Understanding chemical reaction mechanisms in ionic liquids: successes and challenges
Colin D.
Hubbard
,
Peter
Illner
and
Rudi
van Eldik
*
Inorganic Chemistry, Department of Chemistry and Pharmacy, University of Erlangen-Nürnberg, Egerlandstraße 1, 91058 Erlangen, Germany. E-mail: vaneldik@chemie.uni-erlangen.de
First published on 15th November 2010
Abstract
The focus of this article is an examination of chemical reaction mechanisms in ionic liquids. These mechanisms are compared with those pertaining to the same reactions carried out in conventional solvents. In cases where the mechanisms differ, attempts to provide an explanation in terms of the chemical and physicochemical properties of the reactants and of the components of the ionic liquids are described. A wide range of reactions from different branches of chemistry has been selected for this purpose. A sufficient background for student readers has been included. This tutorial review should also be of interest to kineticists, and to both new and experienced investigators in the ionic liquids field.
 Colin D. Hubbard | Colin D. Hubbard earned Chemistry degrees at the University of Sheffield. He carried out postdoctoral work at MIT, Cornell University, and the University of California (Berkeley). He joined the chemistry faculty of the University of New Hampshire in 1967 and became a Professor of Chemistry in 1979, and is now an Emeritus Professor. He joined the group of Rudi van Eldik in Erlangen, Germany, in 1994 and remains as a Visiting Professor. He also served as a Research Manager at Unilever Research (Colworth, UK) (1998–2000). He has published widely in academic journals and has written reviews and book chapters on kinetics and mechanisms. |
 Peter Illner | Peter Illner started his chemistry studies in 1998 at the Friedrich-Alexander-University of Erlangen-Nuremberg in Germany. During his doctoral studies he performed kinetic investigations on fast substitution reactions of metal complexes in ionic liquids to clarify the influence of these new solvents on their mechanistic behavior. He received his PhD in 2009. He stayed in Erlangen as postdoctoral fellow to work on solute–solvent interactions in and solvent–solvent interactions of ionic liquids. The goal of this work is to understand and if possible to predict the behavior of ionic liquids as solvents. He is the co-author of a series of papers on mechanistic studies performed in ionic liquids. |
 Rudi van Eldik | Rudi van Eldik was born in Amsterdam, grew up in Johannesburg, and received his chemistry education at Potchefstroom University (SA) followed by post-doctoral work in Buffalo (USA) and Frankfurt (Germany). After being Professor of Inorganic Chemistry at the Private University of Witten/Herdecke from 1987 to 1994, he was appointed as Chair of Inorganic and Analytical Chemistry at the University of Erlangen-Nürnberg. His research interests cover the elucidation of inorganic and bioinorganic reaction mechanisms, with special emphasis on the application of high pressure thermodynamic and kinetic techniques. He is Editor of Advances in Inorganic Chemistry and author of ca. 800 research papers and reviews (www.chemie.uni-erlangen.de/vaneldik). |
1. Introduction
In the past two to three decades interest has grown in the application of what used to be thought of as non-traditional methods of chemical reaction initiation, and in alternative media for conducting chemical reactions. Microwave dielectric heating has been shown in some cases to be advantageous in chemical synthesis and the basic principles of the method are relatively well understood. Sonochemical transformations are less well understood except that some form of acoustic cavitation is responsible. Although supercritical fluids have been known for over a century their employment as alternative media in both preparative and mechanistic chemistry in any systematic way is more recent. Elevated hydrostatic pressures in selected examples can be of benefit in organic synthesis. Diagnosis of reaction mechanisms in inorganic and bioinorganic chemistry has been dramatically improved by use of elevated pressures in kinetics investigations. All of these developments are driven partly or wholly in pursuit of “greener” chemistry, for greater energy efficiency or labour intensity reduction. Early background1 and more recent representative publications regarding extreme or non-classical conditions within chemistry are cited.2–5 Ionic liquids that once may have been termed as alternative (to traditional organic or other solvents), but are now “mainstream” insofar as an explosion of interest and fascination in their use has occurred, are the focus of this article.1.1 Background of ionic liquids
Ionic liquids are quite distinct from the ionic state of many simple electrolytes that exist as molten salts at elevated temperature, in that many are in the liquid state at ambient temperature. They may be termed RTILs (Room Temperature Ionic Liquids), or interchangeably and thereafter herein as ILs. It does not appear that any firm set of criteria in terms of properties define an IL, and one suggestion is that the specific conductivity should fit a guideline range and the molar conductivity should exceed 0.1 S cm2 mol−1.6 This would be indicative of a true ionic liquid in which ion-pairs do not predominate. The vapour pressure of an IL is low as is the dielectric constant. Early concerns were toxicity, a property that was frequently associated with halogen or halogen containing anions, with moisture sensitivity, and for some potential applications the moderately high viscosity of some ILs. Purification may not be straightforward as the practice of standard distillation traditionally used to purify organic solvents is not normally an option with ILs. Nevertheless, progress has been dramatic. Significant numbers of ILs contain imidazolium, pyridinium or pyrrolidinium ions as the positive ionic component. Numerous academic groups have developed collaborations with industrial partners and investigators have an opportunity to acquire ILs, as they would for other chemicals from suppliers. Sources of ILs would, at present, appear to be specialist suppliers as well as traditional reagent suppliers. There is also opportunity to acquire bespoke, designed ionic liquids from specialists. An overview of the physical properties of typical ionic liquids is presented below.The history of the development of ILs and their applications is accessible through examples in the literature and several authoritative publications,7–12 and this account will be devoted to a selection of uses of ionic liquids in the past few years. To begin to understand how chemical reactions perform in ILs, an examination of the properties of ILs that could be relevant needs to be undertaken. Readers already accustomed to working with ILs could proceed profitably to Sections 4–6.
2. Properties of ionic liquids
The subject of the properties of ILs particularly as they relate to chemical reactivity has attracted many authoritative review articles that proceed from an introductory level6 to more comprehensive levels.13–16 New investigators to the IL field could well benefit from studying these, one of which13 is a valuable, cautionary assessment of practical issues in using ILs. Some reviewers have addressed what may be seen as a need to balance the promotion of the benefits of using ILs with what may be drawbacks in some cases, with toxicity, recycling, disposal and biodegradation as aspects of their use to be considered. Some article titles themselves herald these concerns; for example, “Ionic Liquids: The Neglected Issues”13 and “Understanding Ionic Liquids at the Molecular Level: Facts, Problems, and Controversies”.162.1 Structure
As with other liquids, there is limited scope for establishing the structural arrangement of species. Some neutron scattering experiments on 1,3-dimethylimidazolium salts have shown where the anion, in this case the chloride ion, has a high probability of being located. It would be reasonable to speculate that the aromatic character of imidazolium cations would give rise to π–π stacking interactions among adjacent cations or indeed π–methyl interactions. Computer simulations can be important in modelling the liquid structure and explaining bulk properties, although difficulties remain in simulating properties such as viscosity or electrical conductance. It is also a possibility that non-ionic interactions, such as hydrogen bonds, can occur between the cations and anions of an IL, features not readily tracked experimentally. Possible interactions between ionic solute species and IL components will be discussed. There have been suggestions supported by X-ray scattering experiments and molecular dynamics simulations that mesoscopic structures may exist within ILs and that the mostly homogeneous continuum model of solvation for dipolar solvents may not be appropriate. Thus mesoscopic structural characteristics may be part of the explanation when unpredicted results transpire.2.2 Motions
A range of mostly specialised spectroscopic techniques has been employed to examine the motions of the ionic components. Other approaches have used dissolved ESR spin labels (for example the nitroxide radical) or solvatochromic probes (for example, betaine 30). An emphasis has been placed on characterizing ultrafast motions (femtosecond scale) by optical Kerr effect spectroscopy, because short-time motions have a key role in chemical reaction dynamics and many spectroscopic techniques do not extend to this range.17 An unusually broad spectrum of dynamic processes in ILs was found, unlike that found in molecular liquids; provisionally this could be accounted for by heterogeneity owing to mesoscopic structures formed in ILs.2.3 Bulk properties
2.4 Transport properties
2.5 Polarity
Characterisation of the effective polarity of ILs is an extremely important task, if a comparison or contrast with polarities of molecular solvents is to be developed for an understanding of reaction rates in ILs. However, obtaining a straightforward understanding of polarity and its connection with the dielectric constant, a practice followed for molecular solvents, has proven elusive for ILs. Values that emerge of what may be termed the static dielectric constant (the relative dielectric permittivity) are in the range of 10 to 15, although these appear to be low relative to other approaches to obtain measures of the effective polarity. One difficulty is that the time scale of responses of the particles in an IL does not correlate with frequencies of the measurement techniques. A method involving application of microwave spectroscopy has yielded values of dielectric constants, for example, for imidazolium salts that are for a given anion not very sensitive to the nature of the cation. Simulations lead to the suggestion that the low values of the dielectric constant result from the low dipole densities (the ions of the IL have large molecular volumes).Another approach to determine the solvent polarity of ILs uses the solvatochromic shift of a band in the visible spectrum of the zwitter-ionic dye betaine-30. This shift is expressed as a normalised parameter ENT within a range from zero (for tetramethylsilane) to one (for water). The values of ENT for imidazolium based ILs are those more in the range of ENT for DMSO, ethanol and acetonitrile, and suggest a greater solvent polarity than inferred from dielectric constant values.
Molecular liquids are often subdivided into dipolar aprotic solvents and polar protic solvents; this classification leads to an assignment of contributing components to the polarity. A procedure has been developed for molecular liquids in which different solvatochromic dyes are used for this purpose. Three components were proposed, dipolarity/polarisability (π), H-bond basicity (β), and H-bond acidity (α). When this system was applied to ILs, the values indicate that the ILs investigated possessed a higher dipolarity/polarizability property than alkyl chain alcohols. β reflects the ability of the cation to act as an H-bond donor, and α, the H-bond acidity, is determined by the cation. How this approach pertains to actual reactions in ILs in terms of the Kamlet–Taft scheme of parameters will be considered further below.
3. Preparative chemistry in ionic liquids
ILs have been shown to serve well as host solvents for many preparative processes, some of which are of importance in terms of improvement in efficiency for industrially required reagents. The advantages, especially if the reaction is catalytic, are that the product and catalyst are readily recovered and indeed the IL is often reusable. Examples of reactions carried out and reported up until 2000 are the Friedel–Crafts reaction, cracking and isomerization, dimerization and oligomerization of olefins, Diels–Alder reactions in which significant rate enhancement is achieved, the Heck reaction, Grignard reactions and biotransformations.9 There is always a requirement to assess whether the moisture content of the IL is a factor.Heterocyclic compounds form an important class of chemicals within organic chemistry. Their synthesis in ILs has been comprehensively and systematically reviewed.19 The review also contains general coverage of the properties of ILs, their use as solvents or catalysts and comments on their green aspects and economic perspectives. Attention has been drawn recently to opportunities of applying chiral ILs, and expert reviews of their synthesis as well as applications have appeared.20,21
4. Electrochemistry in ionic liquids
In Sections 4 and 5, the selection of reactions for discussion is subjective and not an attempt to indicate that all are necessarily demonstrably significant. There is no doubt that many important reports are not included but may be accessed through comprehensive review articles. The extent of coverage in terms of detail for each reaction described varies, but an endeavour has been made to provide a sense of the scope of the analyses and interpretations of results undertaken by investigators.There are many reports of electron transfer or other electrochemical processes that take place in an IL being compared with the same process occurring in molecular liquids.22 There are also many studies in which the properties of specific ILs are favourable for an electrochemical application. In the latter case there are a few ILs that have sufficient electrical conductivity and lower than average viscosity, as well as exhibiting chemical stability toward reactive species involved in the application. In general, imidazolium or sulfonium cations combined with complex halide ions are ILs that exhibit useable conductivities. However, tetraalkylammonium-, dialkylpyrrolidinium- and dialkylpiperidinium-based ILs possess superior electrochemical stability compared to the heterocyclic aromatic species because they are more resistant to reduction, but have lower conductivities. Consequently, the selection of an IL for a particular application can involve a compromise between transport properties and stability. Undoubtedly other technical developments have superseded those reported in 2007, but this publication23 includes a wide range of applications with technical details and illustrations regarding the cited applications, and demonstrates admirably the versatility among the range of ILs available. The applications are electrochemical investigations of inorganic chloride complexes and low-valent metal complexes, electrochemical mechanical actuator devices, dye-sensitized photoelectrochemical cells, electrochemical supercapacitors and lithium ion batteries. Herein some illustrative studies of some aspects of electrochemistry or redox chemistry are reported.
4.1 Electron transfer
The acceleration of electron transfer from specific metal complexes to the oxygen molecule in the presence of imidazolium ILs has been reported.24 To an extent the reactions in question mimic aerobic oxidations of metal complexes in biological systems. Consequently, using another medium such as an IL to investigate the kinetics of electron transfer can aid in mechanistic enlightenment, since the oxygen radical intermediate generated had already been shown to be stabilised by coordinating to an acid, resulting in lowering of the energy of activation. One metal complex selected for study was the ferrocene “sandwich” type compound, FeII(C5Me5)2(1). It was predicted that aerobic oxidation of 1 would be facilitated by an imidazolium (Im) IL with the C2-proton of the imidazolium ring functioning as the stabilizing acid, but the magnitude of facilitation depending on the anion, since a basic anion such as Cl−, for example, weakens the acidity of the C2-proton. The reaction carried out in CH2Cl2 and in the absence and presence of imidazolium ILs was monitored spectrophotometrically. Indeed rate acceleration occurred with the magnitude decreasing in the order SbF6− > PF6− > NTf2− ≈ BF4− ≫ Cl−. Corroboration of the role of the C2-proton was obtained by carrying out the same oxidation of 1 in the presence of three SbF6− salts, and finding that [Bu4N][SbF6] caused a sluggish reaction, whereas [N-butylthiazolium][SbF6] gave rise to a very dramatic acceleration relative to the rate in the presence of [Im][SbF6]. This is consistent with the much higher acidity of the relevant proton in the N-butylthiazolium cation. Thus the observations supported the contention that the generated oxygen radical anion can be stabilised by coordinating with the acidic C2-proton of the imidazolium ring, a stabilisation that cannot occur in a conventional solvent. A parallel study24 with the cobalt analogue, CoII(C5Me5)2, yielded similar results, reinforcing the conclusion of the cause for rate acceleration, but also noting that the acceleration can be tuned by the anion of the IL. Supporting experiments involved structural characterisation of the product complex, [FeIII(C5Me5)2](SbF6), and transesterification reactions.4.2 Effect of viscosity on electron transfer kinetics
Some kinetic effects on electron transfer reactions do not depend on a specific stabilisation by a component of an IL (as noted in Section 4.1), but rather are thought to depend on the viscosity of the IL. There are examples where the rate constants exceed those predicted based upon the IL viscosity. In some cases rate constants for reactions in pulse radiolysis studies are lower than for the same reactions in acetonitrile and aqueous solutions.25 Based upon the activation parameters it was suggested that solvent cohesive energy density is a better correlating parameter than solvent polarity. In contrast, electron transfer from the N′-butylpyridinyl radical (BuPy˙) to methylviologen, 4-nitrobenzoic acid and duroquinone (DQ) was characterised by rate constants that were considerably higher than the estimated limits. It was speculated that these rate enhancements arose because the BuPy˙ radical was derived from the solvent cation by a one electron reduction and electron hopping through solvent cations occurred.26Yet other rate enhancements (over the diffusion controlled estimate based upon viscosity) have been found that cannot involve the electron hopping mechanism. One such reaction is that between pyridinyl radicals (bpy˙) and duroquinone in [N4441][Tf2N],27i.e.bpy˙ + DQ → bpy+ + DQ˙−. It was proposed that the viscosity of an IL is a property that does not govern the diffusion of reactants in an IL in a comparable way to the dependence upon viscosity of diffusion of species in molecular solvents. The highly ordered structure of ILs is thought to contain voids that may be occupied by small solute species. It was proposed further that the flexible alkyl moieties on the cation of the IL can facilitate their more rapid motion than that of the whole cation, i.e. rapid diffusion from one void to another. The notion of voids or cavities in ILs has been explored via Monte Carlo simulations and the calculations support this proposition.28
4.3 Photo-induced electron transfer (PET)
This was featured in an investigation involving different acceptors and different donors in both CH3CN and three different ILs.29Laser flash photolysis was applied to monitor the details of the kinetics and spectra of transient species. One purpose of this study was to determine whether a pyridinium IL as used earlier to facilitate rapid intermolecular electron transfer could similarly affect a solvent-mediated pathway of segregating charges generated in PET reactions. From the accumulated data one key result can be reported, viz.[BuPyr][NTf2] (BuPyr is the n-butylpyridinium cation) actively facilitates PET reactions by providing a solvent mediated pathway for electron transfer. However, the authors were unable to distinguish a solvent hopping pathway from another possible mechanism.PET reactions of pyrene and N,N-dimethylaniline in ILs were studied with steady state and time-resolved fluorescence and laser flash photolysis techniques.30 The rate constants for the PET-induced quenching of the fluorescent state of pyrene were between (6.9 and 37) × 107 mol−1 dm3 s−1 at 20 °C, the variation in viscosity of the four ILs used being responsible for the range of values. However, these kinetic parameters exceed those predicted on the basis of diffusion controlled rates by a factor of 2 to 4, but yet are still two orders of magnitude lower than those for the reaction in conventional solvents. Therefore, it was concluded that while IL viscosity is a dominant factor in reducing the rates of fluorescence decay relative to conventional solvents, the microviscosity around the electron donor and acceptor, different from the bulk viscosity of the ILs, was suggested as the cause of the rate constants exceeding those expected based upon diffusion properties. The relevance or consequences of ILs to the steps and species in the overall reaction, viz., the geminate ion pair (GIP), the rate of dissociation from the GIP to the solvent separated ions and the rate of the backward electron transfer were also examined and discussed.
4.4 Electroreduction of CCl4 in [bmim][BF4]
Bond cleavage of a carbon–halogen bond initiated by electron transfer was chosen to explore whether a solvent specific effect pertains.31 The selection of a dialkylimidazolium-based IL, [bmim][BF4], was based upon the appreciable conductivity and air and moisture stability. Different pathways that were proposed were stepwise, concerted, “sticky” dissociative (SD) and almost dissociative electron transfer. In organic solvents the electroreduction of CCl4 has been demonstrated to occur by the SDM (sticky dissociative inner sphere mechanism). Cyclic voltammograms of the reduction in [bmim][BF4] suggested after much deconvolution of the data that electroreduction occurs by a stepwise outer sphere dissociative electron transfer process. Both the electron transfer step to form a radical anion and the subsequent bond cleavage step summarised in reactions (1) and (2), respectively,| CCl4 + e− → −Cl–Cl3C˙, ket | (1) |
| −Cl–Cl3C˙ → Cl3C˙ + Cl−, kc | (2) |
4.5 Heterogeneous electron transfer reactions
The possible technical and interpretative difficulties of using the transient, cyclic voltammetry method commonly used in electrochemical studies as cited herein (vide supra) were discussed.32 Consequently, a steady state high-speed channel electrode method that had been used successfully in several studies employing conventional solvents was used to study the one electron oxidation of several ferrocene compounds in [emim][NTf2]. An additional objective was to ascertain whether the heterogeneous rate constants correlated with expectation for the Marcus model of outer sphere electron transfer. In general the rate constants for electron transfer in the IL were in the range of one half to an order of magnitude lower than for the same reactions studied in acetonitrile. The diffusion coefficients of the ferrocenes in [emim][NTf2] are about two orders of magnitude lower than in CH3CN. Hence it is clear that diffusion alone does not explain the kinetic differences. The authors were unsurprised that there was no correlation between the rate constants and hydrodynamic radii in the IL, as a correlation would be expected on the basis of Marcus theory. But the latter is based upon the reorganisation of solvent dipoles, whereas ILs are formed of ions. Different factors operate in ILs and development of a theory for outer sphere electron transfer is needed. ILs have been successfully applied in many other electrochemical and electron transfer studies and only a small selection can be cited as examples.27,33–36 Attention is now directed to redox reactions in heterogeneous media, with an emphasis on applying high-pressure methods.High-pressure kinetic studies on electron exchange for two redox couples of different charge type, viz.[Fe(bipy)3]3+/2+ and [Fe(cp)2]+/0, at bare Au electrodes within the range 0.1 to 150 MPa, revealed large positive volumes of activation that were found to be virtually the same for the two redox couples, despite the different charge type, in terms of the charge transfer rate constants and diffusion coefficients for the redox process. This is illustrated by the results in Fig. 1. Independent viscosity (fluidity) studies at elevated pressure (up to 175 MPa) were also performed and revealed (as seen from Fig. 2) a similar pressure coefficient closely resembling the kinetically derived data. Complementary temperature-dependent kinetic studies within the range 298 to 358 K also revealed the virtual similarity in activation enthalpies for the same kinetic and diffusion processes, and the viscosity of [bmim][NTf2] as well. This is illustrated by the correlations in Fig. 3, where the pressure and temperature dependences of the heterogeneous electron transfer rate constant and diffusion coefficient are correlated with the viscosity of the ionic liquid. A detailed analysis of the results indicates strongly that dynamic (frictional) control of charge transfer is operative by way of the full adiabatic mechanism. The contribution of the Franck–Condon term to the activation free energy of the kinetic process seems almost diminished due to the high value of electronic coupling and freezing out of the outer-sphere reorganisation energy. A further detailed explanation is contained in a recent report.37
![Effect of pressure on the heterogeneous electron transfer rate constant (left) and diffusion coefficient (right) for the redox couples [Fe(bipy)3]3+/2+ and [Fe(cp)2]+/0 in [bmim][NTf2].](/image/article/2011/CS/c0cs00043d/c0cs00043d-f1.gif) | ||
| Fig. 1 Effect of pressure on the heterogeneous electron transfer rate constant (left) and diffusion coefficient (right) for the redox couples [Fe(bipy)3]3+/2+ and [Fe(cp)2]+/0 in [bmim][NTf2]. | ||
![Effect of pressure on the viscosity of [bmim][NTf2] at 25 °C.](/image/article/2011/CS/c0cs00043d/c0cs00043d-f2.gif) | ||
| Fig. 2 Effect of pressure on the viscosity of [bmim][NTf2] at 25 °C. | ||
![Effect of pressure and temperature on the heterogeneous electron transfer rate constant (left) and diffusion coefficient (right) for the redox couples [Fe(bipy)3]3+/2+ and [Fe(cp)2]+/0 in [bmim][NTf2] as a function of viscosity.](/image/article/2011/CS/c0cs00043d/c0cs00043d-f3.gif) | ||
| Fig. 3 Effect of pressure and temperature on the heterogeneous electron transfer rate constant (left) and diffusion coefficient (right) for the redox couples [Fe(bipy)3]3+/2+ and [Fe(cp)2]+/0 in [bmim][NTf2] as a function of viscosity. | ||
In another study, electrochemical techniques were applied to study heterogeneous electron transfer between selected redox couples and gold electrodes modified with alkanethiol self-assembled monolayers (SAMs), using [bmim][NTf2] as the reaction medium. Ferrocene as a freely diffusing redox probe in the IL was tested for electron transfer through both thin (butanethiol) and thick (dodecanethiol) assemblages at pressures up to 150 MPa, for which the data are shown in Fig. 4.
![Effect of pressure on rate constants (normalized by standard values obtained at 5 MPa) for a [Fe(cp)2]+/0 couple at a gold electrode modified by (1) 1-butanethiol and (2) n-dodecanethiol monolayer films in [bmim][NTf2] at 25 °C.](/image/article/2011/CS/c0cs00043d/c0cs00043d-f4.gif) | ||
| Fig. 4 Effect of pressure on rate constants (normalized by standard values obtained at 5 MPa) for a [Fe(cp)2]+/0 couple at a gold electrode modified by (1) 1-butanethiol and (2) n-dodecanethiol monolayer films in [bmim][NTf2] at 25 °C. | ||
The data yielded large positive and negative values for the volume of activation, for the short-range and long-range electron transfer involving 1-butanethiol and n-dodecanethiol monolayer films, respectively. On the basis of earlier investigations,38 this result can be interpreted as a manifestation of the changeover between the solvent-controlled (adiabatic) and tunnelling-controlled (non-adiabatic) intrinsic electron transfer mechanisms.39 An extended kinetics study was undertaken in which the thickness of the alkanethiol monolayer film was systematically varied for the same IL. In this way the intrinsic electron transfer rate constant could be varied over eight orders of magnitude (from 0.1 to 3 × 107 s−1).40 A schematic presentation of the systematic variation of the thickness of the alkanethiol monolayer film between the [Fe(Cp)2]+/0 couple and a gold electrode has been provided.41
A remarkable interplay of different electron transfer mechanisms was observed accompanied by the stepwise drop in the reorganisation free energy of the medium from 1 to 0.1 eV. This was obtained from detailed analysis of the experimental data as a function of donor–acceptor separation and also based on different theoretical approaches. Different mechanisms for electron transfer at metal/self-assembled monolayer (SAM)/room temperature IL junctions could be obtained, while the collection of results in terms of the reorganisational free energy and the related activation energies as a function of the number of carbon atoms in the SAM could be summarised schematically (see ref. 41).
In the case of thinner alkanethiol films it could be concluded that a thermally activated electron transfer pattern is totally controlled by the viscosity-related slow relaxation mode of the IL acting as the fluctuating environment of the reactant. A correlation could be established between the activation parameters (enthalpy and volume) and the carbon number of the alkanethiol film; these parameters are identical with the corresponding ones for viscous flow (see Fig. 5).41
![The experimental activation enthalpy and activation volume values for different Au/SAM/[bmim][NTf2]/[Fe(Cp)]0/+ composite systems versus the SAM total carbon number. Horizontal dashed lines indicate the respective activation enthalpy and volume values for the fluidity of the room temperature IL.](/image/article/2011/CS/c0cs00043d/c0cs00043d-f5.gif) | ||
| Fig. 5 The experimental activation enthalpy and activation volume values for different Au/SAM/[bmim][NTf2]/[Fe(Cp)]0/+ composite systems versus the SAM total carbon number. Horizontal dashed lines indicate the respective activation enthalpy and volume values for the fluidity of the room temperature IL. | ||
The results of these studies demonstrate the unique properties of ILs in controlling the fundamental mechanism of heterogeneous electron transfer processes and overall mechanistic insight. This insight formed a basis for the understanding of short- and long-range electron transfer for the blue copper protein azurin at Au/SAM junctions in aqueous solution.42
5. Reactions in homogeneous ionic liquids
5.1 Organic reactions
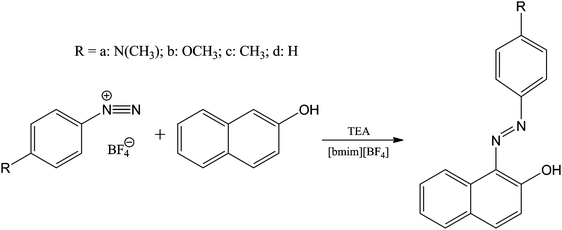 | ||
| Fig. 6 Reaction of benzenediazonium salts with 2-naphthol giving substituted 1-phenylazo-2-naphthols. | ||
The rapidity of the reaction if the substituent is an electron-withdrawing group restricted the investigation to diazonium compounds containing electron-donating substituents. The high viscosity of [bmim][BF4] also limited the compounds that could be employed. Reactions were studied kinetically by UV-Vis spectrophotometry under conditions of excess [2-naphthol] and buffer components (triethylamine and triethylammonium tetrafluoroborate) one order of magnitude higher than the concentration of the diazonium salt. The base, triethylamine (B), is necessary to facilitate reaction and being tertiary prevents coupling of the diazonium salt at the nitrogen atom. Whereas in water, polar aprotic solvents and non-polar solvents, the rate-limiting step is the reaction of 2-naphthoxide with the diazonium salt, in [bmim][BF4] the observed rate constant increased linearly with base concentration. Such a finding would normally be thought to indicate general base catalysis, in which a proton is transferred to the catalyst base in a rate-determining step. In turn this would imply a change in the rate-limiting step from that occurring in other solvents, i.e. dissociation of the tetrahedral intermediate in the IL rather than its formation. The absence of a kinetic isotope effect other than unity and a linear free energy Hammett-type correlation (rate constants and substituent constants) were interpreted to propose that the step affected by the base is the rapid pre-equilibrium between the non-reactive 2-naphthol and the reactive 2-naphthoxide species. Increasing the concentration of triethylamine (B) gives rise to an increase of the activity coefficient, γB, which in turn affects the position of the equilibrium between 2-naphthol and the naphthoxide ion. This could not be tested directly because of a lack of sufficient UV-Vis signals. However, 4-nitrophenol/4-nitrophenoxide ion used as a model confirmed the hypothesis. It was suggested that 1-alkyl-3-methylimidazolium ionic ILs exhibit empirical polarity parameters that imply their polarities are similar to those of short chain alcohols. It was proposed further that ILs do not exhibit super polar solvent properties but act as normal polar solvents, yet acknowledged that ILs may appear to function differently in other applications.
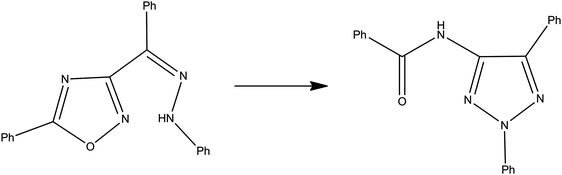 | ||
| Fig. 7 Rearrangement of Z-phenylhydrazone of 3-benzoyl-5-phenyl-1,2,4-oxadiazole into 4-benzoylamino-2,5-diphenyl-1,2,3-triazole. | ||
Under the condition of excess amine, plots of the apparent first-order rate constant versusamine concentration were linear, with a negative intercept. In conventional organic solvents different kinetic laws pertained. The intercepts were related to an acid–base interaction between the cation component of the IL and the amine, and decrease in magnitude from bmim+ to bm2im+ to bmpyrr+, according to the decrease in cation acidity. The acidity property is attributable to the H atom on C2 for bmim+, to the methyl group at the C2 position on bm2im+ and to the hydrogen atom on a carbon α to the quaternary nitrogen for bmpyrr+. The cation acidity is also affected by the anionic component; for a given cation, [bmim], the intercept decreases [BF4−] > [NTf2−] > [SF6−] at least for the bases BuA and Pip. The tertiary TEA lacks interaction between itself and the IL anion. Where direct comparisons are possible, the reaction in ILs is much more rapid than in conventional solvents, with the exception of acetonitrile. The latter solvent facilitates the nucleophilic substitution reaction intra- or intermolecularly and has a favourable solvent polarity. Methanol may also affect reaction acceleration as a result of the presence of the methoxide ion that has a catalytic effect. Polarity effects alone, as manifest in ENT values, cannot account for reactivity trends. This can be aptly illustrated by first-order rate constants for a fixed Pip concentration, at 298 K, of 1.42 × 10−2 and 3.99 × 10−4 s−1 for the reaction in [bmim][BF4] and [bmim][SF6], respectively, ILs that have virtually identical ENT values. There is also a lack of correlation between a kinetic property (more than an order of magnitude difference) and almost identical ENT values, for the reaction in [bm2im][NTf2] and [bmpyrr][NTf2]. Likewise no correlation is obvious for the kinetic parameters and solventπ* parameter, a finding noted earlier also. The kinetic parameters do not show a systematic correlation with IL viscosity values.
In all cases for a given IL the rearrangement reactivity increases from BuA to TEA up to Pip. However, the ratios of reactivity were less marked than for conventional solvents, a trend interpreted on the basis that conventional solvents would differentiate more on the basis of solvation of the base and steric requirements than would ILs.
For each amine the reactivity order was [bmim][BF4] > [bmim][SF6] > [bm2im][NTf2] > [bmim][PF6] > [bmpyrr][NTf2]. Because the reactivity within an IL can arise from contributions from both cation and anion, a further analysis was performed by choosing the bmim-based salts for exploration of the anion effect, while NTf2 salts were selected for examining the cation effect. The reactivity order [BF4−] > [NTf2−] > [PF6−] for the bmim-based salts was explained on the basis of β values of these anions in the following way. When the amine attacks the hydrogen atom of the NH moiety of the phenylhydrazonic chain, the amine begins to develop a positive charge causing an increase in the hydrogen bond donor property of any proton bound to a nitrogen atom. Consequently the stronger (higher β value) hydrogen-accepting anion will preferentially stabilise the transition state relative to the reactant state and in turn increase the nucleophilicity of the NH phenylhydrazonic moiety by interacting with this proton. It was argued that this effect would be greater when more protons are bound to the nitrogen, and this is consistent with the reactivity order TEA < Pip < BuA.
The rearrangement reactivity order for the NTf2 salts, viz.bmim+ > bm2im+ > bmpyrr+, can be explained partly on the basis of the cation ability to participate in π–π interactions, whereby the aromatic imidazolium cations provide a higher stabilising effect than the bmpyrr cation. However, again other factors are extant and different ordering around the transition state was also invoked.44
The activation parameters were readily obtained from excellent linear plots, with ΔH≠ in the range 46–68 kJ mol−1, considerably less than the values for dioxane–water mixtures. Entropies of activation ranged from −91 to −166 J mol−1 K−1, considerably more negative values than obtained for dioxane-water mixtures, showing that the reaction in ILs is principally enthalpy favoured. The unfavourable entropic contribution indicates a significant decrease in molecular freedom, i.e. an increase in structural order, consistent with a proposed zwitterionic transition state stabilised by interaction with the ionic liquid. It was suggested that solvent–amine, substrate–solvent and amine–amine interactions are of limited relevance in ILs, and therefore there is little desolvation accompanying formation of the transition state, that might normally counterbalance to an extent molecular ordering. Further explanation of the ranges and differences of the activation parameters was sought by dissecting the derived values into the separate contributions of the cationic and anionic components of the ILs. The activation parameters obtained for each amine employed were also subject to an analysis and in many cases consistency with arguments advanced regarding reactivity trends was obtained, but the situation for the bmpyr-based ILs was more complex. It should be noted that for a given IL the values of ΔH≠ differed by sometimes only 7 kJ mol−1 and at most 13 kJ mol−1 for the three bases, and while the entropy of activation values differed by as much as 40 J mol−1 K−1 and as little as 20 J mol−1 K−1 (on large negative background values), and precisions reported were reasonably high, these are not major differences. Indeed there is an indication that differences in the energetics of the rearrangement are quite modest and subtle.
The conclusions reported in this publication44 are noteworthy. They include “data collected show that RTILs are very intriguing reaction media”, “…the higher reactivity cannot be ascribed only to the higher polarity of the solvent (IL)”, “but rather due to the scarce relevance of solvation effects”, “…RTILs cannot be considered “simple solvents” but should rather be considered as polymeric supramolecular fluids with regions characterized by “different polar character””. It was also concluded that ILs appear to apply an organizing ability in determining reactivity.
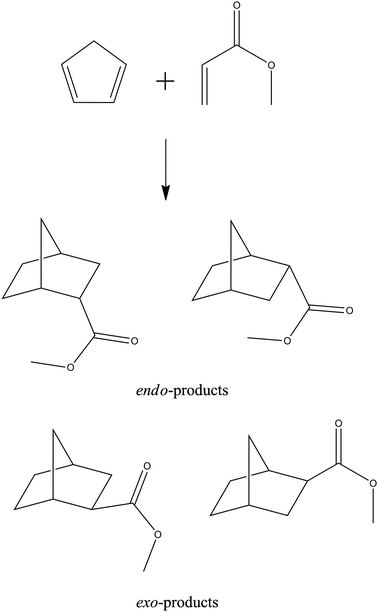 | ||
| Fig. 8 The Diels–Alder cycloaddition of cyclopentadiene to methyl acrylate. | ||
The increase in reaction rate (at ambient pressure) and the acceleration effect upon application of pressure (as manifest in the volume of activation) for the reaction of methyl acrylate in 1[NTf2] is the highest, and is the lowest of the ILs studied in 4[NTf2] (see Fig. 9 for cation structures). Both the reaction rate and pressure acceleration effect were lower in CH2Cl2, and higher in ethanol, than in the ILs. For the reaction of cp with acrolein the reaction rate in CH2Cl2 is again lower than in the ILs studied, whereas the reaction rate in ethanol is intermediate between those for the two ILs cited. However, the acceleration arising from pressure increase is greatest for the reaction in ethanol. These findings were analysed to suggest a non-rigid, poorly ordered transition state in ILs, thought to be attributable to a weakening of the concertedness of the reaction, possibly as a result of steric hindrance from the IL components. There is only a modest improvement of endo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶exo selectivity (obtained from 13C NMR spectroscopic data) in ILs.
∶exo selectivity (obtained from 13C NMR spectroscopic data) in ILs.
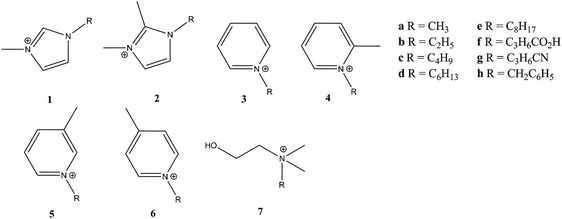 | ||
| Fig. 9 Ionic liquid cation structures from ref. 47. | ||
Fluorinated (fluorous) solvents have been shown to yield large negative volumes of activation for Diels–Alder reactions.48 However, fluorine substituted cationic components of ILs (5[NTf2], 6[NTf2]) and in the anionic component (7[PF3(C2F5)3]) yielded only reduced reaction rates, and significantly less negative volumes of activation. However, the presence of the Lewis acid catalyst ZnI2 for the reaction of cp with methyl acrylate in 3[NTf2] gave rise to reaction rate acceleration, a more negative volume of activation and an improved endo∶exo ratio. This latter finding, therefore, is the most promising from this study. Although the kinetic data yielded parameters of high precision, one issue for further kinetic studies in ILs at elevated pressures is the need for knowledge of the effect of pressure on the properties of the ILs used.
![Reaction between the dimethyl-4-nitrophenylsulfonium ion [(p-NO2PhS(CH3)2]+ and chloride.](/image/article/2011/CS/c0cs00043d/c0cs00043d-f10.gif) | ||
| Fig. 10 Reaction between the dimethyl-4-nitrophenylsulfonium ion [(p-NO2PhS(CH3)2]+ and chloride. | ||
In the latter (for a range of seven solvents) the reaction rate decreases with increasing chloride ion concentration, a finding that can be understood by invoking a stepwise mechanism involving ion pairs. By contrast in ILs (six pyridinium or imidazolium based ILs with the anions [CF3SO3]− or [N(CF3SO3)2]− were employed) the pseudo first-order rate constant (in excess [Cl−]) increases linearly with increasing chloride concentration, i.e. a mechanism that does not involve ion pairing. The implication is that solute ions are screened electrostatically by solvent ions from forming ion pairs. This has been displayed schematically. This model for the explanation of the ionic liquid effect has been tested by theoretical methods, and the calculations support the ideas developed from the experimental findings.50 The authors49 described this as an ionic liquid effect.
5.2 Inorganic reactions
The reaction in the IL showed no exceptional behaviour concerning the observed spectral changes and kinetic traces. The IL seemed to act as a “normal” and innocent solvent. For all nucleophiles and solvents a linear dependence of kobs on the nucleophile concentration was detected. No significant intercept was observed in the concentration dependent kinetic measurements suggesting a straightforward substitution process with no back reaction, owing to the high nucleophilicity of TU and I−. The trend in the derived second-order rate constants (k2) indicated a significant dependence on the polarity of the solvent, namely, k2(H2O) > k2(MeOH) ≈ k2(ionic liquid) (Table 1). It had already been observed52 that a decrease in solvent polarity results in a decrease in the rate of a reaction that involves an increase in dipole moment in going from the reactant to the transition state.
| Nu | Solvent | ε (298 K)/ENT | 103k2/M−1 s−1 at 25 °C | ΔH≠/kJ mol−1 | ΔS≠/J K−1 mol−1 | ΔV≠/cm3 mol−1 |
|---|---|---|---|---|---|---|
| I− | Water | 78.30/1.000 | 253 ± 2 | 58 ± 1 | −62 ± 3 | −6.9 ± 0.3 |
| I− | Methanol | 32.66/0.762 | 15.3 ± 0.2 | 69 ± 1 | −51 ± 4 | −10.0 ± 0.2 |
| I− | [bmim][NTf2] | —/0.642 | 32.1 ± 0.2 | 66 ± 1 | −53 ± 5 | −14.1 ± 0.4 |
| TU | Water | 78.30/1.000 | 1620 ± 10 | 49 ± 1 | −77 ± 4 | −10.4 ± 0.5 |
| TU | Methanol | 32.66/0.762 | 385 ± 2 | 63 ± 1 | −42 ± 4 | −6.6 ± 0.1 |
| TU | [bmim][NTf2] | —/0.642 | 277 ± 3 | 49 ± 1 | −92 ± 2 | −13.9 ± 0.2 |
The negative activation entropies and negative activation volumes clearly support the operation of a compact transition state in terms of an associative mechanism as expected for a square-planar coordinated Pt(II) complex.
The activation volumes for the reactions with TU and iodide in [bmim][NTf2] are indeed nearly identical. The volume changes in this ionic liquid seem to be not influenced by the overall charge of the transition state. This lack of response can be accounted for in terms of strong hydrogen bonding and possibly also van der Waals forces in ILs, leading to specific solvent structures consisting of well aligned cation–anion aggregates. Such aggregates will show a resistance towards solvent structural changes (electrostriction/solvation) as a result of charge neutralisation or charge creation and thus no or only a marginal contribution to the activation volume is expected. The value of about −14 cm3 mol−1 for ΔV≠ points to an associative ligand-substitution mechanism and seems to represent the intrinsic volume collapse associated with the formation of a five-coordinate transition state without any contribution of the solvent. As changes in polarity of the transition state can cause large solvent effects in terms of electrostriction/solvation in conventional solvents, thus complicating the mechanistic assignment on the basis of activation parameters, the activation volume’s virtual independence of solvent contributions in ILs could simplify the accurate assignment of the mechanism. If this unique property is also verifiable in other ILs and with other reactions it may be valuable in resolving mechanistic discrepancies in conventional solvents.
By replacing the apa ligand with ppp (ppp = 2,2′∶6′,2″-terpyridine) the complex becomes much more labile owing to the strong π-back bonding by the three in-plane pyridine rings causing a strong increase in the electrophilicity of the metal centre. The kinetics of substitution of the chloro ligand by TU in [Pt(ppp)Cl]Cl (see Fig. 11) were studied in four ionic liquids containing the common cation [emim]+ where emim = 1-ethyl-3-methylimidazolium and the relevant anions were [NTf2]−, [dca]−, [OTf]− and [EtOSO3]−.53 The formulae and names of the anions are shown in Fig. 12.
![Schematic presentation of the structure of [Pt(ppp)Cl]Cl.](/image/article/2011/CS/c0cs00043d/c0cs00043d-f11.gif) | ||
| Fig. 11 Schematic presentation of the structure of [Pt(ppp)Cl]Cl. | ||
 | ||
| Fig. 12 Schematic presentation of some IL anions. | ||
An earlier study of the reaction of [Pt(ppp)Cl]Cl in a range of conventional solvents established that solvent polarity influenced the reaction rates. Consequently, a necessary, thorough examination of the polarity properties of the ILs, at different temperatures and pressures, was undertaken. Polarity measurements were conducted in an inert atmosphere, employing Reichart’s dye and phenol blue, and exploiting their solvatochromic property. The polarities of three ILs, [emim][tfa], [emim][NTf2] and [emim][dca], were not affected in a significant way (a small decrease) in the range from 10 °C to 50 °C, a trend similar to that in THF. Acetone and methanol showed a somewhat more marked decrease in ET values with temperature increase. The effect of increasing pressure on solvent polarity, based upon results using phenol blue, is a more modest increase than occurs in a roughly linear manner for conventional solvents. The solvent polarity of ILs may also be considered by the Kamlet–Taft scheme of parameters, reiterating from Section 2.5, in which the solvent strength is divided into dipolarity and polarisability (π*), H-bond donating acidity (α), and H-bond accepting basicity (β) components.
The kinetics of substitution of the [Pt(ppp)Cl]Cl complex by TU (eqn (3)) were examined in each of the ILs of Fig. 12.
| [Pt(ppp)Cl]+ + TU ⇄ [Pt(ppp)TU]2+ + Cl−, k1/k−1 | (3) |
The rate constant for the reaction in water is about double that in methanol; a decrease in solvent polarity results in a rate decrease, if there is an increase in dipole moment in progression from the initial to the transition state. In this reaction, a partial lengthening of the Pt–Cl bond in a five-coordinate trigonal bipyramidal transition state causes an increase in dipole moment. This relationship does not apply to ILs that exhibit almost common polarities of about 5% less than methanol, but whose substitution rate constants range from more than a hundred-fold lower ([emim][OTf]) to about 40% ([emim][NTf2]) of the value for methanol (see Table 2).
The Kamlet–Taft parameter β increases with a decrease in the rate constant, in a reasonably systematic manner. However, this finding does not comport with other results in the literature, implying that other factors are needed to explain the relevance of polarity of IL solvent to the rates of reactions in these substitution reactions.
The negative entropy of activation determined for conventional solvents was interpreted as consistent with an associative substitution mechanism, in which the transition state species is highly structured as a result of bond formation and an increase in solvation because of an increase in polarity. While the values of ΔS≠ in ILs differ, they are all distinctly negative and therefore support a mechanism in common with that that pertains in standard solvents. In water a large number of highly structured water molecules including those in the second coordination sphere surround the transition state species, and hydrogen bonding is prominent in this arrangement. The values of ΔS≠ in [emim][NTf2] and [emim][OTf] are similar to the value for the reaction in water and therefore electrostatic interactions would appear to be important in transition state formation in these ILs. By contrast a somewhat less structured transition state can be visualised for methanol, [emim][dca] and [emim][EtOSO3]. These variations in ΔS≠ values mirror, inversely, the values of the enthalpy of activation (representing the ease or relative difficulty of bond reorganisation) as expected for an isokinetic relationship for reactions proceeding by the same mechanism.
Further insight into the special features of ILs that can influence mechanism has been achieved in this substitution reaction of a Pt(II) complex by a combination of 1H and 13C NMR spectroscopy and DFT calculations. The NMR spectra of several imidazolium based ILs (all [emim]+) differing only in their anion were interpreted to show that the strongest interaction between the cation and the anions is between the proton in the C2 position of the cation and, for example, oxygen atoms on the triflate, [OTf] anion. The calculations which were carried out using the methyl analogue [mmim]+ as a working model for [emim]+ were compatible with the results from the analysis of the NMR spectra. The calculated ion-pair stabilisation energies could be correlated with both 1H and 13C chemical shifts, and showed which of the candidate anions were less weakly bound in the ion–ion interaction. This in turn provides a prediction as to which anions would be most likely to have a strong attraction with another positive centre, in this case the Pt metal centre. The ion-pair interaction energies, in the gas phase, were calculated for the [Pt(ppp)Cl]+ cation with the various anions. Differences in the position of occupation of the anions with respect to the cation were found from the computed structures (see ref. 53). [NTf2]− sits on the side of the complex leaving the metal centre vulnerable to nucleophilic attack, consistent with the experimental finding of the highest value of k1. The ppp chelate is planar and [dca]− occupies a position mostly over that plane, providing some shielding of the metal centre from a nucleophile. However, approach by a nucleophile is still quite favourable, therefore k1, although reduced from the value for reaction in [emim][NTf2], is still signifying a rapid reaction. Both [OTf]− and [EtOSO3]− shield the Pt centre and approach closer to it than the other anions, a condition that explains the slower substitution reactions. The Taft–Kamlet β values correlated with the gas stabilisation energies for the interaction between the Pt complex and the anions of the IL, as did the latter with the experimentally determined ΔH≠ for the substitution reaction, as illustrated in ref. 54. In the former case the β value reflects the hydrogen bond acceptor ability of the IL anions and thus their ability to provide electron density to the electrophilic centre. In other words there is an interaction between the anion and the Pt(II) centre. The latter correlation is descriptive of the stronger the Pt(II) complex anion interaction, the higher is the enthalpy term for bond formation by the TU incoming nucleophile in the associative formation of the transition state.
Finally, consideration was given to the possibility of a strongly nucleophilic anion of an IL displacing the coordinated chloro ligand, so that the substitution reaction is actually the displacement of the coordinated IL anion by the reactant nucleophile. It was conjectured based on additional experiments that this may well be the case for a powerful anion such as dicyanamide (dca): i.e. the reaction of the [Pt(ppp)Cl]+ with TU in [emim][dca] would actually be the substitution by TU of dca− from [Pt(ppp)(dca)]+.
Subsequently the kinetics of substitution of [Pt(ppp)Cl]Cl by the thiocyanate ion, employing the same four ILs were investigated (see reaction (4)).54
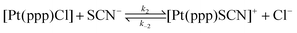 | (4) |
(i) In contrast to the positively charged transition state in the case of thiourea, the transition state of the substitution reaction involving thiocyanate involves charge neutralisation. In a solvent consisting of charged ions this should be energetically disfavoured which could lead to a decrease in the reaction rate. (ii) The solvation shell around the electrophilic Pt(II) complex should consist entirely of negatively charged ions and could thus lead to repulsion of the entering charged nucleophile. (iii) In contrast to thiourea the nucleophile thiocyanate could be highly solvated by the solvent ions of opposite charge, thus decreasing the nucleophilicity of the nucleophile and as a result, the reaction rate.
If one or more of these effects apply, even larger differences compared to aqueous medium or “common” organic solvents than in the case of thiourea should be observed. The reaction rate was studied as a function of the nucleophile (SCN−) concentration and temperature to determine the rate constants (k2 and k−2) and the activation parameters (ΔH≠ and ΔS≠). Consistent with the pseudo-first-order nature of the reaction the kinetic traces were expected to fit to a single-exponential function. This was observed for reactions in ILs containing the [dca]−, [TfO]−, and [EtOSO3]− anions. For reaction in [emim][NTf2] and methanol a subsequent reaction was observed.
The second reaction showed a dependence on the SCN− concentration and was considerably slower than the first reaction; it was assumed a second substitution step was involved, similar to reactions of different Pt(II) complexes with N-donor chelate ligands, where strong sulfur containing nucleophiles such as thiourea or L-methionine are able to substitute the chelate partially or completely. For the first step the pseudo first-order rate constants were plotted against [SCN−]; a linear dependence with a negligible intercept was obtained for all solvents (see examples in Fig. 13).
![Typical concentration dependences for the reaction of [Pt(ppp)Cl]+ with SCN− in [emim][dca] and [emim][NTf2].](/image/article/2011/CS/c0cs00043d/c0cs00043d-f13.gif) | ||
| Fig. 13 Typical concentration dependences for the reaction of [Pt(ppp)Cl]+ with SCN− in [emim][dca] and [emim][NTf2]. | ||
The k2 values determined in the ILs employed differ by 3 orders of magnitude and are at least ten times lower than for reaction in methanol (see Table 3). The decrease in k2 is accompanied by an increase in both ΔH≠ and ΔS≠. Compared to the k2 values reported for the reaction with thiourea in the same solvents, the k2 values obtained for reaction with thiocyanate are at least five times ([emim][NTf2]) and up to 1300 times ([emim][EtOSO3]) smaller. A certain deceleration of the reaction rate on changing from a neutral to a negatively charged nucleophile was expected on the basis of the Hughes–Ingold rules. As the metal complex and the nucleophile are of opposite charge, the transition state of an associative mechanism is neutral and thus less favoured in a solvent that has a high ionic strength, a property of ionic liquids. During the formation of the transition state, partial or complete desolvation of the charged nucleophile and complex, both normally well solvated in solutions of high ionic strength, will occur, leading to a destabilising effect and a deceleration of the reaction. But as the difference between the reaction rate for the “fastest” IL [emim][NTf2] and the “slowest” IL [emim][EtOSO3] was much larger than expected, additional reasons were taken into consideration. The forward reaction rate constant and the enthalpy of activation values can be linked to the hydrogen bond acceptor ability of the IL used, quantified by the Kamlet–Taft parameter β. These correlations are illustrated and deliberated upon further in ref. 54.
The hydrogen bond acceptor ability of the IL affects the activation entropy of the reaction. In the ILs [emim][NTf2], [emim][TfO] and [emim][EtOSO3], an increase of the β-value results in an increase of the activation entropy. In the triad consisting of [emim][NTf2], methanol and [emim][dca], nearly no change in activation entropy can be observed, as illustrated in Fig. 14.
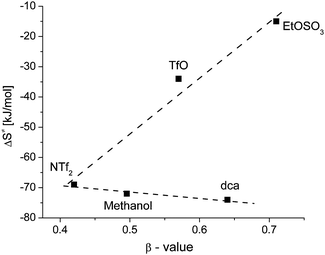 | ||
| Fig. 14 Correlation of the activation entropy versus the β value. | ||
The activation entropy consists of an intrinsic and a solvational term, where the intrinsic term consists of the sum of all entropy changes caused by changes in bond lengths and bond angles on formation of the activated complex. As the complex and the entering nucleophile remain the same, this should be almost the same for all solvents studied. The solvational term is related to changes in the solvation shell caused by differences in electrostriction upon proceeding from the reactant to the transition state. This can originate from charge formation or as in this case charge neutralisation, and also development of a dipole moment or an increase in polarisability. During this substitution reaction two oppositely charged ions form a neutral transition state with complete or at least partial release of the solvating species. In ILs all the solvent species are charged, so this should result in a larger increase in entropy (and volume) during the activation process compared to molecular solvents. Moreover the entropy change is consistent with the number of solvent species bound to the reactant species before formation of the transition state. A larger number of solvent species, reflecting a stronger metal complex–solvent interaction, should result in a larger increase in entropy (and volume) during the activation process.
The ethylsulfate or triflate containing ILs seem to have a stronger interaction with the solutes than the ILs containing [dca]− and [NTf2]−, as a much larger increase in the activation entropy can be observed there. The interaction of [dca]− and [NTf2]− with the metal complex seems to be less favoured, possibly because of structural reasons.
To gain further insight into the solute–solvent interactions between the IL anions and the Pt(II) complex, the ion pair stabilisation energy was calculated as introduced in a previous paper.52 The gas phase energies of the separated ions (metal-complex and IL anion) were compared with the energies of the interacting ion pair. The calculations suggested different interactions for the [dca]− and [NTf2]− anion in contrast to those for the [TfO]− and [EtOSO3]− anions with the Pt(II) centre. Different geometries of the interactions and other arguments led to rationalisation of the relations between the experimental reaction rate, the activation enthalpy and the calculated interaction energy.
Welton and Correia also investigated the reaction of a similar cationic Pt(II) complex to determine if there is a specific ionic liquid effect that influences inorganic reactions in IL solutions.55 They analyzed the kinetics of the substitution reaction of [PtII(dpma)Cl]+ (dpma = di(2-picolyl)amine) with thioacetate in different ionic liquids and organic solvents (see Fig. 15 and 16). They extended their method of analysis to other substitution reactions of Pt(II) complexes in ILs.
![Reaction of [PtII(dpma)Cl]+ with thioacetate.](/image/article/2011/CS/c0cs00043d/c0cs00043d-f15.gif) | ||
| Fig. 15 Reaction of [PtII(dpma)Cl]+ with thioacetate. | ||
 | ||
| Fig. 16 Ionic liquids used by Welton and Correia. | ||
The forward second-order rate constants and activation parameters are tabulated in Table 4.
| Solvent | k 2/M−1 s−1 | ΔH≠/kJ mol−1 | ΔS≠/J mol−1 K−1 |
|---|---|---|---|
| MeOH | 1.90 ± 0.09 | 55 ± 3 | −98 ± 9 |
| DMSO | 5.7 ± 0.2 | 49 ± 1 | −117 ± 4 |
| H2O | 32 ± 1 | 39 ± 2 | −146 ± 8 |
| [C4C1im][NTf2] | 8.4 ± 0.5 | 43.9 ± 0.3 | −129.7 ± 0.9 |
| [C4C1py][NTf2] | 4.6 ± 0.2 | 50 ± 3 | −119 ± 9 |
| [C4C1py][OTf] | 6.7 ± 0.2 | 52 ± 4 | −107 ± 8 |
| [(C1OC2)C1im][NTf2] | 9.00 ± 0.09 | 44.9 ± 0.5 | −127 ± 2 |
There is a limited dependence of k2 on the solvent; a 12-fold increase in rate from the slowest (MeOH) to the fastest solvent (H2O) with the rate constants for reactions in ILs and DMSO between the two limits. The rate constant increases with increasing dielectric constant of the solvent; values are 32.7 (MeOH) < 46.5 (DMSO) < 80.2 (H2O). The dielectric constants of some ILs have been shown to be in the range 10–15, so the reaction should be further decelerated in the ILs if there were a simple correlation of rate constant with apparent dielectric constant of ILs.
In this system other factors such as H-bonding solvation, polarisability of the nucleophile and interactions with the metal ion in the ground and in the activated states appear to contribute to the results obtained. The authors used the approach developed by Kamlet and Taft to understand the solvent dependence of the reaction. A very good correlation between experimental data and the calculated values for lnk2 obtained by a Kamlet–Taft linear solvation and energy relationship fit was obtained with data sets containing only ILs as well as with data sets incorporating all solvents used. Hence, in this reaction, there seems to be no special IL effect, as all significant interactions between the ILs and the solutes can be described by a combination of their Kamlet–Taft parameters. Further analysis considered the latter parameters in terms of specific properties of the cations of the ILs.
The activation parameters point to the same associative to associative interchange mechanism operating in all of the solvents. There is also an isokinetic correlation (a correlation between ΔH≠ and ΔS≠). The rate constant increases on going from methanol to water; this is reflected by a much higher activation enthalpy and a more negative entropy of activation. This originates from a higher number of structured molecules of solvent in the second coordination sphere due to hydrogen bonding effects playing a major role.
While the molecular solvents show a broad range of values for ΔS≠, in ILs a form of cation “control” of the magnitude of this parameter can be observed. For the imidazolium containing ILs, more negative values were obtained suggesting a more ordered structure in these solvents. This can be ascribed to the higher ability to donate H-bonds through the acidic aromatic protons. However, given the precision of derived entropies of activation this cannot be described as a massive difference for the imidazolium containing ILs.
In summary, the anions of the ILs seem to play a key role in the kinetics of substitution reactions, as they differ in their ability to interact with the Pt(II) complex, and thus are differently influential regarding the derived (activation) parameters. These findings should be taken into consideration in the design of other investigations, since rates and possibly the selectivity of reactions can be changed by different solute–solvent interactions.
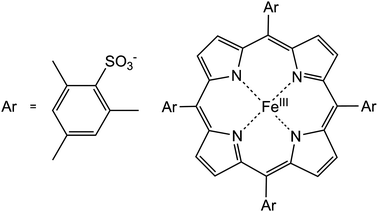 | ||
| Fig. 17 Structure of the FeIII(TMPS) complex. | ||
During initial experiments, depending on the batch of IL used, large kinetic and mechanistic differences for the binding of NO to the FeIII(TMPS) complex were observed. A pre-occupation of the sixth coordination site at the porphyrin iron(III) centre was assumed and a closer look at the UV-vis spectra, especially at the Q-band pattern, clearly showed the formation of two different species of the FeIII((TMPS)(OH)) complex. A HPLC study to determine the purity of the batches of the IL employed verified in one batch a contamination with methylimidazole, MeIm, a starting material in the synthesis of [emim]Br. According to the HPLC-chromatogram the concentration of the contaminant was only about 6 μM, but obviously sufficient for the formation of FeIII(TMPS)(OH)(MeIm). Depending on the presence or absence of the contaminant (MeIm) two different reaction pathways were observed:
(i) Binding of NO to FeIII(TMPS)(OH) (without contaminant): on addition of NO to a deoxygenated solution of FeIII(TMPS)(OH) a low-spin iron(III) porphinatonitrosyl complex is formed, in which the formal charge distribution can be described as FeII(TMPS)(OH)(NO+) (diamagnetic behaviour). The kinetics of the reversible binding of NO to FeIII(TMPS)(OH) were investigated by the stopped-flow technique. The reaction rate showed a linear dependence on excess [NO] and proceeded much slower in [emim][NTf2] than in water, consistent with a higher activation enthalpy (ΔH≠) value. The significantly negative values for ΔS≠ and ΔV≠ suggest that the binding of NO to FeIII(TMPS)(OH) occurs analogously to the reaction in water according to an associative (A) mechanism, in which the NO molecule attacks the free sixth coordination site and coordinates to the iron centre. This is accompanied by a spin state change from a high-spin iron(III) porphyrin (S = 5/2) to a diamagnetic low-spin state (S = 0) explaining the relatively large negative activation volume.
(ii) Binding of NO to FeIII(TMPS)(OH)(MeIm): the spectral changes accompanying the reaction of FeIII(TMPS)(OH)(MeIm) with NO result in the same final product spectrum, assuming the same product, FeII(TMPS)(OH)(NO+), to be formed as in the absence of MeIm. The kinetic measurements showed no dependence on [NO] and a much slower reaction reflected by a very high activation enthalpy, also pointing to a strong coordination of MeIm. The activation entropy was markedly positive suggesting a limiting dissociative (D) mechanism (see reaction (5)), confirmed by the positive activation volume of ΔV≠ = +24 cm3 mol−1.
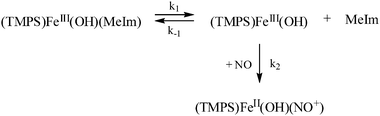 | (5) |
 | (6) |
The presence of and coordination of MeIm to FeIII(TMPS)(OH) has a large influence on the kinetics (marked rate deceleration) and mechanism (change to D) of the binding of NO. Other than deceleration of the reaction rate compared to aqueous solution, the overall reaction mechanism of NO binding seems to be unchanged in pure [emim][NTf2].
This example clearly demonstrates that the purity of the IL is of crucial importance for all investigations performed in ILs, and show that even small traces of impurities could have remarkable effects on the rate and mechanism of a reaction.
5.3 Organometallic reactions
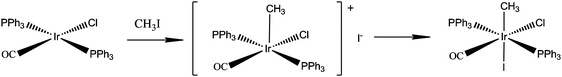 | ||
| Fig. 18 Oxidative addition of methyl iodide to Vaska's complex. | ||
6. Catalysis in ionic liquids
Not unexpectedly a vast array of reactions that are subject to catalysis has been studied in ionic media. In addition to the potential environmental advantage of ILs relative to conventional solvents, the repeated use of ILs in cycles in a catalytic context may be possible. These issues and application of ILs in sonochemical studies, as well as the inclusion of biocatalytic and electrocatalysed reactions, have been cited in a very comprehensive, relatively recent review of catalysis in ILs.59 The preponderance of the more than 400 references therein is from only a few years prior to the date of publication of the review and this emphasises again the phenomenal growth in interest in these media. The composition of the review reflects the overwhelming proportion of studies of homogeneously versus heterogeneously catalysed processes. The history of this topic can be traced in an early review of catalytic reactions in ILs.606.1 Catalysis in homogeneous media
In this section two studies are highlighted.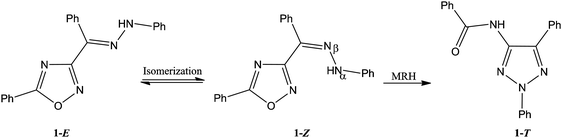 | ||
| Fig. 19 Rearrangement of 3-benzoyl-5-phenyl-1,2,4-oxidiazole. | ||
As a consequence the kinetics of rearrangement of 3-benzoyl-5-phenyl-1,2,4-oxidiazole have been studied in four imidazolium-based ILs in the presence of the catalysts CuCl2 and hexahydrated Cu(ClO4)2.61 Different anions of the Cu(II) species yield different outcomes; different mechanistic patterns arise depending on the nature of the copper salt anion. The initial isomerisation step was promoted by both copper salts, and the rearrangement preferably promoted by CuCl2. The reactions are also differently affected by different IL anions. Conclusions were assembled from analysis of the specific kinetics results. For example, a “high structural order degree favours the rearrangement, stabilising the transition state by means of electrostatic and π–π interactions.” However, the same factor apparently disfavours the isomerisation step. It was further noted that these effects are rationalised because ILs are solvent systems of polymeric supramolecular fluids. In this particular case the situation could be more complicated because methanol was also present, and in the case of the hydrated copper catalyst water will also be present. Nevertheless interesting observations emerge, but as for enzyme catalysed reactions (see Section 6.3) the presence of so many species renders unequivocal interpretation of the results difficult.
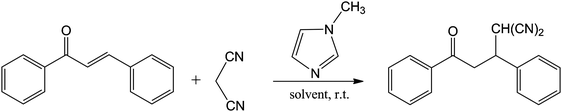 | ||
| Fig. 20 Michael addition of malonodinitrile to chalcone. | ||
The rationale for this study62 seems to be that some Michael addition reactions in acidic ILs did not proceed, yet Michael addition of C nucleophiles in ILs proceed without additional basic catalyst. It had been shown that N-methylimidazole (N-MeIm) is used in the synthesis of many imidazolium-based ILs and is present as a not insignificant impurity in relevant ILs (see also Section 5.2.2).63 The reaction in Fig. 20 does not occur in CH2Cl2 even in the presence of N-MeIm, but does in THF and in CH3CN. The kinetics of the reaction were studied in four ILs and in binary mixtures of ILs. Addition of N-MeIm, further accelerated the reaction rates beyond those occurring from the presence of residual N-MeIm, a finding supporting that extraneous N-MeIm present in the IL can act as a catalyst. The rate constants for reactions in mixed ILs either were lower or higher than those for reaction in a neat IL, in the presence of a fixed quantity of N-MeIm. In the former case it was speculated that trace water could hydrolyse the IL anion [EtOSO3]− to the HSO4− anion that could protonate N-MeIm and thus reduce the rate of the addition reaction. Solvent structure distortion of individual ILs was cited to explain rate increases in mixed ILs. Clearly impurities are key issues, and various properties of ILs are incompletely understood, with both factors contributing to uncertainties in the interpretation of results.
Unfortunately, many important reactions, for example acylation by the Friedel Crafts method and olefin hydroformylation, catalysed by transition metal-based catalysts in ILs are not included herein, but these and countless other named and other catalysed reactions in ILs are included in ref. 59.
6.2 Enzyme catalysis in ionic liquids
In many natural enzyme catalysed reactions the solvent is water containing buffer or other electrolyte species. In examples where substrates are partly or wholly hydrophobic, organic solvents can be used for enzyme catalysis studies. There is a body of literature on the application of organic solvents,64,65 but obvious limitations regarding toxicity and environmental consequences have been noted.66,67 Catalysis by enzymes in ILs can result in reduced activity, if for example the conformational state of the enzyme is changed or the high viscosity of an IL reduces the rate of diffusion controlled steps. In fact in many examples in the literature, any beneficial effects are yielded usually when water or other co-solvent is present. From a point of view of understanding the mechanism of an enzyme catalysed reaction, and how the rates of individual steps are affected, the presence of two solvents can introduce complications. Therefore, although the subject is one of great interest, this article is mostly confined to an endeavour to understand reaction mechanisms of reactions in neat ILs.7. Applications involving ionic liquids
As a result of their unusual properties, ILs have important applications in devices and processes for the production, storage and efficient use of energy and other resources.68 This includes their potential application in solar photo-conversion, solar thermal conversion, biofuel production, fossil fuel production and a cleaner nuclear fuel cycle. ILs can also be used for energy storage in batteries and supercapacitors, and for efficient energy utilization in fuel cells and advanced devices and processes. In addition, the cited review68 also refers to the role of ILs in catalysis and industrial applications. Other recent applications include the development of hydrophobic ILs that contain the hexafluoroacetylacetonate anion and exhibit strong metal-complexing ability.69 In this way the solubility of metal ions can be increased substantially for applications such as the electrodeposition of metals. It was recently reported70 that the liquid and compositional ranges of ILs can be expanded to pharmaceutically active ILs by mixing with a solid acid or base to form oligomeric ions. The latter ions have a tremendous influence on the physical properties of the modified ILs. Although a green agenda has been promoted by some with respect to ILs and clearly there are beneficial applications employing ILs,23 it is worth noting a recent review on the environmental fate and toxicity of ionic liquids.71 In this review the authors conclude that although ILs are green alternatives to traditional organic solvents due to their negligible vapour pressure, studies of the environmental fate and toxicity of ILs have shown that ILs commonly used to date are toxic in nature and their toxicities can vary considerably. In this respect it is interesting to note that the introduction of functional polar groups to the alkyl chain of ILs has been shown to reduce the toxicity of ILs.71 With respect to applications of ILs, the topic of the present report is of fundamental interest since the clarification of the underlying mechanisms of the reactions involved in the mentioned applications is crucial in order to be able to optimize the utilization of ILs.8. Theoretical approach
The employment of computational methods to supplement experimentally derived mechanistic schemes for reactions in ILs has already been undertaken and described briefly in Section 5. DFT computational methods were applied to support experimental findings in platinum complex substitution reactions in ILs.52,53 The whole subject of computational methods with respect to understanding mechanisms of reactions in ILs probably deserves an authoritative review. Examples of the value of computational methods in catalysed reactions in ILs are in a “green” Heck reaction in diethanolamine-based ILs,72 and in a copper(I)-catalysed 1,3-dipolar cycloaddition reaction in ILs, where there is an anion effect on regioselectivity.73 The latter reference is from a journal issue devoted entirely to physical chemistry aspects of ILs and reactions in them.9. Concluding remarks
The work cited in this review focussing principally on mechanistic aspects of chemical reactions in ILs has clearly demonstrated the specific tuning effects that ILs can have on the rate and mechanism of chemical processes in ILs. In many cases the reaction mechanisms are understood reasonably well whether or not the IL functions similarly to conventional solvents (“successes”). In other cases particularly for some redox processes (Section 4) where diffusion rates of species are involved, a comprehensive mechanistic picture is less clear (“challenges”). These aspects were also treated earlier.8 For reactions involving transition metal complexes it is especially the nucleophilicity of the anionic components of the IL that can either weakly interfere with the metal complex or coordinate directly to the metal centre depending on the donor strength of the IL anion. Furthermore, such reactions can also be very sensitive to trace impurities present in the ILs. Thus great care must be taken when dealing with transition metal catalyzed reactions in ILs for the possible role of impurities and coordination ability of the IL anions. A quantification of the donor ability of ILs in terms of different solvent parameters will be very helpful to predict such interference with transition metal species.Finally, overall this account has shown that some reactions are “indifferent” to ILs and operate in them in a systematic way compatible with the same reaction in conventional solvents. On the other hand a complete change of mechanism from that in conventional solvents occurs for some other reactions carried out in ILs and the term ionic liquid effect has been invoked. However, an examination of the properties of the reactants themselves in each case, and of the specific properties of both ionic partners of the ILs in each case, can, at least “after the fact”, explain differences or similarities in mechanisms purely in terms of the chemical and physicochemical properties of the combinations of reactants and components of ILs. Therefore, an ionic liquid effect, although a valid term, is itself not particularly explanatory. The purity issue of ILs has been cited many times in this account and must be considered. What is less clear is a coherent view of the liquid state of different ILs as noted by descriptions by different authors. Hence, this report refers to “known-knowns” (physical properties that have been characterised, Section 2), “known-unknowns” (the various nebulous descriptions of the liquid state of ILs, Sections 4 and 5), and no doubt there are still “unknown-unknowns” of various properties of ILs and of effects attributed to them.74
Acknowledgements
The authors gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft through SPP 1191 (Ionic Liquids) and the Volkswagen Foundation, and the productive collaboration with the group of Peter Wasserscheid (Chair of Chemical Reaction Technology, University of Erlangen-Nürnberg).References
- Chemistry under Extreme or Non-classical Conditions, ed. R. van Eldik and C. D. Hubbard, Wiley-Spektrum, New York, Heidelberg, 1997 Search PubMed.
- Chemistry at Extreme Conditions, ed. M. Riad Manaa, Elsevier, Amsterdam, 2005 Search PubMed.
- Microwave Chemistry: C. O. Kappe and D. Dallinger, Mol. Diversity, 2009, 13, 71 Search PubMed.
- Sonochemistry: J. Gonzalez-Garcia, V. Saez, I. Tudela, M. I. Diez-Garcia, M. D. Esclapez and O. Louisnard, Water, 2010, 2, 28 Search PubMed.
- Supercritical Fluids: A. O. Chapman, G. R. Akien, N. J. Arrowsmith, P. Licence and M. Poliakoff, Green Chem., 2010, 12, 310 Search PubMed.
- K. E. Johnson and C. L. Hussey, Electrochem. Soc. Interface, 2007, 38, 16 Search PubMed.
- P. Walden, Bull. Acad. Imp. Sci. St.-Petersburg, 1914, 1800 Search PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- M. J. Earle and K. R. Seddon, Pure Appl. Chem., 2000, 72, 1391 CrossRef CAS.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2002 Search PubMed.
- An Introduction to Ionic Liquids, ed. M. Freemantle, RSC Publishing, Cambridge, UK, 2009, p. 281 Search PubMed.
- Ionic Liquids, ed. B. Kirchner, Springer, Heidelberg, Germany, 2010, p. 345 Search PubMed.
- P. J. Scammels, J. L. Scott and R. D. Singer, Aust. J. Chem., 2005, 58, 155 CrossRef CAS.
- C. Chiappe and D. Pieraccini, J. Phys. Org. Chem., 2005, 18, 275 CrossRef CAS.
- D. R. MacFarlane and K. R. Seddon, Aust. J. Chem., 2007, 60, 3 CrossRef CAS.
- H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654 CrossRef.
- E. W. Castner, Jr. and J. F. Wishart, J. Chem. Phys., 2010, 132, 120901 CrossRef.
- S. Kern, P. Illner, S. Begel and R. van Eldik, Eur. J. Inorg. Chem., 2010, 4658 CrossRef CAS.
- M. A. P. Martins, C. P. Frizzo, D. N. Moereira, N. Zanatta and H. G. Bonacorso, Chem. Rev., 2008, 108, 2015 CrossRef CAS.
- K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 3235 CrossRef CAS.
- A. Winkel, P. V. G. Reddy and R. Wilhelm, Synthesis, 2008, 999 CAS.
- P. Hapiot and C. Langrost, Chem. Rev., 2008, 108, 2238 CrossRef CAS.
- T. Tsuda and C. L. Hussey, Electrochem. Soc. Interface, 2007, 16, 42 Search PubMed.
- D. S. Choi, D. H. Kim, U. S. Shin, R. R. Deshmukh, S-G. Lee and C. E. Song, Chem. Commun., 2007, 3467 RSC.
- C. Lagrost, D. Carrié, M. Vaultier and P. Hapiot, J. Phys. Chem. A, 2003, 107, 745 CrossRef CAS.
- D. Behar, P. Neta and C. Schultheisz, J. Phys. Chem. A, 2002, 106, 3139 CrossRef CAS.
- A. Skrzypczak and P. Neta, J. Phys. Chem. A, 2003, 107, 7800 CrossRef CAS.
- F. Bresme and J. Alejendre, J. Chem. Phys., 2003, 118, 4134 CrossRef CAS.
- R. C. Vieira and D. E. Falvey, J. Am. Chem. Soc., 2008, 130, 1552 CrossRef CAS.
- A. Paul and A. Samanta, J. Phys. Chem. B, 2007, 111, 1957 CrossRef CAS.
- M. A. Bhat, P. P. Ingole, V. R. Chaudhari and S. K. Haram, J. Phys. Chem. B, 2009, 113, 2848 CrossRef CAS.
- N. Fietkau, A. D. Clegg, R. G. Evans, C. Villagran, C. Hardacre and R. G. Compton, ChemPhysChem, 2006, 7, 1041 CrossRef CAS.
- J. Zhang, A. M. Bond, D. R. MacFarlane, S. A. Forsyth, J. M. Pringle, A. W. A. Mariotti, A. F. Glowinski and A. G. Wedd, Inorg. Chem., 2005, 44, 5123 CrossRef CAS.
- M. Matsumiya, M. Terazono and K. Tokuraku, Electrochim. Acta, 2006, 51, 1178 CrossRef CAS.
- P. Bonhote, A. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Gratzel, Inorg. Chem., 1996, 35, 1168 CrossRef CAS.
- G. R. Evans and R. G. Compton, ChemPhysChem, 2006, 7, 488 CrossRef.
- T. D. Dolidze, D. E. Khoshtariya, P. Illner, L. Kulisiewicz, A. Delgado and R. van Eldik, J. Phys. Chem. B, 2008, 112, 3085 CrossRef CAS.
- D. E. Khoshtariya, T. D. Dolidze, S. Seifert, D. Sarauli, G. Lee and R. van Eldik, Chem.–Eur. J., 2006, 12, 7041 CrossRef CAS.
- T. D. Dolidze, D. E. Koshtariya, P. Illner and R. van Eldik, Chem. Commun., 2008, 2112 RSC.
- D. E. Khoshtariya, T. D. Dolidze and R. van Eldik, Chem.–Eur. J., 2009, 5254 CrossRef CAS.
- D. E. Khoshtariya, T. D. Dolidze and R. van Eldik, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2009, 80, 065101 CrossRef.
- D. E. Koshtariya, T. D. Dolidze, M. Shushanyan, K. L. Davis, D. H. Waldeck and R. van Eldik, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2757 CrossRef CAS.
- J. Hanusek, V. Machček and A. Lyčka, Dyes Pigm., 2007, 73, 326 CrossRef CAS.
- F. D’Anna, V. Frenna, R. Noto, V. Pace and D. Spinelli, J. Org. Chem., 2006, 71, 9637 CrossRef CAS.
- High Pressure Chemistry, ed. R. van Eldik and F.-G. Klärner, Wiley-VCH, 2002, ch. 2 Search PubMed.
- A. Vidis, C. A. Ohlin, G. Laurenczy, E. Küsters, G. Sedelmeier and P. J. Dyson, Adv. Synth. Catal., 2005, 347, 266 CrossRef CAS.
- A. Vidis, G. Laurenczy, E. Küsters, G. Sedelmeier and P. J. Dyson, J. Phys. Org. Chem., 2007, 20, 109 CrossRef CAS.
- K. E. Myers and K. Kumar, J. Am. Chem. Soc., 2000, 122, 12025 CrossRef CAS.
- J. P. Hallett, C. L. Liotta, G. Ranieri and T. Welton, J. Org. Chem., 2009, 74, 1864 CrossRef CAS.
- R. M. Lynden-Bell, Phys. Chem. Chem. Phys., 2010, 12, 1733 RSC.
- C. F. Weber, R. Puchta, N. van Eikema Hommes, P. Wasserscheid and R. van Eldik, Angew. Chem., 2005, 117, 6187 ( Angew. Chem., Int. Ed. , 2005 , 44 , 6033 ) CrossRef.
- C. F. Weber and R. van Eldik, Eur. J. Inorg. Chem., 2005, 4755 CrossRef CAS.
- S. Begel, P. Illner, S. Kern, R. Puchta and R. van Eldik, Inorg. Chem., 2008, 47, 7121 CrossRef CAS.
- P. Illner, S. Begel, S. Kern, R. Puchta and R. van Eldik, Inorg. Chem., 2009, 48, 588 CrossRef CAS.
- I. Correia and T. Welton, Dalton Trans., 2009, 4115 RSC.
- M. Schmeisser and R. van Eldik, Inorg. Chem., 2009, 48, 7466 CrossRef CAS.
- M. Wolak and R. van Eldik, J. Am. Chem. Soc., 2005, 127, 13312 CrossRef CAS.
- S. J. P'Pool, M. A. Klingshirn, R. D. Rogers and K. H. Shaughnessy, J. Organomet. Chem., 2005, 690, 3522 CrossRef CAS.
- V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615 CrossRef CAS.
- R. Sheldon, Chem. Commun., 2001, 2399 RSC.
- F. D'Anna, V. Frenna, S. Marullo, R. Noto and D. Spinelli, Tetrahedron, 2008, 64, 11209 CrossRef CAS , and references loc cit.
- M. Mečiarova, M. Cigň, Š. Toma and A. Gáplovský, Eur. J. Org. Chem., 2008, 4408 CrossRef CAS.
- R. Giernoth and D. Bankmann, Tetrahedron Lett., 2006, 47, 4293 CrossRef CAS.
- A. M. Klibanov, Curr. Opin. Biotechnol., 2003, 14, 427 CrossRef CAS.
- M. N. Gupta and I. Roy, Eur. J. Biochem., 2004, 271, 2575 CrossRef CAS.
- S. Sgalla, G. Fabrizi, S. Cacchi, A. Macone, A. Bonamore and A. Boffi, J. Mol. Catal. B: Enzym., 2007, 44, 144 CrossRef CAS.
- P. C. A. G. Pinto, M. L. M. F. S. Saraiva and J. L. F. C. Lima, Anal. Sci., 2008, 24, 1231 CrossRef CAS.
- J. F. Wishart, Energy Environ. Sci., 2009, 2, 956 RSC.
- H. Mehdi, K. Binnemans, K. Van Hecke, L. Van Meervelt and P. Nockermann, Chem. Commun., 2010, 46, 234 RSC.
- K. Bica and R. D. Rogers, Chem. Commun., 2010, 46, 1215 RSC.
- T. P. T. Pham, C.-W. Cho and Y.-S. Yun, Water Res., 2010, 44, 352 CrossRef.
- Z. D. Petrović, D. Simijonović, V. P. Petrović and S. Marković, J. Mol. Catal. A: Chem., 2010, 327, 45 CrossRef CAS.
- C. Chiappe, B. Mennucci, C. Silvio Pomelli, A. Sanzone and A. Marra, Phys. Chem. Chem. Phys., 2010, 12, 1958 RSC.
- Attributed to Donald Rumsfeld, former Secretary of the US Department of Defense.
| This journal is © The Royal Society of Chemistry 2011 |