, W. M. Hua, H. Y. He and Z. Gao, Top. Catal., 2005, 35, 141–153 CrossRef CAS.
- M. C. Xu, W. Wang and M. Hunger, Chem. Commun., 2003, 722–723 RSC.
- J. Huang, Y. J. Jiang, V. R. R. Marthala, W. Wang, B. Sulikowski and M. Hunger, Microporous Mesoporous Mater., 2007, 99, 86–90 CrossRef CAS.
- J. Huang, Y. J. Jiang, V. R. R. Marthala, Y. S. Ooi and M. Hunger, ChemPhysChem, 2008, 9, 1107–1109 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.