Nanoscale metal–organic materials
Arnau
Carné
,
Carlos
Carbonell
,
Inhar
Imaz
and
Daniel
Maspoch
*
Centre d’Investigació en Nanociencia i Nanotecnologia (CIN2, ICN-CSIC), Esfera UAB, 08193 Bellaterra, Spain. E-mail: daniel.maspoch.icn@uab.es; Fax: +34 93 581 4747
First published on 25th November 2010
Abstract
Metal–organic materials are found to be a fascinating novel class of functional nanomaterials. The limitless combinations between inorganic and organic building blocks enable researchers to synthesize 0- and 1-D metal–organic discrete nanostructures with varied compositions, morphologies and sizes, fabricate 2-D metal–organic thin films and membranes, and even structure them on surfaces at the nanometre length scale. In this tutorial review, the synthetic methodologies for preparing these miniaturized materials as well as their potential properties and future applications are discussed. This review wants to offer a panoramic view of this embryonic class of nanoscale materials that will be of interest to a cross-section of researchers working in chemistry, physics, medicine, nanotechnology, materials chemistry, etc., in the next years.
 Arnau Carné | Arnau Carné Sánchez was born in Manresa (Catalonia, Spain) in 1985. He received his BSc degree in Chemistry at Universitat de Barcelona (UAB) in 2008. In 2009, he got his MSc in Environmental Diagnostics and Management from Cranfield University (UK). He is currently pursuing a PhD in Chemistry at the CIN2 (ICN-CSIC) under the supervision of Dr Daniel Maspoch and Dr Inhar Imaz in the field of metal–organic nanomaterials with special interest in developing new synthetic methodologies. |
 Carlos Carbonell | Carlos Carbonell (Barcelona, Spain, 1975) studied Technical Chemist Engineering (2002), Materials Engineering (2004) and obtained his MSc in Nanotechnology (2008) at the Universitat Autònoma de Barcelona. After working for 7 years at the industry, he started his PhD in 2009 at the CIN2 (ICN-CSIC) under the supervision of Dr Daniel Maspoch, leader of the Supramolecular Nanochemistry & Materials Group. His current field of interest includes the synthesis and patterning of metal–organic materials assisted by soft nanolithographic techniques, such as Dip-Pen Nanolithography. |
 Inhar Imaz | Inhar Imaz was born in Hendaia (Basque Country, France) in 1978. He received his PhD in Materials Science from the Université Bordeaux I in 2005, where he studied the formation of heterometallic metal–organic architectures from tetrahedral building blocks. He joined the CIN-2 (ICN-CSIC) centre in 2005 as a Postdoctoral fellow. At present, he is an associate professor under the ‘‘Ramón y Cajal’’ program at the Institut Català de Nanotecnologia. His research is focused on the design, the synthesis and the study of novel nanostructured metal–organic materials. He is exploring new synthetic methodologies, new properties and new applications of supramolecular nanomaterials. |
 Daniel Maspoch | Daniel Maspoch was born in L’Escala (Catalonia, Spain) in 1976. He received his BSc degree in Chemistry from Universitat de Girona, and he earned his PhD in Materials Science at the Universitat Autònoma de Barcelona and at the Institut de Ciencia de Materials de Barcelona. After postdoctoral work in Prof. Chad A. Mirkin's group at Northwestern University, he moved to the Centre d’Investigació en Nanociència i Nanotecnologia (ICN-CSIC), thanks to the Ramón y Cajal Program financial support. His group is working on controlling the supramolecular assembly of (bio)molecules, metal ions and nanoscale building blocks at the nanometre scale. |
Introduction
Miniaturization to the nanometre scale regime is a very prolific strategy for the development of new materials with novel and often enhanced properties compared to traditional materials, opening up avenues for technological and biomedical applications in many areas, including drug-delivery, catalysis, diagnostics, solar cells, etc. To date, most nanoscale materials are either purely organic or inorganic in composition. However, architectures created from the supramolecular assembly of organic and inorganic components are rapidly growing as a very attractive alternative class of nanoscale materials.But, why do we need another class of nanoscale materials? Traditional metal–organic materials are a fascinating family of solids created from the supramolecular association of inorganic, such as metal ions and metal–organic or inorganic clusters, and organic building blocks, including organic molecules, biomolecules and organic polymers.1 These bulk materials, in which both types of building blocks are assembled through metal coordination, hydrogen bonding, electrostatic interactions or π–π stacking, have the potential to be tailored to show adjustable structures, compositions and properties. As a consequence, they show promise for an impressive number of applications in gas storage, drug-delivery, diagnostics, sensing, catalysis, ion exchange or separation, magnetism, optics, etc. However, metal–organic materials in the form of traditional bulk crystalline materials do not always fulfill all specific needs for these applications. Depending upon the intended application, these materials require to be not only fabricated as bulk crystalline solids, but also miniaturized at the nanometre length scale and immobilized at specific locations on surfaces.
For example, nanoscopic dimensions are typically required to provide materials of sufficiently small sizes for their internalization into cells, and this limits the applications of metal–organic materials as delivery vehicles, diagnostics, etc. But nanoscale metal–organic materials combine these small dimensions with the possibility to design architectures with well-defined and uniform sizes and morphologies, to disperse them in aqueous media or other solvents, and to efficiently coat them for improving their biocompatibility or recognition capabilities. Of particular interest is also the fact that nanomaterials have higher surface areas than their macroscopic counterparts. This property can strongly influence or even improve the catalytic, ion exchange, separation, sensing and sorption properties of these metal–organic materials, which, in turn, can be dependent on their sizes and shapes. A similar situation occurs with some physical properties. It is well known that unique physical properties emerge when at least one dimension of a material is reduced to the nanometre scale. Thus, nanoscale metal–organic materials are also expected to hold highly desirable size-dependent optical, electrical and magnetic properties.
It is obvious, then, that by scaling them down to the nanometre length scale, the scope of metal–organic materials will be expanded. But, how they can be prepared at the nanometre length scale? Like their macroscopic counterparts, they can be basically prepared by using “bottom-up” synthetic methodologies based on self-assembly processes; however, assembly of both inorganic and organic building blocks needs now to be controlled at the nanometre scale. For this, there are two major strategies. One involves a set of synthetic methodologies that induce the controlled precipitation of self-assembled supramolecular metal–organic polymers once they are formed. To date, these methodologies include the use of poor solvents, microwaves, ultrasounds or thermal conditions. The second strategy consists of confining the supramolecular assembly at specific locations. For example, self-assembly processes can be confined into droplets by using micro- or nanoemulsion techniques. Or they can be precisely controlled on the surface of substrates, liquids, membranes or other nanostructures, which act as templates, by using deposition techniques, such as layer-by-layer (LbL) synthesis, Langmuir–Blodgett, etc. A specific case that fits in this second strategy is the use of techniques for their nanostructuration on surfaces. In fact, the growth and positioning control on surfaces of these materials is crucial to start to conceive their integration on supports for the fabrication of complex surface sensors, separation membranes, drug-delivery platforms and catalysts.
The miniaturization of metal–organic materials down to the nanometre length scale is therefore a unique opportunity to develop a new class of highly tailorable nanoscale materials that combine the rich diversity of compositions, structures and properties of classical metal–organic materials with the obvious advantages of nanomaterials. Note that we use the term metal–organic material here to include materials with extended metal–organic structures, crystalline or not, that are built up from the supramolecular assembly of inorganic (metal ions and metal–organic or inorganic clusters) and organic building blocks (organic ligands, polymers, and biomolecules). And the term nanoscale includes these materials with at least one dimension between 1 and 100 nm, although some representative examples with sizes comprised between 100 and 1000 nm have also been included due to their size-dependent characteristics and properties. Therefore, these nanoscale materials have been classified according to their dimensionality: 0-D (particles), 1-D (fibers, tubes, and rods), and 2-D (thin films and membranes). In this review, we describe the recent advances made on the synthesis, growth and nanostructuration of this class of materials, their potential properties and applications, and illustrate their future expectations.
0- and 1-D metal–organic nanomaterials
Synthesis
There are two major strategies for synthesizing 0-D and 1-D nanoscale metal–organic materials (Fig. 1): (i) confinement of the supramolecular assembly at nanoscopic locations by using emulsions or templates; and (ii) the controlled precipitation of self-assembled metal–organic polymers once they are formed, thanks to the use of poor solvents, microwave radiation, ultrasounds or temperature.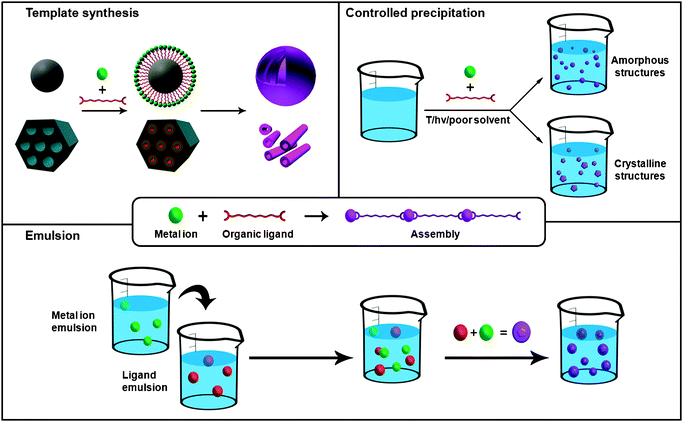 | ||
| Fig. 1 Schematic representation of the principal synthetic strategies used for synthesizing 0- and 1-D metal–organic nanostructures using conventional coordination chemistry, including the LbL growth on templates, the controlled precipitation of metal–organic nanostructures and the microemulsion techniques. | ||
Nanoemulsion is a suspension of small droplets, usually stabilized by a surfactant, with diameters ranging from 50 to 1000 nm, of one liquid in a second liquid in which the first will not mix. Because of these dimensions, these droplets can be used as “nanoreactors” to confine the self-assembly, nucleation and growth of metal–organic nanostructures. Briefly, this approach usually consists of first dissolving each precursor in the solvent that will be dispersed. A water-in-oil, or reverse, emulsion is then prepared from a surfactant, and the collisions between droplets containing those precursors or the application of an external stimulus, such as temperature, light or microwave radiation, spontaneously induce their polymerization, thus delimiting their growth inside the nanodroplet. For example, by dissolving Prussian blue precursors in water, using this solution to create a water-in-oil emulsion and further exposing this emulsion to daylight, Mann et al. synthesized the first examples of nanoscale metal–organic frameworks (NMOFs): Prussian blue cubic nanocrystals.2 Since then, emulsions have been successfully used to produce nanocrystals of heterometallic cyanometalates, Prussian Blue analogues using triazole instead of cyano groups and Gd(III)-based MOFs.
Besides the utility of their internal volume as “nanoreactors”, nanodroplets of these emulsions have also the potential to be used as templates to prepare metal–organic nanoshells. In this case, the polymerization must be concentrated on the interface of the droplets by using, for example, a chemical affinity surfactant. Thus far, this strategy has been used by Wang et al., who synthesized the first examples of Prussian blue nanoshells.3 These shells were synthesized by preparing an oil-in-water emulsion with an organometallic surfactant terminated with pentacyano(4-(dimethylamino)pyridine)ferrate. Then, the addition of Fe(III) ions to the aqueous solution induced the coordinative polymerization between them and the surfactant, thereby creating a metal–organic shell on the interface of each droplet.
Nanomaterials are also excellent candidates to be used as templates for fabricating metal–organic nanostructures. In this methodology, a film of a supramolecular metal–organic polymer is deposited on the template by using a deposition technique, such as LbL deposition. This deposition, followed by the removal of this template using thermal or chemical techniques, can leave behind a metal–organic nanostructure that mimics the shape and size of this template. Martin et al. reported a brilliant example that illustrates how powerful can this approach be.4 The LbL growth of a film composed of Zr(IV) metal ions and diorganophosphonate ligands on the pores of an alumina membrane, followed by the removal of this membrane using phosphoric acid, allowed the fabrication of highly uniform metal–organic nanotubes.
A second family of methodologies that enable the synthesis of 0-D and 1-D metal–organic nanostructures is based on mixing the precursors under certain reaction conditions that favor the fast nucleation (to increase the seed number) of self-assembled metal–organic polymers. In most of the cases, these conditions involve the use of microwave radiation, ultrasounds or temperature. For example, it is well known that at high temperatures, solubility of precursors increases avoiding fast precipitation, and allows crystallization to occur. It is therefore a strategy generally used to produce bulk crystalline metal–organic materials. However, if one controls certain reaction conditions, including solvent, surfactant, temperature and time, solvothermal synthesis can be a very productive strategy for synthesizing a wide variety of NMOFs. Some of them were recently reported by Horcajada, Gref et al., who synthesized a series of NMOFs by simply mixing Fe(III) metal ions with a series of multitopic ligands, such as 1,4-benzenedicarboxylate (BDC), benzene-1,3,5-tricarboxylate (BTC), fumaric acid, etc., in a variety of solvents at temperatures usually above 100 °C.5
The use of microwave radiation and ultrasounds can also be exploited for miniaturizing MOFs. Ni and Masel reported for the first time the use of microwave radiation to induce the precipitation of an already known MOF (IRMOF-1, 2 and 3) based on Zn(II) metal ions and BDC ligands at the nanometre scale.6 This reaction consists of exposing a diethylformamide (DEF) solution of both building blocks at 150 W for only 25 seconds. Similarly, sonochemical synthesis can lead to a homogenous nucleation and a substantial reduction in crystallization time. The well-known MOF-5 was miniaturized at the micrometre scale using this methodology.7 And even more recently, nanoscale arrowheads NMOFs of Prussian blue analogues have been prepared by Wu et al. with the help of ultrasonic radiation.8
However, one of the most extensive approaches used so far for preparing metal–organic nanostructures is based on the use of poor solvents to induce the fast precipitation of metal–organic polymers. This strategy can be divided into two well-established synthetic methodologies: (i) self-assembly takes place in a solution in which the synthesized metal–organic polymers are not soluble, and therefore, the poor solvent is the initial solution used to mix the building blocks; and (ii) the use of an external poor solvent to precipitate the metal–organic polymers previously synthesized in the initial solution, and therefore, the poor solvent is an external one. Wang et al. and Oh and Mirkin simultaneously developed both synthetic methodologies.9,10 The first authors exploited the insolubility of a coordination polymer built up from Pt(IV) metal ions and p-phenylenediamine in water to produce monodisperse spherical particles.9 On the other hand, Oh and Mirkin first prepared a coordination polymer constructed from Zn(II), Cu(II) or Ni(II) metal ions and a carboxylate-functionalized binaphthyl bis-metallotridentate Schiff base (BMSB) in pyridine, and this polymer was then precipitated in the form of amorphous spherical infinite coordination particles (ICPs) by addition of a poor solvent, such as ether or pentane.10 The versatility of this method has been proven by preparing a wide range of spherical amorphous ICPs, multi-shaped NMOFs and even nanoscale fibers.
Shapes and sizes
Even though we are in the early stages of their development,11,12 the abovementioned synthetic methodologies have already facilitated the preparation of several metal–organic nanostructures, ranging from 0-D amorphous ICPs and crystalline NMOFs (with multiple shapes) to 1-D rod-like crystals, fibers and tubes (Fig. 2). In most of the cases, their preparation has allowed researchers to continuously improve these synthetic methodologies, thus starting to understand some of the reaction conditions that one needs to modify in order to control their shapes and sizes. To date, the choice of the synthetic strategy as well as the corresponding reaction conditions seems crucial to prepare a certain type of nanostructure with precise control over its shape and size. | ||
| Fig. 2 Representative SEM images and their corresponding schematic figures of 0- and 1-D metal–organic nanostructures showing the wide diversity of morphologies obtained so far: (a) cubic particles, (b) octahedral particles, (c) arrow-like particles, (d) spheres, (e) hexagonal lumps, (f) plate-like particles, (g) rods, (h) fibers, and (i) tubes. (© The American Chemical Society, Springer and Wiley Interscience, reprinted with permission). | ||
This is the case of amorphous spherical ICPs that have exclusively been prepared by using methods based on fast precipitation induced by poor solvents. This strategy provides further control on the size of the colloidal spheres through modifications in the reaction conditions. There are three known parameters that can be tuned in order to control their diameter. The first one is the characteristics of the poor solvent, such as the polarity. Oh and Mirkin reported that, under identical reaction conditions, the use of pentane as the poor solvent leads to Zn(II)–BMSB spherical ICPs with larger diameters (780 ± 230 nm) than the ones obtained using more polar diethyl ether (190 ± 60 nm).10 The second parameter is the rate of addition of the poor solvent. Generally, larger spheres are obtained by decreasing the addition speed of the poor solvent in the reaction mixture. Finally, the third condition is the concentration of the reactants, which is inversely related to the diameter of the spheres. Maspoch et al. demonstrated that the diameter of ICPs built up from the connection of Zn(II) metal ions through bis(imidazole-1-ylmethyl)benzene (Bix) ligands was controlled from 130 ± 14 nm to 1050 ± 83 nm by decreasing the concentration of both Zn(II) metal ions and Bix components from 1 × 10−1 M to 5 × 10−3 M.13
Crystalline NMOFs are generally synthesized by using emulsions, solvothermal and sonochemical synthesis, poor solvents and microwave-assisted synthesis. In most of these strategies, the size and shape of the resulting NMOFs are closely dependent on the type of solvent, the nature, concentration and stoichiometry of the precursors, and the specific reaction conditions used in each one of these methodologies, such as the applied temperature in solvothermal synthesis, the microwave radiation intensity in microwave-assisted synthesis, etc.
There are several examples where the solvent has an effect on the shape and size of NMOFs. Jung and Oh found that cubic NMOFs built up from Zn(II) metal ions and metallosalen (MS) ligands were prepared with different sizes by performing identical solvothermal reactions under different DMSO/DMF mixtures.14 Here, DMSO/DMF mixtures in proportions 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1, 1∶2, and 2∶1 led to cubes with an average size of 308 ± 36 nm, 600 ± 94 nm, and 4860 ± 780 nm, respectively. The synthesis of NMOFs is also dependent on the nature, concentration and stoichiometry of the reactants. For example, Ni and Masel reported how the dimensions of cubic Zn(II)–BDC NMOFs prepared through microwave-assisted synthesis are reduced by decreasing the concentration of either Zn(II) metal ions or BDC ligands.6
∶1, 1∶2, and 2∶1 led to cubes with an average size of 308 ± 36 nm, 600 ± 94 nm, and 4860 ± 780 nm, respectively. The synthesis of NMOFs is also dependent on the nature, concentration and stoichiometry of the reactants. For example, Ni and Masel reported how the dimensions of cubic Zn(II)–BDC NMOFs prepared through microwave-assisted synthesis are reduced by decreasing the concentration of either Zn(II) metal ions or BDC ligands.6
Of special interest are the specific conditions, such as the temperature, the microwave radiation and the surfactant, used on these synthetic methodologies because they are excellent parameters to control for tailoring NMOFs. Among them, nanoemulsions are interesting because it is possible to tailor their droplet sizes, and therefore, the NMOFs sizes by controlling the composition of these emulsions, such as the relative composition of the surfactant, the oil phase and the aqueous phase. Lin et al. took advantage of this to prove that the dimensions (both diameter and length) of Gd(III)–BDC nanorods can be tailored by simply changing the water to surfactant molar ratio.15 In solvothermal synthesis, the reaction temperature can also affect the dimensions of the produced NMOFs. This dependence was confirmed by Jung and Oh, who reported that smaller cubic Zn(II)–MS NMOFs are generated at lower reaction temperatures.14 While cubes with dimensions of 166 ± 15 nm were obtained at 80 °C, an increase of the temperature up to 120 °C afforded bigger cubes with dimensions of 308 ± 36 nm.
The use of surfactants in methods based on fast precipitation is a further factor to be considered because they can play a critical role not only to control the morphology of NMOFs but also to increase their stability, avoiding agglomeration. For example, Thallapally et al. have recently highlighted the importance of using poly(diallyldimethylammonium chloride) as the surfactant to prepare hexagonally-shaped NMOFs built up from the connection of Zn(II) metal ions through imidazolate ligands.16 The use of other surfactants did not result in the formation of these NMOFs. However, surfactants are not the only type of substances that can help researchers to control the NMOFs shape. Like in purely inorganic nanomaterials, organic ligands also show promise for acting as blocking agents, which can control the growth rate of NMOFs in certain directions. Oh et al. enormously contributed to prove this concept.17 These authors reported that hexagonal rod-like In(III)–BDC NMOFs synthesized by solvothermal synthesis can be tuned to hexagonal lumps or disks by simply adding certain amounts of pyridine to the reaction solution.
Besides the formation of ICPs and NMOFs, there are other attractive nanoscale 1-D metal–organic materials: nanofibers and nanotubes.18 To date, most of the metal–organic nanofibers have been prepared by using fast precipitation methods. Maspoch et al. reported the fabrication of chiral nanofibers by adding a solution of aspartate (Asp) ligands into a solution of Cu(II) metal ions.19 Here, the length of these fibers was controlled by adjusting the addition rate of Asp solution, which is again inversely related to the length of the nanofibers produced. A mixture of both solutions by using a very slow diffusion led to very long (up to one centimetre) nanofibers, whereas a fast mixture decreased this length up to tens of micrometres. Nanotubes are also a very attractive type of nanoscale materials, but only one example made up from metal ions and organic ligands has been reported so far. As stated above, Martin et al. utilized alumina membranes as templates for synthesizing Zn(IV)–phosphonate nanotubes through a LbL synthesis.4 This is a very versatile synthetic methodology, and it will certainly allow the preparation of more metal–organic nanotubes in the near future, opening the door to control their outside diameter and length by selecting the diameter of the pores and the thickness of the alumina membrane, respectively, whereas the pore size and the wall thickness can be controlled by the number of inorganic/organic layers deposited on the pore wall of the template.
Hierarchical metal–organic superstructures
Like pure inorganic nanomaterials, the research on metal–organic nanostructures has recently started to be extended to the assembly of ICPs or NMOFs into ordered hierarchical superstructures that are able to exhibit a rich range of complex architectures (Fig. 3a). For example, Zhang et al. demonstrated the formation of sheaf-, flower-, wheatear-, straw-, bundle-, urchin- and butterfly-like hierarchical architectures from connecting Eu(III), Tb(III) or Ce(III) metal ions through BTC ligands under simple fast precipitation methods (Fig. 3).20 Among other functionalities, hierarchical superstructures can combine micro- and mesoporosity, thus having important prospects in industrial processes, such as catalysis, absorption, etc. Qiu et al. reported a versatile methodology based on the use of micelles as supramolecular templates for synthesizing hierarchically MOFs based on Cu(II) metal ions linked by BTC ligands.21 One of the most important features of these materials is that they exhibit micro- and mesoporous structures with tunable adsorption capabilities.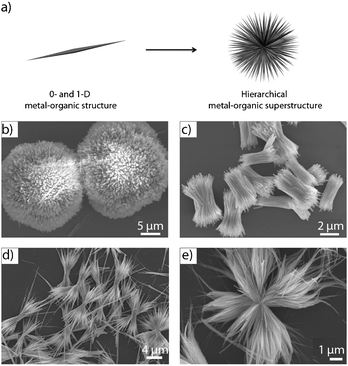 | ||
| Fig. 3 (a) Scheme illustrating the formation of hierarchical metal–organic superstructures. (b–e) SEM images of representative hierarchical metal–organic structures: (b) urchin-like, (c) bundle-like, (d) sheaf-like, and (e) flower-like superstructures. (© The American Chemical Society and The Royal Society of Chemistry, reprinted with permission). | ||
Metal–organic nanostructures constructed from supramolecular non-coordination bonds
The diversity of 0-D and 1-D metal–organic nanostructures can be further expanded by using electrostatic interactions, hydrogen bonds, π–π stacking and van der Waals interactions as the main driving forces for the assembly of the organic and inorganic building blocks. The synthetic methodologies are not so different from those explained above. For example, hybrid spheres of about 150 nm in diameter were assembled from electrostatic interactions between cationic dipeptides and anionic polyoxometalate (POM) clusters by simply mixing both building blocks in the appropriate solvent (Fig. 4).22 The fact that these interactions are weak makes these spheres to be easily disassembled by applying an external stimulus, such as pH (charge neutralization) and temperature. Thus, the sensitivity of electronic interactions to external stimuli could be of interest when thinking of controlled release of guest molecules or any other systems that require triggered disassembly. Based on the same approach, anionic POM clusters were electrostatically assembled with the cationic surfactant dimethyldioctadecylammonium bromide to yield nanostructures with different morphologies depending on the solvents used.23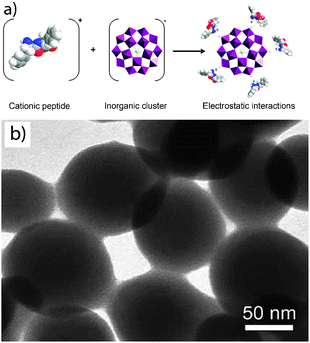 | ||
| Fig. 4 (a) Scheme illustrating the formation of nanospheres through the supramolecular assembly (electrostatic interactions) between a cationic peptide and an anionic inorganic cluster. (b) SEM image of these spheres (© Wiley Interscience, reprinted with permission). | ||
It is known that polyion-neutral diblock copolymers self-assemble into nanoparticles when mixed with an oppositely charged polyelectrolyte in what can be termed complex coacervate core micelles. Particles thus synthesized are stabilized due to the electroneutral segments of the diblock polymer. Stuart et al. followed this approach to assemble a cationic organic polymer with a pre-synthesized coordination polymer with negative charge in its coordination center into nanospheres of about 50 nm in diameter.24 Therefore, a reversible system such as a coordination polymer replaces a conventional polyelectrolyte conferring to the final system more versatility and sensitivity to external stimuli.
Another class of materials held entirely by non-coordination supramolecular interactions is those based on Pt–Pt interactions. It was found that square planar PtII self-assembles into 1-D structures through PtII⋯PtII interactions.25
Properties and applications
A wide range of promising properties can be obtained by taking advantage of the multiple inorganic and organic building blocks that can be combined to prepare nanoscale metal–organic materials. One can use the intrinsic properties of either the organic or inorganic components. The ability of metal ions to interact with phonons and electrons, to show luminescence and strong absorptions and to have interesting magneto-optical properties makes metal–organic materials suitable as functional materials in devices. The organic components can also exhibit their own functionalities. For example, organic chromophores, radicals or biomolecules are excellent building units to prepare optical, magnetic and biocompatible nanoscale materials. New properties also arise from the polymerization of both building blocks. This is the case of the tailorable porous structures constructed from the linkage of inorganic building units through multitopic organic ligands. All these properties can take advantage of the benefits of working at the nanometre length scale, thus opening novel avenues for developing nanomaterials with astonishing properties and applications. Some of these can help to develop novel drug delivery systems, encapsulating matrices, contrast agents, porous catalytic materials, gas storage materials, magnetic and optical nanomaterials, gels, etc. Recent great advances have been made towards these directions.![Representative applications for the nanoscale supramolecular 0- and 1-D metal–organic structures. (a–c) Drug delivery systems. (a) Metal–organic nanospheres built up from the coordination of a Pt(iv) based anticancer drug and Tb(iii) metal ions. (b) Drug release profile obtained by plotting %Pt released against time and (c) its in vitro cytotoxicity assay curves for HT-29 cells obtained by plotting the %cell viability against the Pt concentration of various samples. (d–f) Encapsulating systems. (d) TEM image of amorphous metal–organic ZnBix spheres. (e) TEM image of magnetic iron oxide nanoparticles encapsulated into ZnBix spheres. (f) Fluorescence optical image of fluorescein and quantum dots encapsulated into ZnBix spheres. (g–i) Contrast agents. (g) SEM image of metal–organic nanoparticles of [FeO(H2O)2Cl(fumarate)3] (MIL-88A). (h) Magnetic resonance images of control rats and (i) rats injected with 220 mg kg−1 of MIL-88A nanoparticles (© Wiley Interscience, The American Chemical Society and The Nature Publishing Group, reprinted with permission).](/image/article/2011/CS/c0cs00042f/c0cs00042f-f5.gif) | ||
| Fig. 5 Representative applications for the nanoscale supramolecular 0- and 1-D metal–organic structures. (a–c) Drug delivery systems. (a) Metal–organic nanospheres built up from the coordination of a Pt(IV) based anticancer drug and Tb(III) metal ions. (b) Drug release profile obtained by plotting %Pt released against time and (c) its in vitro cytotoxicity assay curves for HT-29 cells obtained by plotting the %cell viability against the Pt concentration of various samples. (d–f) Encapsulating systems. (d) TEM image of amorphous metal–organic ZnBix spheres. (e) TEM image of magnetic iron oxide nanoparticles encapsulated into ZnBix spheres. (f) Fluorescence optical image of fluorescein and quantum dots encapsulated into ZnBix spheres. (g–i) Contrast agents. (g) SEM image of metal–organic nanoparticles of [FeO(H2O)2Cl(fumarate)3] (MIL-88A). (h) Magnetic resonance images of control rats and (i) rats injected with 220 mg kg−1 of MIL-88A nanoparticles (© Wiley Interscience, The American Chemical Society and The Nature Publishing Group, reprinted with permission). | ||
2-D metal–organic nanomaterials
Beyond the fabrication of discrete 0-D and 1-D nanostructures, another important challenge is to control the growth of these structures directly on surfaces, thus creating functional membranes, thin films, or devices based on these materials. The control of the supramolecular assembly of inorganic and organic building blocks on surfaces is especially of interest to fully exploit the exceptional adsorption properties of metal–organic materials, and create new functional surfaces for applications in electronic, sensing or photonics. An excellent review on this topic has also recently been published by Fischer et al.44Synthesis
Chronologically, three principal strategies have been explored to fabricate metal–organic membranes and thin films (Fig. 6): (i) the LbL synthesis; (ii) the use of Langmuir–Blodgett (LB) monolayer transfer technique; and (iii) the direct assembly and growth of metal–organic nanostructures on surfaces.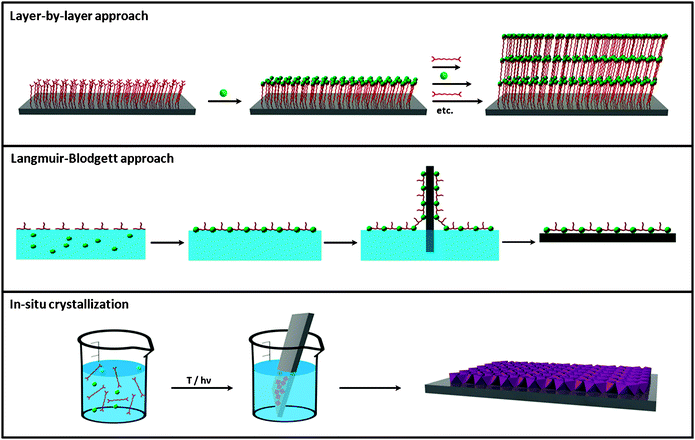 | ||
| Fig. 6 Schematic illustration of the three principal synthetic strategies used for preparing 2-D metal–organic nanomaterials, including the layer-by-layer approach, the Langmuir–Blodgett approach and the in situcrystallization. | ||
Initial efforts to fabricate nanoscale metal–organic surfaces began at the end of 80s decade with the use of the LbL synthesis (Fig. 6). The LbL synthesis is a well-known strategy that allows the step-by-step growth of hybrid organic–inorganic layers on surfaces by sequentially exposing them to solutions of both organic and inorganic building blocks. This approach was first used by Mallouk et al. in 1988 to fabricate an hybrid material composed of multilayers of Zr(IV) metal ions and 1,10-decanebisphosphonate (DBP) on a Si surface.45 In this case, after an initial functionalization of the Si substrate with a phosphonate-terminated monolayer, the substrate was exposed alternatively to aqueous solutions of ZrOCl2 and DBP. By repeating this process eight times, eight layers of Zr–DBP with a height of 13.6 nm (1.7 nm per layer) were fabricated. However, these adsorption steps can in principle be repeated as many times as desired in order to produce multilayer metal–organic films. The sequential dipping can be automated, and films containing more than 100 layers can be grown. Using the LbL synthesis, inorganic–organic multilayers composed of a wide variety of metal ions (e.g.Hf(IV), Ce(IV), Cu(II), etc.) and organic ligands (e.g. different types of diphosphonates, mercaptohexadecanoic acid, etc.) have nowadays been prepared on different surfaces (e.g.gold, silicon, etc.).
Beyond the stepwise coordination of metal ions and organic ligands, ultra thin metal–organic assemblies can also be grown with the electrostatic LbL self-assembly (ELSA) method. This methodology is based on the subsequent deposition of species with opposite charges on the surface. Generally, the inorganic species are anionic inorganic clusters or complexes (e.g.POMs), 2-D inorganic materials (e.g. clays and Zr(IV) phosphonates) and inorganic nanoparticles (e.g.gold nanoparticles). And the vast majority of organic species are oppositely charged organic polymers or large molecules. An illustrative example is the multilayer films of molybdenum oxide reported by Kunitake et al.46 In this case, the inorganic species was the anionic POM [Mo8O26], whereas poly(allylamine hydrochloride) (PAH) was the cationic organic polymer. Alternate electrostatic adsorption cycles of both building blocks on a polymeric surface resulted in films with uniform thicknesses and extremely smooth surfaces.
A second general approach for fabricating nanoscale 2-D metal–organic materials is the LB technique (Fig. 6). LB is a well-known technique that allows the organization of organic molecules on air/liquid interfaces, creating ultra thin layers that can be further deposited on surfaces. To our knowledge, Nagel and Oertel first used this technique to fabricate coordination polymer monolayers on an air/water interface.47 This process usually consists of four steps: (i) a multitopic organic ligand is first functionalized with alkyl chains to allow their organization on the air/water interface; (ii) small volume of a solution of the organic ligand is spread on the surface of an aqueous solution containing the metal ion; (iii) an interfacial coordination reaction occurs between the metal ions in the aqueous subphase and the organic ligands once they are spread or self-organized on the air/water interface; and (iv) the created ultrathin coordination polymer layers are transferred to a flat solid support by immersing the substrate through the compressed layer. Among others, Yuan and Liu followed these steps to fabricate chiral metal–organic monolayers. Initially, the naphtha[2,3]imidazole ligand was functionalized with a long alkyl chain. A chloroform solution of this ligand was then spread on pure water containing Ag(I) metal ions. Immediately, metal–organic monolayers were formed through an in situ coordination between both species, transferring them on mica.48
Both general strategies, the LB technique and the LbL synthesis, can also be coupled for producing metal–organic thin films on solid supports.49 Makiura, Kitagawa et al. recently reported the LbL growth of pre-formed metal–organic sheets using the LB technique. First, these layers were formed by spreading a chloroform/methanol solution of a Co(II) metalloporphyrin complex and pyridine onto an aqueous solution containing Cu(II) metal ions, and instantly transferred on a Si substrate. Sequential sheet formation and deposition on the same substrate allows the LbL stacking, thereby resulting in the generation of MOF thin films of any desired thickness.
The last general strategy so far used for fabricating metal–organic thin films and membranes is the direct nucleation and growth of NMOFs on surfaces. Initial efforts for growing these crystals on bare surfaces (e.g.SiO2, Al2O3, Au, graphite, etc.) were focused on a strategy based on immersing these substrates into a solution containing the precursors. Then, the NMOFs were grown on the surfaces by inducing their nucleation using methods analogous to those used for preparing 0-D and 1-D NMOFs, such as microwave radiation, temperature, etc. With these synthetic approaches, some metal–organic supports were successfully synthesized. However, most of the bare substrates inhibit the nucleation of NMOFs, and thus low crystal density is usually obtained.
A very promising solution to overcome this problem is the use of chemical affinity templates to promote the nucleation of NMOFs. Self-assembled monolayers (SAMs) are excellent candidates to act as such templates because they can be terminated with different functional groups that can direct the nucleation and growth of crystalline materials. The case of the HKUST-1 system is especially appropriate to describe this phenomenon. Bein et al. studied their nucleation and growth by immersing Au substrates previously functionalized with SAMs terminated with COOH–, OH– and CH3– groups into a solution containing Cu(II) metal ions and BTC ligands.50 Under the studied conditions, all terminated SAMs seemed to favor the nucleation, and therefore, increased the crystal density in comparison with bare Au surfaces. In addition, these authors also observed preferential crystal growth orientations depending on the terminal functional group of these SAMs. Indeed, the COOH-terminated SAM favored orientation along the [100] direction and the formation of pyramid-like crystals, whereas the OH-terminated SAM favored the [111] orientation and led to the formation of octahedral crystals. The third surface terminated with CH3– groups did not favor any orientation. These results, even though are quite contradictory to latter exhaustive experiments performed by Fischer et al.,51 confirm the importance of SAMs for controlling not only the density of crystals (homogeneity) deposited on the surface but also their orientation.
More recently, the use of SAMs on surfaces has also shown to be important for selectively growing a certain MOF crystal phase in front of another. This is the case of MIL-88B, a MOF composed of Fe(III) metal ions connected through BDC ligands.52 Scherb et al. demonstrated the preferential formation of crystals of this MOF by placing down a COOH-terminated SAM in a mother liquor solution containing both precursors. Surprisingly, even though the crystals formed in the solution corresponded to MIL-53, another MOF built up from Fe(III) metal ions connected through BDC ligands, the crystals that grew on the surface only corresponded to well-oriented MIL-88B crystals along the [001] direction. Also interesting is the work reported by Wöll et al.,53 who controlled the formation of a non-interpenetrated phase of a MOF built up from the linkage of [Zn4O] clusters through 4,4′-biphenyldicarboxylate ligands. Besides bulk synthetic methods lead to the interpenetrated phase, these authors proved to control the formation of the non-interpenetrated one on surfaces by using SAMs as templates and the LbL synthesis, opening new possibilities to synthesize new types of MOFs on surfaces not accessible by conventional methods.
In addition to the use of SAMs, other ways have been proposed to promote the homogeneous nucleation and growth of NMOFs on surfaces. One of the most interesting approaches is the use of seeds to induce a better crystallization. Similar to the growth of zeolitic membranes, Caro et al. created a homogeneous and dense coating of imidazolate based MOFs (ZIF-7) on alumina supports by first dipping them into an aqueous polyethyleneimine dispersion of pre-synthesized ZIF-7 nanocrystals with dimensions of 30 nm.54 These seeded supports were then immersed vertically into a solution containing the ZIF-7 precursors and heated up to 100 °C using microwave for three hours to obtain a large scale ordered polycrystalline ZIF-7 thin films.
A previous chemical modification of the surface is also an important factor to be considered in order to increase the crystal density on surfaces. One possibility is the “formate” route reported by Caro et al.,55 which consists of pre-oxidation of the surfaces with sodium formate treatment. With this oxidation, the nucleation of [Mn(HCOO)2] was enhanced and rather dense coatings were obtained. Qiu et al. and De Vos et al. further proved this concept.56,57 According to these authors, the oxidation of metallic copper surfaces generates Cu(II) metal ions process, creating more surface nucleation points during the HKUST-1 synthesis, and therefore, enhancing the crystal density on those surfaces (Fig. 7a).
 | ||
| Fig. 7 (a) Optic micrographs of the copper net (left) and the net-supported HKUST-1 membrane (right). (b) SEM top (left) and cross-section (right) views of ZIF-7 membranes obtained after microwave assisted secondary growth (© Wiley Interscience and American Chemical Society, reprinted with permission). | ||
Properties and applications
The exceptional advantages and properties already exhibited by nanoscale 0-D and 1-D metal–organic materials can be integrated on surfaces for fabricating metal–organic functional thin films and membranes with a wide range of properties and applications. Their use can allow the fabrication of electronic or optoelectronic thin films, membranes with gas separation capabilities, new surface sensors, magnetic platforms, etc.![(a) Schematic representation of the LbL film formation from the association of Eu–POM and a cationic polymer, and potential (Pt electrode), current, and absorbance at 700 nm of the LbL-coated ITO electrode during subsequent double-potential steps between ±0.4 and ±1.8 V. (b) Schematic drawing of the sequential assembly of [Fe(pyrazine){M(CN)4}] (M = Ni, Pd, or Pt) films and the temperature dependence of the normalized Raman intensity ratio (I(norm) = I(1025 cm−1)/I(1230 cm−1)) for [Fe(pyrazine){Pt(CN)4}] powder (■) and film samples (○) upon cooling and heating confirming the spin transition in LbL films (© Wiley Interscience, reprinted with permission).](/image/article/2011/CS/c0cs00042f/c0cs00042f-f8.gif) | ||
| Fig. 8 (a) Schematic representation of the LbL film formation from the association of Eu–POM and a cationic polymer, and potential (Pt electrode), current, and absorbance at 700 nm of the LbL-coated ITO electrode during subsequent double-potential steps between ±0.4 and ±1.8 V. (b) Schematic drawing of the sequential assembly of [Fe(pyrazine){M(CN)4}] (M = Ni, Pd, or Pt) films and the temperature dependence of the normalized Raman intensity ratio (I(norm) = I(1025 cm−1)/I(1230 cm−1)) for [Fe(pyrazine){Pt(CN)4}] powder (■) and film samples (○) upon cooling and heating confirming the spin transition in LbL films (© Wiley Interscience, reprinted with permission). | ||
Nanostructuration of metal–organic structures on surfaces
Control over the growth, orientation and positioning of metal–organic materials on surfaces is crucial to start to conceive the integration of these materials on supports for the fabrication of complex surface sensors, separation membranes, drug-delivery platforms and catalysts. There are three potential strategies to achieve this control at the nanometre scale length (Fig. 9): (i) the use of SAM features as chemical affinity templates to selectively control the nucleation and growth of metal–organic materials on them; (ii) the use of lithographic techniques to directly deliver or confine small volumes of the reaction solution containing the precursors, seeds or NMOFs at specific locations of a surface; and (iii) the growth of nanoscale metal–organic materials at specific locations of a surface by locally applying an external factor, such as temperature, addition of an external solvent, etc., that induces their nucleation.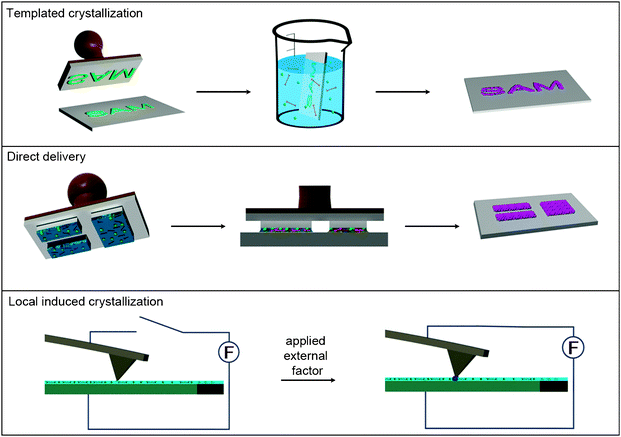 | ||
| Fig. 9 Schematic illustration of the three principal fabrication strategies used for structuring metal–organic nanostructures on surfaces at the nanometre length scale, including the templated crystallization, the direct delivery and the local induced crystallization. | ||
As stated above, it is well known that SAMs terminated with the appropriate functional group are able to coordinate with metal ions. One can then imagine to use patterned SAM features to control the positioning of the nucleation and growth of NMOFs. This approach starts with the fabrication of SAM features on surfaces by using conventional lithographic techniques, such as μ-contact printing (μCP) or photolithography. The NMOFs are then selectively grown on these features by exposing the surfaces to the precursor solution under optimum conditions or using the LbL synthesis. Fischer et al. first proved this concept by controlling the growth of MOF-5 crystals on square-like COOH-terminated SAM features of 40 × 40 μm fabricated by μCP (Fig. 10a–c).66 After patterning these features, the remaining Au surface was passivated with a CF3-terminated SAM in order to prevent the nucleation of MOF-5 in non-specific regions. The substrate was immersed into the mother solution, and this system was heated at 75 °C for 72 h. Interestingly, excellent adsorption properties of the resulting MOF-5 microarrays were successfully confirmed by exposing them to vapors of [(η5-C5H5)Pd(η3-C3H5)]. Using this method, Fischer's group also fabricated MOF-5 and HKUST-1 microarrays on alumina and silica surfaces.51 In this case, however, the SAM features were patterned with standard photolithographic techniques.
 | ||
| Fig. 10 (a–c) Arrays of MOF-5 crystals. (a) Schematic illustration of the concept of anchoring a typical MOF-5 building unit to a COOH-terminated SAM. (b) Optical microscope and (c) AFM images of a selectively grown film of MOF-5 on a patterned SAM of 16-mercaptohecadecanoic acid on a Au surface. The MOF-5-coated squares are 40 × 40 μm, and the crystals are about 100–500 nm in size. (d–f) Arrays of HKUST-1 crystals fabricated by LCW. (d) Large view and (e) details of individual crystals viewed from above (left) and at a 35° angle (right). The arrow indicates intergrowths caused by a second nucleation. (f) Square-like membrane structure composed of HKUST-1 crystals (© The American Chemical Society and Wiley Interscience, reprinted with permission). | ||
In addition to immersing the pre-structured surface into the precursor solution, the LbL synthesis can also be localized on these SAM templates, thus controlling the positioning of the created metal–organic multilayers. For instance, Wöll et al. fabricated HKUST-1 crystalline microarrays on Au surfaces by using the LbL synthesis on COOH-terminated SAM features generated by μCP.67 Similarly, Boom et al. fabricated a metal–organic multilayer pattern by sequentially immersing a glass surface, in which a template consisting of 5 μm OH-terminated SAM lines spaced from each other by 40 μm was created using photopatterning, into solutions of Pt(II) metal ions and 1,4-bis[2-(4-pyridyl)ethenyl]benzene ligands.68
Even though the use of SAM features is the most extended approach, recent advances have been made in using lithographic techniques for delivering small volumes of the precursor solution on surfaces, thus directly controlling the location in which the metal–organic structure is positioned or grown. Thus far, precursor solutions have been successfully patterned through different lithographic techniques, including the lithographically controlled wetting (LCW), microtransfer molding (μTM) and micromolding in capillaries (MIMIC). De Vos et al. controlled the synthesis of HKUST-1 crystals at the single-crystal level on glass substrates through LCW (Fig. 10d–f).69 In this method, a polydimethylsiloxane (PDMS) stamp was initially coated with a DMSO solution containing Cu(II) metal ions and BTC ligands. This stamp was then placed in contact with the glass surface; in this manner, capillary forces drove the liquid to distribute only under the protrusions of the stamp. As the solvent evaporated, the nucleation of HKUST-1 crystals took place under these protrusions, and therefore, the grown crystals were formed in a pattern reproducing the motif of the stamp. Similarly, Vieu et al. used μTM to transfer a colloidal solution containing coordination polymer nanoparticles on glass surfaces.70 It is interesting to note that the resulting nanoparticle structures with dimensions up to 300 nm kept their inherent spin crossover properties. Stamps were also used by Park et al. to structure a coordination polymer on glass surfaces using MIMIC.71 Continuous channels were formed when a PDMS stamp was brought into conformal contact with a solid substrate. These authors used the capillary action to fill the channels with a pyridine solution of a pre-formed coordination polymer composed of Zn(II) metal ions and 4,4′-di(4-pyridyl)cyanostilbene. The selective removal of pyridine by its absorption on the capillary walls drove to the formation while structuration of this coordination polymer.
Conclusions and perspectives
Although at the embryonic stage of their development, the recent advances made on this field, as shown in this review above, assure a brilliant future for this novel type of nanoscale materials. In this review, the advances on the early synthesis of metal–organic nanomaterials up to the initial studies of their properties and potential applications have been discussed. Along the way, there have been a number of achieved “milestones”, including the preparation of the first discrete nanostructures with varied compositions, sizes and shapes, the controlled growth of metal–organic thin films and membranes, new fabrication routes for nanostructuring them in pre-designed features on surfaces, the discovery of interesting structural (e.g. porosity, encapsulation capabilities, ion-exchange, etc.) and physical (e.g. electronic, magnetism, etc.) properties, and the exploration of promising biological-related applications, including drug delivery, bioimaging, sensing, etc.However, these studies are just the tip of the iceberg. The advantages of metal–organic nanomaterials are their high tailorability in terms of compositions, internal structures, morphologies, dimensions and properties, making possible their rational design for any specific application. A major challenge is then to develop novel methodologies and better understand the existing ones to fully exploit the limitless number of possible formulations, sizes and shapes of these materials. As an example of this issue is the possible local induced crystallization approach that shows promise for fabricating and structuring these materials on surfaces (Fig. 9). This strategy has not yet been explored. There is however no doubt that this route as well as other synthetic methodologies (e.g. microfluidics, supercritical conditions, vapour deposition, etc.) and lithographic techniques (e.g. Dip-Pen Nanolithography, Fountain-Pen Lithography, etc.) will be implemented in the next years, expanding the variety of synthesized metal–organic nanomaterials. In the same context, the fundamental understanding of how metal–organic nanomaterials are formed is crucial to begin to conceive of using these methods for controlling the fabrication of nanoscale metal–organic materials with desired properties.
Aside from the development of synthetic routes, there are other challenges which will focus the interest of researchers. One of them is the use of bio-related molecules as new building-blocks. Another direction of development is foreseen in the functionalization of these materials with functional species. Like their inorganic counterparts, this capability is particularly interesting for biological applications because it should allow the biocompatibilization and cell-specific targeting of these nanoscale materials. This future research, which will involve researchers of many different fields, will certainly expand the scope of properties and applications for nanoscale metal–organic materials up to their use for practical applications.
Acknowledgements
This work was supported by projects VALTEC08-2-003 and MAT2009-13977-C03. D. M. and I. I. thank the Ministerio de Ciencia y Tecnología for respective RyC and JdC contracts. A. C. and C. C. thank the Generalitat de Catalunya and the Institut Català de Nanotecnologia (ICN) for research fellowships.Notes and references
- Special issue on metal–organic framework materials. Chem. Soc. Rev. 2009, 38, 1201.
- S. Vaucher, M. Li and S. Mann, Angew. Chem., Int. Ed., 2000, 39, 1793 CrossRef CAS.
- G. D. Liang, J. T. Xu and X. S. Wang, J. Am. Chem. Soc., 2009, 131, 5378 CrossRef CAS.
- S. F. Hou, C. C. Harrell, L. Trofin, P. Kohli and C. R. Martin, J. Am. Chem. Soc., 2004, 126, 5674 CrossRef CAS.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172 CrossRef CAS.
- Z. Ni and R. I. Masel, J. Am. Chem. Soc., 2006, 128, 12394 CrossRef CAS.
- W. J. Son, J. Kim and W. S. Ahn, Chem. Commun., 2008, 6336 RSC.
- S. K. Wu, X. P. Shen, B. S. Cao, L. Lin, K. C. Shen and W. Liu, J. Mater. Sci., 2009, 44, 6447 CrossRef CAS.
- X. P. Sun, S. J. Dong and E. K. Wang, J. Am. Chem. Soc., 2005, 127, 13102 CrossRef CAS.
- M. Oh and C. A. Mirkin, Nature, 2005, 438, 651 CrossRef CAS.
- W. Lin, W. J. Rieter and K. M. L. Taylor, Angew. Chem., Int. Ed., 2009, 48, 650 CrossRef CAS.
- A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218 RSC.
- I. Imaz, J. Hernando, D. Ruiz-Molina and D. Maspoch, Angew. Chem., Int. Ed., 2009, 48, 2325 CAS.
- S. Jung and M. Oh, Angew. Chem., Int. Ed., 2008, 47, 2049 CrossRef CAS.
- W. J. Rieter, K. M. L. Taylor, H. Y. An, W. L. Lin and W. Lin, J. Am. Chem. Soc., 2006, 128, 9024 CrossRef CAS.
- S. K. Nune, P. K. Thallapally, A. Dohnalkova, C. Wang, J. Liu and G. J. Exarhos, Chem. Commun., 2010, 46, 4878 RSC.
- W. Cho, H. J. Lee and M. Oh, J. Am. Chem. Soc., 2008, 130, 16943 CrossRef CAS.
- J. K.-H. Hui and M. J. Maclachlan, Coord. Chem. Rev., 2010, 254, 2363 CrossRef CAS.
- I. Imaz, M. Rubio-Martinez, W. J. Saletra, D. B. Amabilino and D. Maspoch, J. Am. Chem. Soc., 2009, 131, 18222 CrossRef CAS.
- K. Liu, H. You, G. Jia, Y. Zheng, Y. Huang, Y. Song, M. Yang, L. Zhang and H. Zhang, Cryst. Growth Des., 2010, 10, 790 CrossRef CAS.
- L.-G. Qiu, T. Xu, Z.-Q. Li, W. Wang, Y. Wu, X. Jiang, X.-Y. Tian and L.-D. Zhang, Angew. Chem., Int. Ed., 2008, 47, 9487 CrossRef CAS.
- X. H. Yan, P. L. Zhu, J. B. Fei and J. B. Li, Adv. Mater., 2010, 22, 1283 CrossRef CAS.
- A. Nisar, Y. Lu and X. Wang, Chem. Mater., 2010, 22, 3511 CrossRef.
- Y. Yan, N. A. M. Besseling, A. de Keizer, A. T. M. Marcelis, M. Drechsler and M. A. C. Stuart, Angew. Chem., Int. Ed., 2007, 46, 1807 CrossRef CAS.
- W. Lu, V. A. L. Roy and C.-M. Che, Chem. Commun., 2006, 3972 RSC.
- W. J. Rieter, K. M. Pott, K. M. L. Taylor and W. B. Lin, J. Am. Chem. Soc., 2008, 130, 11584 CrossRef CAS.
- K. M. L. Taylor-Pashow, J. D. Rocca, Z. Xie, S. Tran and W. Lin, J. Am. Chem. Soc., 2009, 131, 14261 CrossRef CAS.
- I. Imaz, M. Rubio, L. García, F. García, D. Ruiz-Molina, J. Hernando, V. Puntes and D. Maspoch, Chem. Commun., 2010, 46, 4737 RSC.
- R. Nishiyabu, C. Aime, R. Gondo, T. Noguchi and N. Kimizuka, Angew. Chem., Int. Ed., 2009, 48, 9465 CrossRef CAS.
- K. M. L. Taylor, A. Jin and W. Lin, Angew. Chem., Int. Ed., 2008, 47, 7722 CrossRef CAS.
- K. H. Park, K. Jang, S. U. Son and D. A. Sweigart, J. Am. Chem. Soc., 2006, 128, 8740 CrossRef CAS.
- D. Tanaka, A. Henke, K. Albrecht, M. Moeller, K. Nakagawa, S. Kitagawa and J. Groll, Nat. Chem., 2010, 2, 410 CrossRef CAS.
- N. Kerbellec, L. Catala, C. Daiguebonne, A. Gloter, O. Stephan, J. C. Bunzli, O. Guillou and T. Mallah, New J. Chem., 2008, 32, 584 RSC.
- X. Zhang, Z. K. Chen and K. P. Loh, J. Am. Chem. Soc., 2009, 131, 7210 CrossRef CAS.
- L. Catala, F. Volatron, D. Brinzel and T. Mallah, Inorg. Chem., 2009, 48, 3360 CrossRef CAS.
- E. Coronado, J. R. Galán-Mascaros, M. Monrabal-Capilla, J. García-Martinez and P. Pardo-Ibáñez, Adv. Mater., 2007, 19, 1359 CrossRef CAS.
- I. Imaz, D. Maspoch, C. Rodríguez-Blanco, J. M. Pérez-Falcón, J. Campo and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2008, 47, 1857 CrossRef CAS.
- I. Boldog, A. B. Gaspar, V. Martínez, P. Pardo-Ibañez, V. Ksenofontov, A. Bhattacharjee, P. Gütlich and J. A. Real, Angew. Chem., Int. Ed., 2008, 47, 6433 CrossRef CAS.
- M. Oh and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 5492 CrossRef CAS.
- W. J. Rieter, K. M. L. Taylor and W. Lin, J. Am. Chem. Soc., 2007, 129, 9852 CrossRef CAS.
- W. Cho, Y. H. Lee, H. J. Lee and M. Oh, Chem. Commun., 2009, 4756 RSC.
- S. Hermes, M.-K. Scröter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237 CrossRef CAS.
- M. M. Pires, D. E. Przybyla and J. Chmielewski, Angew. Chem., Int. Ed., 2009, 48, 7813 CrossRef CAS.
- D. Zacher, O. Shekhah, C. Wöll and R. A. Fischer, Chem. Soc. Rev., 2009, 38, 1418 RSC.
- H. Lee, L. J. Kepley, H. G. Hong and T. E. Mallouk, J. Am. Chem. Soc., 1988, 110, 618 CrossRef CAS.
- I. Ichinose, H. Tagawa, S. Muzuki, Y. Lvov and T. Kunitake, Langmuir, 1998, 14, 187 CrossRef CAS.
- J. Nagel and U. Oertel, Thin Solid Films, 1998, 327–329, 495 CrossRef CAS.
- J. Yuan and M. Liu, J. Am. Chem. Soc., 2003, 125, 5051 CAS.
- R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata and H. Kitagawa, Nat. Mater., 2010, 9, 565 CrossRef CAS.
- E. Biemmi, C. Scherb and T. Bein, J. Am. Chem. Soc., 2007, 129, 8054 CrossRef CAS.
- D. Zacher, A. Baunemann, S. Hermes and R. A. Fischer, J. Mater. Chem., 2007, 17, 2785 RSC.
- C. Scherb, A. Schödel and T. Bein, Angew. Chem., Int. Ed., 2008, 47, 5777 CrossRef CAS.
- O. Shekhah, H. Wang, M. Paradinas, C. Ocal, B. Schüpbach, A. Terfort, D. Zacher, R. A. Fischer and C. Wöll, Nat. Mater., 2009, 8, 481 CrossRef CAS.
- Y.-S. Li, F.-Y. Liang, H. Bux, A. Feldhoff, W.-S. Yang and J. Caro, Angew. Chem., Int. Ed., 2010, 49, 548 CAS.
- M. Arnold, P. Kortunov, D. J. Jones, Y. Nedellec, J. Kärger and J. Caro, Eur. J. Inorg. Chem., 2007, 60 CrossRef CAS.
- H. Guo, G. Zhu, I. J. Hewitt and S. Qiu, J. Am. Chem. Soc., 2009, 131, 1646 CrossRef CAS.
- R. Ameloot, L. Stappers, J. Fransaer, L. Alaerts, B. F. Sels and D. E. De Vos, Chem. Mater., 2009, 21, 2580 CrossRef CAS.
- S. Liu, D. G. Kurth, H. Möhwald and D. Volkmer, Adv. Mater., 2002, 14, 225 CrossRef CAS.
- Y. Nishimori, K. Kanaizuka, M. Murata and H. Nishihara, Chem.–Asian J., 2007, 2, 367 CrossRef CAS.
- N. Tuccitto, V. Ferri, M. Cavazzini, S. Quici, G. Zhavnerko, A. Licciardello and M. A. Rampi, Nat. Mater., 2009, 8, 41 CrossRef CAS.
- Y. Pan, B. Tong, J. Shi, W. Zhao, J. Shen, J. Zhi and Y. Dong, J. Phys. Chem. C, 2010, 114, 8040 CrossRef CAS.
- Y.-S. Li, H. Bux, A. Feldhoff, G.-L. Li, W.-S. Yang and J. Caro, Adv. Mater., 2010, 22, 3322 CrossRef CAS.
- E. Biemmi, A. Darga, N. Stock and T. Bein, Microporous Mesoporous Mater., 2008, 114, 380 CrossRef CAS.
- G. Lu and J. T. Hupp, J. Am. Chem. Soc., 2010, 132, 7832 CrossRef CAS.
- S. Cobo, G. Molnar, J.-A. Real and A. Bousseksou, Angew. Chem., Int. Ed., 2006, 45, 5786 CrossRef CAS.
- S. Hermes, F. Schroder, R. Chelmowski, C. Woll and R. A. Fischer, J. Am. Chem. Soc., 2005, 127, 13744 CrossRef CAS.
- O. Shekhah, H. Wang, S. Kowarik, F. Schreiber, M. Paulus, M. Tolan, C. Sternemann, F. Evers, D. Zacher, R. A. Fischer and C. Wöll, J. Am. Chem. Soc., 2007, 129, 15118 CrossRef CAS.
- M. Altman, A. D. Shukla, T. Zubkov, G. Evmenenko, P. Dutta and M. E. van der Boom, J. Am. Chem. Soc., 2006, 128, 7374 CrossRef CAS.
- R. Ameloot, E. Gobechiya, H. Uji-i, J. A. Martens, J. Hofkens, L. Alaerts, B. F. Sels and D. E. D. Vos, Adv. Mater., 2010, 22, 2685 CrossRef CAS.
- C. Thibault, G. Molnár, L. Salmon, A. Bousseksou and C. Vieu, Langmuir, 2009, 26, 1557.
- Y. You, H. Yang, J. W. Chung, J. H. Kim, Y. Jung and S. Y. Park, Angew. Chem., Int. Ed., 2010, 49, 3757 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |