Supramolecular amphiphiles
Xi
Zhang
* and
Chao
Wang
Key Lab of Organic Optoelectronics and Molecular Engineering, Department of Chemistry, Tsinghua University, Beijing, 100084, China. E-mail: xi@mail.tsinghua.edu.cn
First published on 1st October 2010
Abstract
Supramolecular amphiphiles (SA), also named superamphiphiles, refer to amphiphiles that are formed by non-covalent interactions. This tutorial review focuses on the molecular architectures of SAs, including diversified topologies such as single chain, double chain, bolaform, gemini and rotaxane types. Non-covalent syntheses that have been employed to fabricate SAs are driven by hydrogen bonding, electrostatic attraction, host–guest recognition, charge transfer interaction, metal coordination and so on. It should be noted that SAs can be either small organic molecules or polymers. SAs allow for tuning of their amphiphilicity in a reversible fashion, leading to controlled self-assembly and disassembly. This line of research has been enriching traditional colloid chemistry and current supramolecular chemistry, and the application of SAs in the field of functional supramolecular materials is keenly anticipated.
 Xi Zhang | Xi Zhang is a professor of chemistry at Tsinghua University. His research interests include controlled self-assembly and disassembly, organic molecular films, single-molecule force spectroscopy of polymers. In 2007, he was elected Member of Chinese Academy of Sciences. Currently, he serves as Senior Editor of Langmuir and advisory board members of several scientific journals, including Chemical Communications, Polymer Chemistry, Polymer and ACS-Applied Materials & Interfaces. |
 Chao Wang | Chao Wang got his BA in Department of Chemical Engineering, Tsinghua University. In 2007, he joined Prof. Xi Zhang’s group as a PhD student in Department of Chemistry at Tsinghua University. Currently, he is working on supramolecular amphiphiles for controlled self-assembly. |
Introduction
Conventionally, an amphiphile refers to a molecule that contains a hydrophilic part and a hydrophobic part, linked by covalent bonding. There are many kinds of amphiphiles in terms of the different linkages between the hydrophilic and hydrophobic parts. For example, a hydrophilic head group can be covalently linked to single, double or triple hydrophobic alkyl chains. Two hydrophilic head groups covalently linked by one hydrophobic alkyl chain result in so-called bolaform amphiphiles. Two surfactant molecules covalently linked at their charged head groups are termed a gemini amphiphile. Amphiphiles of different structures have different self-assembly behaviours in solution and at interfaces. The interest in these amphiphiles arises from their self-assembly in aqueous solution to form well-defined structures, such as micelles and vesicles, which can find applications in many fields such as nanodevices,1–4drug delivery5–8 and template synthesis.9–11In contrast to conventional amphiphiles, supramolecular amphiphiles (SA) are amphiphiles that are formed by non-covalent interactions.12 The rich topology of SAs is shown in Fig. 1. A hydrophobic building block bearing a hydrogen donor can interact with a hydrophilic hydrogen acceptor to form a single-chain SA on the basis of hydrogen bonding (1). A double-chain SA can be fabricated in the same way (2). Two routes to construction of a bolaform SA can be visualized. One is to hold two building blocks bearing two complementary interacting groups by using either hydrogen bonding or metal coordination (3). The other route is to introduce a bi-functional moiety to bridge the two building blocks (4). Based on electrostatic attraction, cationic surfactants can also be classified as a type of SA (5). A gemini amphiphile interacts with a suitable organic counter ion, leading to the formation of a gemini-type SA (6). Actually, building blocks with suitable molecular design can be used to fabricate gemini-type SAs of different structures on the basis of non-covalent interactions (7). Supramolecular host–guest systems can be intrinsically amphiphilic due to the opposing hydrophilicity of the host and guest (8). In general, the different topologies of covalently bonded amphiphiles can be realized in SAs on the basis of non-covalent bonds. Moreover, SAs can even construct topologies that are not available with conventional amphiphiles.
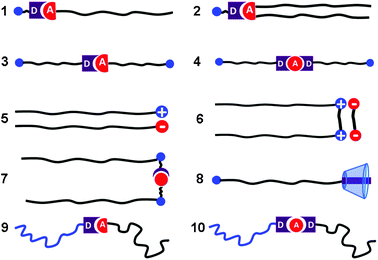 | ||
| Fig. 1 Different topologies of SAs. “D” and “A” refer to donor and acceptor, respectively. | ||
Non-covalent synthesis of SA
Non-covalent synthesis is regarded as a kind of self-assembly method that is very useful in the construction of chemical structures with a high degree of structural complexity.13 One of the advantages of non-covalent synthesis is the avoidance of tedious covalent synthesis procedures. SAs of different topologies and functions can be easily fabricated. As discussed below, various non-covalent interactions can be used for preparation of SAs. For the purposes of the following discussion, the preparation of SAs is classified by the type of driving force involved. It should be pointed out, however, that in most cases the formation of SAs is not driven by a single kind of interaction, but rather by the combined interactions of different forces, though one driving force may play the leading role.Hydrogen bonding
The use of hydrogen bonding for fabricating SA dates from the work of Kunitake and Kimizuka who employed substituted melamines and isocyanuric acid derivatives as molecular building blocks.14 As shown in Fig. 2, melamine and isocyanuric acid were chosen as complementary interacting groups because they can form extended hydrogen bonded arrays. The SA was prepared by mixing equimolar amounts of each building block in ethanol and removing the solvent under reduced pressure. This procedure was repeated three times to ensure complete complexation between the hydrogen bond donors and acceptors. The SA gave a transparent dispersion in water and showed a disk-like aggregate with highly ordered bilayer structures. Double-chain, single-chain15 and dendrimer-like SA16 can be prepared in a similar manner.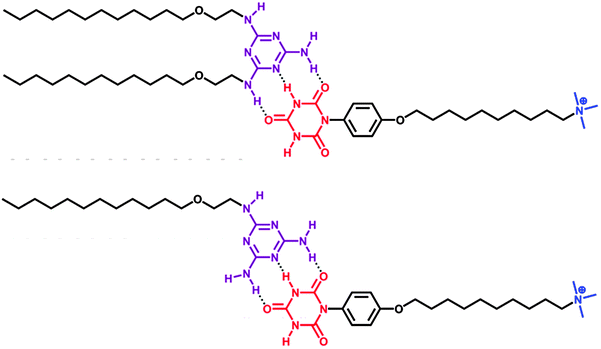 | ||
| Fig. 2 Double chain and single chain SAs based on hydrogen bonding.14–15 | ||
Host–guest recognition
Host–guest recognition can be used for the fabrication of SA. The host component, such as cyclodextrin (CD), is able to form inclusion complexes with guest components such as aliphatic molecules or azobenzene (Fig. 3). A typical example is the inclusion complex of β-CD and fatty alcohol or fatty acid, functioning as a nonionic SA, though it was referred to as a non-covalent amphiphile in the original publication.17 This non-covalent synthesis involved mixing an equimolar suspension of β-CD and fatty alcohol in aqueous solution followed by heating at 55 °C with mechanical stirring. The surface properties of these nonionic SAs are influenced by the length of the hydrocarbon chain, and optimal efficiency is reached for guests having 10–12 carbon atoms. In addition to nonionic SAs, ionic SAs can be prepared in a similar way. For example, β-CD containing nonionic amphiphile can bind adamantane carboxylic acid to form an ionic SA.18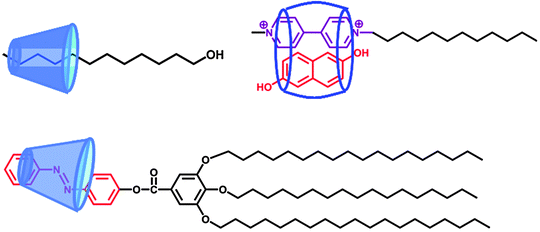 | ||
| Fig. 3 Different types of SAs based on host–guest interactions.17,19,20 | ||
This concept can be extended to fabrication of SA based on photo-controlled inclusion and exclusion of an azobenzene-containing surfactant with α-CD.19 Alternating irradiation with UV and visible light allows for reversible sliding of α-CD along the surfactant, leading to the formation of a rotaxane-like SA. Notably, such a rotaxane-like topology does not exist in the family of conventional surfactants.
Cucurbituril (CB), a well-known macrocyclic cavitand with very similar behaviour to that of CD, can be also used for construction of SA. CB[8] for example, a macrocyclic cavitand bearing eight glycoluril units, can form a stable ternary complex driven by charge transfer interaction between the electron-deficient host and electron-rich guest molecules. Kim et al. have employed the ternary complex to form SA. Their findings have shown, interestingly, that these SAs are able to self-assemble to form vesicles.20
Metal–ligand coordination
Metal–ligand coordination is another driving force for fabricating SAs. This non-covalent bond is stronger than most intermolecular interactions; however, its reversibility makes it function as a type of supramolecular bond. Schubert et al. have demonstrated the use of metal–ligand coordination as a linker for holding hydrophilic and hydrophobic blocks, giving rise to a supramolecular amphiphilic polymer.21 A typical example is shown in Fig. 4. Monochelic polymer end-capped with the 2,2′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6′,2′′-terpyridine ligand was selectively complexed with ruthenium ion and subsequently reacted with other terpyridine-terminated polymer blocks.
:6′,2′′-terpyridine ligand was selectively complexed with ruthenium ion and subsequently reacted with other terpyridine-terminated polymer blocks.
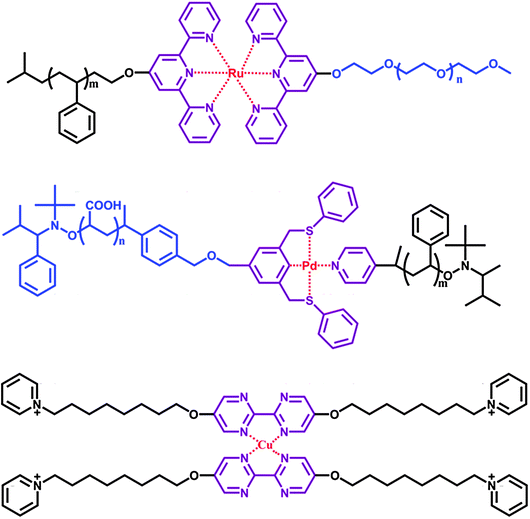
Various metal–ligand coordinations have been used to fabricate SAs, such as SCS “pincer”-Pd(II) complex,22 and bipyrimidine with Cu(II).23 In the latter case, when a ligand is introduced between the bolaform amphiphiles, a kind of H-type SA is constructed by complexing with copper ion. It has been shown that this H-type SA self-assembles to form clustered aggregates in water. When the metal–ligand coordination is ruptured, the clustered aggregates are disassembled simultaneously and form sphere-like structures.
Electrostatic attraction
The double hydrophilic diblock copolymer of poly(ethylene oxide) (PEO) and poly(sodium methacrylate) interacts with various single-tail cationic surfactants to form so-called block ionomer–surfactant complexes. As shown in Fig. 5, this complex should be regarded as a type of SA on the basis of electrostatic attraction, because the ionic head group of the surfactant is bound to the oppositely charged units of the polyion segment.24 It can self-assemble to form micelle-like aggregates in which the insoluble polymer-surfactant parts are stabilized in aqueous media by PEO chains.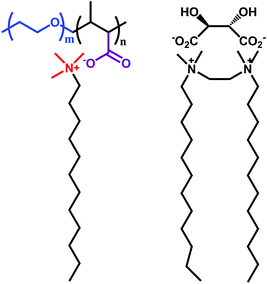 | ||
| Fig. 5 Ionic and gemini types of SAs based on electrostatic interactions.24–25 | ||
A cationic gemini surfactant with a chiral counterion behaves as a supramolecular gemini. When the counterion is L-tartrate, the supramolecular gemini forms gels in both water and some organic solvents by creating extended networks of multilamellar twisted ribbons.25 By introduction of opposite-handed chiral counterions in various proportions, the degree of twist and the pitch of the ribbons can be tuned.
Charge transfer interaction
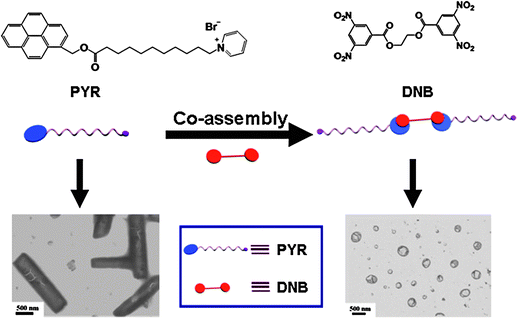
Charge transfer interaction between electron donors and acceptors provides another choice for fabrication of SAs.26 As shown in Fig. 6, two molecules of the amphiphilic electron donor 1-[11-oxo-11-(pyren-1-ylmethoxy)-undecyl]pyridinium bromide (PYR), are held together by the bifunctional hydrophobic electron acceptor ethane-1,2-diylbis(3,5-dinitrobenzoate) (DNB), thus forming a bolaform SA. This bolaform SA was prepared by mixing PYR and DNB in molar ratio 2:1 in THF, a good solvent for both. The solvent was then removed under reduced pressure. The dissolution-evaporation procedure was repeated three times to ensure completed complexation. The pre-assembled SA was readily soluble in water, yielding a yellow transparent solution. The yellow colour is due to the charge transfer-driven complexation between PYR and DNB. PYR itself can form tubular aggregates, however, the bolaform SA self-assembles into vesicles, induced by the curvature-dependent mechanism.
π–π interactions
SAs can be prepared by π–π interaction. Of the many π-conjugated moieties, Wüthner made use of perylene bisimides (PBIs) to synthesize SAs.27 Wedge-shaped and dumbbell-shaped amphiphiles containing the PBI moiety were synthesized. When mixed together, these two amphiphiles co-assembled into an SA with a new topology and formed vesicular structures, governed by the changes of packing parameters.Coiled-coil peptide interaction
Some specific peptide sequences are able to generate precisely defined non-covalent complexes on the basis of so-called coiled-coil peptide interaction, which is a combination of different intermolecular interactions that occur in nature. The coiled-coil motif has been applied to connecting proteins and hydrophilic polymers.28 Kros et al. extended this concept by incorporating a hydrophilic block and a hydrophobic block into a peptide for the non-covalent synthesis of triblock copolymers. More precisely, one peptide was linked with hydrophobic polystyrene (denoted PS-E), and other peptide pair was linked to hydrophilic poly(ethylene glycol) (denoted K-PEG). PS-E and K-PEG underwent two levels of self-assembly upon dispersion in solution, leading to the formation of amphiphilic hybrid triblock polymers. The formation of SA based on coiled-coil complex results in a structural transformation from original nanospheres to rod-like micelles. Moreover, the self-assembled nanorods are thermo-responsive due to the dynamic nature of the coiled-coil peptide interaction.Functions of SA
The increased interest in SAs can be understood from the following two aspects. One concerns controlled self-assembly, for which the chemical basis is amphiphilicity. Non-covalently bonded SAs can be easily prepared and also readily disassembled, allowing for tuning of amphiphilicity. As a result, controlled self-assembly can be realized. The other aspect concerns use of SAs for potential applications. Depending on the nature of the building blocks and their driving forces, SAs can be responsive to various stimuli, including light irradiation, pH, oxidation and reduction. For that reason it is anticipated that SAs can be used for construction of responsive supramolecular materials.Controlled self-assembly
As already discussed, the host–guest interactions between CD and trans- or cis-azobenzene can be significantly different: there is size complementarity between CD and trans-azobenzene, but the size complementarity is lost between CD and cis-azobenzene. We have made use of photo-controlled inclusion and exclusion of an azobenzene-containing surfactant with α-CD for fabricating a dynamic rotaxane-like SA. As shown in Fig. 7, the supramolecule becomes more hydrophilic when the azobenzene head group is included in α-CD. Upon UV irradiation, the trans-azobenzene is photo-isomerized to the cis-form, forcing the α-CD to slide along the alkyl chain. In this way, the supramolecule becomes amphiphilic. Interestingly, upon visible light irradiation the α-CD can move back to interact with transformed azobenzene once again and reverse the amphiphilicity of the supramolecule, leading to disassembly of the aggregates.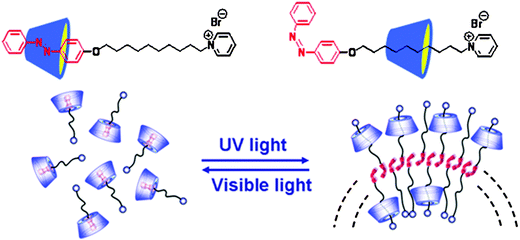
Responsive surface
Rotaxane-like SA can be immobilized on surfaces to fabricate stimuli responsive surfaces. Fig. 8 shows an azobenzene-containing building block with a mercapto end-group pre-assembled with α-CD in water based on the interaction between azobenzene and α-CD, forming a type of SA.29 This SA is able to form a mixed self-assembled monolayer (SAM) with n-butylthiol on a gold substrate. The purpose of the mixed SAM is to ensure that there is enough free space for the movement of the molecular shuttle on the surface. Before UV-light irradiation, α-CD remained above the surface, and the SAM displayed hydrophilic properties. After UV-light irradiation, α-CD slid down onto the alkyl chain, making the SAM hydrophobic. With a water droplet as indicator, a change of nearly 50° for the surface contact angle was found before and after UV-light irradiation. This great change in wettability can be cycled several times by alternating between UV- and visible-light irradiation. Although the response rate is not fast, this responsive surface could be optimized furthermore and find application as a biosurface for controllable adsorption and desorption.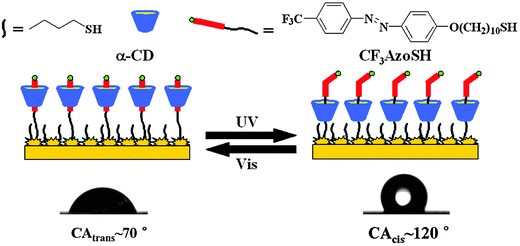
Nanocontainers and controlled delivery
The self-assembly-disassembly feature of SA may be useful for nanocontainers and drug delivery systems. We have developed the concept of a photo-controllable supramolecular polymer nanocontainer through the electrostatic association between an azobenzene-containing surfactant (AzoC10) and the double-hydrophilic block ionomer poly(ethylene glycol)-b-poly(acrylic acid) (PEG43–PAA153).30 The block ionomer complex is a type of polymeric SA, which can self-assemble in aqueous solution and form vesicle-like aggregates (Fig. 9). The vesicle-like aggregates are composed of a poly(ethylene glycol) corona and a poly(acrylic acid) shell associated with azobenzene-containing surfactant. The photoisomerization of azobenzene moieties in the block ionomer complex can reversibly tune the amphiphilicity of the surfactants, inducing disassembly of the vesicles. Such block ionomer complex vesicles are further evaluated as nanocontainers capable of encapsulating and releasing guest solutes on demand, controlled by light irradiation. For example, vesicles encapsulating sodium fluorescein display clear spherical images using fluorescence microscopy. However, such fluorescence-marked images disappear after UV light-triggered release of the solute from the vesicles. Such novel materials have both basic and practical significance, especially as prospective nanocontainers and for cargo delivery. If the stimulus by UV light can be done by near IR, a more friendly light source to the human body, applications in drug delivery could be envisioned.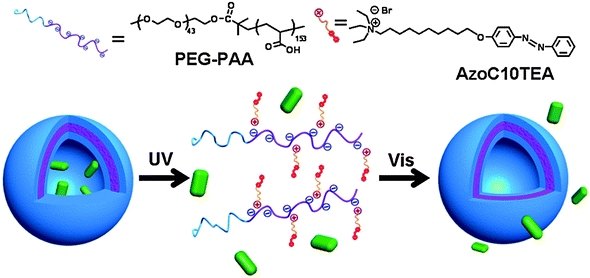
Responsive materials
SAs can be used to fabricate stimuli-responsive materials when suitable functional groups are introduced by non-covalent synthesis. An example of pH responsive one-dimensional nanostructures based on SAs has been reported recently.31 The SA is composed of two components, namely an amphiphile containing an electron acceptor (RV), and the pH responsive electron donor 8-hydroxypyrene-1,3,6-trisulfonic acid (PYR). As shown in Fig. 10, RV and PYR form an SA (RV-PYR), which is driven by electrostatic attraction and charge transfer interaction. In contrast to the vesicular structure made from RV itself, RV-PYR forms ultra-long nanofibers by self-assembly in water. Interestingly, the nanofibers exhibit pH responsiveness. At pH 10 PYR is negatively charged and the adjacent charged moieties would repel each other, making the coiled nanofiber straight; at pH 9 the straight nanofibers become coiled once again. Moreover, the straightness of the nanofibers can be reversibly tuned. At the same time, the physical properties such as conductivity of the nanofibers change upon pH stimulus. Study of ultra-long nanofibers is still under way and potential applications in smart nanodevices may be found.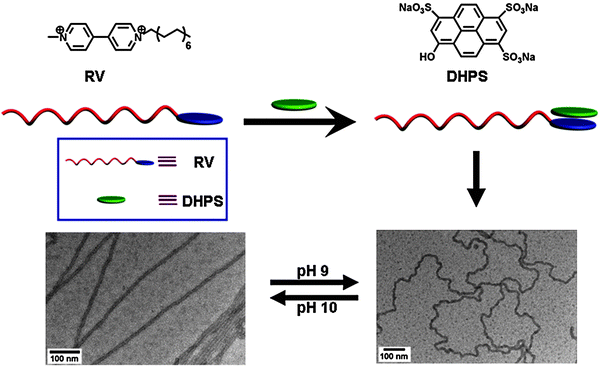 | ||
| Fig. 10 pH responsive self-assembly based on SA.31 | ||
In addition to pH-responsive materials, redox-responsive and photo-responsive materials can also be prepared from SAs. In recent work, we have synthesized an SA in which the redox-responsive viologen molecule is held by charge transfer interaction.32 The self-assembled nanostructures can be reversibly switched between irregular aggregates and vesicles upon oxidative and reductive stimulus. As another example of using SAs for stimuli responsive materials, the photoresponsive malachite green molecule (MGCB) is held with PYR by hydrophobic interactions.33 PYR-MGCB can self-assemble into uniform nanofibers. However, the MGCB molecule can change into a hydrophilic cationic state in response to UV irradiation, and the non-covalent interactions between the MGCB molecule and the pyrenyl group of PYR will decrease drastically. The nanofibers subsequently transform into vesicles. The photo responsive nanofibers could be used as a candidate for stimuli-responsive nanodevices.
Conclusion and outlook
We have summarized different methods of non-covalent synthesis for SA. Moreover, the non-covalent bond in SA can be formed in a designed fashion and it can be broken as well under suitable conditions. Consequently SA can be used for realization of controlled self-assembly and disassembly for functional supramolecular materials. Although the architectures of SA with various topologies are discussed, the molecular engineering of SAs is far from completed, and only limitation is our imagination. It should be kept in mind that beautiful structures are interesting, but should also be functional. Thus the architecture of SAs with tailor-made functions has opened a new avenue for traditional colloid chemistry and is certainly a new direction of supramolecular science as well.Acknowledgements
This work was financially supported by the National Basic Research Program (2007CB808000), NSFC (50973051, 20974059), NSFC-DFG joint grant (TRR 61), and the Tsinghua University Initiative Scientific Research Program (2009THZ02-2).References
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401 CrossRef CAS.
- J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481 CrossRef CAS.
- P. K. Vemula and G. John, Acc. Chem. Res., 2008, 41, 769 CrossRef CAS.
- R. C. Claussen, B. M. Rabatic and S. I. Stupp, J. Am. Chem. Soc., 2003, 125, 12680 CrossRef CAS.
- A. V. Kabanov and V. A. Kabanov, Adv. Drug Delivery Rev., 1998, 30, 49 CrossRef CAS.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3 CrossRef CAS.
- G. S. Kwon and K. Kataoka, Adv. Drug Delivery Rev., 1995, 16, 295 CrossRef CAS.
- A. Rosler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95 CrossRef CAS.
- L. Sierra, B. Lopez, H. Gil and J.-L. Guth, Adv. Mater., 1999, 11, 307 CrossRef CAS.
- E. Ruiz-Hitzky, S. Letaïef and V. Préot, Adv. Mater., 2002, 14, 439 CrossRef CAS.
- Q. Zhang, K. Ariga, A. Okabe and T. Aida, J. Am. Chem. Soc., 2004, 126, 988 CrossRef CAS.
- Y. P. Wang, H. P. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849 CrossRef CAS.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382 CrossRef CAS.
- N. Kimizuka, T. Kawasaki and T. Kunitake, J. Am. Chem. Soc., 1993, 115, 4387–4388 CrossRef CAS.
- N. Kimizuka, T. Kawasaki, K. Hirata and T. Kunitake, J. Am. Chem. Soc., 1998, 120, 4094 CrossRef CAS.
- T. M. Hermans, M. A. C. Broeren, N. Gomopoulos, A. F. Smeijers, B. Mezari, E. N. M. Van Leeuwen, M. R. J. Vos, P. C. M. M. Magusin, P. A. Hilbers, M. H. P. Van Genderen, N. A. J. M. Sommerdijk, G. Fytas and E. W. Meijer, J. Am. Chem. Soc., 2007, 129, 15631 CrossRef CAS.
- T. Bojinova, Y. Coppel, N. L. Viguerie, A. Milius, I. R. Lattes and A. Lattes, Langmuir, 2003, 19, 5233 CrossRef CAS.
- P. Falvey, C. W. Lim, R. Darcy, T. Revermann, U. Karst, M. Giesbers, A. T. M. Marcelis, A. Lazar, A. W. Coleman, D. N. Reinhoudt and B. J. Ravoo, Chem.–Eur. J., 2005, 11, 1171 CrossRef CAS.
- Y. P. Wang, N. Ma, Z. Q. Wang and X. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2823 CrossRef CAS.
- Y. J. Jeon, P. K. Bharadwaj, S. W. Choi, J. W. Lee and K. Kim, Angew. Chem., Int. Ed., 2002, 41, 4474 CrossRef CAS.
- J. F. Gohy, B. G. G. Lohmeijer and U. S. Schubert, Macromolecules, 2002, 35, 4560 CrossRef CAS.
- A. O. Moughton and R. K. O’Reilly, J. Am. Chem. Soc., 2008, 130, 8714 CrossRef CAS.
- B. Song, G. L. Wu, Z. Q. Wang, X. Zhang, M. Smet and W. Dehaen, Langmuir, 2009, 25, 13306 CrossRef CAS.
- A. V. Kabanov, T. K. Bronich, V. A. Kabanov, K. Yu and A. Eisenberg, J. Am. Chem. Soc., 1998, 120, 9941 CrossRef CAS.
- R. Oda, I. Huc, M. Schmutz, S. J. Candau and F. C. MacKintosh, Nature, 1999, 399, 566 CrossRef CAS.
- C. Wang, S. C. Yin, H. P. Xu, Z. Q. Wang and X. Zhang, Angew. Chem., Int. Ed., 2008, 47, 9049 CrossRef CAS.
- X. Zhang, Z. J. Chen and F. Wurthner, J. Am. Chem. Soc., 2007, 129, 4886 CrossRef CAS.
- H. R. Marsden, A. V. Korobko, E. N. M. van Leeuwen, E. M. Pouget, S. J. Veen, N. A. J. M. Sommerdijk and A. Kros, J. Am. Chem. Soc., 2008, 130, 9386 CrossRef CAS.
- P. B. Wan, Y. G. Jiang, Y. P. Wang, Z. Q. Wang and X. Zhang, Chem. Commun., 2008, 5710 RSC.
- Y. P. Wang, P. Han, H. P. Xu, Z. Q. Wang and A. V. Kabanov, Langmuir, 2010, 26, 709 CrossRef CAS.
- C. Wang, Y. S. Guo, Y. P. Wang, H. P. Xu, Z. Q. Wang and X. Zhang, Angew. Chem., Int. Ed., 2009, 48, 8962 CrossRef CAS.
- C. Wang, Y. S. Guo, Y. P. Wang, H. P. Xu, Z. Q. Wang and X. Zhang, Chem. Commun., 2009, 5380 RSC.
- C. Wang, Q. S. Chen, Y. P. Wang, H. P. Xu, Z. Q. Wang and X. Zhang, Adv. Mater., 2010, 22, 2553 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |