Solution-phase counterion effects in supramolecular and mechanostereochemical systems
Travis B.
Gasa
,
Cory
Valente
and
J. Fraser
Stoddart
*
Department of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. E-mail: stoddart@northwestern.edu
First published on 12th October 2010
Abstract
The self-assembly of molecular components into complex superstructures involves the subtle interplay of various noncovalent forces. Charged species are often utilised in self-assembly processes as a result of the favorable π–π, cation–π, electrostatic, and hydrogen bonding interactions that form between these species. Although the counterions associated with these charged species can exert significant effects on the synthesis, stability, and operation of superstructures in solution, rarely are the counterions considered, leading to misinterpretations and misunderstandings of the studied systems. In this tutorial review, we discuss a variety of solution-phase counterion effects, from the fundamental origins to innovative ways in which these effects are exploited for useful functions.
 Travis B. Gasa | Travis B. Gasa received his BS degree from the University of Illinois at Urbana-Champaign, an MS degree from UCLA with the aid of an NSF-IGERT Materials Creation Training Program (MCTP) Traineeship, and a PhD degree in 2010 from Northwestern University. His research has focused on using physical organic chemistry to study the subtle interplay of noncovalent interactions between host and guest species. He now works in Chicago as a Technical Advisor for the intellectual property law firm of Leydig, Voit & Mayer. |
 Cory Valente | Cory Valente is a native of Toronto, Canada. He obtained a HBSc in Biological Chemistry and an NSERC funded PhD in Organic Chemistry with Prof. Michael G. Organ at York University. His PhD research involved the total synthesis of diterpenoids and NHC–Pd-catalyzed cross-couplings. He is currently a postdoctoral fellow with Sir Fraser Stoddart at Northwestern University, where he enjoys working on the synthesis of new carbon allotropes, conformational control in polyrotaxanes, and novel applications of metal-organic frameworks. |
 J. Fraser Stoddart | Sir Fraser Stoddart received all (BSc, PhD, DSc) of his degrees from the University of Edinburgh, UK. Presently, he holds a Board of Trustees Professorship in the Department of Chemistry at Northwestern University. His research has opened up a new materials world of mechanically interlocked molecular compounds and, in doing so, has produced a blueprint for the subsequent growth of functional molecular nanotechnology. |
1. Introduction
The vast majority of molecular components that are designed to assemble into complex supramolecular1 (i.e., inclusion complexes) and mechanostereochemical2,3 (i.e., rotaxanes and catenanes) systems possess typically one or more formally charged atoms in either the host or the guest—or indeed in both. These charged species actively participate—with their associated counterions, largely in the shadows—in the assembly of higher-ordered systems. Such template-directed assembly is often carried out in low polarity media so as to maximise the noncovalent bonding interactions between the components, which include π–π stacking, electrostatic, ion–dipole, and hydrogen bonding (H-bonding) interactions. Historically, counterions were chosen to enhance the solubility of a salt, while possessing weak coordinating abilities so as not to disrupt favourable molecular recognition events. The nonpolar medium in which these host–guest association events are typically performed also increases the ion-pairing strength, such that the counterion can affect significantly the host–guest assembly process both in solution and in the solid-state4 by a variety of means, including steric effects, competition, electrostatics (charge-polarisation), stability of the separated ion-pair, and the ability of the counterion to interact with other species in the system. Unfortunately, these counterion effects have a long history of being overlooked and/or misinterpreted in the assembly of supramolecular and mechanostereochemical systems in solution. In this tutorial review, the potential causes of, inventive uses for, and methods to overcome counterion effects present in supramolecular and mechanostereochemical systems are presented and discussed with the aid of illustrative examples. It is not the intention of this tutorial review to present a comprehensive survey of the literature, but rather to provide an enlightened window into the often overlooked effects of counterions, with the hope that researchers will become more aware of the contributions and caveats from these often promiscuous species —and also perhaps to spark creative ideas that take advantage of counterion effects.2. Counterion effects
2.1 Thermodynamics and kinetics
Counterions have been observed to affect the host–guest association between charged and uncharged species, where typically the guest is charged and the host is neutral, although the same principles described hereafter apply in the reverse case. One of the first known observations of counterions affecting host–guest binding affinities was made in 1977 by Cram and coworkers,5 who noted that the counterion affected the association constant (Ka) between a primary alkylammonium guest and a neutral crown ether host. Since that time, many similar observations have been made, but it was not until 1999 that Bartoli and Roelens6 published a study wherein they systematically varied the constitution of the counterion in a host–guest system to determine the effect on the magnitude of the Ka value. This particular study was the first of a series of publications by various research groups where attempts were made to uncover the origins of counterion effects. The observed counterion effects are most likely a consequence of the interplay of a variety of phenomena and cannot be attributed to any single isolated factor.Although the vast majority of counterion effects have been investigated in terms of thermodynamic association constants between a host and guest—a theme which dominates most of this tutorial review—it has also been observed that the kinetics7,8 of host–guest association and shuttling rates9 in [2]rotaxanes often occur under the influence of associated counterions. The finding that counterions can affect host–guest association and do so with both thermodynamic and kinetic consequences is not surprising, given the fact that the association constant, Ka, a thermodynamic parameter, the rate of association, k1, and the rate of dissociation, k−1, both kinetic parameters, are related mathematically by the following expression:
| Ka = k1/k−1 |
2.2 Ion-pairs and ion-pairing
Ion-pairs consist of two charged species—one positive and one negative—that are held together by an ionic bond. They can exist10 in solution as three different types of complexes: Type (1) solvent-separated ion-pairs, in which the solvation shell of both ions remains largely intact; Type (2) solvent-shared ion-pairs, where a single solvent layer exists between the ions; and Type (3) contact or intimate ion-pairs, where no solvent molecules exist between the ions. Although it is noted that complications on account of higher-order ion-aggregates—which are known11 to exist in organic solvents—remain an intrinsic possibility in each of these cases, the uncertainty surrounding their composition and equilibria precludes discussing them in this tutorial review. In host–guest chemistry, the ion-paired state of the charged host or guest can have significant consequences on the strength of the host–guest interaction (see Sections 3 and 4) and, therefore, it is useful to be able to determine experimentally the extent of ion-pairing. While standard methods for determining the extent of ion-pairing in solution—such as conductometry and potentiometry—are useful for spherical ions in water,10 these methods fall short when studying non-spherical ions in organic solvents, and therefore alternate methods are required.12,13 Both 1H NMR and UV/Vis spectroscopies have been utilised to determine the extent of ion-pairing in solution, and it is generally accepted14 that these methods can only distinguish between contact ion-pairs (Type 3) and solvent-separated ion-pairs (Type 1). It should be noted that, although ion-pairs in solution are in equilibrium between associated and dissociated states, the spectroscopic tools mentioned in this section can help to elucidate whether any significant ion-pairing is present, and thus help to rationalise observed counterion effects (see Section 3).Numerous investigations employing 1H NMR spectroscopy have revealed6,15,16 that, if the limiting chemical shift (δmax) of resonances arising from the cationic guest in a given host–guest complex varies upon changing the counteranion, then the counteranion must remain closely associated to the guest upon host complexation, most likely as a contact ion-pair14 (Type 3). If the counterions themselves possess a spectroscopic probe—such as the protons in tosylate and picrate salts—then their corresponding 1H NMR resonances can be monitored directly during complexation studies to yield extremely useful information. A small (∼0.1 ppm) chemical shift change of the counterion 1H resonances upon host–guest complexation can be interpreted12,17,18 as dissociation of the ion-pair, or conversely, lack of chemical shift changes suggests that the contact ion-pair is retained.14 If the host cavity is large enough to include both a cationic guest and its associated counteranion, then a large upfield shift in the 1H resonances of the counteranion indicates16 that the entire contact ion-pair is included in the host cavity. A large upfield shift of the resonances associated with the counterion can also imply19 that the counterion is interacting specifically with the host through π–π stacking interactions (vide infra), which, of course, can also be observed20 as charge-transfer complexes in the UV/Vis spectra. As such, interpretations must be made carefully based on an in-depth knowledge of the system under examination.
Diffusion ordered spectroscopy (DOSY) has been shown21 to be particularly useful in determining if a counterion remains associated with the host–guest complex, but only for counterions that contain probes visible by NMR spectroscopy. DOSY is a technique that measures the diffusion rate of a particular species in solution, and depends on various factors, including the solution viscosity, temperature, and size of the diffusing species under observation. A comparison of the counterion and host–guest complex diffusion rates can be employed21 to determine if the counterion is associated with the host–guest complex, or if it is diffusing freely in solution.
Additionally, UV/Vis spectroscopy can be used to monitor ion-pairs which interact with each other electronically—i.e., the halide22 or tosylate8 salt of an aromatic cation—to give rise to charge-transfer complexes whose absorptions are indicative of tight ion-pairs in solution. If, in the presence of a host, the absorption maxima of these ion-pairs are unperturbed,22 then the ion-pairs must remain intact upon host–guest complexation. Tight ion-pairs can also be observed in solution if the luminescence of an aromatic ion is quenched in the presence of a counterion.
2.3 Counterion faux pas
When studying the physical processes associated with host–guest complexation, it is of utmost importance to be cognisant of the environment in which these species are examined. It is common practice to note the temperature and solvent in which such investigations are conducted, since these variables are often chosen carefully and optimised for particular systems and specific reasons. When experiments are carried out in buffers or at a specific pH, usually for solubility or pKa reasons, however, the consequences of the presence of these extra salts are often overlooked. For example, molecular recognition studies were carried out23 between a cationic guest with various counterions and a neutral host in phosphate buffer to study, in part, the effect of the counterion on the host–guest binding affinities. The authors found23 that varying the counterions of the guest had no effect on the host–guest binding affinities. These results were misleading, since the excess of PO43− present in the buffer is known24 to exchange with all the counterions that were introduced. Similarly, host–guest binding studies conducted25 in the presence of H2SO4 to achieve pH 2 introduces a large excess of SO42− counterions into solution that are free to exchange with the counterions that had been specifically and systematically introduced into the system. Although the authors determined25 that varying the counterions had no effect on the host–guest binding affinities, counterion exchange processes involving the excess of sulfate would mask any counterion effects intrinsic to each designed system. Clearly, being aware of the media in which molecular recognition studies are performed is essential when carrying out phenomenological investigations.3. The origins of counterion effects
Although it is appreciated that counterions can influence significantly the physical behavior of supramolecular and mechanostereochemical systems, the origins of these counterion effects remain a subject for debate. Many key observations have been made regarding the potential causes of counterion effects, and—from these investigations—it is obvious that not one, but rather the interplay of many factors, contribute to the behavior expressed in a given system. In the sections which follow, we try to isolate, as best as possible, the key factors that give rise to counterion effects.3.1 Counterions and steric effects
In nonpolar organic solvents of low dielectric constant, ion-pairing tends to be significant (Type 3, Section 2.2) in the concentration ranges typically employed (ca. 1–30 mM) in binding studies and in the template-directed syntheses26–28 of mechanically-interlocked molecules. When concentrations29 become high, ion aggregates can even be formed. It is not unreasonable to expect that a tightly associated counterion can have a significant steric effect on the complexation between its ionic partner (guest) and a given host, as a consequence of the counterion's close proximity to the binding event. The steric effect of the counterion is assumed to be most significant when the guest binds to the host as a tightly-associated ion-pair—i.e., the counterion remains bound to the guest after complexation.Böhmer and coworkers16 have investigated the association (Fig. 1) between hosts 1 and 2 and tetramethylammonium (TMA+) and acetylcholine (ACh+) cationic guests with a variety of counteranions in CDCl3 solutions. Under the conditions employed in this investigation, the authors provide evidence that the ion-pairs remain intact upon host complexation, as evidenced by the counterion-dependent 1H NMR chemical shifts of the complexed cations, see Section 2.2. The authors propose (Fig. 2) two possible structures (I and II) for host–cation–anion ternary complexes. In structure I, the steric effect of the counterion is very important to the host binding events, since the cation can only penetrate into the cavity to a somewhat limited extent if it is to remain associated with its counterion. In structure II, however, the anion sits outside the host cavity, and its interaction with the cation is mediated by an aromatic face of the host, as supported by calculations carried out by Dougherty and coworkers,30 and evidenced in this study by upfield chemical shifts on the tosylate and picrate counteranions upon host–guest complexation. Experimentally, the binding affinities of the guests with host 1, when compared to those involving host 2, were larger and more highly sensitive to the nature of the counterion, following the trend Pic− > TFA− > Cl− > TsO− for TMA+, and Pic− > I− > Cl− for ACh+. The authors argue that, while electrostatic polarisation effects (see Section 3.3) from the counteranions are one possible explanation for the different binding affinities expressed by host 1 and 2, another likely cause can be attributed to the host structures—host 1 is bowl-shaped, more rigid, and thus pre-organised, and therefore a deeper penetration of the guest cation is required for effective binding. Since the cavity of host 1 is not large enough to accommodate both species of the tight ion-pair, the steric contributions by the counterions are more pronounced. The authors propose that structure II—either alone or in rapid equilibrium with structure I—provides a good model for the ternary host–cation–anion complex. For the steric consequences of the counterion to be apparent—as they undoubtedly are in this particular investigation—structure I must participate, to at least some extent, in a realistic description of the ternary complex.
 | ||
| Fig. 1 Structural formulae of host and guest compounds studied by Böhmer et al.,16 Arduini et al.,17 and Bartoli and Roelens,6,34 in which the steric and charge-polarisation effects of counterions were found to play an important role in determining the thermodynamic stabilities of host–guest complexes. | ||
![Host–guest–counterion ternary structures proposed by Böhmer and coworkers.16 The bowl-shaped structure represents the [5]calixarene utilised in this investigation, and is composed of five aromatic rings in a macrocyclic arrangement. In structure I, the cation can only penetrate into the host cavity to a somewhat limited extent if it is to remain associated with its counterion. As a consequence, the steric effects of the counterion become very important. In structure II, the anion sits outside the host cavity, and its interaction with the cation is insulated and mediated by one of the five aromatic rings that constitutes the [5]calixarene. Thus, the steric effects of the anion in this structure are not as significant. Reprinted with permission from ref. 16. Copyright 2001 American Chemical Society.](/image/article/2011/CS/c005424k/c005424k-f2.gif) | ||
| Fig. 2 Host–guest–counterion ternary structures proposed by Böhmer and coworkers.16 The bowl-shaped structure represents the [5]calixarene utilised in this investigation, and is composed of five aromatic rings in a macrocyclic arrangement. In structure I, the cation can only penetrate into the host cavity to a somewhat limited extent if it is to remain associated with its counterion. As a consequence, the steric effects of the counterion become very important. In structure II, the anion sits outside the host cavity, and its interaction with the cation is insulated and mediated by one of the five aromatic rings that constitutes the [5]calixarene. Thus, the steric effects of the anion in this structure are not as significant. Reprinted with permission from ref. 16. Copyright 2001 American Chemical Society. | ||
Arduini and coworkers17 have investigated the complexation of the calix[4]arene 3 with N-methylpyridinium (NMP+) and TMA+ cations (Fig. 1) as both the iodide (I−) and tosylate (TsO−) salts. It was found that the association constant in CDCl3 between host 3 and NMP·I was nearly four-fold larger than the Ka value for NMP·OTs complexation. The reported171H NMR chemical shifts are consistent (Section 2.2) with NMP·I binding as a dissociated ion-pair, whereas NMP·OTs binds as a contact ion-pair, and thus has greater steric requirements for complexation. While the spherical shape of the TMA+ cations allows these guests to bind to the cavity of host 3 without any rearrangement of the associated counterion, NMP+, on the other hand, is not spherical, and the position of the counterion may need to shift to facilitate complexation with the host. As such, the complexation between TMA+ and host 3 is unaffected by the association state of the ion-pair, while the association of host 3 with NMP+ is affected by the ion-pairing state, as evidenced by the reported Ka values in this investigation. While polarisation and other electronic effects cannot be excluded as contributing factors to the observed counterion effect, the authors maintain that, in this particular system, it is reasonable to conclude that steric effects control the binding processes to a larger extent, since the cation remains bound to the anion upon host complexation.
In an elegant example demonstrating the role of steric effects involving counterions in complexations, Kubik and coworkers31 examined (Fig. 3) the interaction between a cyclic hexapeptide 5 with salts composed of both chiral cations and chiral anions. Tris(tetrachlorocatecholato)phosphate(V) (TRISPHAT−) salts of quaternary ammonium ions are known32 to form tight ion-pairs in chloroform, and therefore, these ion-pairs express steric consequences upon host–guest complexation. 1H NMR titration experiments revealed that the absolute configuration of the chiral counteranion has a marked effect on the stability of the chiral host–guest complex. The [(R)-6][Δ-7] cation/anion diastereoisomeric complex binds to the chiral host 5 nearly twice as strongly (5210 vs. 2890 M−1) as does the [(R)-6][Λ-7] diastereoisomeric complex. In these binding studies, one cannot rule out the fact that these are diastereoisomeric salts, and thus one would predict that these two ion-pairs would have different physical properties—i.e., charge polarisation within the ion-pair—and thus different intrinsic binding affinities with a given host. The authors note, however, that the differences in charge polarisation within the [(R)-6][Δ-7] and [(R)-6][Λ-7] diastereoisomeric salts would be small, as a consequence of the large size and charge diffusivity of the TRISPHAT− anions. Thus, such a large difference in binding affinities can be attributed to differences in steric effects—here the absolute configuration of the counteranion—over electronic effects.
 | ||
| Fig. 3 Structural formulae of the chiral compounds studied by Kubik and coworkers.31 The absolute configuration of the counteranion was found to effect the strength of the host–guest complex through steric effects. | ||
3.2 Charge diffusivity and salt solubility
The diffusivity of charge on a counterion plays a decisive role in the binding of a charged guest with a neutral host. For example, a small, charge-dense counteranion has a greater polarising effect on its cationic partner (vide infra) than a large, charge-diffuse counteranion, and thus is expected to inhibit host–guest binding to a greater extent by decreasing the charge available on the cation. In addition, if the charge on the counteranion is more diffuse, i.e., more delocalised over the counterion, then the free energy associated with the counteranion charge is lowered. These general trends are valid for counterions that do not interact specifically with the host in a given system. See Sections 3.6 and 4.3 for selected examples of counterions that do interact specifically with hosts.The dependence of counterion charge diffusivity on the binding affinity of a cationic guest with a host can be illustrated by utilising the halide counteranion series Cl−, Br−, and I−. These halides are spherical, monovalent counteranions with ionic radii increasing along the series Cl− < Br− < I−. One of the first investigations where the counterions were varied systematically and their effects observed on host–guest complexation was executed by Bartoli and Roelens.6 When the binding affinities between host 3 and ACh+ (Fig. 1) as Cl−, Br−, and I− salts were investigated by 1H NMR spectroscopy in CDCl3, it was found that the association constant goes from 4 to 5 to 11 M−1 with the increasing ionic radii of Cl−, Br−, and I−. Although these association constants are quite small, the reliability of these measurements was verified33 in a subsequent publication, confirming the observed trend. It should be noted that, although nonspherical, unsymmetrical counterions—such as tosylate and picrate—are expected to follow the same charge diffusivity trends as the spherical halide anions, it is not so straightforward to assign and compare their values, and therefore a discussion of them is not included in this tutorial review.
Bartoli and Roelens6,34 also investigated the use of “assisting ligands” to study the effects of counterion charge-dispersion on host–guest complexation. The authors utilised an organostannane-assisting ligand to convert the small, charge-dense Cl− counteranion into a larger, more charge-diffuse organostannate–chloride complex, and then compared the association constants between TMA+ and host 2 in the presence of different organostannate counteranions. The organostannate–chloride complex was generated in situ by adding an excess of either dimethyldichlorostannane (Me2SnCl2) or dibutyldichlorostannane (Bu2SnCl2) to a stock solution of TMA·Cl, and then performing an 1H NMR titration in CDCl3. A considerable increase in the association constant between TMA+ and host 2 was observed for both stannate complexes compared to TMA·Cl. Specifically, a five-fold and a three-fold increase in Ka value was observed for the Me2SnCl3− and Bu2SnCl3− stannates, respectively, an observation which is consistent with counterion charge dispersion having a significant effect on host–guest binding affinities.
In the course of these investigations which utilised host 2 and various TMA+ and ACh+ salts, a good correlation (r2 = 0.91) was observed6,34 (Fig. 4) between the solubilities (log S) of guest salts and the binding free energy (−ΔG°) of the host–guest complexes. Specifically, as the solubilities of the salts decrease, the stability of the host–guest complexes was observed to increase—i.e., the least soluble salts form the most stable complexes with host 2. This trend was rationalised by considering the charge dispersion on the counterion: the same properties of the cation–counteranion relationship which facilitate less inhibited host–guest binding also suppress solvation—i.e., charge-diffuse counteranions, such as PF6−, do not interact strongly with solvent molecules, thereby decreasing their solvation and lowering the solubilities of the salts. It should be noted that the tosylate and mesylate salts did not follow the trend in solubilities, perhaps as a consequence of specific interactions between these counteranions and host 2. It is not unlikely that this simplistic view is short of being the whole story: rather, it is most likely the interplay between the solvation of the individual ionic species and the solvation of the intact ion-pair which contributes to the observed solubilities.
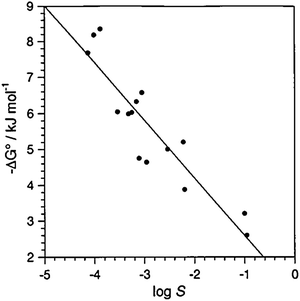 | ||
| Fig. 4 Binding free energies (−ΔG°) plotted against solubilities (log S) for complexes of host 2 with ACh+ and TMA+ salts, excluding tosylate and mesylate.34 As the solubilities of the guest salts decrease, the binding free energies increase. Reprinted with permission from ref. 34. Copyright 2002 American Chemical Society. | ||
3.3 Electrostatic potential and charge polarisation
Proponents of electrostatic causes of counterion effects argue34−36 that many cationic guests are highly polarisable,37 especially in the presence of charge-dense counteranions, and therefore less of the cation's charge is available for binding to an electron-rich host. Theoretical investigations lend38,39 support to this charge-polarisation mechanism, and the investigations which will now be discussed correlate calculated electrostatic parameters of the ion-pairs with experimental solution-state binding data.Bartoli and Roelens34 studied systematically the interaction between neutral host 2 and several TMA+ and ACh+ salts (Fig. 1) of commonly employed counteranions in CDCl3 by 1H NMR spectroscopy. The resulting standard free energies (−ΔG°) of binding were compared to electrostatic ion-pair parameters calculated using density functional theory (DFT). The results are summarised graphically in Fig. 5. For the TMA+ salts, eight counteranions show an excellent correlation (r2 = 0.998) between −ΔG° and the electrostatic potential (EP) on the van der Waals surface (Fig. 6) of the cation that binds to the host. No clear correlation, however, was found for the corresponding ACh+ salts. The authors argue that factors other than pure electrostatics must be involved in the interaction between ACh+ and host 2, and these factors may also account for the fact that not all of the counteranions of the TMA+ salt show a linear correlation between −ΔG° and EP. Extrapolating from Fig. 5 the value of −ΔG° for the free TMA+ cation binding with host 2 in the absence of counteranions, it was found that between 80–100% of binding free energy is lost as a consequence of the charge polarising effect brought about by the presence of counteranions.
 | ||
| Fig. 5 Binding free energies (−ΔG°) plotted against ion-pair electrostatic potentials (EP) for host 2 complexing with the TMA+ salts shown,34 revealing a good correlation (r2 = 0.998) between these parameters. Reprinted with permission from ref. 34. Copyright 2002 American Chemical Society. | ||
 | ||
| Fig. 6 Electrostatic potential mapped onto the van der Waals surface of the TMA·picrate ion-pair, in which the cation (blue) clearly has a polarising effect on the counteranion (yellow, green and red).34 Electron density is represented on a scale from red (negative) to blue (positive). Reprinted with permission from ref. 34. Copyright 2002 American Chemical Society. | ||
Roelens and coworkers35 have repeated a systematic study of the polarising effect of counteranions using the same TMA+ and ACh+ salts, but with a different cyclophane, host 4 (Fig. 1). It was found that the counteranion's inhibiting effect on host–guest complexation follows the same trend as that observed for host 2, at least as judged by the order of binding affinities of the ion-pair to host 4. In addition, the same correlation between −ΔG° and EP for the TMA+ salts was found. Interestingly, host 4 binds most salts roughly 1.9 kcal mol−1 more strongly than host 2, implying that a given counteranion's modulation of cation binding is constant with respect to different hosts. The authors conclude that, as long as electrostatics are the only contributing factor present in a given host–guest–counterion system, the calculated EP values for an ion-pair may be used to predict the binding strengths of ion-pairs to a given host.
In an attempt to determine if the counteranion does indeed polarise a cation's charge, thus making less of the positive charge available for binding to a host, Hunter and coworkers40 measured experimentally the energetics of the cation–π interaction between two complementary binding partners in the presence of three different counteranions using (Fig. 7) a chemical double-mutant cycle41 wherein systematic chemical mutations are introduced into the chemical structures, such that a specific interaction of interest can be singled out and measured. It was found that, within experimental error, the energetics of the cation–π interaction were very similar (0.60 ± 0.096 kcal mol−1) in the presence of BPh4−, PF6−, and I− counteranions. The authors concluded that, although the overall stabilities of the complexes vary with the choice of counteranion, the relatively constant value determined for the cation–π interaction suggests that it is not polarised significantly by counteranions, a conclusion which contradicts the studies by Roelens and coworkers.34,35 Instead, the presence of multiple equilibria causes competition for the binding sites among the cation, anion, and other interacting species present. In summary, a variety of factors contribute to the observed counterion effects, which are likely to be system-dependent, and therefore each subsequent investigation provides a piece to the puzzle, rather than absolute conclusions.
 | ||
| Fig. 7 The chemical double-mutant cycle performed by Hunter and coworkers40 to determine the cation–π interaction in the presence of various counteranions. Specific noncovalent bonding interactions are represented by blue hash marks. The strength of the cation–π interaction was found to be affected hardly at all upon varying the counteranion between BPh4−, PF6−, and I−. | ||
3.4 Host–counterion competitive binding
Competition between the host and counteranion for binding sites on cationic guests can also contribute to the observed counterion effects. If the counterions, such as PF6−, are weakly-coordinating, then rigorous quantitative studies have revealed12 that host–guest complexation is relatively unaffected by the presence of the counterion, indicating that no competition exists between the host and counterion for the guest. Of course, these findings are specific to the host, guest, counterion, and the conditions of the investigation, and so each system needs to be studied individually to determine the competitive effects, if any, of the counterion. Additionally, if a charged guest and its associated counterion bind to a given host as a tight ion-pair, then the steric effects of this ion-pair, as well as charge polarisation of the guest species by its associated counterion, play a dominant role in modulating the host–guest binding. However, if ion-pair dissociation must first take place prior to host–guest binding,42,43 then a competition must exist between the host and the counterion for the guest species, which may then contribute significantly to the observed host–guest binding phenomena.The threading/dethreading of a pseudorotaxane by counterion–host competition has been investigated by Montalti and Prodi.44 Although in a CH2Cl2 solution dibenzo[24]crown-8 (DB24C8) is fluorescent (Fig. 8), the addition of an equimolar amount of the anthracenylammonium derivative 8·PF6 forms a [2]pseudorotaxane, and the fluorescence is quenched completely as a result of excited-state energy transfer from DB24C8 to the anthracene moiety. Adding one equivalent of NBu4Cl exchanges the PF6− counteranion to give 8·Cl, resulting (Fig. 8) in the dethreading of the [2]pseudorotaxane and complete recovery of the DB24C8 fluorescence. This behavior can be explained using eqn (1) and (2).
 | (1) |
 | (2) |
![Fluorescence spectrum changes studied by Montalti and Prodi44 as a result of the threading/dethreading process of guest 8 with host DB24C8. The assembly/disassembly process is triggered by counterion exchange between PF6− and Cl−, respectively. The ability of Cl− to dethread the [2]pseudorotaxane was attributed to competition with DB24C8 for binding with guest 8.44 Reproduced by permission of The Royal Society of Chemistry.](/image/article/2011/CS/c005424k/c005424k-f8.gif) | ||
| Fig. 8 Fluorescence spectrum changes studied by Montalti and Prodi44 as a result of the threading/dethreading process of guest 8 with host DB24C8. The assembly/disassembly process is triggered by counterion exchange between PF6− and Cl−, respectively. The ability of Cl− to dethread the [2]pseudorotaxane was attributed to competition with DB24C8 for binding with guest 8.44 Reproduced by permission of The Royal Society of Chemistry. | ||
Credi, Cort, and coworkers45 examined the pseudorotaxane formation (Fig. 9) between the binaphthyl host 9 and the cationic guest ABH+ as its TRISPHAT− salt in CH2Cl2. Attempts to obtain an association constant between the host and guest using fluorescence spectroscopy produced anomalous results. Specifically, complete quenching of the binaphthyl fluorescence by excited-state energy transfer to ABH+ was observed when titrating host 9 into a dilute solution of ABH·TRISPHAT. During the reverse titration where ABH·TRISPHAT is titrated into a solution of the host 9, however, the fluorescence intensity decreases proportionately with increasing ABH·TRISPHAT concentration. Complete quenching, however, is not observed, even after the addition of four equivalents of guest. This discrepancy can be rationalised by considering that the ABH·TRISPHAT salt exists as a tight ion-pair in solution, and during the titration, both the cation and anion concentrations are increased simultaneously, shifting the equilibrium towards the ion-paired guest, and away from the host–guest complex. Indeed, addition of an excess of the TRISPHAT− counteranion in the form of the tetrabutylammonium salt (TBA·TRISPHAT) to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 solution of host and guest causes a progressive recovery of the binaphthyl fluorescence, indicating that the TRISPHAT− counteranions compete with the host for the cationic guest, causing dethreading of the [2]pseudorotaxane.
∶1 solution of host and guest causes a progressive recovery of the binaphthyl fluorescence, indicating that the TRISPHAT− counteranions compete with the host for the cationic guest, causing dethreading of the [2]pseudorotaxane.
 | ||
| Fig. 9 Structural formulae of the compounds studied by Credi, Cort, and coworkers.45 TRISPHAT− was observed to compete with host 9 for binding with ABH+. | ||
3.5 Stability of the separated ion-pair
An increase in the separation distance between the cation and anion upon host–guest complexation represents46 an unfavourable thermodynamic contribution to the overall stability of a host–guest complex, especially if the counterion is poorly stabilised by the solvent. The energetics of an ion-pair can be tuned46 by three factors: the solvent polarity, the nature of the counterion, and the separation distance of the cation/anion ion-pair.The relationship between these factors was investigated recently by Meijer and coworkers46 in supramolecular assemblies of cyclic guanosine tetramers. In the presence of an alkali metal template (M+X−), such as NaPF6 and KPF6, guanosine monomers (G) self-assemble (Fig. 10a) into cyclic G-quartets, which then further aggregate into G-quadruplexes, all of which is driven by a combination of intermolecular interactions. At monomer concentrations above 30 mM, it was found that the dielectric constant (εs) of the solvent plays a decisive role in determining the size of the G-quadruplexes for compound 10 (Fig. 10b). By increasing the solvent polarity along the series tetrahydrofuran (THF) (εs = 7.8), acetone (εs = 20.7), and acetonitrile (εs = 37.5), the size of the quadruplexes could be tuned to result in the 8-mer, 12-mer, and 16-mer in turn. The authors rationalised this effect by proposing that the increasing charge density, stemming from the metal salts within the G-quadruplexes, is stabilised somewhat better by solvents with higher εs values, as long as the solvents do not disrupt H-bonding. At concentrations below 30 mM in these solvents, it was found that the guanosine monomer is in equilibrium with its corresponding G-quadruplex, without a significant presence of any intermediate species, and that the identity of the counteranion plays a role in affecting the monomer/quadruplex equilibria. For example, quadruplex formation is favoured increasingly along the halide series Cl− < Br− < I−, and it was found that weakly-coordinating anions, such as BF4−, BPh4−, and PF6−, are among the most favorable ones for promoting quadruplex formation. This result reveals that, as the anions become more stable and more charge diffuse, the overall stability of the quadruplexes increases, shifting the equilibrium towards larger aggregates.
 | ||
| Fig. 10 (a) Guanosine monomers (G) self-assemble into G-quartets in the presence of an alkali metal template (MX), which further aggregate into G-quadruplexes. (b) Structural formula of the G monomer 10 utilised in the studies by Meijer and coworkers.46 Reprinted with permission from ref. 46. Copyright 2009 American Chemical Society. | ||
In order to probe the effect that the cation–anion distance has on G-quadruplex formation, the authors introduced46 a substituent at the 8-position of the guanosine monomer 10a, a structural modification which is known to force the ribose groups to adopt an all-syn conformation—as opposed to an all-anti one (10b) in the absence of a substituent. The orientation adopted by the all-syn and all-anti conformations modulates (Fig. 11) the steric environment on the periphery of the quartet, so tuning the distance between the cation and counteranion in the stacked quadruplexes. It was discovered that the ion-pair separation energy can be reduced by decreasing the cation–anion separation, thus increasing the overall stabilisation of the G-quadruplexes and promoting the formation of larger aggregates. Specifically, it was found that, in the presence of KPF6 as the templating agent, the all-syn quartets produced 8- and 12-mers in THF and acetone, respectively, and the all-anti quartets formed 16- and 24-mers in these two solvents. Further evidence of the importance of the cation–anion distance is supported by the observation that the formation of G-quadruplexes from all-anti G-quartets shows a marked dependence on the nature of the counteranion: smaller, non-coordinating anions—i.e., PF6− and BF4−—favoured larger G-quadruplexes, whereas bulkier anions—i.e., BPh4−—which cannot approach the cation as closely possess greater ion-pair separation energy and thus form smaller aggregates.
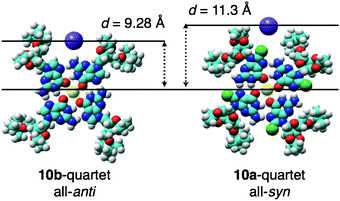 | ||
| Fig. 11 Modulating the steric environment on the periphery of the G-quadruplexes allows the separation distance between the cation (yellow) and anion (purple) to be tuned, a situation which is accomplished by the presence (10a) or absence (10b) of a bromine substituent at the 8-position on the guanosine monomer, causing the ribose unit to adopt an all-syn or all-anti conformation, respectively.46 Reprinted with permission from ref. 46. Copyright 2009 American Chemical Society. | ||
Iwamoto et al.36 have determined the binding thermodynamics between the calix[4]arene host 11 and various N-methylpicolinium salts (Fig. 12) using variable temperature UV/Vis spectroscopy. Although the authors originally attempted to correlate the enthalpy of binding (ΔH°) with the EP on the cation (r2 = 0.548), similar to the work performed by Roelens and coworkers34,35 (vide supra), it was found that a better correlation (r2 = 0.941) exists between ΔH° and the electrostatic stabilisation energy (ΔE) of the ion-pairs. This apparent contradiction to the studies by Roelens and coworkers34,35 can be rationalised in terms of the ion-pair binding modes to the respective hosts. The geometry and flexibility of the binding cavities of hosts 2 and 4 (Fig. 1) allow the charged guests to complex as contact ion-pairs, and therefore the charge polarisation effect from the counteranion is a significant contribution. The size of the cavity in host 11, however, causes the cation to bind as a host-separated ion-pair and, as a result, the polarising effect of the counteranion is less pronounced. Therefore, in the case of host 11 binding with guest 12, the binding enthalpy correlates more appropriately with ΔE than it does with EP.
![Structural formulae of the calix[4]arene host 11 and the N-methylpicolinium guest 12 studied by Iwamoto et al.36 The electrostatic stabilisation of guest 12 ion-pairs was found to correlate with the host–guest binding enthalpies.](/image/article/2011/CS/c005424k/c005424k-f12.gif) | ||
| Fig. 12 Structural formulae of the calix[4]arene host 11 and the N-methylpicolinium guest 12 studied by Iwamoto et al.36 The electrostatic stabilisation of guest 12 ion-pairs was found to correlate with the host–guest binding enthalpies. | ||
3.6 Actively participating counterions
The factors which dictate how a counterion affects the interaction between its ionic partner—i.e., a charged guest—and a neutral host have been shown to follow a particular trend. In general, the more charge diffuse, less charge polarising, and less hydrogen-bond accepting a given counterion, the stronger the complex formed between its charged counterpart and a given host will be in the event. This trend, however, is only valid for hosts which do not interact specifically with the counterion of the guest. When a host possesses this interactive capability with both species of an ion-pair, such as in a heteroditopic receptor (vide infra), the observed trends are typically reversed.One of the first known reports of this trend reversal was that described by Jeong et al.47 in their investigation of the binding properties between the acyclic receptor 13 and a quaternary ammonium guest 14 as three different halide salts (Fig. 13). The association constants in CDCl3 between host 13 and guests 14·Cl, 14·Br and 14·I were found to be 1070, 340, and 56 M−1. This trend, along with concomitant and pronounced downfield chemical shift changes for the amide NH and phenolic OH protons, indicates that the counteranions are interacting with the host through H-bonding interactions. Further proof of the counteranion–host interaction was obtained by converting the OH groups in host 13 to either H, Me, or OMe, thereby eliminating their potential to serve as strong H-bond donors. Subsequent binding studies of these hosts with guest 14 led to extremely small chemical shift changes upon complexation, shifts that were too small to use in the calculation of reliable association constants. The authors ascribed the counteranion effects between host 13 and guest 14 to anion–host H-bonding interactions, which increase the electron density of the aryl moieties, thus enhancing the cation–π interactions, as was proposed and treated computationally by Dougherty et al.48 We would like to propose an additional explanation—namely, that perhaps host 13 is primarily an anion receptor, and thus complexation with the ion-pair is driven more by halide H-bonding than by cation–π interactions, as evidenced by the nearly complete suppression of binding upon modifying the host phenolic OH groups.
 | ||
| Fig. 13 Structural formulae of host and guest compounds examined by Jeong et al.47 These authors investigated the binding affinities between host 13 and guest 14 as the Cl−, Br−, and I− salts, and observed a binding trend reversal in which the more coordinating Cl− salt expresses the highest affinity for host 13. | ||
In an effort to probe potential π–π stacking interactions between the picrate anion and aromatic ring-containing receptors, Talanova et al.19 have evaluated the aqueous/organic extraction selectivities of various benzo-substituted crown ethers with alkali metal picrates and iodides. The study was executed by varying the number of atoms contained in the macrocyclic polyether systematically, as well as by altering the number and location of benzo substituents, and evaluating the extraction selectivities for the metal salts. The authors determined a general trend within their data—namely, that, as the number of benzo substituents suitably located within the crown ether constitution is increased, the extraction selectivity favors the metal picrate salts. This so-called “picrate effect” has been ascribed19 to π–π stacking interactions between the picrate anion and the aromatic rings associated with the macrocycles, leading to their selective complexation in the presence of metal iodides. Importantly, crown ethers in which the benzo substituents are replaced by cyclohexyl rings do not exhibit any enhanced selectivity, further confirming the importance of π–π stacking interactions in the case of the picrate counteranions.
3.7 Kinetic consequences of the presence of counterions
The kinetics of the host–guest self-assembly process can be attributed to a number of factors involving the associated counterions, including steric effects, charge diffusivity, electrostatic polarisation, competitive binding, and the energy of the separated ion-pair. Thus, for clarity's sake, the kinetic consequences attributable to the presence of counterions will be isolated from these other factors and discussed in this section.Credi, Arduini, and coworkers8 have observed that the threading/dethreading rate of a [2]pseudorotaxane composed of a heteroditopic phenylureido-bearing calix[6]arene host 15 and the alkyl bipyridinium guests 16·2PF6 and 16·2OTs (Fig. 14a) depends markedly on the nature of the counteranion present. Stopped-flow absorption experiments, in which the host and guest are mixed rapidly and the absorbance changes measured over time, were conducted in CH2Cl2 at 293 K in order to determine the rate (k1) of the self-assembly process of the [2]pseudorotaxane—namely, the threading of the counterion-dissociated viologen guests into the calix[6]arene host cavity. The rate of dethreading (k−1) could then be calculated using the equation k−1 = k1/Ka by utilising the known association constant (Ka). The data gave a satisfactory fit with a 1∶1 association model and were used to obtain second-order rate constants. It was found that both the rate of threading (280-fold) and dethreading (40-fold) were much faster for the guest as the TsO− salt compared with the PF6− salt. The faster kinetics observed for the self-assembly of 16·2OTs was rationalised by assuming that the transition state (TS‡) leading to (Fig. 14b) the threaded [2]pseudorotaxane, in which the guest must thread without its counteranions as a consequence of steric interactions, involves H-bonding interactions between the TsO− ions and the phenylureido groups on the host, thus stabilising the TS‡ and reducing the activation barrier. The PF6− counteranions, however, are weaker H-bond acceptors, and therefore cannot stabilise the TS‡ as efficiently as do the TsO− ions.
![(a) Structural formulae of the host and guest compounds studied by Credi, Arduini, and coworkers.8 The kinetics of [2]pseudorotaxane formation was investigated by stopped-flow kinetic experiments, and it was found that guest 16 as the TsO− salt threaded faster than the PF6− salt. (b) Energy diagram depicting the proposed transition state for the threading of guest 16 into the cavity of host 15. When TsO− counteranions are employed, the authors propose that the counteranions stabilise the transition state involving [2]pseudorotaxane formation by H-bonding with the phenylureido groups on the host. The PF6− counterions, however, are weaker H-bond acceptors, and therefore cannot stabilise the transition state as efficiently. Reprinted with permission from ref. 8. Copyright 2004 American Chemical Society.](/image/article/2011/CS/c005424k/c005424k-f14.gif) | ||
| Fig. 14 (a) Structural formulae of the host and guest compounds studied by Credi, Arduini, and coworkers.8 The kinetics of [2]pseudorotaxane formation was investigated by stopped-flow kinetic experiments, and it was found that guest 16 as the TsO− salt threaded faster than the PF6− salt. (b) Energy diagram depicting the proposed transition state for the threading of guest 16 into the cavity of host 15. When TsO− counteranions are employed, the authors propose that the counteranions stabilise the transition state involving [2]pseudorotaxane formation by H-bonding with the phenylureido groups on the host. The PF6− counterions, however, are weaker H-bond acceptors, and therefore cannot stabilise the transition state as efficiently. Reprinted with permission from ref. 8. Copyright 2004 American Chemical Society. | ||
Likewise, Sanders and coworkers7 have investigated the kinetic stabilities of [2]pseudorotaxanes (17⊃PmI) composed (Fig. 15) of a crown ether host and a neutral diimide guest. They observed that enhanced host–guest binding occurs in the presence of alkali metal (Li+ and Na+) salts. The alkali metals pre-organise the cavity of the host by coordinating to the ethylene glycol oxygens, as well as coordinating to two imide oxygens in the guest, thus increasing the overall strength of the host–guest complex. The kinetic stabilities of these complexes were examined by 1D NOESY NMR spectroscopy in CD2Cl2 as a function of the alkali metal counteranion at temperatures specifically chosen to render complexation slow on the NMR timescale. Two resonances corresponding to the complexed guest were irradiated in turn, and the resonances associated with the free guest were monitored subsequently for signal transfer from the original irradiation. This procedure allowed the rates of complex dissociation to be extracted. The relative rates of dissociation were then extrapolated to 315 K, a procedure which allows for the comparison of calculated rate constants at various temperatures. The authors observed that for Li+ salts, the order of kinetic stability of the [2]pseudorotaxane increased along the counteranion series CF3SO3− < B(C6F5)4− < Br− < I−—that is, the [2]pseudorotaxane in the presence of Li·CF3SO3 is in faster exchange with the uncomplexed species in solution than is the [2]pseudorotaxane in the presence of Li·I. The authors did not attempt to explain this counteranion effect, but it is reasonable to assume that one cause of the observed kinetic behavior is the competition for binding sites on the Li+ ion between the counteranion, the crown ether oxygens, and the guest imide oxygens. Electrostatic polarisation of Li+, as a consequence of the counteranion, would decrease the oxophilicity of Li+, and thus may also contribute to the observed counteranion effects. It should be noted that meaningful kinetic data could not be obtained for the Na+ complexes, presumably as a result of the complicated multiple equilibria present in solution.
![Structural formulae of host 17 and guest PmI in a metal-templated [2]pseudorotaxane assembly investigated by Sanders and coworkers.7 The authors found that for the Li+ salts, the I− counteranion contributed to a remarkably stable inclusion complex compared to the situation where Br−, B(C6F5)4−, CF3SO3− are the counteranions.](/image/article/2011/CS/c005424k/c005424k-f15.gif) | ||
| Fig. 15 Structural formulae of host 17 and guest PmI in a metal-templated [2]pseudorotaxane assembly investigated by Sanders and coworkers.7 The authors found that for the Li+ salts, the I− counteranion contributed to a remarkably stable inclusion complex compared to the situation where Br−, B(C6F5)4−, CF3SO3− are the counteranions. | ||
The shuttling rate of the ring component in a [2]rotaxane has also been shown9 to depend on the counteranion present. This observation will be discussed in detail in Section 5.2.2.
4. Overcoming counterion effects
In an effort to overcome the often inhibitory effects of counterions discussed in the previous section, various tactics have been devised to deal with these pervasive species. The most commonly employed method to curtail counterion effects in supramolecular and mechanostereochemical systems is to use weakly-coordinating, charge-diffuse counterions49 which interact minimally with their charged counterparts and other species present in the system. By contrast, other researchers have eliminated counterions entirely by making the host and guest counterions of each other and, at the same time, increasing significantly the overall stability of the resulting complex. Alternatively, one can take advantage of the coordinating ability of certain counterions to enhance the host–guest association. This approach is typically accomplished by engineering two separate receptor sites into a host—termed a heteroditopic receptor—such that both species of an ion-pair bind to their corresponding receptor sites on the host.4.1 Non-coordinating counterions
The most common way to minimise the deleterious effects counterions can exert on host–guest complexation is to use weakly-coordinating, charge-diffuse species.49,50 They are often referred to as “non-coordinating,” “passive,” or “spectator” ions, since they are thought to have a minimal effect on the processes involved in host–guest association. Tetrafluoroborate (BF4−), tetraphenylborate (BPh4−), and hexafluorophosphate (PF6−) are the most commonly used weakly-coordinating counteranions when working with charged species in organic solvents, and provide an additional avenue for qualitative analysis through 19F, 11B and/or 31P heteronuclear NMR spectroscopy. A survey of the literature in this area has turned up countless examples of the use of these counteranions. In fact, it is probably safe to say that most supramolecular and mechanostereochemical research groups use these counteranions almost exclusively throughout their routine research unless a specific need or purpose necessitates a change. The reason for the omnipresence of these specific counteranions in host–guest chemistry and mechanostereochemistry is very simple: they work well. They are relatively non-coordinating and non-polarising, and therefore interfere minimally with host–guest association and mechanical movement—i.e., circumrotation in catenanes and shuttling/switching in rotaxanes. Moreover, they improve the solubility of the charged species in organic solvents. Conversely, the most common countercation used51 when studying anion receptors is the tetrabutylammonium cation (Bu4N+) for exactly the same reasons.4.2 Circumventing traditional counterions
Making the host and guest counterions of each other adds a strong electrostatic attractive force to the self-assembly process, in addition to eliminating the traditional counterion effects described in the previous sections. Loeb and coworkers52 have studied the association between the 1,2-bis(pyridinium)ethane cationic guest 18 and the disulfonate macrocycle DB24C8(SO3)2− (Fig. 16) by 1H NMR spectroscopy in a variety of solvents, and have compared their findings with those for the uncharged macrocycle DB24C8. They found that in MeOD, the association constants are greater than 105 M−1 for all four cationic guest derivatives 18a–d with charged macrocycle, DB24C8(SO3)2−, correlating to an 8–45 fold enhancement of the binding constant compared with the association of these guests with the neutral macrocyclic analogue. In the highly H-bond competing and polar solvent system CD3OD/D2O/(CD3)2SO (5∶4∶1), the association constants were lower in general—Ka ≈ 103 M−1—but that for the ion-paired host–guest complex was still approximately five times larger than the Ka value for the complex incorporating the neutral macrocycle, and up to 50 times larger when the tetracationic guest 18d was employed. The authors utilised a chemical double-mutant cycle41 to extract the energetics [Δ(ΔG) = −1.2 kcal mol−1] associated with the stabilising ion–ion interactions and any other accompanying cooperative effects between the ion-paired host–guest complex in a CD3OD/D2O/(CD3)2SO (5∶4∶1) solvent system. This Δ(ΔG) value is significant since it accounts for approximately one quarter of the total stabilisation energy of the complex formed between the guest 18d and the charged macrocycle DB24C8(SO3)2−.
 | ||
| Fig. 16 Structural formulae of compounds studied by Loeb and coworkers.52 When the host and guest are counterions of each other, a significantly more stable complex is observed compared to related host–guest complexes involving guest 18 and a structurally similar neutral host DB24C8 devoid of sulfonate groups. | ||
Nikitin and coworkers53 have investigated (Fig. 17) the association between two anionic bis(p-phenylene)34-crown-10 (BPP34C10) macrocycles—bis(sulfonate) and bis(carboxylate) 20—and the viologen-based guest 19. In agreement with the results obtained by Loeb and coworkers,52 Nikitin et al.53 have observed very stable ion-paired host–guest complexes in organic solvents—reaching up to −9.6 kcal mol−1 (ΔG°) of total stabilisation energy, a ΔG° value which is roughly double that obtained when employing the neutral BPP34C10 macrocycle. In addition, cyclic voltammetry revealed extraordinarily large negative shifts in the reduction potential—up to −450 mV—of the cationic guest in these ion-paired complexes, an observation which is also consistent with strong electrostatic interactions between the guest and the charged macrocycle.
![The association of the ion-paired [2]pseudorotaxane examined by Nikitin and coworkers.53 These authors observed significantly more stable complexes when the host and guest are counterions of each other, compared to host–guest complexes involving guest 19 and a neutral host structurally similar to 20 which is devoid of carboxylate and sulfonate groups.](/image/article/2011/CS/c005424k/c005424k-f17.gif) | ||
| Fig. 17 The association of the ion-paired [2]pseudorotaxane examined by Nikitin and coworkers.53 These authors observed significantly more stable complexes when the host and guest are counterions of each other, compared to host–guest complexes involving guest 19 and a neutral host structurally similar to 20 which is devoid of carboxylate and sulfonate groups. | ||
4.3 Heteroditopic receptors
While the effect of coordinating counterions on host–guest association is typically inhibitory for most supramolecular systems, carefully designed heteroditopic receptors possessing two separate recognition sites—one for the cation and one for the anion—take advantage of the coordinating ability of the counterion to enhance the overall stability of the host/ion-pair complex.54 Although early attempts to stabilise both species of an ion-pair involved using a binary host system55—also called a dual-host or dual-receptor, in which two separate hosts are invoked—there was considerable appeal for engineering both recognition sites into a single receptor, such that selective binding of a target ion-paired salt can be achieved. The field of heteroditopic receptors now dominates the literature. Consequently, binary hosts will not be discussed in this tutorial review. Some receptors, including calix[n]pyrroles56 and calix[n]arenes,57 have been found to possess inherently—and sometimes serendipitously—two recognition sites without specifically engineering these sites into the host structures. Although these receptors and their properties are very interesting, we will focus our attention here on designed heteroditopic receptors specifically. It transpires that the recognition motifs present in most heteroditopic receptors are H-bonding sites suitable for the coordination of H-bond-accepting counteranions—i.e., Cl− and AcO−—leaving electron-rich π-faces and/or crown ether heteroatoms to coordinate to the cation.Sessler and coworkers58 have identified three limiting ion-pair recognition motifs (Fig. 18) for a heteroditopic receptor. Recently they have demonstrated58 all three binding modes using a single host—which is quite rare among heteroditopic receptors—simply by changing the counteranion of the cationic guest. Ion-pairs can bind (Fig. 18) to heteroditopic receptors in one of three different ways: (1) as a contact/intimate ion-pair, in which the cation and anion are in direct contact, (2) as a solvent-bridged ion-pair, in which a solvent molecule mediates the interaction between the cation and anion, or (3) as a host- or ligand-separated ion-pair, in which the host molecule itself bridges the space between the cation and the anion. The binding mode adopted by a particular host/ion-pair complex depends on a number of factors, including—but not limited to—the size of the cation and anion, the distance between the receptor sites on the host, and the polarity of the solvent. Heteroditopic receptor 21, which contains (Fig. 19) both an anion binding calix[4]pyrrole motif and a cation binding calix[4]arene site, was found58 to be a selective host for CsF. For example, in CD3OD∶CDCl3 (9∶1), the addition of either tetrabutylammonium fluoride (TBA·F) or caesium perchlorate (CsClO4) does not result in any shifts in the host 1H resonances, indicating that the host does not bind either Cs+ or F− in isolation. However, mixing these two salts together in one pot resulted in pronounced chemical shift changes for the 1H resonances of host 21. These shifts were similar to those observed on the addition of CsF, a situation which suggests that this ion-pair combination is necessary for binding to host 21 when the respective counterions are TBA+ and ClO4−—as supported by single-crystal X-ray crystallographic data. In addition, combining TBA·F with other metal perchlorate salts, including Li+, Na+, K+, Rb+, and NH4+, produced no change in 1H NMR spectra, once again indicating that F− binds host 21only in the presence of Cs+. By contrast, the Cs+ salts of Cl−, Br−, and NO3− were all found to be complexed by host 21. However, when a mixture of caesium salts (CsF, CsCl, CsBr, and CsNO3) are combined, CsF was found to bind host 21 selectively. The selectivity for CsF is attributed to favourable ion-pairing interactions between Cs+ and F−, as well as to a reduced accessibility of the anion binding site, which would favour a small counteranion like F−. The host/ion-pair binding mode—whether the ion-pair binds as a contact, solvent-bridged, or host-separated ion-pair—can be tuned, simply by changing the counteranion of the Cs+ cation. It was determined by X-ray crystallography that (a) CsBr and CsNO3, (b) CsF, and (c) CsCl bind as (a) contact, (b) solvent-bridged, and (c) host-separated ion-pairs in sequence. We would like to note that many factors can be attributed to this finding, including (i) solvent effects, (ii) the coordinating ability of the counteranion, (iii) the size (steric bulk) of the ions, (iv) the electrostatic polarisation between the ion-pair, and (v) the energy of the separated ion-pair.
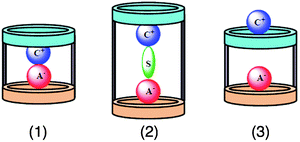 | ||
| Fig. 18 Three limiting ion-pair recognition motifs identified by Sessler and coworkers:58 (1) contact/intimate ion-pair, (2) solvent-bridged ion-pair, and (3) host- or ligand-separated ion-pair. Reprinted with permission from ref. 58. Copyright 2010 American Chemical Society. | ||
 | ||
| Fig. 19 Structural formula of the heteroditopic receptor 21 studied by Sessler and coworkers.58 The receptor was found to bind the Cs+ cation as a contact, solvent-bridged, and host-separated ion-pair (Fig. 18) simply by changing the counteranion to which it is associated. | ||
Another example of an ion-pair heteroditopic receptor has been reported by Smith and coworkers,59,60 who have utilised the macrobicyclic receptor 22 (Fig. 20) to complex with a variety of alkali metal and alkylammonium salts as contact ion-pairs. Receptor 22 is composed of a diazacrown ether macrocycle which coordinates cations through the crown N and O atoms, and an isophthalamide moiety that can bind to anions by forming N–H hydrogen bonds. Smith and coworkers59 first of all examined the binding properties of receptor 22 with various Na+, K+, and Cl− salts in CD3SOCD3—in order to determine the binding affinity of each species—both separately and in the presence of each other. The authors found that Na+ and K+—as their BPh4− salts—have a very low affinity for receptor 22, namely 5 and 8 M−1, respectively, yet, in the presence of Cl−, the association constants increase by 5- and 40-fold, respectively. Similarly, Cl−—as its TBA+ salt—is weakly bound by receptor 22, yet in the presence of K+, the binding affinity increases by over 10-fold. These results suggest that this particular receptor binds ion-paired KCl much more tightly than either K+ or Cl− alone. This is most likely a result of a synergistic effect of the ions coordinating to each other and also to the receptor simultaneously. X-Ray crystallographic evidence supports the conclusion that KCl binds as a contact ion-pair. Using 1H NMR spectroscopy, the binding properties of receptor 22 with the alkylammonium cations n-PrNH3+, i-PrNH3+, and TBA+ as Cl−, TsO−, and AcO− salts were examined60 in CDCl3∶CD3SOCD3 (85∶15). The authors found that the TBA+ salts of all three counteranions gave very low association constants with receptor 22 (≤50 M−1), despite the fact that the n-PrNH3+ and i-PrNH3+ cations—as salts of all three counteranions—were complexed much more tightly with association constants ranging between 102–104 M−1. Evidence supporting the assertion that these alkylammonium salts are being bound as contact ion-pairs can be obtained from the analysis of a single-crystal X-ray structure between receptor 22 and MeNH3·Cl, in which the cooperative effects between the ion-pair and receptor are revealed. The guest RNH3+ group forms two H-bonds to the receptor macrocycle and one H-bond with the Cl− counteranion, an observation which explains the larger association with the n-PrNH3+ and i-PrNH3+ guests compared with TBA+.
 | ||
| Fig. 20 Structural formula of the heteroditopic receptor investigated by Smith and coworkers.59,60 Salts which were observed to bind to host 22 as contact ion-pairs had much higher association constants than salts which do not bind as contact ion-pairs. This situation was attributed to a synergistic effect of the ions coordinating to each other and to the receptor simultaneously. | ||
5. Harnessing counterion effects
In the previous sections of this tutorial review, illustrative examples have been presented which demonstrate both the inhibitory and cooperative effects that counterions can have on various physical processes in host–guest recognition, along with a variety of studies that provide plausible explanations and rationalisations for the observed counterion effects. The goal in this section is to provide an overview of the many examples in the literature where researchers have harnessed the various properties of counterions to perform a desired function.5.1 Anion-templated assembly of mechanically interlocked structures
The anion-templated assembly of mechanically interlocked structures (Fig. 21) is an active field with several excellent reviews on the subject, one of the most recent having been written by Vickers and Beer.61 In it, they focus on the combination of anion templation with ion-pairing for the construction of mechanically interlocked structures. Given this recent coverage, this type of templation strategy will be only briefly discussed in this tutorial review. For the use of anions as templates in macrocyclization strategies, the reader is directed to an earlier review62 on the subject.![The construction of a [2]pseudorotaxane via anion templation is a result of ion-pairing within the thread and orthogonal anion recognition by the macrocycle.63 Reprinted with permission from ref. 63. Copyright 2005 American Chemical Society.](/image/article/2011/CS/c005424k/c005424k-f21.gif) | ||
| Fig. 21 The construction of a [2]pseudorotaxane via anion templation is a result of ion-pairing within the thread and orthogonal anion recognition by the macrocycle.63 Reprinted with permission from ref. 63. Copyright 2005 American Chemical Society. | ||
![Structural formulae of the compounds investigated by Beer and coworkers,63 in which the assembly of a [2]pseudorotaxane between guest 23 and host 24 is templated by the associated counteranion.](/image/article/2011/CS/c005424k/c005424k-f22.gif) | ||
| Fig. 22 Structural formulae of the compounds investigated by Beer and coworkers,63 in which the assembly of a [2]pseudorotaxane between guest 23 and host 24 is templated by the associated counteranion. | ||
5.2 Counterion-controlled mechanical movement in mechanically interlocked structures
The material presented in the following subsections illustrates how various types of mechanical movement within mechanically interlocked molecules, such as (i) circumrotation in [2]catenanes, (ii) translational isomerism in [2]rotaxanes, and (iii) extension/contraction in cage-based [2]rotaxanes, can all be controlled rather precisely simply by changing the identity of the counterion in the system.![Acid/base triggered circumrotation in a [2]catenane.64 Upon counterion exchange from PF6− to Cl−, circumrotation is inhibited as a consequence of tight binding between Cl− and the pocket created by both macrocycles in the [2]catenane.](/image/article/2011/CS/c005424k/c005424k-f23.gif) | ||
| Fig. 23 Acid/base triggered circumrotation in a [2]catenane.64 Upon counterion exchange from PF6− to Cl−, circumrotation is inhibited as a consequence of tight binding between Cl− and the pocket created by both macrocycles in the [2]catenane. | ||
![(a) Structural formula of the [2]rotaxane investigated by Laursen et al.65 (b) Graphical representation of counterion-induced translational isomerism in a [2]rotaxane. Reprinted with permission from ref. 65. Copyright 2004 American Chemical Society.](/image/article/2011/CS/c005424k/c005424k-f24.gif) | ||
| Fig. 24 (a) Structural formula of the [2]rotaxane investigated by Laursen et al.65 (b) Graphical representation of counterion-induced translational isomerism in a [2]rotaxane. Reprinted with permission from ref. 65. Copyright 2004 American Chemical Society. | ||
Counterion-induced switching in a bistable [2]rotaxane (Fig. 25) containing guanidinium and pyridinium stations has been explored by Chiu and coworkers.66 When the counteranion is the weakly-coordinating PF6− ion, the crown ether macrocycle (BPX26C6) encircles the guanidinium unit as a consequence of strong H-bonding interactions, as determined by 1H NMR spectroscopy in CD3NO2 from complexation-induced chemical shifts and from 2D NOESY experiments. Upon the addition of three equivalents of TBA·Cl, Cl− exchanges with PF6− and causes the crown ether to shuttle to the pyridinium recognition site, presumably as a result of competition between the crown ether and the Cl− ion for H-bonding interactions with the guanidinium N–H moieties. Subsequent addition of AgPF6 removes the Cl− ions through precipitation of AgCl, thus reinstating PF6− as the counteranion, and returning the BPX26C6 macrocycle to its original position encircling the guanidinium recognition site.
![Counterion-induced switching in a bistable [2]rotaxane examined by Chiu and coworkers.66 As the PF6− salt, the crown ether preferentially binds to the guanidinium station. When the counteranion is exchanged with Cl−, however, the Cl− anion H-bonds with the guanidinium cation, inducing the macrocyclic polyether to shuttle to the pyridinium station. The addition of AgPF6 causes AgCl to precipitate from solution, and replaces the original PF6− counteranion, resetting the [2]rotaxane to its initial state.](/image/article/2011/CS/c005424k/c005424k-f25.gif) | ||
| Fig. 25 Counterion-induced switching in a bistable [2]rotaxane examined by Chiu and coworkers.66 As the PF6− salt, the crown ether preferentially binds to the guanidinium station. When the counteranion is exchanged with Cl−, however, the Cl− anion H-bonds with the guanidinium cation, inducing the macrocyclic polyether to shuttle to the pyridinium station. The addition of AgPF6 causes AgCl to precipitate from solution, and replaces the original PF6− counteranion, resetting the [2]rotaxane to its initial state. | ||
Chiu and coworkers67 have also investigated the extension/contraction movements in a cage-based [2]rotaxane which can be controlled by counteranion exchange between PF6− and F− (Fig. 26). As the PF6− salt, the stretched isomer of the cage-based [2]rotaxane is found to predominate in CD3CN solution, as determined by the significant upfield 1H NMR chemical shifts of the central n-octyl methylene chain protons compared with proton shifts in the distal n-hexyl chains—as well as the relatively unperturbed chemical shifts of the signals for the pyridinium protons relative to those for the free dumbbell without any macrocycle present. Counteranion exchange from PF6− to F− upon addition of 4 equivalents of TBA·F resulted in a preference for the contracted translational isomer, as verified by the strong shielding of protons in the n-hexyl chains, indicating that they have been drawn into the macrocyclic cavity, as well as the notably less shielded protons on the central n-octyl chain, an observation which suggests it has been extruded through one of the 34-membered ring openings on the macrocycle. Addition of Ca(BF4)2 removes F− anions through precipitation of CaF2 and exchanges the counteranion with the weakly-coordinating BF4−, producing an 1H NMR spectrum similar to that recorded for the original stretched form as the PF6− salt. The authors note that the main driving force leading to the contracted translational isomer upon F− ion addition is disruption of the [N–H⋯O] hydrogen bonds between the dumbbell and the ring, presumably because of strong competition between the ring and F− for H-bonding sites on the thread.
![Counterion-induced extension/contraction in a cage-based [2]rotaxane studied by Chiu and coworkers.67 When the counteranion is BF4−, the [2]rotaxane resides preferentially in a stretched form, as a consequence of H-bonding interactions between the ammonium centres and the crown ether oxygens. When TBA·F is introduced into solution, the F− anion binds to the ammonium centres, disrupting the initially present H-bonds to the crown ether, resulting in the contracted translational isomer. Reprinted with permission from ref. 67. Copyright 2009 American Chemical Society.](/image/article/2011/CS/c005424k/c005424k-f26.gif) | ||
| Fig. 26 Counterion-induced extension/contraction in a cage-based [2]rotaxane studied by Chiu and coworkers.67 When the counteranion is BF4−, the [2]rotaxane resides preferentially in a stretched form, as a consequence of H-bonding interactions between the ammonium centres and the crown ether oxygens. When TBA·F is introduced into solution, the F− anion binds to the ammonium centres, disrupting the initially present H-bonds to the crown ether, resulting in the contracted translational isomer. Reprinted with permission from ref. 67. Copyright 2009 American Chemical Society. | ||
Credi and coworkers9 have observed that the shuttling rate in an acid/base-switchable bistable [2]rotaxane depends on both the shuttling direction and the identity of the counteranion associated with the positively charged recognition sites in the dumbbell component (Fig. 27). Stopped-flow kinetic experiments performed in acetonitrile solution revealed that, at room temperature, the A → B shuttling process is approximately 60-fold slower than the reverse AH ← BH shuttling process, and activation parameters, determined from Erying plots, suggest that the forward and reverse shuttling processes occur by different and complex mechanisms. For example, A → B shuttling is expected to release the two PF6− counteranions associated with the bipyridinium recognition site into the bulk solution upon arrival of the ring, while it is anticipated that the solvent molecules will be highly structured around the naked counteranions, thereby decreasing the entropy of the system. In the reverse AH ← BH shuttle case, only one PF6− will dissociate itself from the ammonium recognition site into the solvent upon complexation with the ring, leading to a concomitant shedding of structured solvent molecules from the two PF6− ions that re-form ion-pairs with the bipyridinium recognition site, from whence it is expected that the entropy change will be smaller. It should also be noted that in the first A → B deprotonation cycle, the counteranions associated with the dumbbell are PF6− ions, whereas upon re-protonation with trifluoroacetic acid, CF3CO2− counteranions are introduced into the system, and they will have the potential to H-bond to the ammonium recognition site and affect the association with the ring, thus contributing to the complexity of the shuttling mechanism. In order to probe the effect of the counteranions on the shuttling dynamics yet further, stopped-flow kinetic measurements were performed in the presence of 100 equivalents of PF6− and TsO− counteranions as their TBA+ and Et4N+ salts, respectively. Compared with the experimental results obtained when no salt is added, an excess of PF6− ions causes the rates of both the forward and reverse shuttling processes to decrease, an observation which the authors note is consistent with an increase in the extent of ion-pairing, which presumably makes it more difficult for the ring to encircle the bipyridinium unit. In the experiment with an excess of TsO−, however, the A → B shuttling rate becomes accelerated two-fold compared to the case where no salt is added, while the reverse AH ← BH shuttling rate was observed to be slightly dampened. The effect of the TsO− counteranions on the shuttling rates has been attributed to its H-bond accepting ability. In particular, the TsO− counteranions may H-bond to the N–H group of the dumbbell, stabilising the transition state of the A → B shuttling process, thus increasing the rate. It should be noted that, although the ion-pairing between TsO− and the bipyridinium recognition site is stronger than with PF6−, a situation which would be expected to inhibit the A → B shuttling, the opposite effect is observed and unexpected. In the reverse process, the TsO− counteranions can H-bond to the ammonium recognition site, thus inhibiting the AH ← BH shuttling.
 | ||
| Fig. 27 Structural formulae and acid/base triggered switching cycle studied by Credi and coworkers.9 The shuttling kinetics depend on the shuttling direction of the macrocyclic ring—i.e., the A → B process is slower than the AH ← BH process. | ||
5.3 Counterion-controlled supramolecular superstructures
The ability to control the size and arrangement of supramolecular superstructures by changing something as simple and seemingly innocent as the nature of the associated counterion demonstrates both the power and the complexity of noncovalent bonding interactions. In this section, the influence of the counterion identity on a variety of different types of supramolecular organisation will be discussed. | ||
| Fig. 28 Counterion-induced supramolecular oligomerisation studied by Parisi and coworkers.21 The number average degree of oligomerisation was found to depend on the identity of the associated counteranion, increasing along the series Cl− < Br− < Pic−. | ||
The solution and solid-state structures of short peptide chains were also found to depend on the nature of the counteranion present in the system. Yang et al.68 have synthesised an amphiphilic peptide chain (Fig. 29) capable of binding Cu2+ ions via a glycine-glycine-histidine residue at the C-terminus of the peptide as a result of copper chelation with the nitrogen atoms. When Cu2+ salts of Cl−, NO3−, and SO42− were added to a solution of the peptide, the extent of α-helix and β-sheet formation was found by Fourier transform infrared spectroscopy (FT-IR) to depend markedly on the nature of the counteranion. Specifically, without any copper salt present, the peptide exists as a mixture of α-helices and β-sheets. However, the addition of Cu2+ salts has been shown to favor β-sheet formation progressively along the series Cl− < NO3− < SO42−, with CuSO4 producing β-sheets all but quantitatively. Additionally, the morphology of nanofibers formed from the peptide and Cu2+ salt mixtures was studied by atomic force microscopy (AFM) in the solid-state. Long fibers (>1000 nm) were formed almost exclusively using CuSO4, but much shorter fibers (<300 nm) on average were observed in the absence of salt, or in the presence of CuCl2 or Cu(NO3)2. The authors attribute the observed differences in β-sheet formation and nanofiber length to the divalent nature of the SO42− counteranion, a feature which may serve as a bridge between two positively charged lysine residues on different peptide chains, thus promoting and stabilising the formation of β-sheets. The monovalent Cl− and NO3− counteranions, however, cannot provide this type of stabilisation. The importance of the countercation for the formation of the nanofiber was also investigated in situations where Na2SO4 and NaCl were used as copper salt surrogates. However, only short fibers were observed in both cases, and thus the presence of both Cu2+ and SO42− are required for the assembly of long nanofibers.
 | ||
| Fig. 29 Structural formula of the amphiphilic peptide chain synthesised and investigated by Yang et al.68 The use of divalent counteranions, such as SO42−, was found to promote and stabilise the formation of β-sheets. The use of monovalent counteranions promoted β-sheet formation to a lesser extent. | ||
The ability to adjust the strengths and stoichiometries of pseudorotaxanes formed between the heteroditopic phenylureido-containing calix[6]arene host 25 and the diazapyrenium guest 26 (Fig. 30) by changing the concentrations and identities of the counteranions was recently demonstrated by Secchi, Silvi, and coworkers.69 UV/Vis spectroscopic titrations in CH2Cl2 of up to 2 equivalents of host 25 into a solution of guest 26 ([26·2PF6]tot = 3.5 × 10−5 M) revealed that both 1∶1 and 2∶1 (host∶guest) complexes are formed under these conditions, with association constants of 3.6 × 105 and 1.9 × 105 M−1, respectively. However, by performing the same titration in the presence of 100 equivalents of tetraethylammonium hexafluorophosphate (Et4N·PF6), an experiment which shifts the equilibrium towards the ion-paired guest, only 1∶1 complexes were observed with association constants which are decreased by a factor of 7. For steric reasons, the cationic guest can only penetrate the cavity of the calix[6]arene after dissociation of its counterions. Therefore, the presence of an excess of PF6− counteranions increases the proportion of ion-paired guest in solution and effectively inhibits pseudorotaxane formation. Interestingly, when the original titration is repeated in the presence of two equivalents of Et4N·TsO, which exchanges PF6− for TsO−, a 1∶1 stoichiometry is observed, and the association constant of the complex is increased to ∼108 M−1. The exceptional stability of this complex can be attributed to the orthogonal coordinating ability of the TsO− counteranion, which can interact strongly with both the cationic guest—presumably through π–π stacking, [C–H⋯O] H-bonding, electrostatics, and cation–π interactions—as well as with the phenylureido moieties on the host as a result of [N–H⋯O] H-bonding and π–π stacking interactions. The authors note that the stoichiometries and strengths of the complexes in CH2Cl2 solution are affected by the presence of the counteranion. Ion-pairing with the guest modulates the observed behavior as a consequence of steric effects—wherein the guest can only bind to the host when dissociated from at least one counteranion—as well as competition effects, wherein the host 25 competes with the guest for the counteranion. It is instructive to note that, although the counteranion effects are inhibitive in the case of PF6−, the ability of the TsO− counteranion to bind to both the guest and host species actively facilitates host–guest binding and the resulting complex is extremely stable. This investigation constitutes an excellent example which illustrates the fact that the stoichiometry and strength of a host–guest complex can be modulated simply by changing the concentration and nature of the associated counteranion in the system.

6. Conclusions
In this tutorial review, counterion effects have been shown to affect both the thermodynamic and kinetic stabilities of host–guest complexes, as well as the threading and shuttling rates in [2]pseudorotaxanes and [2]rotaxanes, respectively. These effects have been attributed to the properties of the counterion, and have been shown to operate under the influence of a variety of phenomena relating to steric effects, charge polarisation, competition, stability of the separated ion-pair, and the counterion coordinating ability. Weakly-coordinating counterions are typically employed to minimise any inhibitory effects that they may impart. As the coordinating ability of a counterion associated with a charged guest increases, the inhibitory effects become more pronounced. An exception to this rule is observed when a host can interact specifically with both a guest and its associated counterion. In this case, a synergistic effect is observed which typically increases the overall stability of the ternary complex. Some researchers have demonstrated how the nature of the counterion can be harnessed—for example, (i) to template the syntheses of interpenetrated superstructures and mechanically interlocked structures, (ii) to influence the circumrotation in [2]catenanes and translational isomerism in [2]rotaxanes, and (iii) to exert control over the solution state organisation of complex supramolecular systems. It is our hope that, through a better understanding of counterion effects, novel stimuli-responsive supramolecular and mechanostereochemical systems will be constructed that utilize the predictable effects of counterions for applications spanning molecular electronics to molecular muscles to untold intellectual fancies.7. Note
Since this review was written and submitted for publication, it has come to our attention that a themed issue on anions in supramolecular chemistry, edited by Professors Philip A. Gale and Thorfinnur Gunnlaugsson, has appeared in Chemical Society Reviews. We refer readers of this article on counterion effects in supramolecular and mechanostereochemical systems to the Editorial70 of the above mentioned themed issue, and in particular the article that highlights progress on anion receptor chemistry from 2008 and 2009,71 in addition to the articles detailing anion effects in crystal engineering,72 heteroditopic receptors,73 energetics involved in host-anion complexation,74 anion-tuned supramolecular gels75 and de novo structure-based design methods for anion receptors.76Acknowledgements
This work was supported by the US Air Force Office of Scientific Research (AFOSR: FA9550-07-1-0534) and by the National Science Foundation (CHE-0924620), and the Microelectronics Advanced Research Corporation (MARCO) and its Focus Center Research Program (FCRP) on Functional Engineered NanoArchitectonics (FENA). The authors thank Aleksandr Bosoy for preparing the graphical abstract.References
- J. F. Stoddart, Nat. Chem., 2009, 1, 14–15 Search PubMed.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- M. A. Olson, Y. Y. Botros and J. F. Stoddart, Pure Appl. Chem, 2010, 82, 1569–1574 CrossRef CAS.
- The current tutorial review concentrates on counterion effects in supramolecular and mechanostereochemical systems in solution. However, counterion effects in the solid-state are known, and noncovalent intermolecular bonding interactions in supramolecular systems that are absent or too weak to be observed in solution may play a dramatic role in the assembly of the crystal lattice during crystallization. For example, a threaded 1∶1 complex between dibenzylammonium hexafluorophoshate and tetrabenzo[24]crown-8 (TB24C8) does not form in solution under the same conditions that lead efficiently to the [2]pseudorotaxane when using dibenzo[24]crown-8 (DB24C8). However, on crystallization, a network of [2]pseudorotaxanes is formed and is stabilized by a multitude of enthalpically favorable [C–H⋯F] hydrogen bonds with a highly ordered matrix of PF6− anions—the unusual lack of disorder in the anions suggests that they are playing an intrinsic role in the kinetically controlled crystallization. For further details, see:
(a) P. R. Ashton, S. J. Cantrill, J. A. Preece, J. F. Stoddart, Z.-H. Wang, A. J. P. White and D. J. Williams, Org. Lett., 1999, 1, 1917–1920 CrossRef CAS. For an early example, see:
(b) P. R. Ashton, M. C. T. Fyfe, P. T. Glink, S. Menzer, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1997, 119, 12514–12524. CrossRef CAS For some recent examples, see:
(c) B. H. Northrop, S. J. Khan and J. F. Stoddart, Org. Lett., 2006, 8, 2159–2162 CrossRef CAS;
(d) Y. Yang, H.-Y. Hu and C.-F. Chen, Tetrahedron Lett., 2007, 48, 3505–3509 CrossRef CAS;
(e) H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 Search PubMed.
- S. S. Moore, T. L. Tarnowski, M. Newcomb and D. J. Cram, J. Am. Chem. Soc., 1977, 99, 6398–6405 CrossRef CAS.
- S. Bartoli and S. Roelens, J. Am. Chem. Soc., 1999, 121, 11908–11909 CrossRef CAS.
- S. I. Pascu, T. Jarrosson, C. Naumann, S. Otto, G. Kaiser and J. K. M. Sanders, New J. Chem., 2005, 29, 80–89 RSC.
- A. Credi, S. Dumas, S. Silvi, M. Venturi, A. Arduini, A. Pochini and A. Secchi, J. Org. Chem., 2004, 69, 5881–5887 CrossRef CAS.
- S. Garaudée, S. Silvi, M. Venturi, A. Credi, A. H. Flood and J. F. Stoddart, ChemPhysChem, 2005, 6, 2145–2152 CrossRef CAS.
- Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585–4621 CrossRef CAS.
- K. Koszinowski and P. Böhrer, Organometallics, 2009, 28, 100–110 CrossRef CAS.
- T. B. Gasa, J. M. Spruell, W. R. Dichtel, T. J. Sorensen, D. Philp, J. F. Stoddart and P. Kuzmic, Chem.–Eur. J., 2009, 15, 106–116 CrossRef CAS.
- S. Roelens, A. Vacca and C. Venturi, Chem.–Eur. J., 2009, 15, 2635–2644 CrossRef CAS.
- M. Krell, M. C. R. Symons and J. Barthel, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 3419–3427 RSC.
- R. Arnecke, V. Böhmer, R. Cacciapaglia, A. Dalla Cort and L. Mandolini, Tetrahedron, 1997, 53, 4901–4908 CrossRef CAS.
- V. Böhmer, A. Dalla Cort and L. Mandolini, J. Org. Chem., 2001, 66, 1900–1902 CrossRef CAS.
- A. Arduini, A. Pochini and A. Secchi, Eur. J. Org. Chem., 2000, 2325–2334 CrossRef CAS.
- A. Arduini, W. M. McGregor, D. Paganuzzi, A. Pochini, A. Secchi, F. Ugozzoli and R. Ungaro, J. Chem. Soc., Perkin Trans. 2, 1996, 839–846 RSC.
- G. G. Talanova, N. S. A. Elkarim, V. S. Talanov, R. E. Hanes Jr., H. -S. Hwang, R. A. Bartsch and R. D. Rogers, J. Am. Chem. Soc., 1999, 121, 11281–11290 CrossRef CAS.
- S. M. Doughty, J. F. Stoddart, H. M. Colquhoun, A. M. Z. Slawin and D. J. Williams, Polyhedron, 1985, 4, 567–575 CrossRef CAS.
- S. Pappalardo, V. Villari, S. Slovak, Y. Cohen, G. Gattuso, A. Notti, A. Pappalardo, I. Pisagatti and M. F. Parisi, Chem.–Eur. J., 2007, 13, 8164–8173 CrossRef CAS.
- S. Roelens and R. Torriti, J. Am. Chem. Soc., 1998, 120, 12443–12452 CrossRef CAS.
- A. L. Whiting, N. M. Neufeld and F. Hof, Tetrahedron Lett., 2009, 50, 7035–7037 CrossRef CAS.
- P. C. Kearney, L. S. Mizoue, R. A. Kumpf, J. E. Forman, A. McCurdy and D. A. Dougherty, J. Am. Chem. Soc., 1993, 115, 9907–9919 CrossRef CAS.
- Z. Miskolczy and L. Biczók, Chem. Phys. Lett., 2009, 477, 80–84 CrossRef CAS.
- C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705–1723 RSC.
- D. H. Busch and N. A. Stephenson, Coord. Chem. Rev., 1990, 100, 119–154 CrossRef CAS.
- S. Anderson, H. L. Anderson and J. K. M. Sanders, Acc. Chem. Res., 1993, 26, 469–475 CrossRef CAS.
- H. Mo, A. Wang, P. S. Wilkinson and T. C. Pochapsky, J. Am. Chem. Soc., 1997, 119, 11666–11673 CrossRef CAS.
- S. M. Ngola, P. C. Kearney, S. Mecozzi, K. Russell and D. A. Dougherty, J. Am. Chem. Soc., 1999, 121, 1192–1201 CrossRef CAS.
- G. Heinrichs, S. Kubik, J. Lacour and L. Vial, J. Org. Chem., 2005, 70, 4498–4501 CrossRef CAS.
- E. Martínez-Viviente, P. S. Pregosin, L. Vial, C. Herse and J. Lacour, Chem.–Eur. J., 2004, 10, 2912–2918 CrossRef CAS.
- S. Roelens and R. Torriti, Supramol. Chem., 1999, 10, 225–232 CAS.
- S. Bartoli and S. Roelens, J. Am. Chem. Soc., 2002, 124, 8307–8315 CrossRef CAS.
- P. Sarri, F. Venturi, F. Cuda and S. Roelens, J. Org. Chem., 2004, 69, 3654–3661 CrossRef CAS.
- H. Iwamoto, K. Niimi, T. Haino and Y. Fukazawa, Tetrahedron, 2009, 65, 7259–7267 CrossRef CAS.
- M. Szwarc, Ions and Ion Pairs in Organic Reactions, Wiley-Interscience, New York, 1974 Search PubMed.
- P.-O. Norrby and T. Liljefors, J. Am. Chem. Soc., 1999, 121, 2303–2306 CrossRef CAS.
- D. Kim, E. C. Lee, K. S. Kim and P. Tarakeshwar, J. Phys. Chem. A, 2007, 111, 7980–7986 CrossRef CAS.
- C. A. Hunter, C. M. R. Low, C. Rotger, J. G. Vinter and C. Zonta, Chem. Commun., 2003, 834–835 RSC.
- S. L. Cockroft and C. A. Hunter, Chem. Soc. Rev., 2007, 36, 172–188 RSC.
- F. Huang, J. W. Jones, C. Slebodnick and H. W. Gibson, J. Am. Chem. Soc., 2003, 125, 14458–14464 CrossRef CAS.
- J. W. Jones and H. W. Gibson, J. Am. Chem. Soc., 2003, 125, 7001–7004 CrossRef CAS.
- M. Montalti and L. Prodi, Chem. Commun., 1998, 1461–1462 RSC.
- M. Clemente-León, C. Pasquini, V. Hebbe-Viton, J. Lacour, A. Dalla Cort and A. Credi, Eur. J. Org. Chem., 2006, 105–112 CrossRef CAS.
- D. González-Rodríguez, J. L. J. v. Dongen, M. Lutz, A. L. Spek, A. P. H. J. Schenning and E. W. Meijer, Nat. Chem., 2009, 1, 151–155 Search PubMed.
- K.-S. Jeong, K.-M. Hahn and Y. L. Cho, Tetrahedron Lett., 1998, 39, 3779–3782 CrossRef CAS.
- S. Mecozzi, A. P. West and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10566–10571 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS.
- S. H. Strauss, Chem. Rev., 1993, 93, 927–942 CrossRef CAS.
- C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520–563 RSC.
- D. J. Hoffart, J. Tiburcio, A. de la Torre, L. K. Knight and S. J. Loeb, Angew. Chem., Int. Ed., 2008, 47, 97–101 CrossRef CAS.
- E. Lestini, K. Nikitin, H. Müller-Bunz and D. Fitzmaurice, Chem.–Eur. J., 2008, 14, 1095–1106 CrossRef CAS.
- S. Roelens, A. Vacca, O. Francesconi and C. Venturi, Chem.–Eur. J., 2009, 15, 8296–8302 CrossRef CAS.
- G. Cafeo, G. Gattuso, F. H. Kohnke, A. Notti, S. Occhipinti, S. Pappalardo and M. F. Parisi, Angew. Chem., Int. Ed., 2002, 41, 2122–2126 CrossRef CAS.
- D. E. Gross, F. P. Schmidtchen, W. Antonius, P. A. Gale, V. M. Lynch and J. L. Sessler, Chem.–Eur. J., 2008, 14, 7822–7827 CrossRef CAS.
- A. Arduini, G. Giorgi, A. Pochini, A. Secchi and F. Ugozzoli, J. Org. Chem., 2001, 66, 8302–8308 CrossRef CAS.
- S. K. Kim, J. L. Sessler, D. E. Gross, C.-H. Lee, J. S. Kim, V. M. Lynch, L. H. Delmau and B. P. Hay, J. Am. Chem. Soc., 2010, 132, 5827–5836 CrossRef CAS.
- J. M. Mahoney, A. M. Beatty and B. D. Smith, J. Am. Chem. Soc., 2001, 123, 5847–5848 CrossRef CAS.
- J. M. Mahoney, J. P. Davis, A. M. Beatty and B. D. Smith, J. Org. Chem., 2003, 68, 9819–9820 CrossRef CAS.
- M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211–225 RSC.
- N. Gimeno and R. Vilar, Coord. Chem. Rev., 2006, 250, 3161–3189 CrossRef CAS.
- M. R. Sambrook, P. D. Beer, J. A. Wisner, R. L. Paul, A. R. Cowley, F. Szemes and M. G. B. Drew, J. Am. Chem. Soc., 2005, 127, 2292–2302 CrossRef CAS.
- K.-Y. Ng, V. Felix, S. M. Santos, N. H. Rees and P. D. Beer, Chem. Commun., 2008, 1281–1283 RSC.
- B. W. Laursen, S. Nygaard, J. O. Jeppesen and J. F. Stoddart, Org. Lett., 2004, 6, 4167–4170 CrossRef CAS.
- T.-C. Lin, C.-C. Lai and S.-H. Chiu, Org. Lett., 2009, 11, 613–616 CrossRef CAS.
- C.-J. Chuang, W.-S. Li, C.-C. Lai, Y.-H. Liu, S.-M. Peng, I. Chao and S. H. Chiu, Org. Lett., 2009, 11, 385–388 CrossRef CAS.
- H. Yang, M. Pritzker, S. Y. Fung, Y. Sheng, W. Wang and P. Chen, Langmuir, 2006, 22, 8553–8562 CrossRef CAS.
- M. Semeraro, A. Arduini, M. Baroncini, R. Battelli, A. Credi, M. Venturi, A. Pochini, A. Secchi and S. Silvi, Chem.–Eur. J., 2010, 16, 3467–3475 CrossRef CAS.
- P. A. Gale and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3595 RSC.
- P. A. Gale, Chem. Soc. Rev., 2010, 39, 3746 RSC.
- R. Custelacean, Chem. Soc. Rev., 2010, 39, 3675 RSC.
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784 RSC.
- F. P. Schmidtchen, Chem. Soc. Rev., 2010, 39, 3916 RSC.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686 RSC.
- B. P. Hay, Chem. Soc. Rev., 2010, 39, 3700 RSC.
| This journal is © The Royal Society of Chemistry 2011 |