Applications of vectorized gold nanoparticles to the diagnosis and therapy of cancer
Audrey
Llevot
a and
Didier
Astruc
*b
aENSCPB, Université Bordeaux 1, 33607 Pessac, France
bInstitut des Sciences Moléculaires, UMR CNRS No5255, Université Bordeaux 1, 33405 Talence Cedex, France. E-mail: d.astruc@ism.u-bordeaux1.fr
First published on 22nd July 2011
Abstract
This critical review focuses on the anti-cancer fight using gold nanoparticles (AuNPs) functionalized with chemotherapeutic drugs in so-called “complexes” (supramolecular assemblies) and “conjugates” (covalent assemblies) as vectors. There is a considerable body of recent literature on various tumor-imaging techniques using the surface plasmon band (SPB) and the “passive” and “active” vectorization of anti-cancer drugs. This article reviews the main concepts and the most recent literature data with emphasis on AuNP preparation, cytotoxicities and use in selective targeting of cancer cells with over-expressed receptors for diagnosis and therapy (108 references).
 Audrey Llevot | Audrey Llevot studied in Bayonne where she attended the “classes préparatoires” of physics and chemistry for two years before her admission at the National Superior School of Biology and Physics of Bordeaux. Presently in her third year, she chose the polymer engineering section and passed a Master degree directed towards research at the University of Bordeaux 1 in the area of polymers and colloids. |
 Didier Astruc | Didier Astruc is Professor of Chemistry at the University Bordeaux 1 and Member of the Institut Universitaire de France. He was born in Versailles and studied in Rennes, Britanny, where he passed his PhD under the guidance of René Dabard before a post-doc at MIT with Dick Schrock. In 1990–1991, he spent a sabbatical year in U.C. Berkeley with K. Peter C. Vollhardt. His research interests are in inorganic chemistry and nanosciences and their applications in molecular recognition, molecular electronics, catalysis and nanomedicine. http://astruc.didier.free.fr/welcome.htm |
1. Introduction
Cancer is one of the main causes of mortality all over the world.1 It is characterized by the uncontrolled proliferation of cells that find their origin in genetic mutations. The main treatment is surgery that is limited, however, to accessible tumors. Chemotherapeutic drugs are also used, but their action provokes damageable side effects, because it is not limited to cancerous cells. The drugs could be selectively transported into the cancerous cells using vectors in order to avoid this problem. Therefore the use of nanotechnologies in the medical field is growing exponentially.2Nanoparticles that are most used for cancer nanotechnology include polymers, dendrimers, liposomes, perfluorocarbons, quantum dots, iron oxides, nanotubes, nanowires and gold nanoparticles (AuNPs).3 In particular AuNPs have been the subject of considerable recent studies. It has been shown that they can selectively recognize cancer cells.4 Their properties render them increasingly attractive vectors of diagnostic and chemotherapeutic substances.5–10 Their well-known optical absorption in the visible region (surface Plasmon band, SPB), first scientifically studied by Faraday, renders them ideal nano-objects for medical imaging and even for photothermal therapy. This short review is dedicated to the applications of functional vectorized AuNPs for cancer diagnosis and treatment.2. Vectorization
2.1. Need and characteristics of vectors
Tumors include regions that are highly vascularized to supply nutrients and oxygen leading to tumor growth (angiogenesis). Nanoparticles can accumulate by diffusion or convection across the leaky permeable tumor vasculature. Passive targeting using in particular polyethylene glycol can also be achieved by retention in the tumor interstitium (enhanced permeability and retention, EPR). Drug vectors should not only fit physiological and physicochemical properties for transport into the interstitium, but also take into account the multi-drug resistance at the cellular level.11–13 A good vector must be soluble in aqueous media, be directed solely towards cancerous cells, and its degradation products should not be toxic for the organism.132.2. Major anti-cancer drugs
The diterpenoid taxoid (or taxane), natural product paclitaxel (taxol) and semi-synthetic analog docetaxel (taxoter) are the most important anti-cancer drugs presently used (Fig. 1).13 Such taxoids bind to a tubulin polymer, promoting its polymerization, which leads to arrest of the cell cycle and to apoptosis. These taxoids are also administrated in combination with drugs functioning according to a distinct mechanism such as cis-platin (cis-PtCl2(NH3)2) and doxorubicin (vide infra). Other new discovered anti-cancer natural products having a taxoid-like mechanism include epothilones, discodermolide, eleutherobin, laulimalide and peloruside. Severe limitations of the taxoid-type drugs, however, are their poor water solubility, non-selective toxicity to tumor cells and inactivity against drug-resistant cell lines.13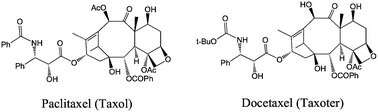 | ||
| Fig. 1 Structures of the two main taxoids used in cancer chemotherapy. | ||
The solubility of these taxoids can be increased upon addition of a surfactant, Cremophor EL (a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 mixture of polyethoxylated castor oil and ethanol), but it can provoke allergy problems due to histamine release and hypersensitivity reactions.13 In a newer formulation, it is bound to albumin. The vector should increase the solubility in the organism. The lack of selectivity is another major problem. Indeed, it can have toxic effects on the non-targeted organs and provoke ulcers, cardiac anomalies, neurotoxicity and suppression of the bone marrow. The doses provided are then restricted, and the tumor is not totally suppressed.14 Then, the vector would encapsulate the drug during its circulation in the blood and would free it only near the cancer cells. Thus, a taxoid associated with a vector is ten times more efficient than the taxoid alone.13
∶1 mixture of polyethoxylated castor oil and ethanol), but it can provoke allergy problems due to histamine release and hypersensitivity reactions.13 In a newer formulation, it is bound to albumin. The vector should increase the solubility in the organism. The lack of selectivity is another major problem. Indeed, it can have toxic effects on the non-targeted organs and provoke ulcers, cardiac anomalies, neurotoxicity and suppression of the bone marrow. The doses provided are then restricted, and the tumor is not totally suppressed.14 Then, the vector would encapsulate the drug during its circulation in the blood and would free it only near the cancer cells. Thus, a taxoid associated with a vector is ten times more efficient than the taxoid alone.13
Doxorubicin (DOX), an anthracycline antibiotic, is another chemotherapeutic substance, commonly used in the treatment of a wide variety of cancers. It is related to the natural product daunomycin (or daunorubicin) used especially against acute myeloid leukemia and acute lymphatic leukemia. These drugs are DNA intercalating agents that rapidly bind the nuclear structures of the cell and block the DNA and RNA syntheses as well as the proliferation of cells. When DOX is delivered alone, it has a number of undesirable side effects, and in particular life-threatening heart damage.
Fluorouracil (Fig. 2), also called 5FU (anti-metabolite family), is an anti-cancer drug that is involved in the synthesis of nucleic acids. It is usually given to treat bowel, breast, skin, stomach and gullet (oesophagus) cancer. Its toxicity is also reduced when it is associated to a vector, and a larger amount can reach the cancer cells.
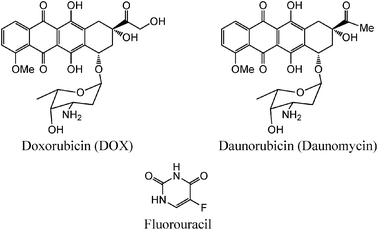 | ||
| Fig. 2 Structures of doxorubicin, daunorubicin and fluorouracil. | ||
2.3. Functioning of the vectors
The working principle of a vector consists in using the affinity of certain substrates for metabolite receptors. Indeed, cancer cells are subjected to higher metabolite rates compared to normal cells, leading to hypoxic environment inducing anaerobic metabolism that lowers the pH of cancer cells. Therefore cancer cells over-express receptors that attract primary metabolisms (peptides, polysaccharides, fatty acids, etc.). The association between the vector and the drug consists in binding the drug to a vector that recognizes the cancer cells (Fig. 3).13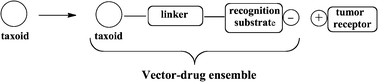 | ||
| Fig. 3 Formation of the drug–vector nanoconjugate. | ||
The vector bound to the drug must have a selective affinity for cancer cells, a good stability in blood circulation and not be toxic for normal cells. The bonding with the taxoid proceeds by an esterification or related reaction. It occurs on site C2′ (Fig. 4). The ester bond is easily hydrolysable or depredated by enzymes to free the drug.
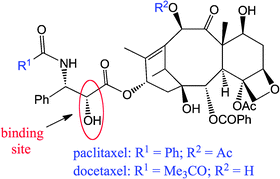 | ||
| Fig. 4 Binding site between the taxoid and the vector. | ||
The AuNPs and dendrimers are often used as vectors, because they allow increasing the blood circulation time. These vectors protect the drug from potential transformations in the organism, and they can be specific, biodegradable and non-toxic (“magic bullet”).
3. Synthesis of the AuNPs
The two most well known methods of synthesis are the citrate method reported by Turkevitch et al. in 195115 (and improved since then16) for large AuNPs up to 150 nm and the biphasic Schiffrin–Brust method reported in 1994 that allows the synthesis of small AuNPs including very small AuNPs (1–2 nm) for which the SPB is not observed.17,183.1. The citrate (Turkevitch) method
The citrate method uses tetrachloroauric acid (HAuIIICl4), AuIII being reduced by citrate in water at 100 °C to Au atoms that assemble in citrate-stabilized AuNPs ranging from 9 to 120 nm.15,16 The histogram of size distribution is determined by transmission electron microscopy (TEM), and the AuNP size depends on the temperature, concentration and reagents ratio. Recent modifications have improved the preparation procedure.16 It has been shown that the size of the AuNPs is independent of the absolute reagent concentrations if the latter are inferior to 1 mM, the optimum concentration being 0.8 mM. Then the dispersion is weaker, and the shape is better defined. The sizes of the AuNPs obtained in this way are between 12 and 40 nm. A new approach consists in adding traces of Ag in the mixture of citrate/HAuCl4 and to mix the reagents first at ambient temperature before heating up to the boiling point. Almost spherical AuNPs, between 5 and 30 nm are reproducibly obtained, and the nucleation step is accelerated.19 This citrate method is more and more often used, because the citrate ligands are conveniently substituted by biologically related thiol ligands.20 Thus, the peptides, proteins, DNA, dyes, etc. are functionalized with a terminal thiol group, then mixing with citrate-stabilized AuNPs displaces the citrate ligands that are substituted by the functional thiolate on the AuNP surface. The stabilization is due to the relatively strong gold–sulfur bond, this strength being related to the fact that both Au and S atoms are soft (diffuse d orbitals). Thus in most cases, the AuNPs are stabilized by thiolate ligands upon reactions of AuNP–citrate with thiols (the H atom of RSH is released during the synthesis). In relation with these seminal studies, it was shown that the inter-AuNP distance depends on the length of the alkyl chain of the alkylthiolate ligands.183.2. The Schiffrin–Brust method
The second popular procedure, the Schiffrin–Brust method,17 uses a biphasic toluene–water system under ambient conditions. First, the reaction of tetra(octyl)ammonium bromide with reduction of HAuCl4 leads to the transfer of the AuCl4− anion into the organic phase in which it forms an ion pair with the ammonium cation. Then, reduction of this AuCl4− salt by NaBH4 in the organic phase forms the AuNPs on which the surfactant stabilizer is replaced by the thiolate ligand upon reaction with the thiol. The diameter of the AuNPs synthesized by this method is usually between 1 and 3 nm, and their dispersity is rather low. The AuNPs are all the smaller as the thiol/gold ratio is larger, and their dispersity is all the smaller as NaBH4 is added more slowly. Some functional thiols can be introduced into AuNPs using this method, but their functional group must then be compatible with the aqueous medium and the reductive conditions.173.3. The seeding-growth method
The seeding-growth method is an old method that has been more recently renewed by controlling the size distribution to 10–15% and that allows synthesizing AuNPs with diameters between 5 and 40 nm. This size control is carried out by varying the seed-to-metal salt ratio. The step-by-step AuNP enlargement is more efficient than in the single-step seed growth.213.4. Other methods
A variety of other sulfur ligands such as xanthates, dithiocarbamates, disulfides, di- and trithiols, and resorcinarenes, polythioethers have been used to stabilize and synthesize AuNPs upon reaction with a AuIII source in the presence of a reductant, but they are generally not used for medicinal applications.20 Likewise, microemulsions, reversed micelles, surfactants, membranes and other amphiphiles are common stabilizers of AuNPs.203.5. Polymer and dendrimer stabilization of AuNPs
Various polymers and dendrimers were also shown to stabilize AuNPs. Dendrimers have been most used recently due to their precise nature and functions compared with polydisperse polymers. Stabilization of AuNPs by dendrimers, pioneered by Crooks group,22,23 proceeds either by encapsulating the AuNP if the dendrimer is large enough22–26 or upon surrounding the AuNP if the dendrimer is small (zeroth generation).23,24 In these cases, the addition of HAuCl4 to the dendrimer containing ligands leads to AuIII–dendrimer complexes, then further reduction with NaBH4 or other reductants provides the dendrimer-stabilized AuNPs of various sizes that are controlled by the reaction parameters.25,263.6. Non-spherical AuNPs
AuNPs are also known with non-spherical shapes such as rods, shells, cages, prisms, triangles, tetrapods, dogbones and cubes. For instance, Au nanocubes with surface pores were synthesized by redox reaction (galvanic method) between Ag nanocubes39,40 and HAuCl4, AuIII being a stronger oxidant than AgI.27Nanorods 30 were synthesized by various means including electrochemical deposition using polycarbonate,41 mica42 or alumina membrane templates,43 other electrochemical methods,31 bioreduction,32 photochemistry33 or seed mediation.30,34 The latter method has been developed by the Murphy group and is the most well-established one, whereby Au seeds are added to HAuCl4 and a weak reductant such as sodium ascorbate in the presence of hexadecyltrimethylammonium bromide.34–37Silver nitrate is also used.35
4. Functionalization of AuNPs
4.1. Functionalization of pre-coordinated thiolate ligands
Another possible method for the functionalization of AuNPs is the reaction of pre-functionalized thiolate ligands on AuNPs. The classic peptide type coupling has been used with dicyclohexylcarbodiimide (DCC). In another more recent example, bromoalkylthiolate–AuNPs44 can be transformed into azidoalkylthiolate ligands, and further “click” reactions with alkyne-terminated substrates can be achieved in high yield under carefully controlled conditions. The “click” reaction in this way is a convenient method to introduce polyethylene glycol (PEG) tethers and a variety of other functional groups.384.2. Functionalization by thiolate ligand substitution on AuNPs
If this is not the case, it is also possible to exchange alkylthiolate ligands with functional alkylthiolates upon reactions of the functional thiols with the alkylthiolate–AuNPs without AuNP size change at the core. This useful substitution reaction highlighted by Murray's group39 follows an associative mechanism whereby the hydrogen atom of the incoming thiol group is transferred onto the leaving alkylthiolate ligand at the AuNP surface.24–29 For instance, amine-terminated alkylthiolate ligands have been introduced using this procedure in AuNPs. In this way, oligonucleotides, peptides and polymers have been covalently bound to AuNPs.4.3. Synthesis of AuNP–oligonucleotide conjugates
The synthesis of AuNPs functionalized with synthetic oligonucleotides has been carried out by Mirkin's group upon reaction between alkanethiol-terminated oligonucleotides and citrate–AuNPs. The addition of NaCl to such a reaction medium shields charge repulsion, which allows a greater number of oligonucleotides to bind the AuNP surface, yielding a dense ligand layer. For instance approximately 250 oligonucleotides could be bound to the 15 nm AuNP surface.45–47 The AuNP–DNA bioconjugates bind complementary nucleic acids with a high affinity, for instance with binding constants that are as large as two orders of magnitude greater than the analogous oligonucleotides of the same sequence. They are also resistant to degradation by enzymes such as DNase and are able to enter a wide variety of cell types with potential applications in gene transfection.474.4. Functionalization of AuNPs with antibodies
AuNP–antibody conjugates have been used in immunohistochemistry for 40 years. These conjugates are synthesized by hydrophobic adsorption, ionic binding, using N-hydroxysuccinimide ester-mediated covalent linkage, immobilization with oligonucleotides, or by ligand binding to the AuNP surface if a thiol fragment has been attached to them. Another astute way consists in hybridization of AuNP–DNA conjugates with antibodies that have been conjugated to complementary oligonucleotides (Fig. 9).48 See Section 6.2 for their use.5. Advantages of the AuNPs for the vectorization
5.1. Biocompatibility
In order to be useful for cancer treatment, the AuNPs must be non-cytotoxic (i.e. non toxic for cells) for normal cells. This cytotoxicity has been much studied, and several parameters are involved: the concentration, the nature of the ligands bonded to the AuNP core and the targeted cells.| Reference | Size/nm | Shape | AuNP surface group | Cell line | Toxicity results |
|---|---|---|---|---|---|
| 50, 2005 | 3.5 ± 0.7 | Sphere | Lysine, poly(lysine) | RAW64.7 mice macrophages | 85% de viability of the cells after 72 h exposure to 100 μM AuNPs |
| 51, 2005 | 4, 12, 18 | Sphere | Citrate, cysteine, glucose, biotin, CTAB | K562 cells of human leukemia | To toxic AuNPs at concentrations of the order of μM |
| 49, 2004 | 2 | Sphere | Ammonium, carboxylic acid | COS-1 mammary cells, mammary red blood cells, Escherichia coli | Cationic AuNPs much more cytotoxic than anionic AuNPs of the same size |
| 52, 2006 | 65 ± 5, | Rod | CTAB, PEG | Hela cells | 80% of the cells die with 0.05 mM AuNPs covered with CTAB, only 10% die at 0.5 mM AuNPs covered with PEG |
| 11 ± 1 | |||||
| 53, 2007 | Rod | CTAB | KB cells of human tumor | Rapidly internalized AuNPs form aggregates but remain healthy | |
| 54, 2007 | 33 | Sphere | CTAB citrate | BHK cells baby hamster kidneys, Hep2G cancer cells of human liver, A549 cancer cells of human lungs | Not toxic for BHK21 and HepG2 cells, but toxic for A549 cells |
| 55, 2006 | 65 × 11 | Rod | Phosphatidylcholine | Hela cells | AuNPs modified with phosphatidylcholine are much less toxic than those covered with CTAB |
| 56, 2008 | 20 | Sphere | Citrate | Lung fibroblast | Decrease of cell proliferation |
| 57, 2007 | 18 | Sphere | Citrate | Hela cells | AuNPs do not modify gene expression and do not present cytotoxicity although they are internalized in cells |
| 58, 2010 | 13 ± 1 | Sphere | DNA | Pancreatic islet | Introduction of AuNP–DNA inside cells, no cytotoxicity |
| 59, 2010 | 34 ± 6 | Sphere | Keratinocyte | Low concentrations (about 5 ppm) AuNP can stimulate proliferation of keratines. Not cytotoxic till concentrations lower than 10 ppm ⇒ low concentrations can be used for biomedical applications | |
| 60, 2010 | 40 ± 15 | Rod | CTAB treated with HCl | Hela | 95% viability ⇒ cytotoxicity was due to CTAB only |
| 61, 2010 | 10 | Sphere | Citrate | Dendritic cells of mice bone marrow | No cytotoxicity but accumulation inside cells. Cytokynes secretion modified by internalization |
5.2. Detection by imaging: the surface plasmon band (SPB)
The color of AuNPs is related to their plasmon absorption leading to the surface plasmon band (SPB) usually around 520 nm and is long known.20,65 After Faraday' seminal work,65 the SPB has been subjected to theoretical interpretation by Mie.66 The SPB is due to the collective oscillation of free electrons of the conduction band at the surfaces of the AuNPs provoked by the electromagnetic field of the incoming light.20,66,67 It is influenced by the shape (vide infra), size, nature of the nanoparticles, the medium dielectric constant, the inter-particle distance (vide infra) and the temperature. For instance, when the AuNP size increases, the SPB shifts to a longer wavelength. As a consequence, the width of this SPB depends on the range of size distribution. The SPB is very useful for cancer diagnosis using various imaging techniques (vide infra) and even for cancer therapy using the photo-thermal technique for the destruction of cancer cells (vide infra).6–8The sensitivity of the SPB in the visible region to physical and chemical parameters is extremely useful for tumor imaging. Besides TEM, the main imaging techniques used are optical microscopy that involves light diffusion in a dark microscopic field and photo-thermal imaging, DIC or photo-acoustic microscopy, because light diffusion provokes warming of the neighborhood. Other techniques are fluorescence,76 optical coherence tomography, SPR multi-photon microscopy using a scanning laser microscope to measure the multi-photon increase of the AuNPs, when the plasmon frequency is in resonance with the laser light and X-ray micrography (Fig. 5).10 The association of these detection techniques with the functionalization of AuNPs makes a support for the detection of proteins, nucleic acids and other biomolecules. Altogether, these biodiagnostic methods represent a significant step forward in the fight against cancer.68,69
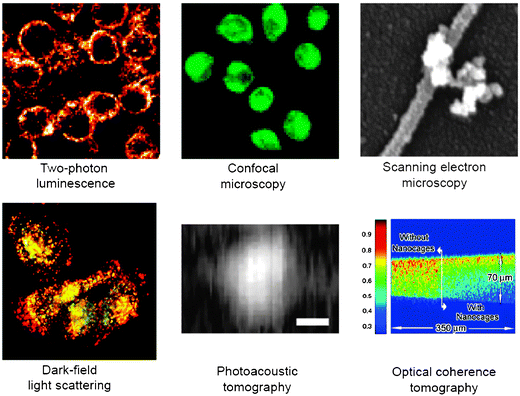 | ||
| Fig. 5 AuNP imaging using various techniques. Reprinted with permission of Dove Medical Press (ref. 3, Cai's group). | ||
5.3. Influence of the AuNP size and shape
The association of a drug to AuNP allows overtaking the systems of resistance in the cells. First, the nanometric size of the AuNPs allows them to diffuse through the interstitial tissues and to easily circulate in the blood vessels. Then, in the cases of normal or inflamed tissues, the molecules are removed from the tissues by the lymphatic system. This is not the case for the tumors for which the molecules flow in lymphatic capillaries through the endothelial cells, because cancerous tissues are more permeable, more heterogeneous and of larger size than normal ones. This phenomenon is called the Enhanced Permeability and Retention (EPR) effect and is used for passive vectorization, in particular with PEG.70In addition, the internalization of the AuNPs depends on their size and shape. Chan et al. have examined Hela cells and shown in 2006 that the internalization time of AuNPs measuring between 14 and 74 nm is independent of their size. However, this difference modifies the number of internalized particles.71
Two surface plasmon bands, a longitudinal SPB along the length and a transverse SPB along the width, are observed, their position varying with the nanorod aspect ratio (width/length). A great interest of the Au nanorod SPBs is that the longitudinal band is located in the near-infrared region, with position control at will by the aspect ratio, which allows penetration into tissues, whereas the bands in the visible region do not. This property has been largely exploited for cancer diagnosis and therapy in particular by the Murphy6 and El-Sayed8groups.
This very useful SPB absorption property of some AuNPs in the near infrared is shared by gold nanospheres made of a silica, microgel or polymer core surrounded by a gold shell. The SPB is controlled by the ratio between the core size and the thickness of the gold shell and can be tuned in the near-infrared region in which the tissues do not absorb.72–74 The seed technique is also mostly used, the shell thickness being controlled by the Au amount. The Halas group pioneered this area and showed that the SPB is all the more red shifted as the ratio between the shell thickness and the core size is smaller.75–77
6. AuNPs for the diagnosis of cancer
6.1. Use of the SPB in cancer diagnosis
AuNPs have long been used in cancer diagnosis, with the advantages over quantum dots and organic dyes of low toxicity, much better contrast than organic dyes and surface enhanced optical properties.12 The most frequent biodiagnostic approach involves the knowledge of the nucleotide sequences that is crucial for the diagnosis of genetic illnesses such as cancer. AuNP–oligonucleotide conjugates are used for such diagnosis due to their optical properties, an area for which Mirkin's group has carried out important and numerous contributions78–81 (Fig. 6).78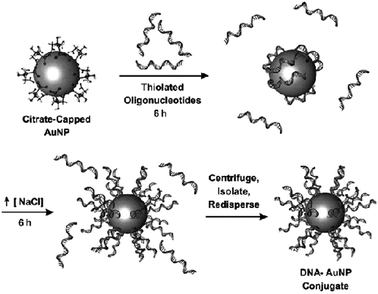 | ||
| Fig. 6 AuNP functionalization with DNA. Reprinted with permission of Dove Medical Press (ref. 3, Cai's group). | ||
The hybridization is not limited to the binding of the nucleotides to the targeted sequence, but consists in the formation of an extended polymeric network and is detected by colorimetry. Indeed, the AuNPs are red, but inter-AuNP nucleotide hybridization shortens the inter-AuNP distance, and the hybridized AuNPs become blue (Fig. 7).80 Moreover, the hybridized AuNP-nucleotides exhibit a lower transition temperature when they are denaturized. This feature allows us to differentiate imperfect targeted sequences and thus to detect sickness. Part of the AuNPs is functionalized with half of the complementary sequence of the targeted sequence, and the other part with the other half. When the AuNPs are aligned tail-to-tail, imperfections such as insertions are detected (Fig. 8).80
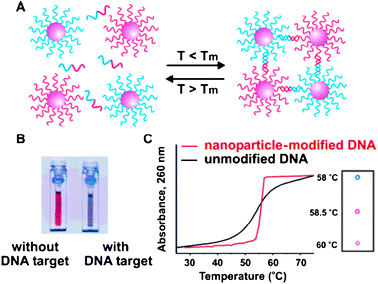 | ||
| Fig. 7 (A) Color change upon formation of a polymer–AuNP network. (B) Observation of the aggregation with naked eye or by UV-vis spectroscopy. Reprinted with permission of the American Association for the Advancement of Science (ref. 47, Mirkin's group). | ||
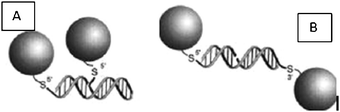 | ||
| Fig. 8 (A) Head-to-tail alignment. (B) Tail-to-tail alignment. Reprinted with permission of the American Association for the Advancement of Science (ref. 80, Mirkin's and Letsinger's groups). | ||
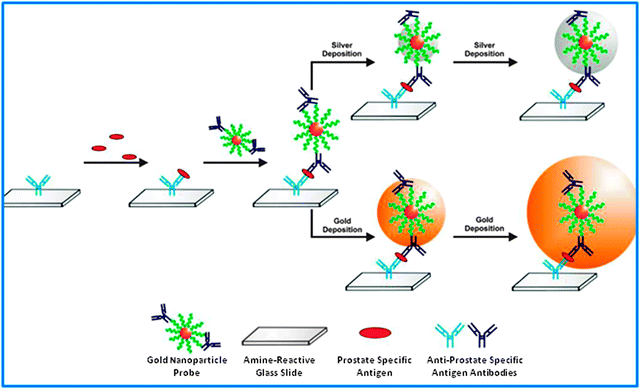 | ||
| Fig. 9 Use of antibody–AuNP conjugates for cancer diagnosis. Reprinted with permission of the American Chemical Society (ref. 80, Mirkin's group). | ||
The detection can be improved when the AuNPs are covered with silver. The addition of silver allows increasing the Surface Enhanced Raman Scattering Effect (SERS). A high sensitivity is obtained in this way.82
6.2. The use of AuNP–antibody conjugates
The expression of some antigens is important in cancers, and AuNPs functionalized with antibodies can then allow the diagnosis of cancer. Experiments using prostate-cancer-specific antigens (PSA) have been carried out by Mirkin et al. The AuNPs that are functionalized with antibodies for PSA are incubated with PSA antigens. The detection is carried out using a scanometric method. In the preceding section, it has been shown that the presence of silver allowed increasing the detection signal. Mirkin's group showed that a gold layer around the functionalized nanoparticle was more efficient than a silver layer, and the sensitivity is still increased.817. PEGylated AuNPs: a major vectorization improvement
Conventional AuNPs associated with drugs are rapidly removed by macrophages of organs such as lungs, bladder, liver and bone marrow. Their association with polymers is increased in this way, and they can reach other organs.12The improvement of chemotherapy requires the use of hydrophilic vectors. One of the solutions to make vectors hydrophilic is to modify their surface with polyethylene glycol (PEG, Fig. 10). PEGs bind AuNPs by coordination of thiolate or disulfide groups at their surface or by formation of copolymers. It can for instance contain pentaethylenehexamine termini. In synergy with the cations of the salts of the physiological conditions, it strengthens the formation, stabilization and lack of aggregation of the AuNPs. The half-life time of the AuNPs in blood is increased in this way, for instance one hour instead of a few minutes. Their stability depends on the length of the PEG, its molar fraction and the decrease of the AuNP core size. It also modifies the biodistribution.83
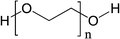 | ||
| Fig. 10 Structure of PEG. Molecular weights usually are 2000 or 5000 D. | ||
Paciotti's group has compared two vectors: Au–TNF-α and PT–Au–TNF-α.40. PT is a thiolated PEG and TNF (Tumor Necrosis Factor) is a labeling of tumors that also has therapeutic effects. The AuNPs used have been obtained by reduction of citrate and have a size of 33 nm. The PT and TNF share the AuNP binding sites. These bonds are independent. Cancer cells have been implanted at mice colon, and the vectors are injected in their tail. One group is sacrificed, and the liver and bladder are then collected. This animal protocol was reviewed and approved by the University of Maryland, College Park, IACUC committee (protocol number R-2001-3).84 The accumulation of AuNPs can then be observed upon analysis of the organ colors (Fig. 11). Indeed, aggregation of the AuNPs produces a black color, whereas they remain red when they are monodispersed. When the vector does not contain PT, the AuNPs are accumulated in liver and bladder already 5 minutes after injection. The accumulation is still present after one month, but the mice behavior is normal, and no toxicity is observed. When the vector is PT–Au–TNF-α, the AuNPs do not accumulate in liver and bladder, but they remain in a monodisperse form. Two hours after the injection, the vector is sequestered in the tumor. In the presence of PEG derivatives, the vector concentration progressively increases with time, whereas the concentration in blood and other organs progressively decreases. The PEG acts there as a hydrophilic blocker screening the Reticulo-Endothelial System (RES). The latter can then no longer inhibit the vector circulation.83
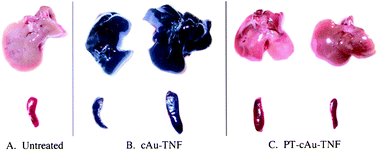 | ||
| Fig. 11 Mice bladder and liver treated with various vectors, 5 h after injection. Reprinted with permission of Taylor and Francis (ref. 84, Paciotti's group). | ||
Usually, the polycationic materials are cytotoxic. The AuNPs that are covered with PEG present a neutral surface that decreases this toxicity as well as the possibility of binding with the blood components.84
8. Use of the AuNP properties: passive vectorization
8.1. Protection of hydrophobic drugs within the thiolated layer of the AuNPs
The controlled delivery of drugs under their active forms is a key challenge. A possible solution resides in the incorporation of drugs in a non-covalent way inside the ligand shell surrounding the AuNP. This type of vector–drug ensemble is called a “complex”. The structure of thiolated AuNPs is similar to that of unimolecular micelles, with a hydrophobic interior and a hydrophilic surface. The thiolated zwitterionic layer in Fig. 12 allows us to encapsulate drugs and to deliver them efficiently. The number of encapsulated drug molecules depends on the AuNP size, its hydrophobicity and the structure of hydrophobic molecules.85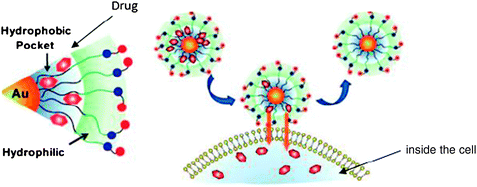 | ||
| Fig. 12 Delivery of the drug trapped inside the hydrophobic layer surrounding the AuNP. Reprinted with permission of the American Chemical Society (ref. 83, Rotello's group). | ||
The accumulation of AuNPs near cancer cells is occurring because of the EPR effect, and the vectorization is called a “passive” one. In 2008, Rotello has reported the synthesis and use of AuNPs having a core size of 2.5 nm encapsulating tamoxifen (TAF) and β-lapachone (LAP), two anti-cancer drugs. The toxicity results are presented in Fig. 13.
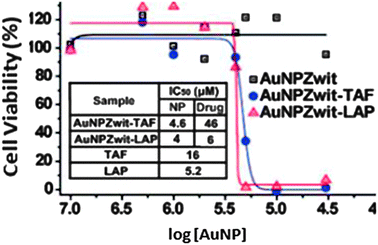 | ||
| Fig. 13 Cytotoxicity on MCF-7 cancer cells of AuNPs functionalized with anti-cancer drugs. Reprinted with permission of the American Chemical Society (ref. 83, Rotello's group). | ||
The delivery of drugs inside the cell proceeds by diffusion across the membrane, hence, the vector is not internalized inside the cell.85
8.2. Photoregulated drug delivery using AuNPs
This approach requires an outsider action, that of light as a stimulus, in order to deliver the drug. The drug is bound to the AuNP through a photosensitive bond such as that involving o-nitrobenzyl. The AuNP surface is thus functionalized both by photocleavable ligands and zwitterionic thiolated ligands providing biocompatibility, solubility and avoiding cell internalization. The accumulation of AuNPs near the cancer cells proceeds with the EPR effect. The o-nitrobenzyl group is stable under the light of the biological environment, but allows photo-cleavage when it is exposed to UV light at 365 nm, liberating the drug.69 Experiments have been carried out with 2 nm AuNPs conjugated with fluorouracil (Fig. 14). After irradiating for 10 minutes, 82% of the fluorouracil molecules have been delivered. Various conditions have been tested. In particular irradiation of the cells before or after incubation with functionalized AuNPs and the lifetime of the cells have been analyzed in all the cases. The data (Fig. 15) show that the cell lifetime decreases when the irradiation time increases, and the fluorouracil lifetime is indeed due to the irradiation of the AuNPs.83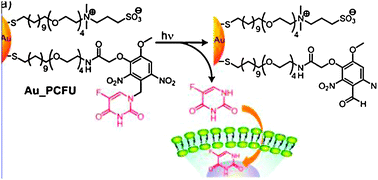 | ||
| Fig. 14 Photoregulation of fluorouracil. Reprinted with permission of the American Chemical Society (ref. 83, Rotello's group). | ||
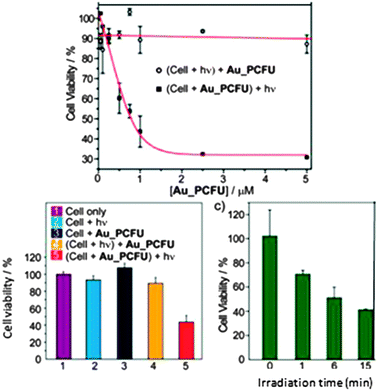 | ||
| Fig. 15 Cytotoxicity on MCF-7 cancer cells of AuNPs functionalized with fluorouracil. Reprinted with permission of the American Chemical Society (ref. 83, Rotello's group). | ||
8.3. Intracellular thiolated drug/glutathione ligand exchange
Glutathione (GSH) is used as an endogen agent for drug delivery. This mechanism is based on the difference between the intracellular glutathione concentration (1–10 nM) and the extracellular thiol concentration (cysteine: 8 μM and GSH: 2 μM). The thiolate/thiol exchange reactions between the thiolate AuNP ligands and intracellular GSH allow delivering the thiolated drug (Fig. 16).85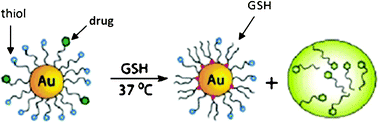 | ||
| Fig. 16 Intracellular ligand exchange between AuNP-coordinated thiolates and glutathione. Reprinted with permission of the American Chemical Society (ref. 85, Rotello's group). | ||
Rotello's group has shown that GSH was responsible for drug delivery. The AuNP surface charge and the layer structure around the AuNP are influential on this exchange.86
9. Bonding with cancer cells: active vectorization
9.1. Folate receptors
Folate receptors are glycophosphatidylinositols. They are membrane proteins that can bind amino acidsvia their terminal carbonyl group. They allow internalizing folic acids or components that are bound to folic acid by endocytosis. Folic acid (Fig. 17) is a component regulating the cell that participates in cell growth, genetic replication and protein synthesis. Their affinity for folate receptors is so important that even therapeutic agents that are little charged in folic acid can be easily captured.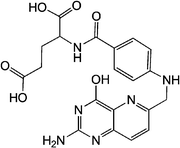 | ||
| Fig. 17 Structure of folic acid. | ||
On normal cells, there is no proliferation of folate receptors. On the other hand, they are over-expressed in the cases of cancer cells and in particular for ovarian, brain, breast and lung cancers. They become accessible via vascular epithelium only after the cells become cancerous. Dixit has carried out syntheses of PEGylated AuNPs, bound or not to folic acid, in order to compare their accumulation at the level of tumors, their binding to normal cells and the importance of folic acid as a specific recognition agent. 10 nm sized AuNPs are obtained by reduction using citrate, then they are suspended with Folate–NH–PEG–NH–thioctamide (F–PEG–T) on one hand and with mPEG–thioctamide (mPEG–T) on the other hand. The synthetic scheme is shown in Fig. 18. The vectors synthesized in this way are contacted with KB cells (cells of tumor cell line), on which the folate membrane receptors are over-expressed, and with WI 38 cells (normal cells). The accumulation of functionalized AuNPs is observed by TEM, and the pictures are shown in Fig. 19. The observation of Fig. 19B shows the presence of AuNPs that are internalized by cells when the vector is F–PEG–T. Indeed, 20% of the KB cells show internalization in the presence of F–PEG–T. However, the aggregation of AuNPs prevents the determination of their densities. Fig. 19C shows that the WI 38 cells cannot internalize AuNPs because of the lack of folate receptors. These experiments show that folic acid allows the specific recognition of cancer cells.87
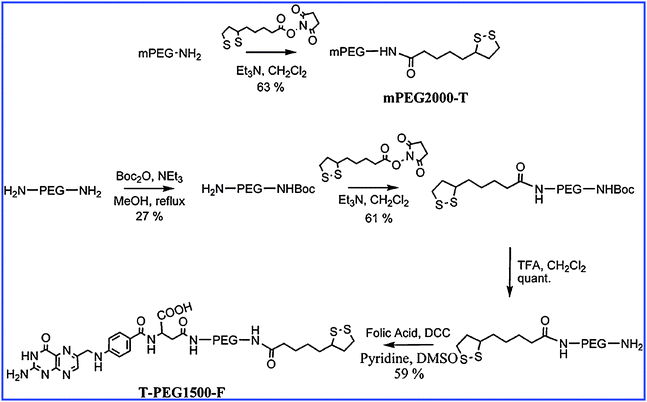 | ||
| Fig. 18 Synthesis of F–PEG–T and mPEG–T. Reprinted with permission of the American Chemical Society (ref. 86, Thompson's and Andres' group). | ||
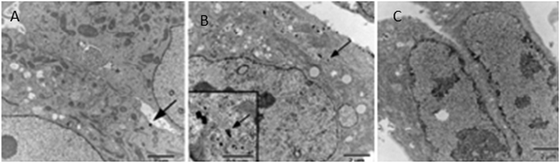 | ||
| Fig. 19 TEM images of cells incubated for 2 h with various AuNP conjugates. (A) KB cells with T–mPEG. (B) KB cells with T–PEG–F. (C) WI 38 cells with T–PEG–F. Reprinted with permission of the American Chemical Society (ref. 86, Thompson's and Andres' group). | ||
Very recently, other experiments using folic acid as the recognition agent have been reported. The goal was to verify that doxorubicin was specifically delivered to cancer cells.88 10 nm AuNPs were charged with PEG, folic acid and doxorubicin. The steps of the syntheses are shown in Fig. 20. The sequence: DOX–FOL–PEG–SHAuNPs was obtained in this way.
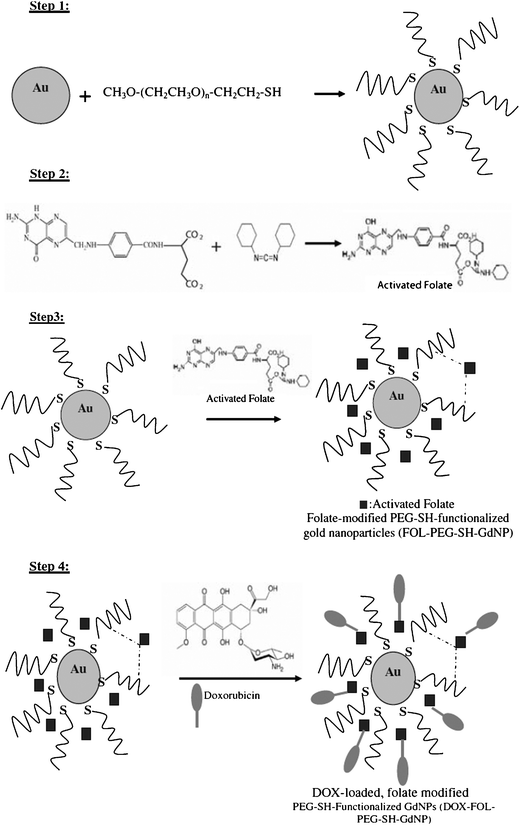 | ||
| Fig. 20 Synthesis of the conjugated AuNPs. Reprinted with permission of Springer (ref. 87, Alamzadeh's group). | ||
Toxicity tests were carried out on three cell types: KB cells (over-expression of positive folate receptors), A459 cells (negative folate receptors), and HFF (normal cells). These are MTT tests that allow determining the number of living cells. Injections of DOX–FOL–PEG–SHAuNPs and of DOX alone were carried out. The results are shown in Fig. 21. When the DOX concentration increased, the number of living cells decreased irrespective of whether DOX was conjugated or not. However, the effect of DOX alone was more important on the HFF cells than when it was conjugated. The association of DOX to a vector allows reducing the toxicity of normal cells. Moreover, conjugated DOX has less effect on A59 cells than on KB cells, which illustrates the vector specificity due to folic acid.
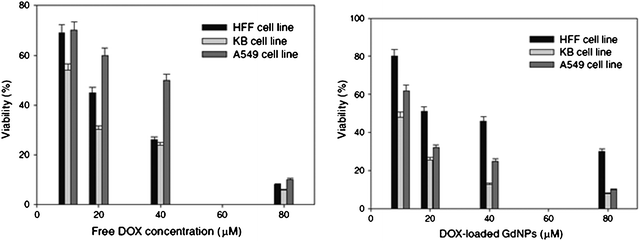 | ||
| Fig. 21 Cytotoxicity of DOX alone, then loaded with an AuNP on various cells. Reprinted with permission of Springer (ref. 87, Alamzadeh's group). | ||
9.2. Transferrin receptors
Transferrin is a glycoprotein that can be recognized by receptors that are also endocytosis mediators (Fig. 22). Many cancer cells present a large amount of transferrin receptors because of the fast cellular division and the large energy needs. AuNPs that are conjugated to transferrin are favorable assemblies for vectorization. Transferrins are bonded to AuNPs using mercaptoacetic acid.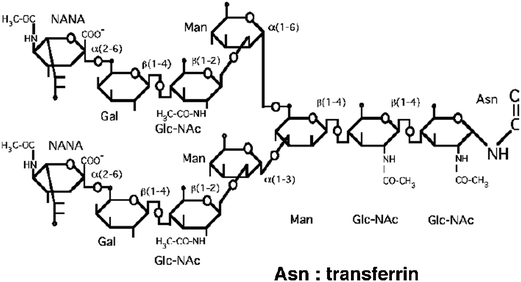 | ||
| Fig. 22 Structure of transferrin. | ||
Experiments have shown that cancer shells using AuNPs with a 20 nm diameter for the core and 90 nm overall size can internalize vectors based on AuNPs with a 20 nm core diameter and 90 nm overall size. The vectors were incubated in physiological conditions with NCP cancer cells. Internalization of the AuNPs was observed by AFM imaging and confocal microscopy (excitation at 488 nm and emission at 512 nm) (Fig. 23).
 | ||
| Fig. 23 AFM image and confocal microscopy showing the internalization of AuNPs. Reprinted with permission of the American Chemical Society (ref. 88, He's group). AFM picture: (A) NCP cells alone, (B) AuNP–TF at the surface of cells at the beginning of the incubation, (C) surface of the NPC cells 5 h after the beginning of incubation, (D) growing of picture C. Confocal microscopy: (E) cells line, (F) cells treated with AuNPs AuTF, (G) cells treated with AuNPs alone, (H) cells treated with AuNPs Au-albumin. | ||
Picture B of Fig. 23 shows the presence of AuNPs at the cell surface. Five hours after incubation, the pictures C and D of Fig. 23 indicate that internalization has started and reveal the endocytosis in this way. Also the strong florescence of picture F compared to pictures E, G and H (Fig. 23) indicates that internalization in cells occurs because of transferrins that are bonded to be AuNPs. It is an “active” targeting between the AuNPs and the transferring receptors that are present at the cell surface.88
Very recently, other studies have shown that AuNPs functionalized with PEGylated transferrins are easily internalized by cancer cells and thus deliver more drugs than non-functionalized AuNPs. These results underline the importance of the ligand content on the AuNP. They suggest that functionalized AuNPs allow better delivery of drugs to cancer cells than non-functionalized AuNPs do.90
9.3. Tumor-Necrosis Factor (TNF-α)
The Tumor-Necrosis Factor (TNF-α) discovered in 1975 is a cytokine that causes the necrosis of cancer cells. This excellent anticancer agent presents a high toxicity, however. The problem can be circumvented by associating it with PEGylated AuNPs. The optimal vector is PT–AuNP–TNF in which the PT (PEG) and the TNF are directly bound to the AuNP surface. It is called CYT-6091. The circulation time of this vector and its biodistribution have been treated in Section 5.1. A comparison between the vector and TNF alone has been reported. First, when the same amount of vector and of TNF alone is injected, mice treated with the vector present, 3 to 6 hours after injection, concentrations of TNF in blood that are 20 times larger than that of mice treated with TNF alone. In the case of TNF alone, the same phenomenon is involved as that of TNF–Au (without PT) discussed in Section 5. TNF plays both recognition and therapeutic functions. The recognition role allows that TNF specifically reaches the tumor. Fig. 24 shows the evolution of the TNF concentration as a function of time after injection of the vector and TNF alone.89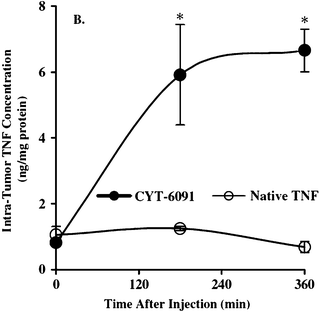 | ||
| Fig. 24 Variation of the TNF concentration in the tumor as a function of the injection time for injections of the vector and of TNF alone. Reprinted with permission of Taylor and Francis (ref. 89, Paciotti's group). | ||
The TNF concentration is 7 times higher when it is delivered via a vector. Indeed, with 15 μg TNF alone, 25 to 30% of mice present an incomplete response, and 33% of mice die, whereas for similar vector concentrations no negative response is obtained with the mice. The bond between the vector and the tumor can be shown by creating a new vector with murine albumin serum. As shown in Fig. 25, the murine albumin serum vector is present in the tumor 48 h after injection due to the EPR effect.91 This is a “passive” accumulation. The vector with TNF is already present 2 h after the injection. This is an “active” accumulation whereby TNF binds its receptors. Thus the AuNPs associated with TNF serve as binders, and the AuNPs remain inside the tumor. 400 TNF molecules bind to the surface of a 33 nm AuNP. Only one of them is necessary for recognition, and the others serve as therapeutic agents. Edemas have appeared, however, upon implanting human cancer cells in mice.91
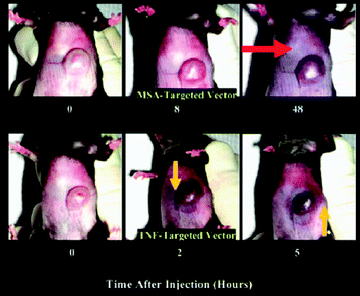 | ||
| Fig. 25 Active targeting of the TNF vector and passive accumulation of the MSA vector. Reprinted with permission of Wiley Interscience (ref. 91, Paciotti's group). | ||
Much interest is devoted to multifunctional AuNPs. The idea is to utilize TNF as a ligand for recognition and to bind paclitaxel to the AuNPs. The synthesis of thiolated paclitaxel was required in order to bind it to AuNPs to which PEG and TNF are also bound (Fig. 26). The thiolation of paclitaxel reduces its activity, however. Thus the vector has been designed in order to keep paclitaxel inactive during transport and to deliver it under the effect of a reducing agent such as dithiothreitol (DTT) or cysteamine.
 | ||
| Fig. 26 Functionalization of paclitaxel with a thiol group for conjugation with an AuNP: SIM 284. Reprinted with permission of Wiley Interscience (ref. 91, Paciotti's group). | ||
Attempts have been made on C57BL/6 mice with B16/F10 tumors. They receive injections of TNF alone, SIM-284 or CYT-21001, then two hours later, some of them receive an injection of cysteamine. The tumor volume is measured as a function of time (Fig. 27). Only the association of the CYT21001 vector and cysteamine allows decreasing the tumor volume.92
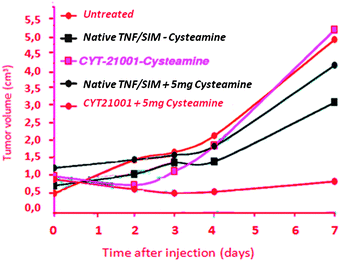 | ||
| Fig. 27 Efficiency of the CYT 21001 vector. Reprinted with permission of Wiley Interscience (ref. 91, Paciotti's group). | ||
10. Other AuNP-based anti-cancer therapies
10.1. Phototherapy
Phototherapy uses the optical heating to destroy cancer cells.93 The irradiation of AuNPs with visible light in the SPB leads to light energy absorption that is relaxed thermally within one picosecond.8 The AuNPs are efficient because they present a large surface for absorption and require little irradiation energy. The ideal size is in the 10–30 nm range. They are delivered to cancer cells either under physiological conditions or by recognition. Whereas classic (spherical or polygonal-shaped) AuNPs absorb near 520 nm, rod-shaped AuNPs absorb in the near-IR region, the red shift of the SPB being all the larger, as the length/width ratio is larger. Thus the SPB of Au nanorods is located around 700–1000 nm, i.e. in the near IR region. Infrared lasers penetrate under the skin more easily than visible-light lasers and reach deeper tumors. For the “biological window” of 650–900 nm that can be obtained by Au nanorods, nanospheres and nanocubes, the depth of near-infrared light penetration can reach one cm. The photo-thermal destruction of cancer cells has been demonstrated both in vitro and in vivo. The Halas group has carried out experiments under physiological conditions upon functionalizing AuNPs only with PEG,7,75–77,93 and the El-Sayed group has used AuNPs functionalized with antibodies.8,92,94,9510.2. Gene therapy
Gene therapy is the use of one or several genes as drugs to compensate the deficiency of a gene. DNA can be bound to AuNPs, as indicated in Section 5, and oligonucleotides that are present at the surface of AuNPs have a greater affinity for their complementary nucleic acids than for free oligonucleotides.97 The efficiency of gene transfection depends on the ratio of positive charges of the amines that are bonded to the AuNPs vs. the negative charges of the phosphate groups of the oligonucleotides and the length of the thiolated chain. The AuNPs have a great capacity to bind plasmid DNA and to deliver it inside the cell. Thus AuNPs are a valuable support for gene transfection. For instance, the Mirkin group has reported locked nucleic acid–AuNP conjugates that incorporate bridged sugars and form remarkably stable duplexes with complementary nucleic acids. These locked nucleic acids increase the effectiveness of gene knockdown compared to analogous DNA-modified AuNPs.98 This group also reported the synthesis and introduction in cells of RNA–AuNPs without the use of transfection agents, these conjugated RNA–AuNPs being 6 times more stable than molecular RNA in serum-containing media. RNA–AuNPs have a more persistent ability to silence genes, which should find therapeutic applications for RNAi.9911. Conclusion
Vectorization is a key technique in anti-cancer fight. It allows us to decrease side effects of the chemotherapeutic agents and to better target the cancer cells. The AuNPs present numerous advantages for vectorization: their biocompatibility, size, great affinity for thiolates and other sulfur ligands and surface plasmon band. Various synthetic protocols allow controlling their size, shape and nature of ligands. The most common syntheses are the Turkevitch method using citrate or its improved procedure and the Schiffrin–Brust using a biphasic synthesis. Functionalization can be carried out by direct synthesis with functional thiols, ligand substitution of alkyl thiolate with functional thiols or functionalization of pre-coordinated thiolate ligands.An important aspect is the check of the selective cytotoxicity of the AuNPs for cancer cells. It was found that it depends on the doses, the AuNP ligand nature and charges (positively charged ligands are usually cytotoxic for normal cells as well as some synthesis precursors), the purification of the AuNPs and the means of administration.
The surface plasmon band, due to the collective oscillation of electrons at the AuNP surface upon irradiation, depends very much on various parameters, which allows using it with imaging techniques for cancer diagnosis. Functionalization of AuNPs allows detecting abnormal amino acid sequences using hybridizing procedures. Antibodies can also bind AuNPs, thus antibodies over-expressed by cancer cells allow accumulating AuNPs near cancer cells for scanometric detection.
Drug vectorization using AuNPs can proceed either in “passive” or “active” way.96
In “passive” vectorization, the drug is either entrapped inside micellar AuNPs (supramolecular interaction involving a “complex”) or covalently bound in a “conjugate” to a cleavable group of the AuNPs (acidic cleavage or photo-cleavage near the cancer cell). Another solution for passive vectorization is exchange of the thiolate-drug ligand with glutathione, an endogen thiol. In this type of vectorization, the driving force is the EPR effect provided by thiolated polyethylene glycol ligands.
Active vectorization involves over-expression of receptors at the surface of cancer cells by inter aliafolate, transferrin and α-tumor necrosis factor. Upon binding, the AuNPs are then subjected to endocytosis.
Another promising therapy technique is photo-thermal therapy that uses the transformation of light energy into heat destroying the cancer cells, and can be extended to near-infrared light when the SPB is red-shifted by using rod-shaped, nanocubes or coating nanospherical AuNPs.
Finally, gene therapy that allows delivering DNA into cancer cells can be envisaged, although it requires polycationic AuNP ligands that cause damage to cytotoxicity.
In conclusion, AuNPs are at the forefront of nanoparticle therapeutics as an emerging treatment modality for cancer.2–13,62,100–106 Multifunctional AuNPs that will allow us to both image and treat cancer thus combining targeting, diagnosis and therapeutics functions, recently called theranostic, are very promising.54,107,108
Acknowledgements
Financial support from the Université Bordeaux 1, the Centre National de la Recherche Scientifique (CNRS), and the Agence Nationale de la Recherche (ANR) is gratefully acknowledged.References
- ACS, Cancer Facts & Figures. American Cancer Society, 2010. http://www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-and-figures-2010.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, C. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- W. Cai, T. Gao, H. Hong and J. Sun, Nanotechnol., Sci. Appl., 2008, 1, 17–32 CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- U. Dreshler, B. Erdogan and V. M. Rotello, Chem.–Eur. J., 2004, 10, 5570–5079 CrossRef.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–1730 CrossRef CAS.
- S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842–1851 CrossRef CAS.
- P. Jain, I. H. El-Sayed and M. A. El-Sayed, Nano Today, 2007, 2, 18–29 CrossRef.
- G. F. Pascioti, D. G. I. Kinston and L. Tamarkin, Drug Dev. Res., 2006, 5, 2255–2262 Search PubMed.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- R. K. Jain, Cancer Res., 1987, 47, 3039–3051 CAS.
- I. Brigger, C. Bubernet and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54, 631–651 CrossRef CAS.
- T. Ganesh, Bioorg. Med. Chem., 2007, 15, 3597–3623 CrossRef CAS.
- L. B. Michaud, V. Valero and G. Hortobagyi, Drug Saf., 2000, 23, 401–428 CrossRef CAS.
- J. Turkevitch, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- J. Kimling, M. Maier, B. Ovenke, H. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS.
- M. Brust, M. Walker, D. Bethel, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- M. Giersig and P. Mulvaney, Langmuir, 1993, 9, 3408–3413 CrossRef CAS.
- H. Xia, S. Bai and J. Hartmann, Langmuir, 2010, 26, 3585–3589 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef.
- S. Meltzer, R. Resch, B. E. Koel, M. E. Thompson, A. Madhukar, A. A. G. Requicha and P. Will, Langmuir, 2001, 17, 1713–1718 CrossRef CAS.
- R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS.
- R. W. J. Scott, O. M. Wilson and R. M. Crooks, J. Phys. Chem. B, 2005, 109, 692–704 CrossRef CAS.
- E. Boisselier, A. K. Diallo, L. Salmon, J. Ruiz and D. Astruc, Chem. Commun., 2008, 4819–4821 RSC.
- E. Boisselier, A. K. Diallo, L. Salmon, C. Ornelas, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2010, 132, 2729–2742 CrossRef CAS.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS.
- J. Chen, F. Saeki, B. J. Wiley, H. Cang, M. J. Cobb, L. Au, H. Zhang, M. B. Kimmey, X. Li and Y. Xia, Nano Lett., 2005, 5, 473–477 CrossRef CAS.
- C. R. Martin, Science, 1994, 266, 1961–1966 CAS.
- B. M. I. van der Zande, M. R. Boehmer, L. G. J. Fokkink and C. Schönenberger, J. Phys. Chem. B, 1997, 101, 852–854 CrossRef CAS.
- A. J. Mieszawska and F. P. Zamborini, Chem. Mater., 2005, 17, 3415–3420 CrossRef CAS.
- Y. Y. Yu, S. S. Chang, C. L. Lee and C. C. R. Wang, J. Phys. Chem. B, 1997, 101, 6661–6664 CrossRef CAS.
- G. Canizal, J. A. Ascencio and J. Gardea-Torresday, J. Nanopart. Res., 2001, 3, 475–481 CrossRef CAS.
- F. Kim, J. H. Song and P. Yang, J. Am. Chem. Soc., 2002, 124, 14316–14317 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Adv. Mater., 2001, 13, 1389–1393 CrossRef CAS.
- N. R. Jana, L. Gearheart, S. O. Obare and C. J. Murphy, Langmuir, 2002, 18, 922–927 CrossRef CAS.
- B. D. Busbee, S. O. Obare and C. J. Murphy, Adv. Mater., 2003, 15, 414–416 CrossRef CAS.
- E. Boisselier, L. Salmon, J. Ruiz and D. Astruc, Chem. Commun., 2008, 5788–5790 RSC.
- A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS.
- Y.-S. Shon and H. Choo, C. R. Chim., 2003, 6, 1009–1018 CrossRef CAS.
- M. Yamada, T. Takera, K. Kubo and H. Nishihara, Langmuir, 2001, 17, 2363 CrossRef CAS.
- M. Yamada and H. Nishihara, C. R. Chim., 2003, 6, 919–934 CrossRef CAS.
- A. Labande, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2002, 124, 1782–1789 CrossRef CAS.
- M.-C. Daniel, J. Ruiz, S. Nlate, J.-C. Blais and D. Astruc, J. Am. Chem. Soc., 2003, 125, 2617–2628 CrossRef CAS.
- S. J. Hurst, A. K. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313 CrossRef CAS.
- J.-S. Lee, D. S. Seferos, D. A. Gilhoyann and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 5430 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CAS.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS.
- A. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS.
- R. K. Delong, C. M. Reynolds and Y. Malcom, Nanotechnol., Sci. Appl., 2010, 3, 53–63 CAS.
- T. B. Huff, M. N. Hansen, Y. Zhao, J.-X. Cheng and A. Wei, Langmuir, 2007, 23, 1596–1599 CrossRef CAS.
- H. K. Patra, S. Banerjee, U. Chaudhuri, P. Lahiri and A. K. Dascupta, Nanomedicine, 2007, 3, 111–119 CAS.
- H. Takahashi, Y. Niidome, T. Niidome, H. K. Kaneko, H. Kawasaki and S. Yamada, Langmuir, 2006, 22, 2–5 CrossRef CAS.
- J. J. Li, L. Zou, D. Hartono, C.-N. Hong, B.-H. Bay and L.-Y. L. Yung, Adv. Mater., 2008, 20, 138–142 CrossRef CAS.
- J. A. Khan, B. Pillai, T. K. Da Y. Singh and S. Maiti, ChemBioChem, 2007, 8, 1237–1240 CrossRef CAS.
- J. S. Rink, K. M. McMahon, X. C. Chen, C. A. Mirkin, C. S. Thakston and D. B. Kaufman, Surgery, 2010, 148, 335–345 CrossRef.
- S. Lu, D. Xia, G. Huang, H. Jing, Y. Wang and H. Gu, Colloids Surf., B, 2010, 81, 406–411 CrossRef CAS.
- Z. Wang, S. Zong, J. Yang, C. Song, J. Li and Y. Cui, Biosens. Bioelectron., 2010, 26, 241–247 CrossRef CAS.
- C. L. Villiers, H. Freitas, R. Couderc, M.-B. Villiers and P. N. Marche, J. Nanopart. Res., 2010, 12, 55–60 CrossRef CAS.
- A. E. Hawleya, S. Davis and L. Illum, Adv. Drug Delivery Rev., 1995, 17, 129–148 CrossRef.
- S. G. Sonavane, K. Tomoda and K. Makino, Colloids Surf., B, 2008, 66, 274–280 CrossRef.
- J. H. Jong, W. I. Hagen, P. Krystek, M. C. Burger, A. J. A. M. Sips and R. E. Geertsma, Biomaterials, 2008, 29, 1912–1919 CrossRef.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- G. Mie, Ann. Phys., 1908, 25, 377–445 CrossRef CAS.
- L. M. Liz Marvan, Langmuir, 2006, 22, 32–41 CrossRef.
- B. Kim, G. Han, B. J. Toley, C.-k Kim, V. M. Rotello and N. S. Forbes, Nat. Nanotechnol., 2010, 5, 465–472 CrossRef CAS.
- S. S. Agasti, S. R. Rana, M. H. Park and V. M. Rotello, Adv. Drug Delivery Rev., 2010, 62, 316–328 CrossRef CAS.
- S. H. Jang, M. G. Wientjes and D. Lu, Pharm. Res., 2003, 20, 1337–1350 CrossRef CAS.
- D. Chithranib, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef.
- S. J. Oldenburg, R. D. Averitt, S. L. Wescott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS.
- S. J. Oldenburg, J. B. Jackson, S. L. Wescott and N. J. Halas, Appl. Phys. Lett., 1999, 75, 2897–2899 CrossRef CAS.
- S. J. Oldenburg, S. L. Wescott, R. D. Averitt and N. J. Halas, J. Chem. Phys., 1999, 111, 4729–4735 CrossRef CAS.
- R. Bardhan, S. Lal, A. Joshi and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842–1851 CrossRef.
- B. E. Brinson, J. B. Lassiter, C. S. Levin, R. Bardhan, N. Mirin and N. J. Halas, Langmuir, 2008, 24, 14166–14177 CrossRef CAS.
- A. M. Gobin, M. H. Lee, N. J. Halas, W. D. James, R. A. Drezek and J. L. West, Nano Lett., 2007, 7, 1929–1934 CrossRef CAS.
- W. Cai, T. Gao, H. Hong and J. Sun, Nanotechnol., Sci. Appl., 2008, 1, 17–32 CAS.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CAS.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, Science, 1997, 277, 1078–1081 CrossRef CAS; J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef.
- Y. W. C. Cao, R. C. Jin and C. A. Mirkin, Science, 2002, 297, 1536–1540 CrossRef CAS.
- T. Niidome, M. Yamagata, Y. Okamoto, Y. Akiyama, H. Takahashi, T. Kawano, Y. Katayama and Y. Niidome, J. Controlled Release, 2006, 114, 343–347 CrossRef CAS.
- S. S. Agasti, A. Chompoosor, C. C. You, P. Ghosh, C. K. Kimand and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 5728–5729 CrossRef CAS.
- G. F. Paciotti, L. Myer, D. Weinreich, D. Goia, N. Pavel, R. E. McLaughlin and L. Tamarkin, Drug Delivery, 2004, 11, 169–183 CrossRef CAS.
- B. Duncan, C. Kim and V. M. Rotello, J. Controlled Release, 2010, 148, 122–127 CrossRef CAS.
- V. Dixit, J. Van den Bossche, D. M. Sherman, D. H. Thomson and R. P. Andres, Bioconjugate Chem., 2006, 17, 603–609 CrossRef CAS.
- B. Asadishad, M. Vossoughi and I. Alamzadeh, Biotechnol. Lett., 2010, 32, 649–654 CrossRef CAS.
- P. H. Yang, S. Xuesond, J. F. Chiu, H. Sun and Q.-Y. He, Bioconjugate Chem., 2005, 16, 494–496 CrossRef CAS.
- G. F. Paciotti, L. Myer, D. Weinreich, D. Goia, N. Pavel, R. E. McLaughlin and L. Tamarkin, Drug Delivery, 2004, 11, 169–183 CrossRef CAS.
- C. H. J. Choi, C. A. Alabi, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1235–1240 CrossRef CAS.
- G. F. Paciotti, D. G. I. Kingston and L. Tamarkin, Drug Dev. Res., 2006, 67, 47–54 CrossRef CAS.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409–453 CrossRef CAS.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS.
- X. Huang, P. K. Jain and I. H. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS.
- S. Link and M. A. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409–453 CrossRef CAS.
- B. Duncan, C. Kim and V. M. Rotello, J. Controlled Release, 2010, 148, 122–127 CrossRef CAS.
- D. S. Seferos, D. A. Giljohann, N. L. Rosi and C. A. Mirkin, ChemBioChem, 2007, 8, 1230–1232 CrossRef CAS.
- D. A. Giljohann, D. S. Seferos, A. E. Prigodich, P. C. Patel and C. A. Mirkin, J. Am. Chem. Soc., 2009, 131, 2072 CrossRef CAS.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug. Discovery, 2008, 7, 771–782 CrossRef CAS.
- R. A. Sperling, P. Rivera Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- R. Wilson, Chem. Soc. Rev., 2008, 37, 2028–2045 RSC.
- D. Astruc, C. R. Acad. Sci., 1996, 322 (Sér. IIb), 757–766 Search PubMed.
- C. J. Murphy, A. M. Gole, S. E. Hunyadi, J. W. Stone, P. N. Sisco, A. Alkilany, B. E. Kinard and P. Ankins, Chem. Commun., 2008, 544–557 RSC.
- X. C. Yang, B. Samanta, S. S. Agasti, Y. Jeong, Z. J. Zhu, S. Rana, O. R. Miranda and V. M. Rotello, Angew. Chem., Int. Ed., 2011, 50, 477–481 CrossRef CAS.
- X. H. Huang, X. H. Peng, Y. Q. Wang, Y. X. Wang, D. M. Shin and M. A. El-Sayed, ACS Nano, 2010, 4, 5887–5896 CrossRef CAS.
- B. Sumer and J. M. Gao, Nanomedicine, 2008, 3, 137–140 CrossRef.
- J. R. McCarthy, Nanomedicine, 2009, 4, 693–695 CrossRef.
- W. Chen, R. Bardhan, M. Bartes, C. Perez-Torres, R. Pautler, N. J. Halas and A. Joshi, Mol. Cancer Ther., 2010, 9, 1028–1038 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |