Recent progress on polymer-based fluorescent and colorimetric chemosensors
Ha Na
Kim†
a,
Zhiqian
Guo†
b,
Weihong
Zhu
b,
Juyoung
Yoon
*ac and
He
Tian
*b
aDepartment of Chemistry and Nano Science BK21, Ewha Womans University, Seoul 120-750, Republic of Korea. Fax: +82 2 3277 2400; Tel: +82 2 3277 2384
bKey Laboratory for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, Shanghai 200237, P. R. China. E-mail: tianhe@ecust.edu.cn; Fax: (+86) 21-6425-2758
cDepartment of Bioinspired Science, Ewha Womans University, Seoul 120-750, Republic of Korea. E-mail: jyoon@ewha.ac.kr
First published on 25th November 2010
Abstract
Recently, fluorescent or colorimetric chemosensors based on polymers have attracted great attention due to several important advantages, such as their simplicity of use, signal amplification, easy fabrication into devices, and combination of different outputs, etc. This tutorial review will cover polymer-based optical chemosensors from 2007 to 2010.
 Ha Na Kim | Ha Na Kim was born in Seoul, in 1980 and received her BS degree from the department of chemistry at Ewha Womans University. She was then given an MS degree in medical science from Seoul National University in 2006. She is on a doctoral course within Prof. Juyoung Yoon's research group in Ewha Womans University. |
 Weihong Zhu, He Tian and Zhiqian Guo | Prof. Weihong Zhu received his PhD degree in 1999 from the East China University of Science & Technology (ECUST, Shanghai). He worked with Dr Nobutsugu Minami in AIST Central 5, Tsukuba, Japan (Postdoctoral) from 2001 to 2003 and worked with Prof. Akasaka in Tsukuba University, Japan (visiting professor) from 2004 to 2005. He became a full professor in 2004 and deputy director in 2006. His current research interests include the syntheses of novel functional organic or copolymer dyes and dendrimers. Prof. He Tian received his PhD degree in 1989 from the East China University of Science & Technology (ECUST, Shanghai). In 1999, he was appointed Cheung Kong Distinguished Professor by the Education Ministry of China. His current research interests include the syntheses of novel functional organic dyes or copolymers and the development of inter-disciplinary materials science that determines the electronic and optical properties of materials. Prof. Tian has published over 240 papers in international journals and academic books (in Chinese). He has also been awarded 46 Chinese patents. Since 2006, he has been an Associate Editor of Dyes and Pigments. Zhiqian Guo was born in 1978 and completed his BSc studies in Fine Chemicals (2002) at Zhengzhou University, China. In 2010, he received his PhD degree in Applied Chemistry from the East China University of Science and Technology (ECUST, Shanghai) under the supervision of Prof. Weihong Zhu. Now he works at the Institute of Fine Chemicals of ECUST. His research interests mainly focus on fluorescent chemosensors and molecular logic devices. |
 Juyoung Yoon | Juyoung Yoon received his PhD (1994) from The Ohio State University. After completing postdoctoral research at UCLA and at Scripps Research Institute, he joined the faculty at Silla University in 1998. In 2002, he moved to Ewha Womans University, where he is currently a Professor at the Department of Chemistry and Nano Science and Department of Bioinspired Science. His research interests include investigations of fluorescent chemosensors, molecular recognition and organo EL materials. |
1. Introduction
Chemosensors utilizing color and/or fluorescence intensity have been developed to be useful tools for sensing various analytes.1–3 A typical optical chemosensor contains a receptor (the recognition site), linked to a fluorophore (the signal source), which translates the recognition event into the fluorescence signal.4 Compared to small organic compounds, polymer based optical sensors display several important advantages. For instance, signal amplification could be one of the most important advantages of polymer-based sensors. Moreover, conjugated polymer chains with multiple recognition elements can increase both the binding efficiency and recognition selectivity for specific analytes. In contrast with molecular sensors, film sensors based on polymers, are easily fabricated into devices. In addition, incorporating several different recognition units into a functional polymer can produce a valuable conclusion based on the combination of different outputs.There are numerous reviews available for polymer-based sensors. For example, in 2007, Swager et al. wrote a good review on “Chemical Sensors Based on Amplifying Fluorescent Conjugated Polymers”, which covered the papers on conjugated polymer based sensors published up to mid 2006.5 Since 2006, there have been many exciting results from various groups for polymer based chemosensors. Recently, water-soluble fluorescent conjugated polymers were discussed by Wang and Zhu et al.,6 and polydiacetylenes (PDAs) based sensors were discussed by Ahn and Kim.7
However, to our knowledge, this is the first review to cover optical sensors based on polymers. In this tutorial review, we focus on recent contributions (2007–2010) for polymer-based optical sensors, including polymer sensors bearing pendant ligands for recognition, conjugated polymer based sensors, dendrimer based sensors and imprinted polymer based sensors.
This tutorial review will classify polymer chemosensors based on polymer backbone and receptor types and help to elucidate the design and application of polymer based optical chemosensors in an assessable manner to those new to the field.
2. Polymer sensors bearing pendant ligands for recognition
2.1 Polymer sensors bearing pendant ligands
Optical recognition units existing as pendant ligands via covalent bonding to the polymer matrix present a diverse and highly functional approach to construct chemosensors.Lu et al.8 designed and synthesized P1 (Fig. 1) that copolymerized quinoline derivatives in the side chain, with soft methacrylate segments as the main chain, to improve its solubility in organic solvents and film-forming ability for application in fluorescent devices. P1 displayed high selectivity and sensitivity towards Fe3+ with significant quenching of fluorescence compared to all other tested transition-metal ions such as Zn2+, Ni2+, Cu2+, and Co2+ in different organic solvents. Moreover, Suet al.9 utilized free radical copolymerization to prepare a water-soluble coumarin-bearing polymer (P2 in Fig. 1) for sensing proton and Ni2+. P2 was found to be an efficient off–on switcher in the pH range of 3.02–12.08, and exhibited high selectivity to turn-on fluorescence with Ni2+ in the range of 0.33 × 10−5−7.67 × 10−5 M−1 in aqueous solution. The vinylpyrrolidone unit was selected as the functional monomer due to its advantages of excellent water-solubility, good film forming ability and biocompatibility. The coumarin fluorophore was covalently bonded to the main chain, thus avoiding any migration and crystallization with respect to physical doping. The piperazine ring in the pendant coumarin moiety of P2 performed a dual role in the sensing process: recognition ligands for metal ion and a photo-induced electron transfer (PET) switch.
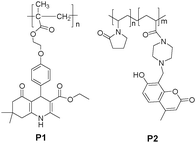 | ||
| Fig. 1 Chemical structures of P1 and P2. | ||
Although the strategy of using a metal ion complex for anions as a binding site has been successfully utilized to design molecular sensors, few fluorescent film sensors for anion based hydrophilic polymers have been constructed. Tian et al.10 reported a hydrophilic copolymer bearing a dicyanomethylene-4H-pyran (DCM) moiety as a fluorescent film sensor for Cu2+ and pyrophosphate anion. For hydrophilic copolymer P3 (Fig. 2), poly(2-hydroxyethyl methacrylate) (PHEMA), which exhibits a high hydrophilicity, but is insoluble in water, has been chosen as the hydrophilic chain segment to improve the permeability of ions into the polymer backbone, and a DCM derivative fluorophore was grafted into the polymer backbone as the metal ion-sensing unit. Intriguingly, copolymer P3 not only exhibited turn-off fluorescence to the selectively targeted Cu2+, but the relative metal complex, P3-Cu2+, displayed off–on fluorescence, with high sensitivity to PPi both in solution and in thin films over other anions such as adenosine monophosphate (AMP), adenosine phosphate (ADP), adenosine triphosphate (ATP) and phosphate (Pi) in a solution of water–ethanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, v/v). Furthermore, the low-cost hydrophilic copolymer film of P3-Cu2+ on a quartz plate showed a rapid response to PPi, with turn-on orange-red fluorescence due to high permeability of its side chains (Fig. 2).
:5, v/v). Furthermore, the low-cost hydrophilic copolymer film of P3-Cu2+ on a quartz plate showed a rapid response to PPi, with turn-on orange-red fluorescence due to high permeability of its side chains (Fig. 2).
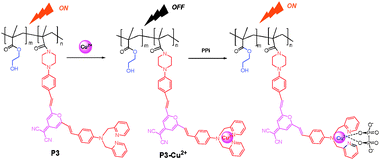 | ||
| Fig. 2 Scheme of Cu2+ and PPi sensors based on the fluorescence “on–off” and “off–on” of P3 and P3-Cu2+, respectively. | ||
Liu et al. constructed a type of responsive double hydrophilic block copolymer as a multifunctional chemosensor for Hg2+ ions, pH, and temperature, using reversible addition-fragmentation chain transfer (RAFT) polymerization.11Water-soluble poly(ethylene oxide)-b-poly(N-isopropyl acrylamide-co-RhBHA) (P4, PEO-b-P(NIPAM-co-RhBHA)), contained rhodamine derivative as Hg2+-sensing moiety in the thermo-sensitive block. Based on the rhodamine-B process from spirolactam (non-fluorescent) to ring-open (fluorescent) process, P4 exhibited a significant 77- or 82-fold enhancement in fluorescence at 584 nm upon adding 4 equiv Hg2+ or decreasing pH from 6.0 to 1.0, respectively. Interestingly, upon heating above the lower critical solution temperature (LCST, about 36°C) in the PNIPAM block, the detection sensitivity for Hg2+ ions and pH could be dramatically improved at elevated temperatures by embedding the acyclic ring-open rhodamine-B form within hydrophobic cores in thermo-induced micellar aggregates (Fig. 3). This was a proof-of-concept example for multifunctional fluorescent polymer sensors that integrated the stimuli-responsive block copolymers with small molecule-based sensing moieties, using the optimized RAFT polymerization process.
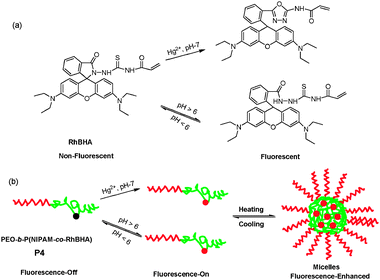 | ||
| Fig. 3 (a) Scheme of rhodamine B-based spirolactam monomer (RhBHA) in the presence of Hg2+ ions or acid media; (b) proposed ring-open mechanism of P4 in the presence of Hg2+ or acid media. | ||
Tian et al.12,13 have recently reported a series of highly sensitive, colorimetric, and fluorescent polymer sensors (P5 and P6 in Fig. 4) for fluoride anion utilizing naphthalimide as the signal moiety, and incorporating amide or thiourea moieties as recognition units at the side pendant viaRAFT polymerization. The range of molecular weights and a narrow polydispersity index of P5 were efficiently controlled by use of a chain transfer agent in RAFT. Its sensitivity to F− was investigated by the growth in polymeric molecular weight, revealing that the higher the molecular weight, the greater the sensitivity to F−. Upon the addition of F−, P5 and its monomer NAP-1, showed a progressive decrease in fluorescence at shorter wavelength (λex = 360 nm, λem = 468 nm), while new red shift wavelength bands (λex = 490 nm, λem = 580 nm) were formed, and increased in the solvent mixture of CH2Cl2-DMSO (9:1, v/v). The deprotonation of an amino moiety can enhance the charge intensity on the amino nitrogen, thus increasing the push–pull effect on the intramolecular charge transfer (ICT) process. The thin films of P5 and P6 on quartz plates displayed quite similar selectivity and sensitivity to F−versus other halide anions, showing a promising fluoride sensor (naked eye) in practice.
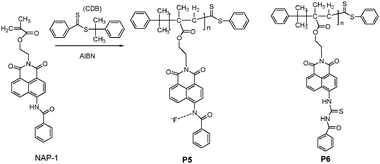 | ||
| Fig. 4 Scheme of synthetic procedure for P5 viaRAFT polymerization and chemical structure of P6. | ||
Moreover, a particular interest is the development of polymeric indicating materials as amine sensors, which display excellent stability and potential for reuse. Ghosh and Dey14 utilized azobenzene derivatives as a “push–pull” type of versatile-responsive system to the epoxy-based polymer (P7) in Fig. 5. The azobenzene moiety was grafted onto the backbone of P7 containing electron-donating and electron-withdrawing groups at 4- and 4-positions, which exhibited a characteristic reversible colorimetric response to aminesvia the protonation-deprotonation mode (Fig. 5).
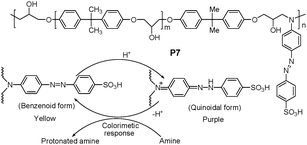 | ||
| Fig. 5 Chemical structure of P7, and mechanism mode of protonation-deprotonation of P7. | ||
The concept of dual-mode optical oxygen sensors, containing indicators incorporated into a single coating matrix with high oxygen sensing performance at atmospheric pressure, was reported by Nishide et al.15 In this strategy, both the cobalt porphyrin (CoP) ligated with the polymer matrix and the luminescent pyrene derivative (Py) were utilized in the double-layer film sensing system. The dual-mode oxygen-sensing was based on the oxygen-adduct formation at the cobaltporphyrin-polymer and the luminescence quenching of pyrene. Terpolymers (P8, P9, and P10 in Fig. 6) were successfully developed as optical oxygen sensors, with significantly high sensitivity, under the practical atmospheric oxygen pressure of 10–21 kPa, based on luminescence quenching. The dual-mode oxygen pressure-sensitive films were fabricated by combining terpolymers (P8, P9, and P10) and the pyrene derivative (Py) via stepwise coating as shown in Fig. 6. Here, three functional moieties were involved in these terpolymers: methyl methacrylate for increasing the coating flexibility; vinylidenechloride as an excellent gas-barrier unit, and a nitrogenous residue for bonding the cobaltporphyrin (CoP, in Fig. 6) to rapidly and reversibly form oxygen adducts. In double layer polymer films, the first layer of luminescent pyrene derivative was designed as a luminesence-quenching signal unit with oxygen, and the second coating of terpolymers bonding with the cobaltporphyrin, specifically provided both an oxygen-barrier for the pyrene derivative and an improvement of the oxygen-adduct formation equilibrium. As a consequence, the film luminescence intensity was decreased along with an increase in the oxygen partial pressure, yielding a non-Stern–Volmer response with high sensitivity to the oxygen partial pressure.
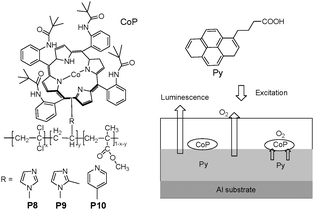 | ||
| Fig. 6 Chemical structures of P8, P9 and P10, and schematic diagram for sensing oxygen. | ||
The oxygen response of optical film sensors can be measured with two parameters, such as luminescence decay time and intensity. The former, in contrast to the latter, is independent of the concentration of indicator dyes, and only depends on the concentration of oxygen as the dynamic quencher. Therefore, the measurement of luminescence decay time versus luminescence intensity is advantageous. Demas et al.16 designed a star polymer by covalently attaching ruthenium tris(bispyridine)-centrated polystyrene complex (P11 in Fig. 7) as a novel quenchometric oxygen sensor, by measuring sensitive luminescence lifetime. However, the above strategy suffered from the multi-exponentials of luminescence decays due to the heterogeneity issue in the polymerization process. For the sensing moiety in a polymeric system, a homogeneous environment can avoid or minimize problems associated with multicomponent sensors and the leaching of metal complex. P11 was obtained using chain growth methods (atom transfer radical polymerization, ATRP) to realize a very narrow polydispersity (PDI), as near as a single distinct species, for solving the heterogeneity issue based on the star polymer strategy.
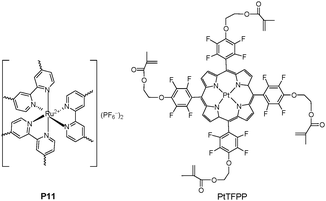 | ||
| Fig. 7 Chemical structure of P11 and PtTFFP. | ||
An alternative strategy for constructing polymer films can be realized by chemically anchoring the sensing moiety with acrylate onto the quartz surface to form membranes by in situpolymerization. Tian et al.17 fabricated sensing films to dissolved oxygen (DO) via direct immobilization on the quartz substrate using a modified popular oxygen sensor consisting of platinum(II)-5,10,15,20-tetrakis-(2,3,4,5,6-pentafluorophenyl)-porphyrin (PtTFPP) and biocompatible hydrophilic poly(2-hydroxyethyl methacrylate)-co-polyacrylamide (PHEMA) as a matrix in Fig. 7. PtTFPP was an excellent phosphorescence-based oxygen sensor due to its good response to oxygen concentration, high photostability with long triplet state lifetime, and sufficient triplet energy transfer to oxygen molecules. Moreover, the utilization of hydrophilic PHEMA as a matrix showed much faster response and higher sensitivity to oxygen than achieved with the hydrophobic polystyrene (PS).
2.2 Mesoporous and nanoparticle silica bearing pendant ligands and optical sources
There has been increasing interest in utilizing supramolecular concepts for inorganic mesoporous materials to form organic–inorganic hybrids for supramolecular recognition or sensing protocols. Receptor-immobilized inorganic mesoporous materials have several important advantages as solid chemosensors: high sensitivity resulting from large surface areas and well-defined pores, easy bead-preparation and recyclability, and improved solubility of functionalized hybrid nanoparticles. Using anthraquinone as a signal unit, Kim and Jung et al.18 anchored the Cu2+-induced desulfurization chemodosimeter onto mesoporous silica (P12, Fig. 8), indicating high sensitivity to Cu2+ with an instantaneous visible color change from red to pale yellow, to give P12-closed arising from the Cu2+-induced guanylation. Moreover, in the presence of Cu2+, the suspension of P12 exhibited a distinct fluorescent change from red to yellow with a remarkable enhancement centered at 560 nm. P12 has been demonstrated to be a promising heterogeneous sensor for the detection of Cu2+, with high selectivity over other cations in the environmental field.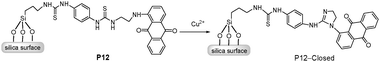 | ||
| Fig. 8 Scheme of P12 in presence of Cu2+ to form P12-Closed. | ||
More recently, Tian et al.19,20 have reported two Hg2+ chemodosimeters based on gold nanoparticles and mesoporous silica, respectively. The well-developed naphthalimide-based chemodosimeter units were synthesized and grafted onto mesoporous silica (P13, Fig. 9).19 The mesoporous silica materials showed fluorescent ratiometric and characteristics recognizable by the naked eye, with a detection limit of 2 μM for Hg2+. In addition, the thin films based on the ordered mesoporus silica materials showed the distinct signal changes with Hg2+, further strengthening the advantages for their practical environmental application due to their characteristic of using easily prepared materials and simple operational techniques.
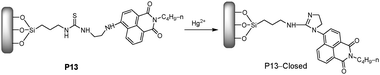 | ||
| Fig. 9 Scheme of P13 in the presence of Cu2+ to form P13-Closed. | ||
One convenient approach to prepare silica-hybrid nanomaterials is to attach silica nanoparticles to functional organic molecules using a grafting reaction. Yoon and Jung et al.21 have recently reported using fluorescein-functionalized silica nanoparticles P14 (Fig. 10) as a new type of synthetic fluorogenic chemosensor for imaging Cu2+ in living cells, showing a high sensitivity and selective characteristics for Cu2+versus other metal ions. Moreover, the nanoparticles of P14 can be used to detect intracellular Cu2+ in living cells.
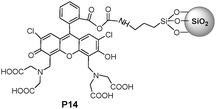 | ||
| Fig. 10 Chemical structure of P14. | ||
SBA-15, a well-known ordered mesoporous silica material, was directly covalently-coupled via the silylation of 1,8-naphthalimide-based receptor for detection and adsorption of Cu2+ in aqueous solution (Fig. 11).22 Bearing two imine-pyridyl arms to form a proper cavity configuration, the 1,8-naphthalimide derivative in P15 was suitable for Cu2+ coordination in the square planar geometry. Importantly, P15 displayed an extremely high fluorescence selectivity and sensitivity to Cu2+, with a very low detection limit (ca. 0.65 ppb) in aqueous solution, which can be ascribed to the significant fluorescence quenching induced by a PET and/or a d–d electron paramagnetism.
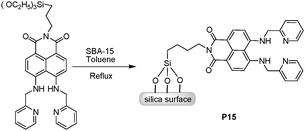 | ||
| Fig. 11 Scheme of synthetic procedure for P15. | ||
Banerjee et al.23 reported a selective fluorescent Zn2+ sensor (P16), in which 4-methyl-2,6-diformyl phenol was covalently grafted onto 3-aminopropyl triethoxysilane-functionalized, 2D-hexagonally ordered mesoporous silica (MCM-41, Fig. 12). P16 exhibited significant enhancement of fluorescence upon titration with Zn2+, while other biologically relevant cations (Ca2+, Mg2+, Na+, and K+) and environmentally hazardous transition-metal ions with subtle interference. As demonstrated, the mesoporous material P16 could be developed as a fluorescent turn-on Zn2+ sensor with high sensitivity and selectivity (5 μM–1.4 mM). Moreover, the intracellular Zn2+ concentration could be monitored using the mesoporous silicavia microscopic imaging, stimulating the development of a new category of tailor-made biocompatible systems based on nanomaterials.
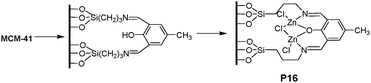 | ||
| Fig. 12 Synthetic procedure for functionalized material P16 and binding with Zn2+. | ||
Martínez-Máñez et al. adopted the suitable mixing of functional mesoporous materials and polymeric materials using supramolecular concepts.24 The composite was prepared by mixing a hydrophilic polyurethane polymer (Hydromed D4) with the mesoporous material (P17 in Fig. 13) by stirring 1 h in chloroform, and then transferring onto a polyester layer. Both X-ray patterns and TEM images demonstrated a homogeneous dispersion of the nanometric mesoporous particles within the polymeric matrix, similar to P17. The composite polymer film exhibited an enhanced response to n-octylamine and n-decylamine, while giving no response to smaller hydrophilic amines or larger aliphatic amines. The attractive aspect of this system is that several building blocks, i.e. mesoporous materials, hydrophobic binding pockets, reversibly responding amine indicator dyes and a polymeric backbone, can be combined in the design of tailor-made optical sensors with improved features.
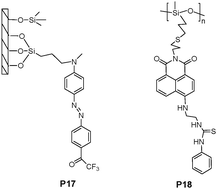 | ||
| Fig. 13 Chemical structures of P17 and P18. | ||
As the most widely applied biomaterials, various pending functionalities have been incorporated into silicon systems containing a siloxane (Si–O–Si) backbone. Callan et al.25 grafted a naphthalimide signal moiety, with a thiourea recognition unit, into poly(mercaptopropylmethyl)siloxane (PMPMS) as a backbone, and developed the structure as a fluoride anion sensing polymer P18 (Fig. 13). The sensing mechanism was attributed to the co-operative binding of a DMSO molecule and a fluoride ion with thiourea groups on adjacent residues.
3. Conjugated polymer based sensors
In comparison to their small molecule counterparts, conjugated polymers (CPs) coordinate the action of a large number of absorbing units with efficient intrachain and interchain energy transfer mechanisms.5 The excitation energy along the whole backbone of the conjugated polymers, transferring to the chromophore reporter, results in the amplification of fluorescent signals, and makes them useful as the optical platforms in highly sensitive chemical and biological sensors.3.1 Polyfluorenes (PFs) backbone
Wang and coworkers demonstrated that via electrostatic interactions, complexes of conjugated polymers/enzymatic substrates could be utilized as probes for continuous and sensitive fluorescence-based enzyme assays. (Fig. 14).26 The charged substrates and enzymes used for proof of concept were anionic ATP and alkaline phosphatase (ALP), as well as cationic polyarginine peptide (Arg6) and trypsin. ALP is known to remove the 5′-phosphate groups from ATP, ADP, and AMP to convert each of these species into adenosine. The emission maximum of P19 in buffer solution appeared at ∼410 nm and no emission from the BT unit at 540 nm was observed. Addition of ATP produced significant quenching of the fluorene unit emission at 410 nm, and the appearance of the BT unit emission at 540 nm, with a detection limit of 40 nM. P19 demonstrated an obvious emission color change from blue to green. This distinct shift in emission color is characteristic of the aggregation of the main chains of P19. P20 displayed similar changes upon the addition of Arg6 with a detection limit of 10 nM.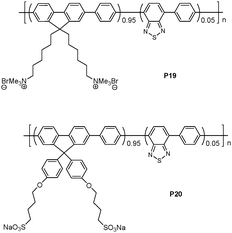 | ||
| Fig. 14 Chemical structures of P19 and P20. | ||
A new water-soluble, cationic polyfluorene bearing, boronate-protected fluorescein (peroxyfluor-1) was synthesized as a fluorescence probe to optically detect hydrogen peroxide (H2O2) and glucose in serum (Fig. 15).27 The peroxyfluor-1 exists as a lactone form, which is colorless and non-fluorescent. Since the fluorescence resonance energy transfer (FRET) from fluorene units (donor) to peroxyfluor-1 (acceptor) was absent, without addition of H2O2, only blue donor emission was observed upon excitation of the fluorene units. In the presence of H2O2, the peroxyfluor-1 reacted with H2O2 to deprotect the boronate protecting groups, and generated green fluorescent fluoresceinvia the efficient FRET from the fluorene units to the fluorescein. The P21 probe could detect H2O2 in the range from 4.4 to 530 mM. Since glucose oxidases (GOx) can specifically catalyze the oxidation of β-D-(+)-glucose to generate H2O2, glucose could be detected using the PF-FB probe.
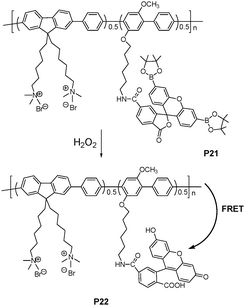 | ||
| Fig. 15 Schematic representation of H2O2 assay. | ||
A highly selective assay method was developed to detect Hg2+ using cationic conjugated polymer (CCP) (Fig. 16) by Wang et al.28 In the absence of Hg2+ ions, the CCP can form a complex with an anionic 1,3-dithiole-2-thione derivative through electrostatic interactions. The fluorescence of CCP was efficiently quenched by 1,3-dithiole-2-thione derivative via an electron transfer process. Upon adding Hg2+ ions, the transformation of 1,3-dithiole-2-thione into 1,3-dithiole-2-one inhibited the quenching, and the fluorescence of CCP was recovered. Distinguishing aspects of this assay included the signal amplification of CCPs and a specific Hg2+ promoted reaction. The detection limit was reported as 0.5 μM.
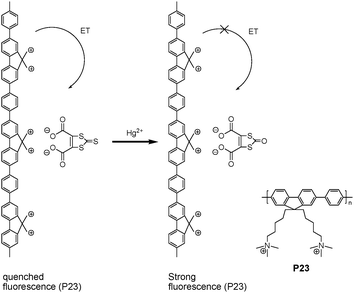 | ||
| Fig. 16 Representation of the Hg2+ assay and structure of P23. | ||
The incorporation of porphyrin into PFE backbone (P24a-d) displayed dual emissive polyelectrolytes with both blue and red emission bands resulting from the incomplete intramolecular energy transfer from the fluoreneethynylene moieties to the porphyrin units (Fig. 17).29 Upon the addition of Hg2+, blue emission from the fluoreneethynylene moieties and red emission from the porphyrin units were quenched, and the quenching in red emission was significantly larger than that of blue emission. The fluorescence quenching of porphyrin for P24c showed a linear response to Hg2+ in the concentration range of 0–100 μM, with a detection limit of 0.1 μM. In addition, the fluorescent color change of the polymer solution allows naked-eye detection of Hg2+ with a detection limit of 10 μM.
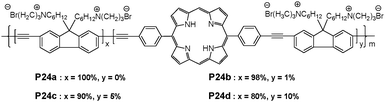 | ||
| Fig. 17 Chemical structures of porphyrin-containing poly-fluoreneethynylene. | ||
An anionic water-soluble polymer P25 (Fig. 18) has been synthesized by Suzuki coupling polymerization, and displays an extremely high solubility in water (100 mg mL−1) and high PL quantum yield (89%).30 P25 displayed a highly sensitive and selective fluorescence quenching effect with Hg2+ at pH 6.2, with a detection limit of 10 nM, which is the EPA limit of Hg2+ ions in drinking water. An 1H NMR study implied that all the amide groups in the two neighboring fluorene units were equally coordinated with Hg2+.
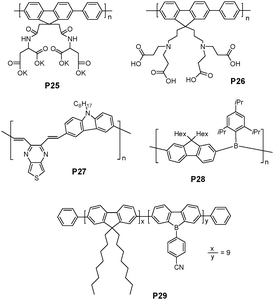 | ||
| Fig. 18 Chemical structures of P25-29. | ||
A highly pH sensitive water-soluble polyfluorene derivative (P26 in Fig. 18) was recently reported by Wang et al.31 The polymer charge could control electrostatic repulsion between polymer chains, leading to different levels of aggregation behaviors. At low pH, the protonation of carboxylic groups made the P26 charge positive, which leads to the red-shift in the absorption band due to interchain π–π interactions. However, the aggregates were broken upon increasing pH of the aqueous polymer solution. The decreased interchain contacts could lead to a decrease in the interchain π–π interactions, thus resulting in a blue shift in the absorption maximum. Different fluorescent responses of P26 were demonstrated when the environmental pH was changed from 3 to 12. Since boronic acid is a weak Lewis acid, which is more basic than boronic ester, the resulting pH is decreased when sugars bind to boronic acid to form boronic esters. D-Fructose could be detected with a detection limit of 2 mM.
Li et al. has recently reported that a donor-π-acceptor (D-π-A) alternative copolymer of carbazole and thieno[3,4b]-pyrazine (P27 in Fig. 18) displayed two absorption peaks at 306 and 452 nm, respectively, and the PL spectrum of the polymer solution displays its maximum at 543 nm in dilute THF solution.32 Upon addition of Hg2+ into its THF solution (containing 0.3% CH3CN), P27 exhibited a selective new absorption peak at 560–600 nm and its emission was dramatically quenched. The Hg2+ detection limit with the polymer solution using emission quenching was found to be 0.1 μM. P27 also showed a selective chromogenic behavior toward Hg2+ in solution, with a color change from yellow to blue dark, which can be detected with the naked eye.
Jäkle et al. reported a new blue-violet luminescent poly(fluorenylene borane) P28 (Fig. 18), in which electron-deficient borane moieties alternate with dihexylfluorene moieties in the polymer main chain.33 Binding studies with P28 indicate that both fluoride and cyanide, but not other anions such as chloride or bromide, can effectively bind to P28, leading to distinct changes in the absorption and emission spectra in THF. The authors suggested a two-step binding process based on the changes in absorption and emission. Upon the addition of these anions, the emission intensity at 403 and 425 nm was initially decreased, and a broad red-shifted band was developed at 475 nm, which could be attributed to the charge-transfer structure with alternating electron-rich borate (e.g.fluoroborate moieties) and electron-deficient borane moieties in the polymer main chain.
Recently, a random conjugated dialkylfluorene/dibenzoborole copolymer P29 (Fig. 18), containing dibenzoborole building blocks has been reported.34 Among a variety of anions, fluoride/cyanide and iodide addition induced photoluminescence quenching effects both in THF solutions and drop-cast thin films. In the solid-state quenching experiments, fluoride and cyanide could be detected down to a concentration of ∼0.1 mM. The lack of selectivity between cyanide and fluoride may be of secondary importance when concerning a potential application in the detection of toxic gases and their decomposition products (e.g., sarinvs. tabun).
The affinity of cyanide anion to copper has attracted special attention.35 Generally, cyanide can react with copper ions to form stable [Cu(CN)x]n− species. Li and coworkers extended this strategy in the design of imidazole-functionalized polyfluorene P30 (Fig. 19) as a selective cyanide chemosensor.36 The fluorescence of P30 was quenched completely by Cu2+ at concentrations as low as 0.20 ppm. The quenched fluorescence in a solution of P30 and Cu2+ could be recovered upon the addition of cyanide, with a minimum detection limit of 0.31 ppm.
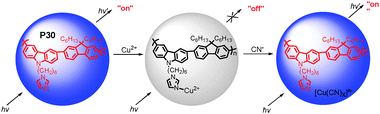 | ||
| Fig. 19 Schematic representation of cyanide sensor based on the fluorescence “turn-off” and “turn-on” of P30. | ||
3.2 Poly(p-phenyleneethynylene) (PPE) backbone
Polyamines were proposed to be possible biochemical markers for malignant tumors. Swager et al. reported an anthryl-doped, polyanionic poly(p-phenylene ethynylene) and its spectroscopic behavior under various aggregation conditions (Fig. 20).37 Multicationic amines (spermine, spermidine, and neomycin) were found to effectively induce the formation of tightly associated aggregates between PPE chains in 50:50 EtOH–H2O. The induced aggregation of conjugated polymer chains resulted in enhanced exciton migration from the blue-emitting PPE segments to the green-emitting anthryl units, leading to a visually noticeable blue-to-green fluorescence color change. The normalized fluorescence spectra clearly exhibited the aggregation-induced enhancement in the green emission band (510 nm) with respect to the blue emission band (430–445 nm). The threshold concentrations of spermine (0.69 μM) and spermidine (1.6 μM) to induce a fluorescence color change in anionic-PPE might be low enough to develop an assay for detecting the urinary concentration differences between cancer patients and healthy volunteers.
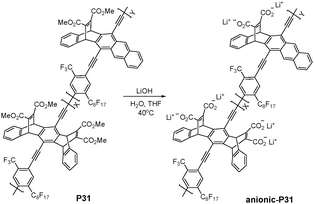 | ||
| Fig. 20 Chemical structure of P31. | ||
Bunz and Rotello et al. have recently developed a conjugated fluorescent polymer-based sensor array using PPE polymers (Fig. 21), and demonstrated its utility in cell sensing.38 Fluorescent conjugated polymers with pendant charged residues provided multivalent interactions with cell membranes, allowing the detection of subtle differences between different cell types on the basis of cell surface features. Analysis of the combined results indicated that four polymers (P32, P33, P34a, and P35a) out of the original eight, could effectively distinguish different mammalian cells. Since the cationic characteristics of P32, P33 and P34 were expected to interact with the negatively charged cell membrane, the least-charged anionic polymer (P35a) was demonstrated to be the most effective at detecting cell differentiation among P35–P37. Using this sensor array, they could distinguish between several cancer cell types as well as between isogenic healthy, cancerous, and metastatic cells that possess the same genetic background.
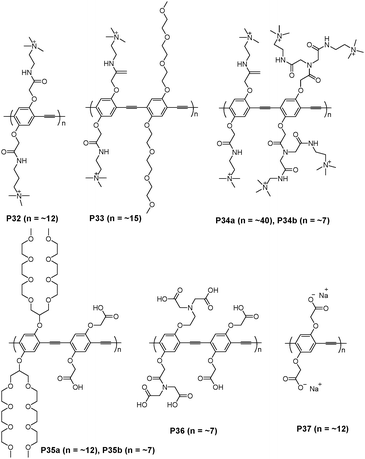 | ||
| Fig. 21 Chemical structures of P32–P37. | ||
Rotello and Bunz et al. have also demonstrated that noncovalent conjugates of gold nanoparticles and PPE can effectively identify bacteria (Fig. 22).39Nanoparticle-bacteria interactions could release the bound fluorescent polymer from the gold-nanoparticle quencher, thus turning on of the polymer fluorescence. The differential fluorescence responses generated by the bacterial surfaces provided a potentially powerful biodiagnostic tool, in which the functional nanoparticles and the fluorescent polymer could serve as the recognition elements and the transducer, respectively. The efficient quenching ability of gold nanoparticles, coupled with the molecular-wire effect of conjugated polymers, magnified the pronounced fluorescence response, and is indicative of the binding strength of the bacterium to the gold nanoparticle.
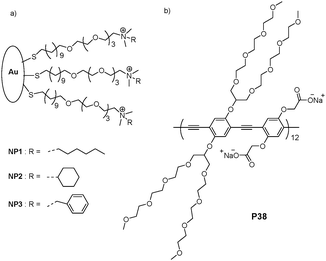 | ||
| Fig. 22 Receptor and transducer components of the Bacteria sensor. (a) Three cationic gold nanoparticles (NP1–NP3). (b) Chemical structure of the conjugated polymer (P38) featuring a branched oligo(ethylene glycol) side chain. | ||
Zhu et al. reported a dipyrido-[3,2-a:2,3-c]phenazine (dppz)-based metal ion-sensing conjugated polymer with high sensitivity and selectivity (P39 in Fig. 23).40 A selective fluorescent quenching effect was observed with Ni2+ among various metal ions in THF. The ability to completely quench the fluorescence of P39 by Ni2+ was attributed to the electron-transfer interactions between the conjugated polymer backbone and metal ion. In particular, a detection limit of 6.5 × 10−12 M has been achieved for Ni2+. An unprecedented picomolar sensitivity with P39 was attributed to a rigidly planar structure. Moreover, an instant color change from light red to colorless upon the addition of Ni2+ was observed for a THF solution of P39.
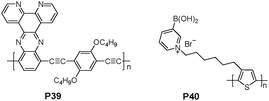 | ||
| Fig. 23 Chemical structures of P39 and P40. | ||
3.3 Polythiophene backbone
Boronic acids are well known to bind diol species with high affinities via reversible boronate formation in aqueous solution. Water-soluble regioregular head-to-tail zwitterionic fluorescent conjugated boronic acid-bearing polythiophene P40 (Fig. 23) was reported by Liu et al.41 In 4.0 mM of P40, buffered with 0.1 M phosphate buffer (pH 7.4), titration of monosaccharides, lactose, ascorbic acid, or dopamine resulted in significant concentration-dependent quenching of the polymer fluorescence.Semifluorinated polymer (P41 in Fig. 24), incorporating π-conjugated diphenylbithiophene and a distyrylbenzene derivative as chromophoric main chain subunits, has been explored as an anion sensor.42P41 exhibited visually identifiable colorimetric changes and photoluminescence enhancement in response to fluoride or cyanideversus other common anions in THF. A strong colorimetric change of P41 solutions from pale yellow to orange was observed only in the presence of fluoride or cyanide. It was reported that a rigid, electron-withdrawing FP and a chromophore capable of acting as a hydrogen bond donor were required for efficient sensing. Modest dissociation constants of 4.5 × 10−5 and 6.7 × 10−3 M were determined for binding of fluoride and cyanide, respectively.
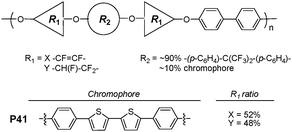 | ||
| Fig. 24 Chemical structure of P41. | ||
Lee and co-workers demonstrated that conjugated polymeric materials, with boron–oxygen linkages (P42a and P42b), can be used to detect different aliphatic and aromatic amines (Fig. 25).43 An efficient cyclocondensation with dihydroxysilane converted simple arylboronic acids to bifunctional borasiloxane cage molecules, which were subsequently electropolymerized to furnish air-stable thin films. This boron-containing polymer displayed a rapid and reversible color change from green to orange upon exposure to volatile amine samples under ambient conditions. This direct, naked-eye detection was explained by invoking the reversible B–N bond formation that could profoundly influence the p–π* orbital overlap. The process was reversed by addition of B(C6F5)3 as a stronger Lewis acid, which was able to effectively compete for amine binding.
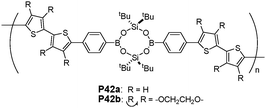 | ||
| Fig. 25 Chemical structures of P42a and P42b. | ||
Tang et al. reported hyperbranched poly(2,5-silole)s [hb-P1(m), m = 1, 6] (Fig. 26)44 which showed unusual phenomena of aggregation-induced (AIE) and -enhanced emissions (AEE). The hyperbranched polymers cam function as sensitive fluorescent chemosensors for the detection of explosives, with a superamplification effect observed in the emission quenching of the polymer nanoaggregates by picric acid. A related review paper regarding aggregation-induced emission was also reported by the same group.45
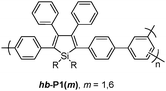 | ||
| Fig. 26 Chemical structures of hb-P1(m). | ||
3.4 PDA backbone
Polydiacetylenes (PDAs), a family of conjugated polymers, are becoming very intriguing materials. PDAs, in many cases, display an intense blue color, which can be generally prepared by UV irradiation of self-assembled diacetylene (DA) supramolecules. These blue PDAs undergo a color shift to a red phase upon environmental stimulation, which is known to be accompanied by the generation of fluorescence. The stimulus-induced apparent blue-to-red transition of PDAs has led to the development of a variety of PDA-based chemosensors.7Yoon and Kim et al. have developed a conceptually novel PDA-based chemosensor system for the detection of cationic surfactants (CS) in aqueous solution (Fig. 27).46 Among the various analytes examined, cetyltrimethylammonium chloride (CTAC) induced a unique colorimetric change (blue to red) and large fluorescent enhancement. The colorimetric and fluorometric responses of conjugated polymers were attributed to the disruption of the headgroup hydrogen bonding, for which both the ammonium group and long alkyl chain were required.
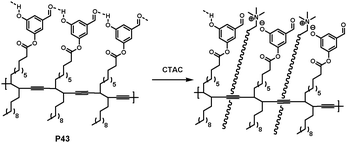 | ||
| Fig. 27 Proposed headgroup structures of PDAs derived from P43 in the presence of CTAC. | ||
Yoon and Lee et al. reported an anionic surfactant-selective sensor (P44 in Fig. 28) based on PDA and imidazolium, which could exhibit fluorescent enhancements and colorimetric changes in the presence of a variety of anionic surfactants.47 Remarkably, using the simple colorimetric changes, this system easily distinguished two different anionic surfactants, sodium dodecyl sulfate (SDS) and sodium dodecylbenzenesulfonic acid (SDBS). P44 is also the first example to show dual blue-to-yellow and blue-to-red transitions, which can be tuned by headgroup shape and alkyl length of analytes. A blue to red transition is observed when the extent of binding and penetration is low, on the other hand, a larger extent of binding and penetration leads to a blue-to-yellow transition. Using the fluorogenic method, the detection limit for SDS was reported to be 56 ppb.
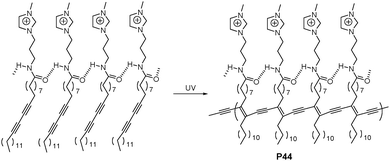 | ||
| Fig. 28 Chemical scheme of P44. | ||
König et al. reported that blue PDA vesicles of a uniform size of ∼200 nm were obtained upon UV irradiation from mono-(P45) and dinuclear Zn2+-cyclen (P46) (cyclen = 1,4,7,10-tetraazacyclododecane) and Cu2+-IDA modified diacetylenes (P47) (IDA = iminodiacetato), embedded in amphiphilic diacetylene monomers (Fig. 29).48 The Zn2+-cyclen based particles responded to the presence of ATP and PPi (pyrophosphate) in buffered aqueous solution (pH 7.2) by visible changes of their color (blue to red) and fluorescence enhancements. On the other hand, the Cu2+-IDA modified vesicular PDA receptors showed a selective response to PPi, allowing a simple colorimetric and visible discrimination between ATP and PPi. The induced aggregation of liposomes by the specific binding of ATP or PPi analytes to the surface receptors could be confirmed from the origin of the intensive changes in their optical properties.
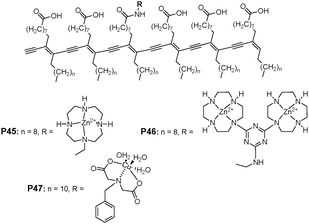 | ||
| Fig. 29 Structure of mono- (P45) and dinuclear Zn2+-cyclen (P46) and Cu2+-IDA modified diacetylenes (P47). | ||
König et al. reported self-assembled poly diacetylene (PDA) based blue vesicles LS-Terpy, LS-DPA, LS-DP and LS-DEA with metal chelating sites (Fig. 30).49 Among the various metal ions, the addition of Zn2+, Mn2+, Cd2+, Hg2+ or Ag+ salts to solutions of the vesicles induced a color change from blue to red, observable by the naked eye at pH 7.2. The order of metal ion selectivity for blue vesicle LS-Terpy was Zn2+ > Cd2+ > Hg2+. In vesicle solution, Zn2+ ions displayed a fast response time (∼1 min) with a detection limit of 2.1 μM. On the other hand, LS-DPA displayed selectivity for Mn2+ and Zn2+, and LS-DEA displayed selectivity for Hg2+ and Ag+. The metal ion coordination selectivity to ligands in the vesicle surface immobilized ligands was slightly different from the reported metal cation binding of the corresponding ligands in solution, which could be attributed to the specific environment at the lipid-solution interface.
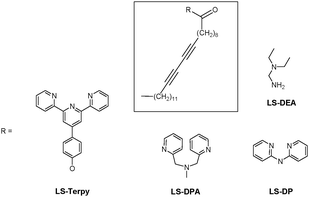 | ||
| Fig. 30 Chemical structure of LS-Terpy, LS-DPA, LS-DP and LS-DEA. | ||
Kim et al. have developed a highly selective PDA liposome-based sensory system to detect K+, even in the presence of Na+, via the color change from the blue phase to the red phase of the PDA liposome and red fluorescence emission (Fig. 31).50 The tethered G-rich ssDNAs at the liposome surface provided a high response by selectively wrapping around K+ to form quadruplexes. The steric hindrance of bulky quadruplexes induced ene-yne backbone perturbation of PDA liposomes. They also demonstrated the possible quantitative analysis of K+ concentration by using the PDA liposome microarray
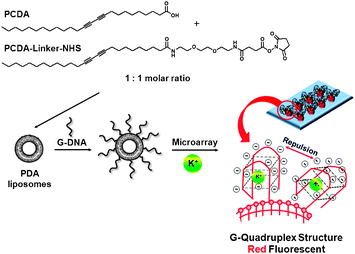 | ||
| Fig. 31 Chemical structure of the diacetylene monomers and a schematic representation of the PDA liposome-based microarray for K+ detection. | ||
Kim and Song et al. have demonstrated that α-cyclodextrin (α-CD) can form inclusion complexes with PDAs in PDA-based sensor systems (P48 in Fig. 32).51 Among α-, β- and γ-CDs, only α-CD can induce a selective blue to red colorimetric transition. They demonstrated that the molecular recognition event(s) occurring between CD and PDA served as good model for ligand–receptor interactions in PDA-based microfluidic sensor systems. Relatively strong fluorescence bands were observed when the flows of PDA and α-CD solutions come into contact.
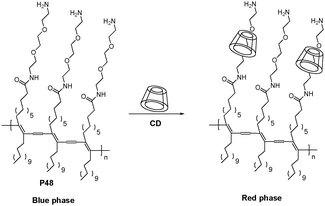 | ||
| Fig. 32 Schematic representation of the interaction between P48 and cyclodextrin. | ||
Li et al. reported the light-emitting polyacetylene bearing imidazole moieties as a cyanide sensor (Fig. 33), in which the affinity of this anion for copper was utilized.52 The polymeric sensor exhibited selective fluorescence quenching in the presence of Cu2+. The Cu2+-promoted luminescence quenching could be inhibited by addition of cyanide.
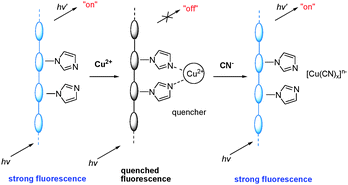 | ||
| Fig. 33 Schematic representation of Cu2+ and cyanide sensors based on the fluorescence ‘‘turn-off’’ and ‘‘turn-on’’ of polyacetylenes. | ||
Li et al. also reported a new imidazole-functionalized disubstituted polyacetylene(P49, Fig. 34)53 which was utilized to sense copper ions and α-amino acids. The quenching of the fluorescence of the imidazole-functionalized disubstituted polyacetylene was observed at a very low level of Cu2+ (0.7 μM). The addition of α-amino acids to the solution of the polyacetylene/Cu2+ complex could turn on the fluorescence of the polyacetylene, in which α-amino acids could remove the copper ions from the complex. For example, on the addition of glycine, the quenched fluorescence of P1 turned on immediately with a detection limit of 60 μM.
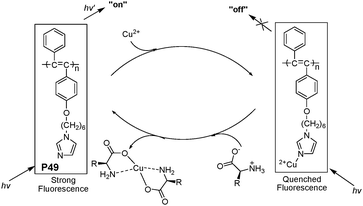 | ||
| Fig. 34 The speculated conversion cycle of P49 in the presence of Cu2+ and α-amino acids. | ||
3.5 Others
Cheng et al. recently reported a chiral polymer P50 (Fig. 35), which displayed orange fluorescence due to the extended π-electronic structure between a binaphthyl unit and a benzo[2,1,3]thiadiazole (BT) groupviaethynyl bridge.54 The fluorescence of the chiral polymer can produce a highly sensitive and selective enhancement with Hg2+ in THF.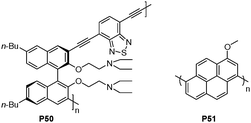 | ||
| Fig. 35 Chemical structures of P50 and P51. | ||
A highly fluorescent conjugated oligomer, oligo(1-methoxyl pyrene) (P51 in Fig. 35), was synthesized via electrochemical polymerization by Shi et al.55 The organic solutions of P51 emit strong blue lights and show a solvatochromic effect, while these fluorescent emissions are quenched upon chain aggregation in its dry films. On the basis of these phenomena, the turn-on fluorescence of a P51 thin film was used for detecting volatile aromatic compounds (VACs), such as toluene, which was attributed to the strong π–π interaction between the pyrenyl rings of P51 and the aromatic solvent molecules.
Tang et al. reported TPE-containing polytriazoles(P52, Fig. 36)56 with high molecular weights in high yields by the Cu(I)-catalyzed click polymerization. Utilizing the novel AIE effect, the polytriazoles were used as chemosensors for the detection of explosives in the aggregate and solid states.
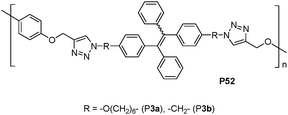 | ||
| Fig. 36 Chemical structures of P52. | ||
4. Dendrimer-based sensors
Dendrimers, the most recently recognized members of the polymer family, have become fascinating molecular probes due to their distinct morphology and unique characteristics. Dendrimers are composed of multiple perfectly-branched monomers that emanate from a central core with predictable three-dimensional shapes. The dendrimer properties can be controlled by rational design of functional units in predetermined sites on their nanometric dimension structures.57Calixarenes have been chosen as one of the building blocks to prepare hyperbranched supramolecules and dendrimers as fluorescent probes.58 Due to the specific core-shell architecture of dendrimers, the separation of receptors and signalling moieties can enable further sophisticated systems, through the construction of various photoreactive fluorescent antennae. As shown in Fig. 37, Kim and Vicens et al.58 have recently extended a series of luminescent dendrimer-based sensors derived from pyreneamide-appended calix[4]arenes. Multidentate ligands based on a tripodal tris(2-aminoethyl)amine moiety (tren) were branched with calixarenes to construct Y-shaped calix-dendrimers. As was the case with the response to two metal cations (Al3+ and Hg2+), a rhodamine-dipyrenyl N-tren-di-calix fluorescent sensor P53 tuned its photoproperties through different processes, such as photoinduced electron transfer (PET) and fluorescence resonance energy transfer (FRET).59 Upon addition of Hg2+ to CH3CN solution, P53 exhibited a significantly enhanced fluorescence of the rhodamine unit (FRET-ON) by the energy transfer from the pyrene excimer to the ring-open rhodamine-B, while the addition of Al3+ induced a distinct increase of pyrene excimer emission with no FRET-ON phenomena (Fig. 37).
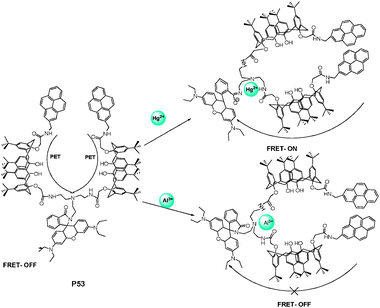 | ||
| Fig. 37 Scheme of different complexation behavior of P53 with Hg2+ and Al3+. | ||
The architecture of hyperbranched polymers provides a greater number of possible exciton migration pathways to enhance the sensitivity of a receptor to an analyte. Yang et al.60 reported a novel hyperbranched copolymer P54 produced by Heck coupling reaction of divinyl bipyridyl with tris(4-bromophenyl)amine (Fig. 38). The coordination ability of metal ions with bipyridyl moieties had great effect on emission spectra. The energy or electron transfer between metal complexes and polymer backbone was dependent upon the coordination ability of metal ions with bipyridyl moieties. The interaction of P54 with Co2+ and Ni2+ resulted in complete quenching of fluorescence due to their strong affinity with bipyridyl moieties, while partially decreasing fluorescence with Cr2+, Mn2+ and Fe3+ in THF solution.
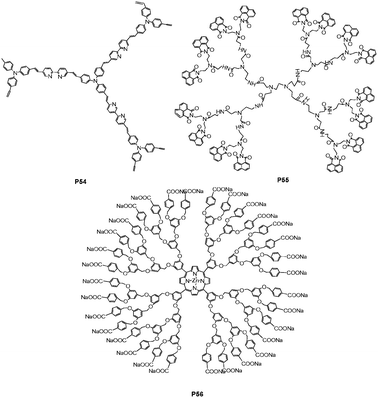 | ||
| Fig. 38 Chemical structures of P54, P55 and P56. | ||
The uniqueness of poly(amidoamine) dendrimers is their interiors, containing both secondary (amide) and tertiary amines functions, while their peripheries are functionalized with various functional fluorophore units. Yang et al.61 reported the second generation of polyamidoamine dendrimer P54 (Fig. 38), functionalized in the periphery using 1,8-naphthalimide as luminescent units for sensing rare earth ions. Emission spectra of P55 exhibited two maximum peaks at ca. 478 and 384 nm. The former, at the peak of 478 nm, was attributed to the excimer formed from adjacent parallel-oriented naphthalimide chromophores of dendrimers, and the latter, (ca. 384 nm), was assigned to the emission of naphthalimide. The internal amido groups in the dendrimer can coordinate with the earth metals (Er3+, Tb3+, Nb3+, Eu3+, Yb3+ and Gd3+) and H+ ion, resulting in the fluorescence enhancement at 478 nm, due to the formation change of naphthalimide chromophores.
Dendrimers have a number of beneficial characteristics for biological applications, such as being drug carriers and imaging agents. Koh et al.62 reported a new type of multi-functional dendrimer-coated surface for protein immobilization and detection of protein activity. The silicon surface was first modified with positively charged amino groups using 3-aminopropyltriethoxysilane (APTES), and subsequently coated with ionic dendrimer porphyrin (P56) by electrostatic interaction (Fig. 38). On dendrimer-modified surfaces, three dimensional architectures with numerous functional groups can exhibit a high capacity for the covalent binding of protein and have the flexible nature of branched arms on the dendrimer for minimizing protein denaturation. Large dendritic wedges of P56 can prevent the collision quenching of the core porphyrin, while avoiding porphyrin self-aggregation from the strong π–π and hydrophobic interaction. More importantly, by using the fluorescent property of the porphyrin core, the relative amounts and activities of proteins immobilized on the dendrimer-coated surface can be examined by fluorescence microscopy.
5. Imprinted polymer-based sensors
Imprinted polymer materials are prepared by template-directed synthesis. The templates (imprinted molecules) with attached (covalently or noncovalently) functional monomers are copolymerized in the presence of cross-linking agents, resulting in the formation of a polymer network around the template. The imprinted molecules are subsequently removed from binding sites that are complementary in size, shape and functionality to analytes. The intermolecular forces among the template molecule, functional monomer, and polymer matrix can develop a microenvironment to retain the imprint process. The “molecular memory” character can exhibit specific binding sites to develop imprinted polymer-based sensors.Narayanaswamy and Ng63 reported a molecularly imprinted polymer (MIP, P57 in Fig. 39) containing a 8-hydroxyquinoline sulfonic acid ligand as the fluorescence tag for sensing Al3+ in an aqueous medium. The template molecule of Al3+ can be leached from the MIP by NaF to form specific sensing sites. P55 showed a selective off–on fluorescent signal with Al3+ by flow-cell analysis, indicative of linear response up to 1.0 × 10−4 M, with a detection limit of 3.62 μM in aqueous media.
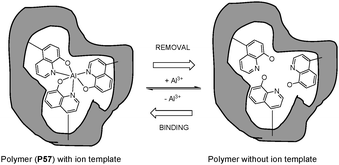 | ||
| Fig. 39 Scheme of imprinted process for Al-imprinted polymer. | ||
Imprinted materials possessing relatively low affinity and high levels of cross-reactivity toward similar species can become advantageous for differential sensor arrays. Signals are generated for an analyte from one or more imprinted materials, thus obtaining a characteristic finger print for pattern recognition using mathematical multivariate analyses. Yan et al.64 designed a series of fluorescent indicator-displacement molecular imprinting sensor arrays based on functionalized mesoporous silica. In the imprinting sensor array, two imprinted materials (P58 and P59), based on phenylboronic acid functionalized mesoporous silica, and a control blank nonimprinted material, were applied to distinguish and identify 10 saccharides by their unique fingerprints (Fig. 40). The synthetic processes for the above imprinted mesoporous silica used for saccharide sensor arrays were very convenient. The saccharide-imprinted mesoporous materials used for sensor array can successfully distinguish 10 saccharides at 100 mM by identifying unique characteristic fingerprints for individual saccharides based on the patterns of fluorescent signals.
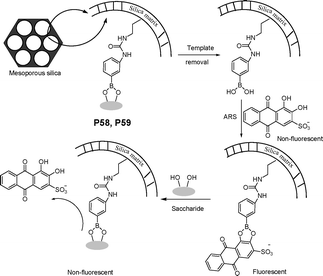 | ||
| Fig. 40 Scheme of molecularly imprinted mesoporous silica as indicator-displacement sensor for saccharides. | ||
6. Concluding remarks
In this review, recent progress in the field of fluorescent and colorimetric polymer chemosensors (mainly from 2007 to 2010) based on various polymer backbone and receptor types has been described. Polymer sensors bearing fluorescent recognition units as pendant ligands present a diverse and highly functional approach to the design of more active and specific hydrophilic film polymer sensors, and make possible a new heterogeneous sensor based on the blending of supramolecular concepts with mesoporous inorganic solids. However, instant identification and production of sensitive target components for film polymer sensors and miniature devices for mesoporous polymeric sensors remains a great challenge. More research efforts are needed to accelerate the availability of durable and cheap artificial polymer sensors for human consumption.Due to signal amplification abilities of CPs, water soluble conjugated polymers have been intensively studied as new fluorescent platforms for sensitive biosensors. The excitation energy along the entire backbone of CPs, while transferring to lower energy electron/energy acceptor sites over long distances, results in an amplified fluorescence signal, and makes them ideal materials for chemical and biological sensors.
Even though dendrimer size being conformation controllable, high structural and chemical homogeneity, and high functional ligand density are beneficial attributes for their biomedical applications, the multistep syntheses and purification of dendrimers still present obstacles for dendritic polymer applications. Imprinted polymer sensors have obvious advantages in fabricating sensor arrays due to low over all affinity and high level of cross-reactivity toward structurally similar species. Great efforts towards identifying combinations of MIP arrays with appropriate chemometrics for developing integrated sensors (an artificial nose and an artificial tongue) is of paramount importance for pattern recognition.
Considering the polymer sensors reviewed here, combining different sensing concepts, based on more than one analyte-recognition principle for the design of polymer sensors, may be a well-practiced approach to improve sensor performance. Clearly, the field of polymer-based optical chemosensors is far from mature, and great collaboration of polymer and supramolecular scientists will provide impetus for exciting new discoveries and commercial technologies in the fields of environmental sensing and healthcare.
Acknowledgements
J. Yoon acknowledges National Research Foundation (NRF) grant (2010-0018895, F-01-2008-000-10026-0) and WCU (R31-2008-000-10010-0). H. Tian and W. Zhu gratefully acknowledge the financial support from NSFC/China, National Basic Research 973 Program (2006CB806200) and the Program for Professor of Special Appointment at Shanghai Institutions of Higher Learning. Thanks goes to all coworkers who worked on chemosensors at ECUST and Ewha.Notes and references
- T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094 CrossRef CAS.
- X. Chen, Y. Zhou, X. Peng and J. Yoon, Chem. Soc. Rev., 2010, 39, 2120 RSC.
- Z. Xu, S. K. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 1457 RSC.
- A. P. de Silva, H. Q. N. Gunaratne, T. A. Gunnlaugsson, T. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef.
- S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS.
- X. Feng, L. Liu, S. Wang and D. Zhu, Chem. Soc. Rev., 2010, 39, 2411 RSC.
- D. J. Ahn and J. M. Kim, Acc. Chem. Res., 2008, 41, 805 CrossRef CAS.
- N. Li, Q. Xu, X. Xia, L. Wang, J. Lu and X. Wen, Mater. Chem. Phys., 2009, 114, 339 CrossRef CAS.
- B. Wang, X. Liu, Y. Hu and Z. Su, Polym. Int., 2009, 58, 703 CrossRef CAS.
- Z. Q. Guo, W. H. Zhu and H. Tian, Macromolecules, 2010, 43, 739 CrossRef CAS.
- J. Hu, C. Li and S. Liu, Langmuir, 2010, 26, 724 CrossRef CAS.
- J. Jiang, X. Xiao, P. Zhao and H. Tian, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1551 CrossRef CAS.
- P. Zhao, J. Jiang, B. Leng and H. Tian, Macromol. Rapid Commun., 2009, 30, 1715 CrossRef CAS.
- S. Ghosh and C. K. Dey, Supramol. Chem., 2009, 21, 591 CrossRef CAS.
- T. Hyakutake, I. Okura, K. Asai and H. Nishide, J. Mater. Chem., 2008, 18, 917 RSC.
- S. J. Payne, G. L. Fiore, C. L. Fraser and J. N. Demas, Anal. Chem., 2010, 82, 917 CrossRef CAS.
- Y. Tian, B. R. Shumway and D. R. Meldrum, Chem. Mater., 2010, 22, 2069 CrossRef CAS.
- H. J. Kim, S. J. Lee, S. Y. Park, J. H. Jung and J. S. Kim, Adv. Mater., 2008, 20, 3229 CrossRef CAS.
- B. Leng, J. Jiang and H. Tian, AIChE J., 2010, 56, 2957 CrossRef CAS.
- B. Leng, L. Zou, J. Jiang and H. Tian, Sens. Actuators, B, 2009, 140, 162 CrossRef.
- S. Seo, H. Y. Lee, M. Park, J. M. Lim, D. Kang, J. Yoon and J. H. Jung, Eur. J. Inorg. Chem., 2010, 843 CrossRef CAS.
- Q. Meng, X. Zhang, C. He, G. He, P. Zhou and C. Duan, Adv. Funct. Mater., 2010, 20, 1903 CrossRef CAS.
- K. Sarkar, K. Dhara, M. Nandi, P. Roy, A. Bhaumik and P. Banerjee, Adv. Funct. Mater., 2009, 19, 223 CrossRef CAS.
- M. Comes, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, L. A. Villaescusa, A. Graefeb and G. J. Mohr, J. Mater. Chem., 2008, 18, 5815 RSC.
- N. Singh, N. Kaur, J. Dunn, R. Behan, R. C. Mulrooney and J. F. Callan, Eur. Polym. J., 2009, 45, 272 CrossRef CAS.
- L. An, Y. Tang, F. Feng, F. He and S. Wang, J. Mater. Chem., 2007, 17, 4147 RSC.
- F. He, F. Feng, S. Wang, Y. Li and D. Zhu, J. Mater. Chem., 2007, 17, 3702 RSC.
- H. Lu, Y. Tang, W. Xu, D. Zhang, S. Wang and D. Zhu, Macromol. Rapid Commun., 2008, 29, 1467 CrossRef CAS.
- Z. Fang, K. Y. Pu and B. Liu, Macromolecules, 2008, 41, 8380 CrossRef CAS.
- C. Qin, X. Wu, B. Gao, H. Tong and L. Wang, Macromolecules, 2009, 42, 5427 CrossRef CAS.
- Q. Xu, L. An, M. Yu and S. Wang, Macromol. Rapid Commun., 2008, 29, 390 CrossRef CAS.
- Y. Zou, M. Wan, G. Sang, M. Ye and Y. Li, Adv. Funct. Mater., 2008, 18, 2724 CrossRef CAS.
- H. Li and F. Jäkle, Angew. Chem., Int. Ed., 2009, 48, 2313 CAS.
- V. D. B. Bonifácio, J. Morgado and U. Scherf, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2878 CrossRef CAS.
- Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127 RSC.
- Z. Li, X. Lou, H. Yu, Z. Li and J. Qin, Macromolecules, 2008, 41, 7433 CrossRef CAS.
- A. Satrijo and T. M. Swager, J. Am. Chem. Soc., 2007, 129, 16020 CrossRef CAS.
- A. Bajaj, O. R. Miranda, R. Phillips, I.-B. Kim, D. J. Jerry, U. H. F. Bunz and V. M. Rotello, J. Am. Chem. Soc., 2010, 132, 1018 CrossRef CAS.
- R. L. Phillips, O. R. Miranda, C. C. You, V. M. Rotello and U. H. F. Bunz, Angew. Chem., Int. Ed., 2008, 47, 2590 CrossRef.
- X. Liu, X. Zhou, X. Shu and J. Zhu, Macromolecules, 2009, 42, 7634 CrossRef CAS.
- C. Xue, F. Cai and H. Liu, Chem.–Eur. J., 2008, 14, 1648 CrossRef CAS.
- R. J. Gilliard Jr., S. T. Iacono, S. M. Budy, J. D. Moody, D. W. Smith Jr. and R. C. Smith, Sens. Actuators, B, 2009, 143, 1 CrossRef.
- W. Liu, M. Pink and D. Lee, J. Am. Chem. Soc., 2009, 131, 8703 CrossRef CAS.
- J. Liu, Y. Zhong, J. W. Y. Lam, P. Lu, Y. Hong, Y. Yu, Y. Yue, M. Faisal, H. H. Y. Sung, I. D. Williams, K. S. Wong and B. Z. Tang, Macromolecules, 2010, 43, 4921 CrossRef CAS.
- J. Liu, J. W. Y. Lam and B. Z. Tang, J. Inorg. Organomet. Polym. Mater., 2009, 19, 249 CrossRef CAS.
- X. Chen, J. Lee, M. J. Jou, J.-M. Kim and J. Yoon, Chem. Commun., 2009, 3434 RSC.
- X. Chen, S. Kang, M. J. Kim, J. Kim, Y. S. Kim, H. Kim, B. Chi, S.-J. Kim, J. Y. Lee and J. Yoon, Angew. Chem. Int. Ed., 2010, 49, 1422 CAS.
- A. D. Jose, S. Stadlbauer and B. König, Chem.–Eur. J., 2009, 15, 7404 CrossRef CAS.
- A. D. Jose and B. König, Org. Biomol. Chem., 2010, 8, 655 RSC.
- J. Lee, H.-J. Kim and J. Kim, J. Am. Chem. Soc., 2008, 130, 5010 CrossRef CAS.
- S.-H. Eo, S. Song, B. Yoon and J.-M. Kim, Adv. Mater., 2008, 20, 1690 CrossRef CAS.
- Q. Zeng, P. Cai, Z. Li, J. Qina and B. Z. Tang, Chem. Commun., 2008, 1094 RSC.
- Q. Zeng, C. K. W. Jim, J. W. Y. Lam, Y. Dong, Z. Li, J. Qin and B. Z. Tang, Macromol. Rapid Commun., 2009, 30, 170 CrossRef CAS.
- X. Huang, Y. Xu, L. Zheng, J. Meng and Y. Cheng, Polymer, 2009, 50, 5996 CrossRef CAS.
- Y. Gao, H. Bai and G. Shi, J. Mater. Chem., 2010, 20, 2993 RSC.
- A. Qin, J. W. Y. Lam, L. Tang, C. K. W. Jim, H. Zhao, J. Sun and B. Z. Tang, Macromolecules, 2009, 42, 1421 CrossRef CAS.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857 CrossRef CAS.
- J. S. Kim, S. Y. Lee, J. Yoon and J. Vicens, Chem. Commun., 2009, 4791 RSC.
- A. B. Othman, J. W. Lee, J. Wu, J. S. Kim, R. Abidi, P. Thuéry, J. M. Strub, A. V. Dorsselaer and J. Vicens, J. Org. Chem., 2007, 72, 7634 CrossRef CAS.
- J. Feng, Y. Li and M. Yang, Eur. Polym. J., 2008, 44, 3314 CrossRef CAS.
- S. Yang, L. Lin, L. Yang, J. Chen, Q. Chen, D. Cao and X. Yu, J. Lumin., 2007, 126, 515 CrossRef CAS.
- Y. Lee, J. Kim, S. Kim, W. Jang, S. Parka and W. Koh, J. Mater. Chem., 2009, 19, 5643 RSC.
- S. M. Ng and R. Narayanaswamy, Anal. Bioanal. Chem., 2006, 386, 1235 CrossRef CAS.
- J. Tan, H. Wang and X. Yan, Anal. Chem., 2009, 81, 5273 CrossRef CAS.
Footnote |
| † Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |