Water-soluble, self-assembling container molecules: an update
Zachary
Laughrey
a and
Bruce C.
Gibb
*b
aProtein and Proteomics Laboratory, Department of Chemistry and Biochemistry, Arizona State University, Tempe, AZ, USA. E-mail: bgibb@uno.edu
bDepartment of Chemistry, University of New Orleans, New Orleans, LA 70148, USA
First published on 15th November 2010
Abstract
Over the past five years, an important development in the area of self-assembling containers has been the increase in interest in those containers that function in aqueous solution. This progress is a reflection of a similar trend within supramolecular chemistry in general, and is driven in part by the need to address issues and challenges within the biological sciences, as well as a desire to develop new strategies for greener chemistries carried out in water. It is also an opportunity to learn more about fundamental topics such as the hydrophobic effect. In this critical review we discuss progress in aqueous-based self-assembling container molecules since 2005 (177 references).
 Zachary Laughrey | Zachary Laughrey was born in Denver, Colorado, in 1972. His BA and PhD degrees were received from the University of New Orleans where he examined the host–guest chemistry and the reactivity of deep cavity cavitands with Prof. Bruce Gibb. His post-doctoral research was conducted at the University of North Carolina–Chapel Hill with Prof. Marcey Waters. Research there primarily concerned carbohydrate–π interactions in model β-hairpins. He is currently director of the Protein and Proteomics Laboratory at Arizona State University in Tempe, Arizona. |
 Bruce C. Gibb | Professor Bruce Gibb was born in 1965 and hails from Aberdeen, Scotland. He received both his BSc and PhD degrees from Robert Gordon's University. In 1992, he relocated to the North American continent, carrying out his first post-doctoral research with Prof. John Sherman at the University of British Columbia, and subsequently working with Prof. James Canary at New York University. His research in the Sherman group centered on novel cavitands and cavitand–peptide conjugates, whilst metallo-enzyme mimics were the main topic of research in New York. His independent career began in 1996 at the University of New Orleans. He was promoted to associate professor with tenure in 2002, full professor in 2005, and University Research Professor in 2007. The over-arching theme of his research is aqueous-based supramolecular chemistry, with particular emphasis on self-assembly and compartmentalization. |
Introduction
This review is an update on the state-of-affairs of water-soluble, self-assembling container molecules. As the papers highlighted suggest, such containers are capable of displaying many different phenomena that arise as a result of compartmentalization. Frequently overlooked because it is so conceptually simple and ubiquitous, the phenomenon of compartmentalization is of immense importance to any chemical system. For example, in complex systems as big as the Earth's biosphere or as small as a bacterium, compartmentalization is required to maintain the non-equilibrium conditions required for existence. Chemists are not yet adept at constructing complex systems of this level of sophistication,1,2 however, as is highlighted here, even in systems that cannot by any stretch of the imagination be defined as complex, compartmentalization engenders many interesting phenomenon; phenomenon that arise through a combination of the kinetic barrier between the inner and outer environments, how the outer surface of the compartment interacts with the external environment, and how the inner surface interacts with the contents.For chemists, the most familiar example of compartmentalization is the humble round-bottomed flask; an excellently high kinetic barrier that is capable of keeping reactions confined inside, and species such as adventitious water out. By and large however, round-bottomed flasks do not do much more than that; they interact minimally with both the exterior environment and the contents. Moving down in scale, the development of liposomes, particularly the pioneering work of Gregoriadis' regarding liposomes as drug delivery devices,3 took compartmentalization to a new level; a micrometre one to be more precise. Lacking a permanent portal, liposomal studies highlighted the importance of how changes to the wall of the container could control the kinetic barrier between inside and out. In addition, their application to drug delivery highlighted the importance of the interactions between the outer surface of the compartment and the external environment. The pioneering work of Cram moved containers into the molecular and nano-scale4,5 and pointed towards a further level of compartmental control; with a container molecule not only could the kinetic barrier between inner and outer domains and the interactions between the outer surface and the external domain be controlled, but so could the interactions of the contents with the walls of the container.
Originally, container molecules4,5 existed as one-piece shells and possessed little in the way of portals for guest entry and egression. This led to permanent entrapment, an intrinsically interesting phenomenon for the study of normally highly reactive species,6–9 but one that is unfortunately also intrinsically undynamic. The two obvious ways around this lack of dynamism were to make holes in the side of the containers,10–12 or use multiple copies of one or more carefully sculpted subunits and assemble dynamical containers; in which case guest exchange13,14 can be intimately tied to the kinetics of host assembly and disassembly. This review concerns the latter dynamical assembly strategy. The initial targets for such self-assembling containers were small guests such as methane,15 but as we highlight here contemporary hosts are capable of sequestering more structurally interesting guests, and consequently can engender many fascinating phenomena via compartmentalization.
There are now many different examples of dynamic container molecules,16,17 but the focus here is on containers that assemble and function in aqueous solution rather than organic solvents. Supramolecular chemistry is slowly and inexorably moving into the aqueous realm, it necessarily must do so in order to affect biochemistry or engender new fields of greener organic chemistry.18,19 However, in addition to these important motivators there are some interesting fundamental reasons for studying dynamic containers in water, not least of which is the difference in the environments between the inside and the outside of a container. As we discuss in the next section, the vast differences between bulk water and the inside of a molecular container gives a thermodynamic boost to guest complexation. In other words, it is not only structural complementarity between container and guest that drives complexation, but also the ‘dislike’ the majority of organic molecules have for aqueous solution. As a result, complexation events are generally stronger, and more importantly, the range of guests that can be complexed by a particular host wider. One more caveat beyond self-assembly in water defines the boundaries of this review. The focus here is on containers that largely envelop a guest or guest molecules. The definition of ‘largely envelop’ is naturally subjective, but here we focus on hosts that shield >50% of the surface of a guest from the bulk aqueous environment. The reader should note that the focus on self-assembling systems that largely envelop a guest or guests means that we will not specifically cover many fascinating pieces of work involving cyclodextrins20–24 and cucurbiturils,25–27 nor water soluble hosts that are synthesized19,28–33 or assembled in organic solvent and ultimately moved into water.34 Readers interested in the broader topic of host–guest chemistry in water should not neglect these seminal areas.
The organization of this review is as follows. We begin with a brief summary of water and its solvation properties, before going on to highlight the strategies that can be used in water to affect assembly of a molecular container. In the subsequent sections we detail the papers that have appeared in the literature since 2005, breaking this compendium down according to the different driving forces that lie behind the self-assembly of the containers.
A word about water
Water is a classic example of a complex system. It possesses a very simple structure, yet possesses emergent (and beguiling) properties intimately tied to its dynamical structure. Indeed, the innate complexity is probably why in the past water has been associated with controversial ideas such as polywater35 and memory-water,36 and why such controversies continue with, for example, the idea that water can store charge.37,38 Regarding water-based self-assembled containers and their binding properties, supramolecular chemists are primarily concerned with the range of forces that they can use to drive assembly of the container, and the forces that can subsequently be harnessed to drive guest complexation into the container. We discuss assembly in the next section, but one important factor in either assembly or guest binding is unique to water, and that is the hydrophobic effect.39–47 The basic principle of the hydrophobic effect, i.e., that water and oil don't mix, is one emergent phenomenon of bulk water that is not debated. However, there is still much to learn regarding how this phenomenon comes about, and in this regard supramolecular chemists are perfectly poised to contribute to current understanding. The reason why this is so is that extensive in situ and in silico studies have determined that the root cause of the hydrophobic effect is the interfacial region between bulk water and solute and how this varies as a function of solute size, shape and polarity, as well as the nature and concentration of any cosolutes present. For example, in situ studies have provided a good understanding of the solvation of small entities such as protons48–51 and methane,52 as well as how the solvation shell around larger organic molecules—and hence their structure—is influenced by co-solute salts.53–56 Complementing this research, in silico studies40 have identified the differences in the solvation shells of small hydrophobic solutes versus large ones, and the differences between the solvation of concave versus convex surfaces.57,58 Who better to examine how two molecules (or three when a cosolute is present) interact non-covalently than supramolecular chemists!Strategies for assembly in water
The roots of synthetic self-assemblies are in non-polar solvents, and under such conditions self-assembly is dominated by two strategies: metal coordination and hydrogen bonding. These are powerful strategies, and have been used to form dynamic containers capable of selective binding (including enantioselectivity),59,60 redirecting chemical reactivity (away from that observed in solution),17,61–65 catalysis,29,66–75 kinetic systems with unusual feed-back loops,76 memory effects,77 and many other unusual phenomena that can only be engendered by compartmentalization.78–90 Both approaches utilize relatively strong and highly directional non-covalent interactions to drive product formation. Note that both, “strong” and “directional” are descriptors for the enthalpy of interaction. A strongly favorable enthalpy change is a prerequisite because bringing many subunits together is more often than not entropically penalizing. Likewise, “highly directional” refers to how the enthalpy of interaction in the non-covalent bond varies as a function of bond angle between, for example, a metal ion and ligand. Strong directionality helps to ensure an assembly with a well-defined architecture.The rules of assembly in water are very different. Its polar nature and strong hydrogen bonding ability impose a considerable handicap on hydrogen bonding as an assembly strategy. Shielding the hydrogen bonding arrays can allow them to function as a driving force for assembly, for example in the assembly or RNA, DNA and G-quadruplexes.91–93 However, to the authors' knowledge shielding has not been utilized to engender large encapsulating hosts that self-assemble in water via hydrogen bonding. In contrast, metal ion coordination has proven to be extremely useful in both organic solvents and water. ‘Simply’ tuning the strength of coordination, i.e., increasing the electrostatic component of the coordination bond, by careful choice of metal and ligand allowed this strategy to be successfully ‘translated’ into aqueous solution. Indeed, as is apparent in this review, metal ion coordination is by far the most common approach to self-assembly in water. There is however an alternative, and this is to do what Nature has done since time immemorial and harness the hydrophobic effect. This allows the opportunity to use either assembly motifs that are enthalpically strong and directional, or, motifs that are entropically strong and directional.94 This latter possibility comes about because of the hydrophobic effect; because in water a large part of the free energy of assembly arises from the difference in solvating the subunits versus the assembly product. In other words, bringing many components together in water to form an assembly product may not be entropically penalizing. Indeed, it is theoretically possible in such assemblies to be entirely driven by entropy and endothermic. These notions raise an interesting question. What assembly motifs are to entropy, what metal coordination or hydrogen bonds motifs are to enthalpy? As yet, there is no comprehensive answer to this question. However, future studies on self-assembling hosts, similar work upon proteinaceous assemblies, and computational studies of both will help address this important question.
Systems utilizing metal coordination
As discussed above, metal coordination has proven to be the most commonly applied strategy for self-assembly in aqueous solution. The Fujita group has been important pioneers in this regard, and has devised a number of self-assembling hosts that have found utility in a diverse range of applications. One of the most successful hosts from the Fujita group is tetrahedral M6L4 coordination cage 1, assembled from four tridentate ligands, six metal ions, and six ancillary ligands for capping the metal corners (Scheme 1). Most of Fujita's work has focused on the use of palladium or platinum ions, but others have also been studied (vide infra). Likewise, the principle ancillary ligands used have been ethylenediamine derivatives, but other ligands have also been examined. First reported in 1995,95 host 1 is now commercial available, a fact that has increased its accessibility dramatically.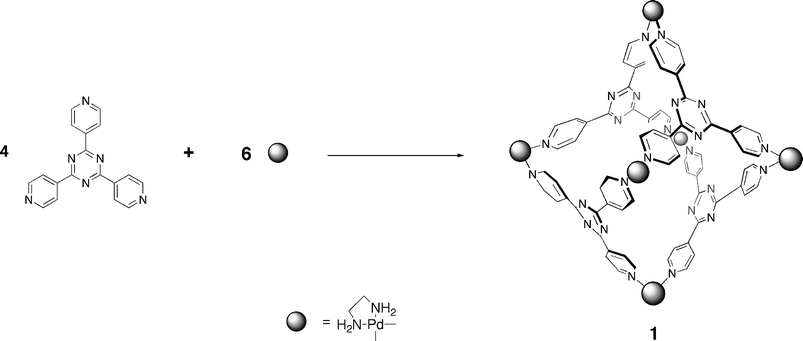 | ||
| Scheme 1 Self-assembly of coordination cage 1. | ||
Beginning with a biochemical slant, the ever-expanding proteome, and with it the increased need to isolate proteins from complex mixtures, has mostly relied on recombinant technologies to insert for example biotin or His6 tags into a target. On the other hand, supramolecular chemistry has a lot to potentially offer protein recognition,96 and with this idea in mind, cage 1 has been examined for its ability to selectively bind peptides.97,98 The first studies examined the binding of tripeptides of the general structure Ac-XXX-NH2. The strongest binder to the coordination cage 1 was the Ac-Trp-Trp-Ala-NH2 (Ka > 106), which bound in the cavity via a series of π⋯π and C–H⋯π intermolecular host–guest and intramolecular guest–guest interactions. Many of these non-covalent interactions involved charge transfer, and as a result color changes were observed upon internalization of the guest. In general, as the size of the amino acid residues in the sequence decreased so the affinity to the host was observed to decrease, but of even more interest was the observation that different sequences comprised of the same three residues were readily differentiated between by the host. For example, the binding constant for Ac-Trp-Ala-Trp-NH2 and Ac-Ala-Trp-Trp-NH2 were Ka = 2.5 × 105 M−1 and 2.1 × 104 M−1, respectively, both considerably weaker than the complexation of Ac-Trp-Trp-Ala-NH2.
Moving onto materials topics, one thrust from the Fujita group has been to examine how compartmentalization can be used to bring two or more radicals into close proximity so that their intermolecular interactions can be examined. One potential application of the confinement of multiple radicals is the design of molecular magnetic devices whose properties arise from carefully controlled spin–spin interactions. For example, the Fujita group reported the encapsulation of two copies of amine radical 2 within the compartment of host 1, and how the resulting spin–spin interactions are responsive to the external environment.99 In the complex, the radical centers of these guests were approximately 6.3 Å distant, and as a result strong intermolecular triplet-state spin–spin interactions were observed. However, when the pH of the solution was reduced, the amino groups of the guests were protonated and the guests released into free solution. As a result, the ESR spectrum of the guests changed considerably, with the loss of a forbidden transition seen in the complex, and the appearance of a spin-allowed doublet rather than a triplet. This switching process could be reversed by the addition of base. In more recent related work, Fujita and coworkers also examined how these spin–spin interactions vary as a function of the structure of the guest.100 These results demonstrated that the goodness of fit that the guest pair has with the cage highly influenced the spin–spin interactions, and that other factors such as temperature could also be used to control the radical interactions.
In a interesting twist on the idea of entrapping radicals, the Fujita group recently revealed the tetrahedral M6L4 self-assembling cage made from the assembly of four verdazyl radical tridentate ligands 3.101 Because of the C2v symmetry of this ligand the assembly product consisted of a mixture of ten isomers. Hence, in the obtained X-ray structure the verdazyl panels were disordered. Nevertheless, ESR spectrometry did show intramolecular interactions between the four spin centers of the nano-cage, and intermolecular interactions between the host and the encapsulated radical guest(s).
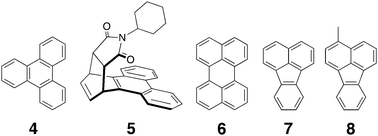
As alluded to in the introduction, one common application of container molecules is as nano-sized (yoctolitre) reaction flasks. The Fujita group has been one of the pioneers in this field, and have recently used host 1 to bring about reaction of normally unreactive species,102 induce asymmetric reactions in 1 possessing remote chiral auxiliaries attached to the metal centers,103 and bring focus to a photochemical process such that only one product is observed.65 The entrapment of two molecules within such a small volume leads to a high concentration of the guests and hence an increased likelihood of reaction between them. This phenomenon is beautifully illustrated by the Diels–Alder reaction between triphenylene 4 and maleimides.102 The former is a highly stable aromatic compound and had not previously been observed to undergo any pericyclic reactions. However, heating of the quantitatively formed ternary complex between this diene, the dienophile N-cyclohexylmaleimide, and host 1 resulted in the quantitative formation of the endo adduct at one of the benzene rings of the product (5). Indeed the authors noted that several ‘non-reactive’ aromatics underwent similar reactions. In some of these cases, such as the reaction of perylene 6 and N-cyclohexylmaleimide, the products were only stable in the confines of the host; in the free state they rapidly underwent air-induced aromatization. An extension of this work revealed that when chiral diamines replaced the normally utilized ethylenediamine ancillary ligands in host 1, enantioselective reactions could be carried out within the confines of the host. For example, when either fluoranthene 7 or 3-Me-fluoranthrene 8 was bound with N-cyclohexylmaleimide in chiral 1, the chiral [2+2] cross photo-addition products were isolated in 40 and 50% ee respectively.103 The steric bulk of the N-alkyl group on the maleimide was essential to obtaining good enantiomeric excesses. When the cyclohexyl group was replaced with a methyl group or a hydrogen atom, the ee values dropped to 20 and 5% respectively. These remote inductions of chirality via a tight supramolecular complex are reminiscent of the long range (>20 bond lengths) induction of chirality observed by Clayden et al.104 within very, sterically-demanding molecules.
In addition to leading to high concentrations, confinement within yoctolitre flasks often leads to complexes of very precise geometry. As a result, when two guests are encapsulated and subsequently reacted together it is often the case that selective product formation is observed. Such is the case when o-quinone 9 and 4-(1-adamantyl) toluene 10 are entrapped within 1 (Scheme 2). Irradiation of this complex resulted in the formation of only one product, the 1,4-adduct (11), rather than a range of compounds expected when the photochemistry is carried out in free solution.65 In this particular case, an X-ray structure of the ternary complex revealed the precise relative orientation of the two guests and hence the reason why only one product was formed.
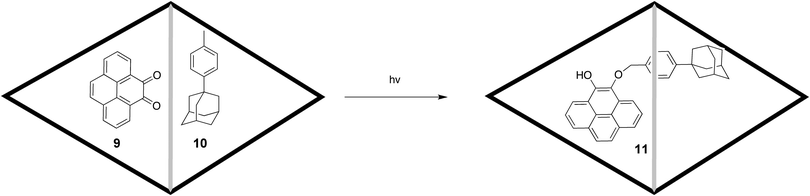 | ||
| Scheme 2 Encapsulation reaction of o-quinone 9 and 4-(1-adamantyl) toluene 10. | ||
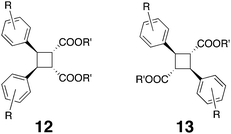
The Ramamurthy group has also used the Fujita host 1 to study photo-dimerizations in enclosed cavities. For example, nano-cage 1 was investigated for its ability to template the photo-dimerization of trans-cinnamic acid esters.105 Typically, irradiation of trans-cinnamic acid esters results in isomerization to the cis isomer, as well as formation of limited or trace amounts of dimers. In the presence of 1, the twelve esters examined produced both the monomeric cis-isomer in considerably reduced yield, and dimeric products in a range of 21–63% yield. The formation of the cis-isomer suggested a relatively weak ternary complex and that 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 host–guest complexes and free guest were present in solution. The dimeric products observed were mostly of syn head–head geometry (12), although two of the cinnamic acid investigated also produced the anti head–tail dimer (13), indicating that the substituents could control the packing of the guests and hence the photochemical outcome of these templated reactions.
∶1 host–guest complexes and free guest were present in solution. The dimeric products observed were mostly of syn head–head geometry (12), although two of the cinnamic acid investigated also produced the anti head–tail dimer (13), indicating that the substituents could control the packing of the guests and hence the photochemical outcome of these templated reactions.
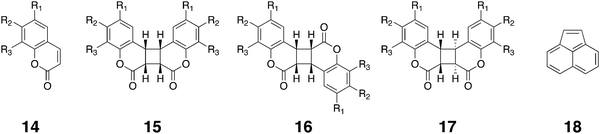
The Ramamurthy group has also examined how host 1 can template the exclusive formation of syn head–head dimers of a number of coumarin derivatives, e.g.14.106 In free aqueous solution the four coumarins examined produced different yields of the syn head–head (15), syn head–tail (16), and anti head–head dimers (17). In the confines of the host however, only the syn head–head isomers were observed. The power of host 1 to template photo-dimerizations was also evident when the sensitized dimerization of encapsulated acenaphthylene (18) was examined. In free solution, acenaphthylene produces primarily the anti dimer in the presence of a sensitizer. However, in the presence of either of the sensitizers Eosin Y or Rose bengal, the ternary complex formed between two molecules of acenaphthylene and host 1 yielded only the syn dimer;107 although the sensitized reaction goes through the triplet state, the confining nature of the cavity forced the formation of the ‘wrong’ isomer.
In addition to promoting reactions, the Fujita group have followed the pioneering work of Cram,4–6 and identified how 1 can be used to stop reactions from occurring. One particularly intriguing example of storing a reactive species is the observation of a coordinatively unsaturated transition-metal complex using X-ray crystallography.108 Crystallographic analysis revealed that the complex between Cp′Mn(CO)3 (Cp′ = methylcyclopentadiene) and 1 consisted of four copies of the manganese complex within the host. Photo-irradiation of this entity led to the quantitative dissociation of a CO ligand from one of these guests, and the formation of a new complex that could also be analyzed by X-ray crystallography. The structure of this complex clearly revealed the CO ligand in the central void of the cage, and more interestingly, a pyramidal (rather than planar) geometry around the metal of the 16-electron Cp′Mn(CO)2 complex. This observation was of particular interest because conventional matrix isolation IR spectroscopy has not been able to determine whether 16-electron Mn complexes are pyrimadal, or whether their d6-orbitals are rehybridized to give a planar geometry. Close inspection of the cavity and its contents suggested that the reason only one of the four complexes lost a CO ligand is that the void in the center of the guest cluster is only large enough to accommodate one CO ligand. In other words, the complex represented an interesting balance between space sufficient for (one) decarbonylation, and sufficient crowding such that the CO group was kinetically trapped; it could neither exit the host–guest complex, nor recombine with the unsaturated complex.
These aforementioned studies might suggest that beyond simple host–guest interactions host 1 is in large part an inert and passive component, but this is not necessarily the case. For example, replacement of the six palladium or platinum metal centers with ruthenium metal ions produces a host that undergoes color change upon guest binding.109 Hypothesized to occur via conformational changes that induce a bathochromic shift in the metal to ligand charge-transfer band, these color changes were shown to occur with the inclusion of 1-adamantol. The palladium-based host 1 has also been shown to be more than just an inert carrier.110 Thus, a combination of fluorescence spectroscopy and picosecond time-resolved measurements indicates that irradiation of the complex formed between 1 and bisanthracene 19 results in efficient energy transfer from the guest to the host. Three decay paths were observed after irradiation at 470 nm; the relaxation of the initially populated excited state guest; guest decay from a long-lived twisted charge transfer state; and decay from a host–guest exiplex. The yield of the energy transfer resulting in the latter was calculated to be ΦET = 0.82.
The above examples have all involved organic guests binding to nano-cage 1. But what is inside the cavity when no such guest is present? The short answer is water. Nature abhors a vacuum. The longer answer is that a combination of low temperature X-ray crystallography and neutron diffraction reveals an adamantoid (H2O)10 cluster reminiscent of naturally occurring IC-type ice.111 Surprisingly, the adamantoid framework of ten waters could still be located at room temperature, suggesting that encapsulation greatly stabilizes the cluster. The crystal structure demonstrated that this stability was not as a result of metal or anion coordination, but rather—as neutron diffraction showed—that D2O⋯π interactions were the principle non-covalent interaction between host and the adamantoid cluster. These unusual interactions were hypothesized to exist because ligand coordination to the metal centers of the host means that each of the four tris-pyridyl ligands is relatively electron deficient. These results, along with computational studies of water in restricted environments (carbon nanotubes,112,113 deep-cavity cavitands,58 as well as artificial concave pockets)57 are beginning to shed light on water at the interface of concave hydrophobic surfaces.
The Fujita group has also extensively examined hosts of the general structure 20 shown in Fig. 1. These assemblies use the same components as those used to form 1, but with the addition of bidentate ‘struts’ that control the ‘height’ of the host. As this in turn controls the type of guests that bind, it is the bidentate struts that are the main variable in these assemblies, whereas most of the systems reported involve the well utilized tridentate triazine ligands (although verdazyl panels have also been investigated), six Pd or Pt ions, and ethylenediamine derivatives as ancillary ligands. That these components are also used in the formation of 1 is reflected in the observation that ensuring quantitative formation of the host type 20 sometimes requires a templating guest, otherwise host 1 and polymeric materials are also formed. All of the guests thus far examined have been planar species that are capable of slipping between the two tridentate panels.114
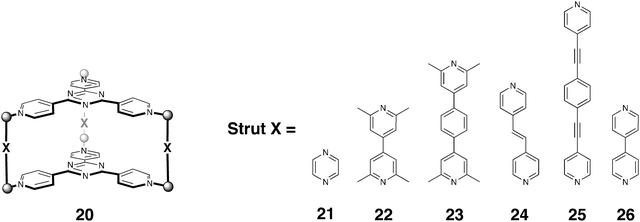 | ||
| Fig. 1 Structures of hosts 21–26. | ||
Simple aromatic guests were among the first guests examined in the assembly of 21. Previous work had shown that with three pyrazine struts the assembly of the host in the presence of electron rich coronene (27) led to an acceptor–donor–acceptor (A–D–A) structure with the donor guest sandwiched between the electron deficient panels of the host.115 Consequently, a natural extension of this work was to examine assembly with a longer bipyridine strut 22.114 With this longer strut the height of the cavity is approximately 7.5 Å, roughly twice the width of a planar aromatic compound. As a result, assembly in the presence of coronene 27 led to the host–guest complex containing two stacked guests. The X-ray structure of this complex clearly showed that its quadruple-stacking structure replete with aromatic stacking interactions between the guests, and between the host and the guests. Taking the notion of strut lengthening one step further, when the longer strut 23 was utilized a remarkable, quintuple-stacked, host–guest complex was formed with two coronenes sandwiching one tridentate triazine panel within the host. The preference for two coronenes and one ligand as guests, and not three coronene molecules as guests, was attributed to the thermodynamic preference for an A–D–A–D–A motif rather than an A–D–D–D–A motif obtained if three coronenes were bound.
In separate work, bipyridine host (22) was also shown by MS to complex two copies of tetrathiafulvalene (TTF).116 When excess TTF was added to a colorless solution of the host the solution turned deep green, a color change that was attributed to a charge transfer complex formed between the electron deficient host and electron rich guests. The strong charge transfer interaction was indicative of the TTF possessing radical cationic character, and this was confirmed by cyclic and square-wave voltammogram studies. These experiments, along with in situ electronic absorption analysis identified a sequence of oxidations in which the 1∶2 host–guest complex first loses an electron to form the mixed valance pair (TTF2)+˙ (152 mV), which in turn can be oxidized to the radical cation dimer (TTF+˙)2 (304 mV). This latter complex is unstable and so the two radical cations are ejected from the host into free solution, where they can subsequently each undergo a further single electron oxidation to form TTF2+. This fascinating redox chemistry is made all the more interesting by being the first instance in which a mixed valance TTF dimer has been described.
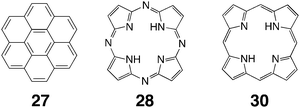
With the idea of developing new fluorescent materials, the encapsulation of azaporphine 28 has also been studied. The electron-deficient nature of this guest allows it to retain its fluorescence when complexed with host 21.117 This is in contrast to the majority of emissive guests which form non-emissive complexes because of charge transfer interactions between the triazine panels of the host and the guest(s). Azaporphine itself is not very water-soluble and is prone to quenching because of aggregation. However the 21–azaporphine complex was shown to have good water solubility and emit red fluorescence. Indeed, its fluorescence properties were shown to be very similar to that observed in chloroform, indicating that that the host itself was not involved in the emission process; there were few, if any, charge-transfer interactions between host and guest. Nevertheless, entrapped azaporphine did not behave the same way as it does in organic solution. For example, when known quenchers DMSO and DMA were added to solutions of the host–guest complex emission was reduced. This is in contrast to free azaporphine in chloroform, which is efficiently quenched by these common solvents. More interestingly, Et3N, which does not quench the fluorescence of azaporphine in chloroform, did quench the fluorescence of the host–guest complex. NMR and UV-vis spectrometry revealed that in the cationic host, the internal protons of the azaporphine are more acidic and that the base is capable of removing one of these protons to form the corresponding (non-emissive) anion. The addition of acid resulted in the reestablishment of fluorescence, and it was shown that the switching off and on of fluorescence was fully reversible via the sequential addition of base and acid.
These examples demonstrate that this family of hosts is capable of orienting guest molecules in a supramolecular stack. Building on this, it is interesting to consider if the guests in such a stack can be organized in a rotation sense around the stack axis. To examine for this possibility the Fujita group formed a 1∶3 host–guest complex between 24 and pyrene-4,5-dione 9.118 This guest has a large dipole moment (6.1–6.7 D) that lies in the plane of the molecule. Thus, when three of these guests are stacked in the host there are considerable dipole–dipole interactions between the guests. The X-ray structure of this complex confirms this. Although there are no specific host–guest interactions that can control how a guest is oriented, the three guests are observed to be rotated 120° relative to each other around the main rotation axis of the host. Adopting these highly specific relative orientations within the stack the complex avoids a large net dipole.
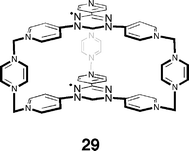
Recently, the aforementioned pyrazine struts have been utilized in the assembly of verdazyl-paneled host 29.119 The solution ESR spectrum of this host encapsulating triphenylene demonstrated that there were magnetic interactions between the two panels even with a bound guest. Furthermore, ESR of a frozen solution of the complex showed fine-structure splitting indicative of magnetic dipole interactions. To observe host–guest spin–spin interactions the host was complexed with Cu(acac)2. The square-planer Cu(II) center of the guest was shown, again using ESR, to readily interact with the host panels.
The stacking of two or more guests that contain metal ions offers many possibilities for studying metal–metal interactions, and in this regard an examination of the complex of porphyrin or porphyrin-like guests is particularly interesting. Fujita has for example examined the binding of porphine 30 to hosts 22 and 23.120 In the smaller host, two porphines are observed to bind to the cavity to form an A–D–D–A stack. The structure of the 1∶2 complex was confirmed by NMR, X-ray crystallography, and UV-vis spectrometry; the latter confirming exciton coupling between co-facial porphine nuclei. When the larger host was examined, rather than three porphine guests being accommodated within the host, two porphines and one triazine ligand were seen (NMR, MS and UV-VIS) to bind to give an overall D–A–D–A–D stacking motif. In this complex the porphine guests, being separated by a triazine ligand, only weakly interacted with each other. The same type of packing motifs were observed when porphine was replaced with Cu(II) porphines. In these complexes, ESR demonstrated that there was spin exchange in the smaller complex with only two bound porphine complexes, but that the triazine ligand between the Cu(II) porphines of the latter complex strongly suppresses any spin–spin exchange interaction.
Continuing this foray into the development of new molecular-based materials, the Fujita group have also demonstrated the formation of linear arrays of Cu(II)–M–Cu(II) atoms (M = Cu(II), Pd(II) and Co(II)) using host 23 and porphyrin-like guests.121 To do this required some ingenuity to circumvent the issue that three porphines had not been observed to stack in these hosts. The answer to induce this motif was to switch the porphines for azaporphines 28. The latter has a polarizable periphery that was expected to induce quadrupole interactions when stacked with other guests, and that this was the case was demonstrated by the formation of the 1∶3 complex between host 23 and azaporphine 28 (NMR, MS and X-ray crystallography). The replacement of azaporphine with Cu(II) azaporphine leads to a similar complex, and exciton coupling between the three stacked guests was evident from UV-vis spectrometry. Furthermore, ESR spectrometry confirmed that the three copper complexes couple ferromagnetically to give the quartet state within the host. Expanding on these successes, the hetero-guest complex formed between host 23, porphine, and azaporphine was also demonstrated. By mixing the components in the correct stoichiometry, the 1∶1∶2 host–porphine–azaporphine complex with the overall A–D–A–D–A motif could be formed in 31% yield. This advance opened the way to the formation of similar complexes containing two different metal ions. More specifically, it allowed the assembly of complexes containing Cu(II)–Pd(II)–Cu(II) and Cu(II)–Co(II)–Cu(II) metal arrays, as well as complexes in which some of the guests were metallated and some were not. Again, ESR confirmed the formation of these different species.
Stacking guests within host 22 has also allowed metal–metal interactions to be observed between guests.122 For example, both Pt(acac)2 and the corresponding Pd(II) complex form 1∶2 host–guest complexes with 22. Focusing on the former, this orange complex was shown to possess a 1∶2 stoichiometry by a combination of NMR, UV-vis, and MS. UV-vis was particularly instructive; the spectrum showed an adsorption band around 500 nm indicative of d–d interactions between Pt(II) complexes. This was confirmed by an X-ray crystal structure of the complex, which showed that the two guests stacked in such a manner as to have their Pt(II) centers lying within the common C2 rotation axis of the guests and separated by 3.32 Å. The corresponding Cu(acac)2 complex also formed a 1∶2 host–guest complex, the d–d interaction of which was noted by ESR spectrometry. In this case however the ESR data suggested that the 1∶1 host–guest complex was also formed as a minor component. In all three cases, the formation of discrete d–d interactions between guests suggests interesting opportunities for new materials possessing unique optical, electroconductive and magnetic properties.
Spin crossover, the phenomenon in which a transition metal ion switches between high and low spin states as a result of an external stimuli, is frequently observed in Fe(II), Fe(III) and Co(II) complexes. Fujita et al. recently demonstrated that Ni(II) and Co(II) complexes contained within hosts 21, 22 and 23 also exhibit this interesting phenomenon.123 For example, when low spin Ni(II)(acen, acen = N,N′-ethylenebis(acetylacetoneiminato)) is encapsulated within 21, the complex was observed to be in the high spin state. The first indication of this was that mixing the colorless host and the red Ni(II) complex gave a dark green complex. NMR also supported this notion, but it was SQUID measurements that demonstrated that the complex had paramagnetic behavior. When the same guest was complexed to larger 22, two copies of the guest were observed to bind to the host (NMR, X-ray), and again the complex was a dark green color rather than red. SQUID again confirmed the high spin state of the complex. Importantly, X-ray crystallography revealed that the two guests did not undergo any geometry changes upon complexation. Normally, spin crossover in Ni(II) complexes arise through changes in geometry and/or coordination number. In the case at hand however, it is host–guest and guest–guest interactions that induce crossover by perturbing the dz2 orbitals of the metal. Spin crossover was also observed when Co(II)(tap, tap = tertaazaporphinato) was encapsulated. With this guest however, no spin crossover was observed when two copies of this guest were encapsulated within 22. Rather, it was host 23, which accommodated two Co(II)(tap) complexes and one coronene (27), where this phenomenon was observed. The intensity of the ESR signal from this complex as a function of temperature revealed thermal promotion from the low (S = 1/2) to the high (S = 3/2) spin state, whilst the 2D-ESTN (electron spin transient nutation) spectrum revealed that the complex is best described as a mixture of the low and high spin states. Again, host–guest and guest–guest interactions were attributed to the observed crossover.
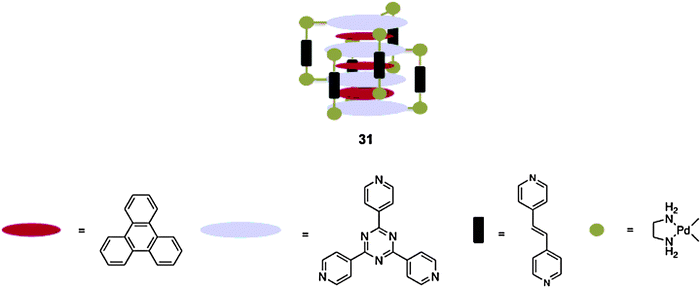
The Fujita group has also demonstrated that these types of host can assemble in an interpenetrating manner to form assemblies composed of up to nine planar aromatic rings.80 For example, using linker 24 it is possible to assemble twenty-five components to form complex 31 containing three triphenylene guests sandwiched between four triazine panels. Both NMR and MS confirmed the formation of 31, and although this complex was not amenable to single crystal X-ray crystallography, its counterpart with N,N,N,′N′-tetramethylethylenediamine ancillary ligands was. The resulting structure demonstrates a 2.1 nm high septet aromatic stacking structure with efficiently overlapping aromatic rings with inter-plane distances of 3.3 Å. The presence of the guest was found to be essential for assembly. In the absence of triphenylene other assemblies, such as 1 were noted to form. Building on this advance, the Fujita group subsequently assembled host–guest complexes comprised of eight and nine stacked aromatic systems. The latter complex was formed using long strutted host 25, and was composed of four (triazine) acceptor panels from two interpenetrating hosts, and five pyrene (donor) guests to give an overall A–D–A–D–D–D–A–D–A motif.
Finishing our discussion on this family of hosts on a biochemical note, it has been noted that in water, mono-, di-, and tri-nucleotides do not undergo duplex formation because hydrogen bonding between the nucleotide and water is far more prevalent than hydrogen bonding between nucleotides. However in the presence of host 21, 5′-adenosine monophosphate and 5′-uridine monophosphate were shown to form a 1∶2 host–guest complex.124 This complex could not be induced to grow crystals suitable for X-ray crystallography, however the corresponding host with (S,S)-1,2-diaminocyclohexane ancillary ligands proved more amenable. The structure revealed that the purine and pyrimidine bases were bound within the host, whilst the sugar and phosphate moieties remained outside the cavity. Furthermore, the X-ray structure demonstrated that shielded from the bulk water, the bases formed an anti-Hoogsteen-type base pair interaction. Hence, both hydrogen bonding between the guests and aromatic stacking interactions between the host and guests contribute to the stability of the complex. A similar result was obtained with larger host 26 and the di-nucleotide thymidylyl-(3′-5′)-2′-deoxyadenosine (A–T). A combination of 1H NMR and NOESY experiments strongly suggested that when combined together these molecules formed a 1∶2 host–guest complex, and this was confirmed when an X-ray crystal structure of the complex was solved. Again, as was the case with the complexation of the mono-nucleotides, the hydrogen bonds between the A–T pairs were of the anti-Hoogsteen-type.
Continuing on the genetic material theme, a variation on these types of host was recently realized by Therrien et al.125 Host 33 (Scheme 3), assembled from the triazine ligand and bis-ruthenium complex 32, was found to bind Pd(acac)2 and Pt(acac)2 (acac = acetylacetone). Platinum complexes, and to a lesser extend palladium and ruthenium complexes, have a strong record of anti-cancer properties. Hence, Therrien et al. investigated the cytotoxicity of the free guests, the free host, and the corresponding complexes against an ovarian cancer cell line. As expected, the water insoluble free complexes did not display any activity. In contrast, the free host did possess some activity (IC50 = 23 μM), whilst the host–guest complexes possessed stronger activity (IC50 = 12 and 1 μM for the Pt and Pd complexes respectively). VT NMR demonstrated that the palladium guest is less tightly bound in this host, and this is one possibility as to why the palladium complex was more active. However, further studies are underway to ascertain why this host–guest complex possesses such high cytotoxicity.
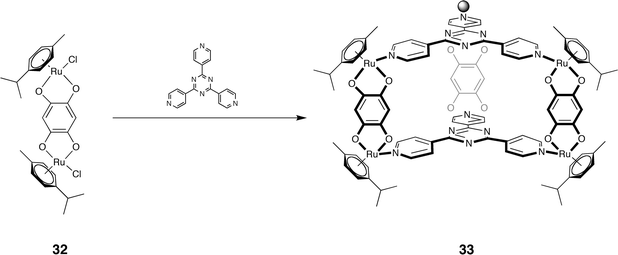 | ||
| Scheme 3 Self-assembly of host 33 from strut 32 and triazine ligand. | ||
In each of the two aforementioned families of hosts, the three pyridine nitrogen atoms are para with respect to the central triazine ring. Fujita's group have also been examining the assembly of another family of tridentate triazine ligands in which the pyridine nitrogens are meta to the triazine ring. In such cases, square bowl-shaped host 34 is formed by the assembly of four copies of the ligand, and six palladium ions (and their ancillary ligands). The cavity of 34 has been shown to bind a range of different guests, and in doing so a number of interesting properties have been identified. For example, the host preferentially encapsulates the homo-chiral pair of 1,1′-bi-2-naphthol (BINOL) 35 and is therefore capable of bringing about enantiomeric enrichment.126,127 Thus, when a two-phase system was established with 34 in water and the (S)-BIONOL (50% ee) in hexane, the homo-chiral R–S pair bound preferentially to the host leaving the hexane phase containing S-diol in 81% ee. The importance of the OH groups to enrichment was emphasized by the fact that there was no enrichment when they were replaced by methyl groups, and weaker enrichment (50% ee up to 71% ee) when amino groups were present. These results were interpreted as the phenol groups being essential for stabilizing the complex by being positioned at the portal of the cavity, i.e., at the water interface. An extension of this work was to examine for the possibility of enriching a racemate compound by co-complexation with another chiral molecule. In this experiment, the expectation was that racemate octahydro-2,2′-binaphthol 36 could be enriched using host 34 and enantiopure (R)-BINOL via the selective complexation of either the (R) or the (S)-octahydro-2,2′-binaphthol with the (R)-BINOL. In fact, NMR strongly suggested that the complex formed in this experiment was a capsular complex (built from two copies of the host) containing two copies of the (R)-BINOL and one of (S)-octahydro-2,2′-binaphthol.
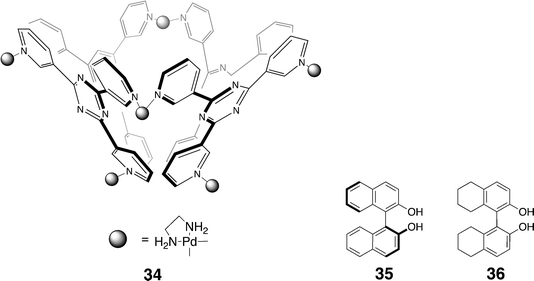
Host 34 has also been utilized for peptide recognition.126,127 Thus, a combination of the host and the 9-mer peptide Ac-Trp-Ala-Glu-Ala-Ala-Ala-Glu-Ala-Trp-NH2 led to complex NMR data that were difficult to interpret. However, the addition of a trace of chloroform led to interpretable spectra that in combination revealed a helical structured peptide with its two terminal tryptophan residues bound deeply into the pocket of the host. Modeling suggested that in a helical conformation Ala5 was also buried into the hydrophobic pocket, whilst the Glu3 and Glu7 residues lay on the solvent exposed surface of the helix.126 In contrast, a 2∶1 host–guest complex was observed to form when the 9-mer bound to 34 in the presence of NaNO3.128 In this instance the higher ionic strength of the medium led to the capping of both ends of the helical peptide by the host.
The host–guest chemistry from the Raymond group has focused on a series of tetrahedral assemblies formed from the combination of four metals and six catecholamide ligands. The resulting chiral assembly 37 (T symmetry) possesses a tetrahedrally shaped hydrophobic pocket for guest binding. Varying the metal used in these assemblies, e.g., Ga(III), Al(III), In(III), Fe(III), Ti(IV), or Ge(IV) controls their rate of formation and disassembly, but in all cases the use of the bis-catechols as ligands gives the host an overall negative charge. Hence, the preference for these hosts is for hydrophobic and positively charged guests, although recently neutral hydrophobic guests have been observed to bind (vide infra). The hosts themselves are chiral by virtue of a chiral twist to the three ligating groups around each metal center, and only homochiral assemblies are ever observed (ΔΔΔΔ and ΛΛΛΛ). During the assembly, once one twist sense has been set at a metal center, this is mechanically transmitted to the other metal centers in the assembling product to ultimately yield a racemate of the two enantiomers. If desired, these racemates can be separated. Thus, by way of example, the two enantiomers of the Ga(III), Al(III) and Fe(III) assemblies can be readily resolved using chiral guest (S)-N-methyl-nicotinium; when the assembly is carried out in methanol and in the presence of this guest, the host–guest complex with the ΔΔΔΔ host selectively precipitates out of solution.129 Interestingly, the isolated complexes could then undergo guest exchange with an achiral but strongly associating guest such as NEt4+ to yield stable enantiomeric complexes. The fact that the enantiomers were stable even when containing the achiral guest allowed an examination of resolution in the presence of both more strongly binding NEt4+ and (S)-N-methyl-nicotinium. This again led to successful resolution, demonstrating that enantiomer separation did not require encapsulation of the chiral guest but instead was dependent on the interaction between the (S)-N-methyl-nicotinium and the outside of the container. CD spectroscopy demonstrated that the racemization rate of the chiral hosts was dependent on the nature of the guest inside the host, as well as the type of metal ion used in the assembly.
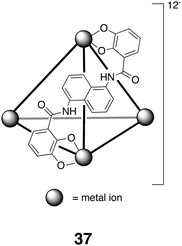
Returning to the binding preferences of these hosts, the Raymond group have observed that a wide range of saturated compounds, including n-alkanes, cyclic alkanes, polycyclics,130 and enantiopure diterpenoids131 form stable complexes with the gallium host [Ga4L6]12−37. The largest guests observed to bind were cyclodecane, adamantane, and decalin, whilst the complexation of the diterpenoids to the racemate host led to modest diastereomeric recognition. A wide range of benzene derivatives also form complexes with 37,131 and one interesting trend observed with this series of guests is that ortho-derivatives formed 1∶2 host–guest complexes, whilst meta- and para-isomers formed 1∶1 complexes.
These tetrahedral hosts have fewer and smaller holes in their shells than the aforementioned Fujita hosts, which raises the issue as to how guests get into and out of these more integral assemblies. The initial assumption was that one catechol–metal ligation must break in order that the ligand—pivoting on its second catechol–metal linkage—could swing out of the way and allow the guest to escape. However, more recent studies have demonstrated that this is not in fact the case.132 The first piece of evidence that this is not so was a comparison between the guest exchange rates of the assemblies constructed from Ga(III), Ti(IV), or Ge(IV). Because of their 4+ charges, the latter two ions exchange much more slowly that Ga(III), and yet the exchange of PEt4+ for NEt4+ into and out of the container was noted to be independent of the nature of the metal center. Instead, computational studies revealed that guests enters or egresses the host via one of the four C3 apertures at the center of each face of the tetrahedron. For this to occur, the host must deform somewhat in order for the aperture to dilate, with the extent of deformation dependent on the size of the guest.133 Thus, the relatively small NEt4+ must force a 7.6 Å opening, whilst the largest guest to be observed to bind, CoCp*2+ (Cp* = pentamethylcyclopentadenyl), requires a 9.8 Å opening and correspondingly had an activation energy twice that of NEt4+. Building on this work, more recent diffusion NMR studies of the Ga(III) host 37 reveal that latent guests such as NEt4+ associate with the exterior of the capsule via a combination of Coulombic, cation–π and van der Waals interactions.134 These results suggest that in general, before a guest is internalized by the host, there is an initial weak binding step in which the guest associates with the outside of the host.
As just mentioned, the preference of this type of host is positively charged guests, and this includes examples of species that are not normally stable in water. For example, iminium ions only have a transient existence in neutral of basic water, but when host 37 was added to an aqueous mixture of ketone and an imine the corresponding iminium ion product was observed inside the host.135 Thus, using pyrrolidine as amine, ketones as small as acetone and as large as 2-nonanone formed the corresponding iminium ion. Many of these iminium products were noted to be too large to fit inside the host in their extended conformations, and so it is likely that the n-alkyl chain of the larger guests is coiled up inside the host. Competition experiments in which different amines and one ketone (or different ketones and one amine) were placed in solution with the host revealed the best iminium guests for the host.
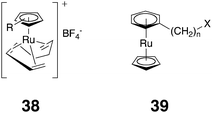
Building on the idea of encapsulating water-sensitive guests, the Raymond group also examined the C–C bond-forming catalyst CpRuCl(cod) (Cp = cylopentadiene, cod = 1,5-cyclooctadiene) as a potential guest inside gallium host 37.136 It was anticipated that the resulting aquo complex CpRu(cod)(H2O) would be encapsulated, but when the precursor was added to a solution of the host the NMR spectrum of the resulting complex showed a doubling of all the expected signals. Being chiral, host 37 exists as a racemate of homochiral stereoisomers (ΔΔΔΔ and ΛΛΛΛ), and so this complicated NMR could potentially be explained by the inclusion of a chiral guest and the formation of diastereomeric complexes. But which chiral guest? An extensive study revealed that encapsulation led in fact to a complex between 37 and chiral CpRu(cis-1,3,7-octatriene) (38). This complex was stable in aqueous solution, even though the free guest in dichloromethane reacted rapidly when water was added. Although water did not apparently enter the cavity to react with the encapsulated guest, the guest could however react with carbon monoxide; the addition of which led to the slow (five days) formation of the corresponding achiral CpR(cod)(CO) complex.
What are the effects of encapsulation within these negatively charged hosts? A recent study on the binding of basic amines, phosphines, and (super basic) azaphosphatranes (31P[1H] NMR, MS) revealed that the internalized guests are in the protonated state, and that they remained so even above solution pH values greater than the pKa of each guest examined.137 In other words, electrostatic interactions between host and guests caused shifts in the pKa of the guest. Computational studies further revealed that these protonated guests are not stabilized by interactions with the catecholate oxygens at the apexes of the host. In order to prove that the amine/ammonium guests were exchanging and that the host was not acting as a kinetic trap, a series of selective inversion recovery NMR experiments were carried out. These experiments confirmed this, demonstrated that guest exchange was fast on the NMR timescale, and also confirmed that the rate-limiting barrier for guest ejection was primarily the size of the guest and not its thermodynamic binding affinity. Finally, to determine the effective basicity of the encapsulated guests, thermodynamic cycles were constructed involving each putative protonated and unprotonated guest and their corresponding complexes. These showed that pKa shits of up to 4.5 units were occurring as a result of encapsulation. Such shifts are close to the largest pKa shifts of functional groups observed within the active sites of enzymes when the shift is caused by electrostatic interactions. In a related, and more recent paper, the Raymond group has measured the hydrogen bond breaking and subsequent nitrogen inversion/rotation of protonated diamine guests.138 A combination of 1H NMR experiments (in methanol to give a broader range of temperature for analysis) and computational studies revealed that the nitrogen inversion/rotation process occurs inside the host rather than via a guest ejecting, nitrogen inversion/rotation, and re-encapsulation process.
Continuing the theme of how guest properties are changed by encapsulation, the Raymond group has also noted how encapsulation influences the rotation barrier of amide bonds.139 By examining amide bond rotations of nine amide guests within Ga(III) complex 37, and comparing this data to that obtained when the same guests were examined in free water, it was determined that the hydrophobic cavity of 37 does stabilize the (hydrophobic) transition state of rotation. However, in comparison to results obtained in toluene, the barrier to rotation within the host was higher in all instances. 13C[1H] NMR studies revealed that the probable reason for this difference is that within the host the guest can access a stabilized twisted conformation.
The thermodynamic driving forces that lead to guest complexation have been examined using the Ga(III) host 37.140 In this study, Raymond and co-workers examined the complexation of a series of mono-cationic iridium complexes of general structure Cp*(PMe3)Ir(CO)(R)+ where R was either a small alkyl group or a small perfluoro-alkyl group. Using 1H NMR the complexation of these guests to the host was examined over a range of temperatures to provide the free energy, enthalpy and entropy changes upon guest binding. For binding in water—complexations in methanol and DMSO were also carried out—the authors noted a strong enthalpy–entropy compensation; one that could not be attributed to any statistical correlation artifact inherent in the van't Hoff analysis utilized. The dramatic differences observed between the results from water and methanol suggested that solvent reorganization upon guest complexation, rather than specific host–guest interactions, played the dominant role in these binding events; desolvation of the guest and an emptying of the cavity of the host lead to a favorable entropy change.
Guests of a size such that there is a relatively small energy difference between a partially encapsulated guest binding motif and a fully encapsulated motif have been examined with Ga(III) host 37.141 For example, in the three ruthenium complexes 39 (n = 4, 6 or 8), the smallest guest is completely encapsulated within the host whereas the largest guest is too large so that the alkyl chain must protrude through one of the four apertures in the side of the container. Guest 39 (n = 6) represents the grey area between these two extremes. Variable temperature 1H NMR experiments clearly show that the guest exits in two states, one in which an extended chain protrudes through one of the apertures, and one in which the chain of the guest is retracted and the guest is fully encapsulated. Furthermore, a comparison to the NMR spectra of the complexes with the smallest and largest guests suggested that the methyl group at the end of the chain resides inside the host 24% of the time, i.e., that the complex with the extended alkyl chain is 0.6 kcal mol−1 lower in energy than the fully encapsulated state. As a result of this low energy difference, and a low kinetic barrier between the different entities, the complex exists as a dynamic ensemble of four degenerate C3 symmetric species with the chain extending through an aperture, and the T symmetric fully encapsulated complex. The complex is therefore analogous to a common example of second-order Jahn–Teller distortion, i.e., the gas phase structure of XeF6. In this molecule, the octahedral structure is desymmetrized by the sterically active lone pair on the Xe that must reside in one of the eight degenerate faces of the molecule.
A large portion of the recent supramolecular research from the Raymond group has focused on host 37 as a nano-scale (yoctolitre) reaction vessel, and in this regard a number of non-catalytic and catalytic processes have been reported. We begin with the former, and a report on the reactivity of a series of encapsulated mono-cationic half-sandwich iridium guests such as 40 (Scheme 4).142 The association constants for these chiral complexes—which as the racemate host and guest were employed were weighed averages of the diastereomers formed—were measured to be in the order of 103 M−1, and noted to be largely entropically driven. An examination of the reactivity of this complex revealed highly specific size and shape selectivities in the C–H bond activations of aldehydes and ethers (Scheme 4); a product of the limited space available for the substrate to cohabit the host along with the half-sandwich complex. A detailed kinetic analysis further revealed that dissociation of the iridium complex is slower than C–H bond activation, and occurred via a two step process in which the guest exits the cavity but remains associated with (the outside of) the host, before fully dissociating from the host.
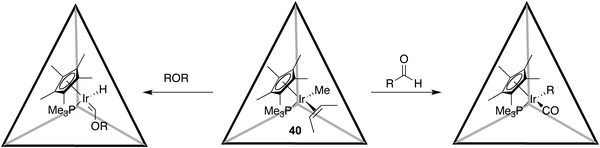
As just mentioned, host 37 has been observed to perform a range of catalytic processes. For example, the Raymond group has investigated the ability of host 37 to catalyze the 3-aza Cope rearrangement (Scheme 5); a reaction chosen because product inhibition is avoided by virtue of the cationic enammonium salt substrate binding more strongly than the neutral product.143 A range of enammonium salts were observed to bind to the host, and acceleration of the reaction by encapsulation was determined to range from 5–854. Within this range, a general observation was that mid-sized optimally fitting substrates reacted faster than the smaller or larger guests. Experiments revealed that these accelerations did not arise through the hydrophobicity of the cavity, or the anionic nature of the host. Rather, the entropy of activation for the rearrangement was noted to be considerably lower in the cavity of the host; NOESY NMR studies revealed that the host preorganized each guest into chair-like ground-state conformations more suited for rearrangement. Kinetic studies also revealed that, as expected, the host could accelerate the 3-aza Cope rearrangement catalytically, and that the strongly binding NEt4+ effectively inhibited catalysis. Further detailed kinetic analysis revealed an overall mechanism in which the enammonium ion rearranges inside the host, before the resulting iminium ion reversibly dissociated from the interior to the exterior of the container. Under basic conditions, hydroxide cannot nucleophilically attack the iminium cation product whilst it is associated with the large, negatively charge host. Hence hydrolysis can only occur once the iminium ion dissociates fully from the host. However, at neutral pH the weaker nucleophile water can react with the iminium ion whilst it is associated with the host.
 | ||
| Scheme 5 The 3-aza Cope rearrangement. | ||
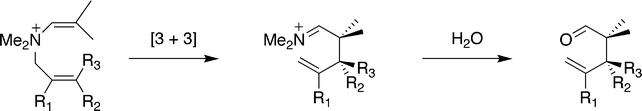
More recently, catalysis of the aza-Cope rearrangement of propargyl enammonium ions (Scheme 6) has also been examined.144 These derivatives undergo rearrangement more slowly and therefore represent a more challenging target. In this study a range of guests from as small as 41 to 42 were examined. Only the guests with R = t-Bu, n-pentyl or Ph were observed not to bind. Interestingly, the other butyl isomers (n-butyl, 2-butyl and i-butyl) did form complexes with the host. A comparison of the rearrangement of these propargyl enammonium ions in free solution and encapsulated within the host revealed rate accelerations of up to 184. Again, mid-sized, optimally fitting guests gave the greatest rate accelerations. Furthermore, an extensive kinetic analysis revealed that the catalytic process followed the Michaelis–Menton model of enzyme kinetics. It also revealed the activation parameters for the uncatalyzed and catalyzed reactions, and that the rate accelerations for the catalyzed processes were due to a more positive ΔS‡ arising through a preferred C-shaped guest binding motif.
 | ||
| Scheme 6 The 3-aza Cope rearrangement of propargyl enammoniums. | ||
Encapsulated rhodium complexes of the general formula [(P–P)Rh(diene)][BF4], where P–P = 2 × PR3 or 1,2-bis(dimethylphosphino)ethane, and diene = 1,5-cyclooctadiene (COD) or norbornadiene (NBD), have also been encapsulated with host 37.66 These form relatively weak complexes (Ka ≈ 102 M−1), which upon treatment with hydrogen gas, formed reactive (P–P)Rh(OD2)2 complexes that catalyzed the isomerization of allylic alcohols. In the case of the (PMe3)2Rh(OD2)2 complex, the host–guest complex was shown to be thermodynamically unstable; the guest is sufficiently lipophilic such that it prefers bulk water over the host. However, the kinetics of dissociation were sufficiently slow such that the allyl alcohol isomerization of a series of guests could be examined. As expected, high shape and size selectivity were observed when the reactivity of the catalysis inside the host were compared to those in free solution.
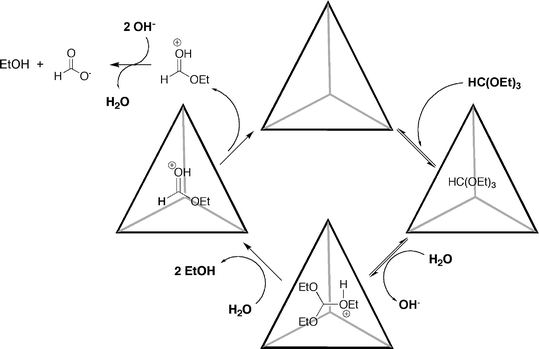
Building on their understanding of how host 37 can shift the pKa of an encapsulated guest,137 the Raymond group recently reported an example of acid catalysis in basic solution.67 The group investigated the hydrolysis of formate esters HC(OR)3, where R = Me, Et, Pr, i-Pr, n-Bu, i-Bu and n-pentyl. Normally stable in neutral and basic solution, with the exception of the n-pentyl derivative all of these substrates were found to be rapidly hydrolyzed at pH 11 in the presence of a catalytic amount of host Ga(III) 37. As anticipated, the addition of strongly binding NEt4+ completely inhibited these reactions. Probing the reaction kinetics of the hydrolysis of triethyl orthoformate gave a mechanism (Scheme 7) in which the orthoformate first entered the cavity of the host. This is subsequently protonated to give a thermodynamically more stable complex, but one that is kinetically unstable and reacts when water enters the cavity. The resulting (protonated) ester guest then egresses the cavity and is ultimately hydrolyzed to formate ion. This mechanism meant that the reactions could be fitted to Michaelis–Menton kinetics, and correspondingly it was possible to determine the rate acceleration of the reaction in the presence of the host. Thus, the kcat/kuncat for the ethyl and isopropyl orthoformates were calculated to be 650 and 890 respectively. In addition, it was noted that not only did the isopropyl guest bind slightly more strongly than the ethyl derivative, but that it was also hydrolyzed more efficiently. Recently, a follow-on full paper on this topic has provided a better understanding of this catalysis.69 In binding experiments with the aforementioned ortho esters (and additionally HC(OPh)3), the six smallest guests resulted in significant shifts of the host 1H NMR signals indicating fast guest exchange, whilst the two largest guests (R = n-pentyl and Ph) induced only slight broadening of the signals from the host. Furthermore, the shifts in the signals of the host were not observed when strongly binding NEt4+ was present, indicating that those guests that appeared to form complexes were not associating with the outside of the host. A further analysis of the Michaelis–Menton like kinetics for these catalytic hydrolyses revealed that the addition of organic solvents greatly reduced the rate of product formation; an observation that is consistent with the idea that the hydrophobic effect is an important factor in substrate encapsulation. Although under normal conditions it was not possible to observe the encapsulated substrate, when a near saturated solution of host 37 was treated with a stoichiometric amount of ethyl orthoformate, bound guest signals in the 1H NMR spectrum were observed. This allowed the probing of the different encapsulated species using 13C[1H] chemical shift and 1JCH coupling constant data with a suitable labeled orthoformate (H13C(OEt)3). Combined with calculated shift and coupling constant data, these NMR experiments revealed that the complex of the neutral orthoformate guest was the resting state in the catalytic cycle. Finally, an expanded analysis of the kinetics of hydrolysis of four substrates (HC(OR)3, where R = Me, Et, Pr, i-Pr) revealed an apparent A-SE2 mechanism of hydrolysis (rather than the A-1 mechanism that generally occurs in free solution), and that the n-propyl guest underwent the most accelerated reaction (kcat/kuncat = 3900).
 | ||
| Scheme 7 The catalytic cycle for orthoformate ester hydrolysis within host 37. | ||
Extending the idea of stabilizing positively charged reaction intermediates whose formation is the rate determining step in a conversion, the Raymond group have also determined that Ga(III) host 37 can catalyze the hydrolysis of acetals and ketals under basic conditions.68 As protecting groups, these derivatives are of course an integral part of synthetic chemistry, and normally their removal requires acidic conditions to efficiently bring about their protonation as a first step towards hydrolysis. However, in the presence of 5 mole% host hydrolysis at pH 10 was observed for a variety of substrates examined. The results for fifteen substrates painted an all or nothing picture. 1H NMR revealed that most guests examined underwent complete hydrolysis, but guests that were too large for the cavity—specifically the dimethyl acetals of undec-2-one and nonanal—underwent no reaction. Expanding on these early results, the Raymond group further established that acetal complexation was driven primarily by the hydrophobic effect, and that rate accelerations as high as 103 were possible.145 Furthermore, the determination of activation parameters and an analysis of isotope effects pointed to a change in the A-1 mechanism usually seen in solution to an A-2 mechanism.
If assembly via metal coordination allows the straightforward synthesis of large hosts, then a combination of dynamic covalent chemistry146 and metal coordination—an example of a subcomponent self-assembly approach147—has the potential to make such syntheses even more straightforward. By way of example, Nitschke and colleagues recently reported on the formation and properties of anionic tetrahedral host 43.148 Constructed from commercially available and inexpensive components, the host was shown to bind the hydrophobic guests cyclopentane and cyclohexane. More polar guests such as NMe4+ (and other small tetraalkylammonium species) and t-BuOH, despite being of ideal size and shape, were not observed to complex with the host. The lack of binding of the cationic salts is an interesting contrast to the binding ability of host 37 from the Raymond group. Guest exchange was also noted to be much slower than host 37. However, the host could be disassembled irreversibly or reversibly by the addition of tris(2-ethylamino)amine or p-toluenesulfonic acid respectively. In the latter case, addition of base reformed the host.
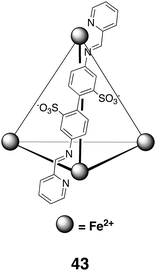
Building on the pioneering work of Cram et al. and the stabilization of cyclobutadiene,6 host 43 has also been shown to bind and stabilize white phosphorus (P4).149 A combination of the weak bonds of tetrahedral P4 and the very strong P–O bonds upon oxidation is behind the pyrophoric nature of white phosphorus. However, within the confines of host 43 (1H, 31P NMR and X-ray) white phosphorus is air stable, both in aqueous solution and in the solid state. In a biphasic mixture of water and benzene or cyclohexane, the P4 guest was displaced by both of these organic solvents. This did not occur in n-heptane, illustrating that guest exchange required both good solubility of the white phosphorus in the organic solvent, and strong binding of the solvent in the tetrahedral host. When guest exchange did occur and the white phosphorus was solubilized in the organic phase, the phosphorus quickly underwent reaction to form phosphoric acid when the solution was exposed to air.
Systems utilizing the hydrophobic effect
Although to date less utilized, the hydrophobic effect can also be used to drive the assembly of supramolecular hosts. Assembly is driven by the desolvation of hydrophobic surfaces built into the subunits that come together to form the supramolecular host. Inevitably, the synthesis of such subunits is more intensive than is the case when metal coordination is used to drive assembly, and the task of subunit design is made all the more complicated because our understanding of the hydrophobic effect is not yet fully developed. Consequently, to the authors' knowledge only one family of self-assembling host driven by the hydrophobic effect has been reported in the literature. These water-soluble deep-cavity cavitands of general structure shown in Fig. 2 were first reported by Gibb and Gibb in 2004,94 and undergo dimerization in the presence of a wide range of guests.150,151 Within this family, host 44—the so-called “octaacid”—has proven to be particularly useful with at least six research groups independently exploring the unusual properties of the nano-capsule that results from its dimerization.33,152–160 Although the octaacid 44 is the most studied, it is relatively straightforward to change the outer coating of the cavitand. To take a structurally extreme example, Gibb and Grayson have reported on the synthesis of G3 dendritic cavitand 45, the first neutral, water-soluble, self-assembling host.161–163 The coating of this cavitand was efficiently built up in a divergent synthesis using high yielding steps and no chromatography. An analysis of the solubility of the first and second-generation derivatives, those with sixteen and thirty-two hydroxy groups respectively, revealed that sixty-four hydroxy groups were necessary for solubility in pure water. That caveat noted, with the exception of self-inclusion of the long coating chains, the properties of the dendritic host are qualitatively very similar to octaacid 44 (vide infra).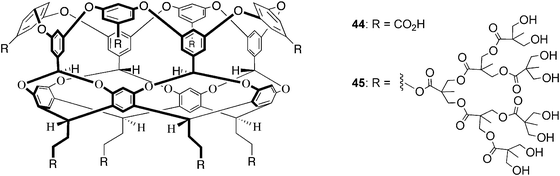
An ideal prelude to discussing the hosting properties of 44 was recently reported by Rick et al., who carried out in silico studies to evaluate water binding to the empty host.58 These dynamics studies revealed that the cavity of the host can contain between 0–7 water molecules (4.4 on average), and that they possess less hydrogen bonds than the bulk. In combination with the restricting concave surface, this means that the solvating waters possess slower translational motion and faster orientational motion. Approximately 5 kcal mol−1 is gained by solvating the cavity, a process that is exothermic and entropically penalized. In general terms, the water molecules in the cavity can be described in terms of 3 layers. Thus, a lone water molecule constitutes the lowest layer occupying the narrowest base of the cavity and has an average of 1.3 hydrogen bonds (in which case it predominantly acts as a hydrogen bond acceptor). Those waters in the middle layer have on average 2.6 hydrogen bonds, whilst those in the upper layer are involved in an average 3.3 hydrogen bonds. Bulk water has on average 3.6 hydrogen bonds. Interestingly, water within the cavity is stabilized by its interactions with the bulk. Thus, when a small hydrophobic guest such as ethane is placed within 4 Å of the cavity portal, the cavity is dewetted and the probability of forming an empty cavity increases to near unity. This unusual exchange mechanism, a “triggered dissociative” process, lies somewhat between the associative mechanism typically seen with cyclodextrins in which a new guest coming in one portal of the binding pocket pushes the resident guest out the other, and a dissociative mechanism where the resident guest(s) must exit before a new guest can enter.
The maximal capacity of the nano-capsule formed by 44 was indicated in the initial report of this host and how steroids could be trapped within its interior.94 Subsequent to this Gibb and Gibb observed that the same capsule also formed in the presence of propane and butane; the former constituting the lower limit of occupancy within the dimeric host.164 Even in these cases 1H NMR of the resulting 2∶2 host–guest complex revealed guest exchange to be slow on the NMR timescale unless there was an excess of gas. Indeed, the affinity of propane and butane for the capsule was such that a host solution could extract the gas directly from the gas phase and into the aqueous phase. The quaternary complex of propane is an interesting entity. If viewed in terms of packing coefficient, the cavity is only 28% full. Alternatively, if the inappropriate assumption is made that the contents of the capsule is an ideal gas, the pressure of propane in the quaternary complex is in excess of 100 atmospheres. Finally, it is worth noting that although propane has a high affinity for the capsule, its binding constant is one order of magnitude weaker than butane. As a result, the capsule formed by 44 can affect the separation of the two gases by selectively extracting butane from a propane/butane mixture in contact with the aqueous phase.
Between the extreme of steroids and hydrocarbon gases, n-alkanes are also seen to bind to 44 and trigger dimerization.165 Thus, the alkanes heptadecane through nonane were shown (1H NMR) to form kinetically stable 2∶1 host–guest complexes. In these entities, the methyl 1H NMR signal of the guest was the most shifted, indicating that each was located deep in the polar region of a hemisphere. That said, guests longer than decane cannot adopt a fully extended conformation in the nano-capsule, and therefore must fold up in some manner to be accommodated. For example, NOESY NMR revealed long range interactions down the chain length of dodecane, indicating that it adopts a helical conformation within the host. Octane as a guest represented a change in binding mode; it formed a mixture of ternary and quaternary complexes, presumably because one octane guest results in a rather low occupancy, whereas two represents a packing coefficient that is on the high side. In contrast, with the guests heptane through pentane only quaternary complexes were observed. Most recently,156 Ramamurthy et al. have also examined how guest size affects the assembly product, have reconfirmed that the binding of amphiphiles such as adamantane carboxylic acid form 1∶1 complexes,166 and have further reconfirmed that the host tends to aggregate at higher concentrations.151
In related work examining how guest polarity affected capsule formation, the binding of a series of approximately isosteric guests (46–51) was examined.167 In this series, guest 50 proved to signify the boundary between assembly to form 2∶1 complexes and simple 1∶1 complexation. Thus, guests 46–49 formed kinetically stable capsules, guest 50 formed a capsule that was unstable on the NMR timescale, and guest 51 formed a simple 1∶1 complex. The binding constant of this most hydrophilic guest was still 2500 M−1, suggesting perhaps that it is more appropriate to discuss polyethylene glycol derivatives as hydrophobic rather than hydrophilic.
Kaifer has examined how encapsulation with the nano-capsule formed by 44 influences the electrochemical properties of a guest. For example, 1H NMR and NMR diffusion studies revealed that one equivalent of ferrocene is bound within the nano-capsule, and under these conditions instead of demonstrating its normally fast one-electron oxidation, ferrocene is voltammetrically silent.168 The detection of faradaic currents in the presence of excess ferrocene argued against passivation of the electrochemical surface. Instead, encapsulation results in very slow heterogeneous electron transfer. In contrast, similar NMR studies revealed that viologen guests were bound to the outside of the capsule. Two copies were observed to bind, suggesting that each is associated with the four carboxylates at each pole of the capsule. It is interesting to note that this was the first observation of an empty dimeric capsule; presumably dimerization in the absence of a guest is promoted by the 40 mM NaCl concentrations used in the experiments. Furthermore, this unusual quaternary complex could internalize one equivalent of ferrocene to form a quinternary capsular complex with one guest inside and two guests outside the host. Returning to the 2∶2 host–viologen complex, voltametric studies revealed that the guest could be reduced whilst associated with the host. The heterogeneous electron transfer rate was attenuated relative to viologens in free solution, but the host did help solubilize the fully reduced and uncharged product of the two sequential one-electron reductions. In an interesting extension to this study, Kaifer and colleagues recently revealed that suitable charged guests on the outside of the capsule can mediate electrochemical oxidation of encapsulated ferrocene.153 As just discussed, encapsulated ferrocene is electrochemically silent, but when cationic ferrocene 52+ was added to a solution of the 442–ferrocene complex the cationic guest became associated with the outside of the capsule. In contrast to the binding of viologen, only one cationic guest was observed to bind. Cyclic voltammetry revealed that cationic guest 52+ could be reversibly oxidized to the corresponding ferrocenium derivative when an electrochemically inactive guest (stilbene) was encapsulated within the capsule. However, when stilbene was replaced with ferrocene a number of interesting changes were observed in the cyclic voltammogram. On the forward scan there was no evidence that the internalized ferrocene could be oxidized, whilst at more positive potential the outer guest 52+ was readily oxidized. However, on the reverse scan the reduction of 522+ was observed as well as a second reduction peak in the potential range for the reduction of free ferrocenium. These observations clearly indicate that oxidized 522+ on the outside of the capsule accepts an electron from encapsulated ferrocene in a homogeneous electron transfer process thus regenerating 52+ on the outside and yielding ferrocenium inside the capsule. The untenable situation of an internalized water-soluble guest results in the capsule breaking apart and liberation of ferrocenium. Additionally, further cycling revealed that this whole process is reversible. In contrast, when cationic guest 52+ was replaced with anionic guest 53−, this behavior was not observed; as the anionic guest was unable to associate with the ferrocene-containing capsule, the homogeneous electron transfer process could not occur. Likewise, when a cucurbituril host that strongly bound 52+ was added to the experiment the observed electrochemistry was shut down.
Another example of guest molecules associating with the outside of the capsule formed by 44 was recently described by Ramamurthy and Turro.152 In this study NMR and EPR spectroscopy was used to examine electron spin–spin interactions (super-exchange) between an encapsulated nitroxide guest and a free N-nitroxide, as well as derive direct information on the motion and environment polarity of the free and bound species. More specifically, the encapsulation of paramagnetic 14N-nitroxide 54 leads to broadening of all the 1H NMR signals of the inner protons of the capsule but left the external protons unaffected. As encapsulation was ambiguous, it was confirmed by 1H NMR analysis of the encapsulation complex formed by structurally similar but diamagnetic 55. A comparison of the 1H NMR spectrum of the complex with the paramagnetic guest 54 to the 1H NMR spectra of the complex with the diamagnetic guest 55, in the presence of cationic and anionic 14N-nitroxides 56 and 57, revealed association with the outside of the host. Thus, broadening of the 1H NMR signals was greatest when the paramagnetic guest was encapsulated, intermediate when the cationic 14N-nitroxide 56 was added to the diamagnetic guest complex, and minimal in the presence of anionic 14N-nitroxide. This strongly supported the notion that the cationic 14N-nitroxide 56 was associating with the outside of the capsule, whereas the anionic 14N-nitroxide 57 was not. Further support came from fitting the obtained EPR data to computer simulations to give the rotational correlation time (τc, a measure of the rotational mobility of the probe) and the hyperfine coupling (aN, a measure of the polarity of the environment around the probe) for the same three samples. The τc and aN values for the encapsulated paramagnetic guest 54 were significantly different that those obtained for 54 in free solution, confirming that the bound guest resides in a restrictive and non-polar environment. In contrast, the τc and aN values of the anionic 14N-nitroxide 57 were independent of the presence of the capsule. However, in the case of the cationic 14N-nitroxide 56 the aN value was the same as that obtained in the absence of the capsule, i.e., it resides in free solution in the presence of the capsule, but its τc value was considerably increased. Thus, its interaction with the anionic coat of the capsule does hinder its rotational options. That the cationic (external) guest can spin–spin super-exchange with a guest inside the capsule was demonstrated by examining the EPR spectrum of the complex between host 44 and the 15N-nitroxide guest 54 in the presence of cationic 14N-nitroxide external guest 56. When this was compared to the sum of the EPR spectra of the complex with the diamagnetic guest 55 in the presence of cationic 56, and the 15N-nitroxide guest 55 complex on its own, it was apparent that the “true” spectrum was considerably broadened and of reduced signal intensity relative to the combined data. The extent of broadening and signal reduction was indicative of strong electron–electron spin interaction between external and internal guests via host 44. In contrast, there was no evidence of such an interaction with the anionic 57.
Tolbert et al. have investigated how mimics of the chromophores found in fluorescent proteins are affected by encapsulation within the capsule formed by 44.155 The authors observed that a series of benzylidene-3-methylimidazolidinones of general structure 58 readily formed 2∶1 host–guest complexes with the octaacid. By varying the nature and position of R1 and the nature of R2, it was possible to control the extent to which the R groups anchored the guests inside the host by forming strong non-covalent interactions with the “polar” regions of the cavity. Measuring the cis–trans ratio of each guest after irradiation of the corresponding complex, as well as the emission properties of each encapsulated guest, revealed that neither meta- nor para-alkyl groups ameliorated internal conversions. However, in contrast ortho-alkyl groups increased the emission quantum yield by one order of magnitude. Additionally, it was noted that irradiation of the free guests gave cis–trans ratios of between 40∶60 and 60∶40, whereas the ratios obtained for the encapsulated guests ranged from between 24∶76 and 2∶98. These latter results suggest that the trans-isomers have a better goodness-of-fit to the capsule than the corresponding cis. The combined data indicated that of the different compounds examined only ortho-substitution prevents rotation around the single bond (indicated in 58), inhibiting internal conversion without necessarily inhibiting cis–trans isomerization, and hence engendering enhanced fluorescence.
In a more general study on the effects of encapsulation upon the fluorescence of guests, Ramamurthy et al. have examined nine fluorescent dyes within octaacid 44.158 The complexation of these guests, which ranged from the large hydrophobe pyrene aldehyde 59 to the much small and amphiphilic 2-naphthoic acid 60, confirmed that the larger guests formed 2∶1 host–guest complexes, smaller hydrophobes formed 2∶2 complexes, and amphiphilic guests formed weaker 1∶1 (non-capsular) complexes. More importantly, all of the emission properties of the dyes that formed capsular complexes were reminiscent of spectra recorded in dry aromatic solvents. In other words, the inside of these capsule formed by 44 is essentially dry.
Since the first report of octaacid 44 acting as a nano-scale (yoctolitre) reaction flask,169 the host has found considerable utility for inducing unique photochemical conversions. Before reviewing this topic, it is however worth pointing out that the capsule formed by 44 can also be used to stop photochemical reactions from occurring.170 For example, Gibb and Ramamurthy have reported on how encapsulation affects the fluorescence properties of naphthalene 61, anthracene 62, and tetracene 63 (Fig. 3). Only monomer emission is observed when naphthalene is examined in free solution. In contrast, two copies of naphthalene 61 are observed to bind into the host (NMR), and as a result the fluorescence spectrum of the encapsulated guest demonstrates both monomer and excimer emissions. Monomer emission is presumably detected because the size of the guest is such that even with two molecules inside the host there is still a relatively large amount of free space in the capsule. With the almost twice as large guest tetracene 63, the ‘reverse’ situation is observed. In free solution tetracene 63 shows both monomer and excimer emissions; indeed tetracene 63 readily undergoes photochemical-induced dimerization. However, 1H NMR clearly demonstrates that only one guest can fit with the confines of the nano-capsule formed by 44. As a result, the guest is photochemically stable and only undergoes monomer emission. In between these two examples is the interesting case of anthracene 62. This guest normally undergoes efficient photochemical-induced dimerization, and related to this, shows monomer emission but only the weakest of excimer emission in free solution. Consequently, the study of the anthracene excimer has previously been carried out at <77 K in either rigid glass or the crystalline state. Interestingly, the complex formed with host 44 is a tight 2∶2 complex (1H, COSY, and NOESY NMR). These experiments reveal that the two guest are stacked upon one another, but are forced out of register because of the shape of the cavity (Fig. 3). Held in this packing motif, the two guests cannot react when irradiated; the guest is recovered after irradiation for 10 hours. Instead, the complex shows a strong excimer emission in the fluorescence spectrum. Thus, the two guests are held in a π-stacked excimer state that can neither dissociate nor lead to anthracene dimer.
 | ||
| Fig. 3 Guests 61–63 and the packing structure of anthracene (62) within the dimeric capsule formed by 44. | ||
A collaborative effort from the Gibb, Turro and Ramamurthy groups led to an encapsulated photochemical conversion involving the transfer of chemical information—in the form of singlet oxygen (1O2)—from one capsule containing a sensitizer for generating it, to another containing a reactive substrate.171,172 In this system (Scheme 8) the substrates were a series of 1-methyl cycloalkenes. Two guests were bound inside the capsule, and because of the methyl substitution, both guests were oriented specifically with their methyl groups anchored into the narrowest polar regions of the capsule (1H, diffusion and NOESY NMR). Irradiation of a mixture of substrate complex and a capsular complex of the sensitizer dimethylbenzil lead to efficient and highly selective hydroperoxide formation. Extensive photophysical studies revealed the generation of 1O2 by the encapsulated sensitizer, and that 1O2 resided primarily in solution. Additionally, monitoring the phosphorescence and T–T absorption of encapsulated *3DMB as a function of oxygen concentration revealed that 1O2 generation occurred through the entry and egress of oxygen molecules at a rate controlled by the opening and closing of the capsule. The generated 1O2 then enters the capsule containing the substrate and abstracts a hydrogen atom from the most accessible of three allylic methylenes in the guest.
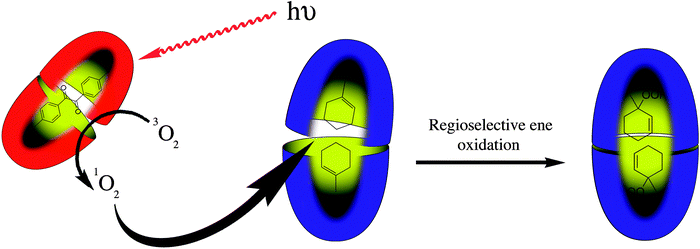 | ||
| Scheme 8 Regioselective ene oxidation of encapsulated allylic guests. | ||
In most instances, the rate at which the capsule formed by 44 assembles and disassembles is slower than the rate of a photochemical reaction. However, this is not always the case. In a study of the photodimerization of acenaphthylene 18 (Scheme 9) within the nano-capsule, Kaanumalle and Ramamurthy173 noted by NMR that the two acenaphthylene guests are stacked in a syn manner with the long axis of each guest oriented along the long axis of the capsule. Irradiation of this complex gave essentially only the syn-dimer 64. No anti-dimer 65 was observed. In contrast a 2∶3 ratio of syn- and anti-dimer was observed upon irradiation of an aqueous suspension of acenaphthylene 18. Additionally it was noted that irradiation of the acenaphthylene complex in the presence of the triplet sensitizer eosin-Y gave a 3∶2 ratio of syn- and anti-dimer; with the anti-dimer product being deposited on the walls of the reaction flask because it did not fit inside the capsule. This result demonstrated that even without direct contact between eosin-Y and acenaphthylene the latter can be sensitized. It was concluded that subsequent to through-host excitation the relatively long-lived (ms) triplet acenaphthylene exits the cavity as an excimer, re-orientates into an anti-type excimer, and then combines.
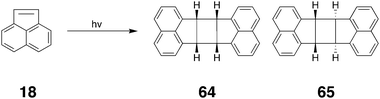 | ||
| Scheme 9 Photodimerization of acenaphthylene. | ||
Recently Sundaresan and Ramamurthy160 have built on early work noting the rearrangement of dibenzylketone when irradiated within the nano-capsule formed by 44.169 This unusual rearrangement, shown in Scheme 10, occurs efficiently in part because the photochemical timescale for this reaction is much faster than the assembly and disassembly timescale of the capsule and the generated radicals are therefore trapped within the capsule. In addition, there appears to be a thermodynamic preference for the benzyl radical to re-orientate relative to the phenylacyl radical so that the incipient methyl group of the former efficiently packs the narrow confines of the “polar” region of the cavity. With this chemistry noted Sundaresan and Ramamurthy160 synthesized a series of guests with different R groups (R = Me, Et, n-Pr, n-Bu, n-pentyl and n-hexyl). The last of these was not observed to complex to the capsule, but the others formed 2∶1 complexes (NMR). Irradiation of these complexes allowed an examination of how the rearrangement shown in Scheme 10, and competing reactions involving decarbonylation of the phenylacyl radicals, varied as a function of R group. Briefly, as the size of the R group increases so the yield of the rearrangement products decreases. This trend suggested that as the size of the phenylacyl radical increases the amount of space afforded to the benzyl radical decreases and its ability to rotate is impaired. The extreme case was the n-pentyl guest that resulted in none of the rearranged benzylphenyl ketone. Apparently, in this case the space within the container is so limited that the benzyl radical is kinetically “frozen” in the time frame of the photochemical reaction.
 | ||
| Scheme 10 The rearrangement of encapsulated dibenzylketones within the capsule 442. | ||
In related work, Gibb et al. investigated how encapsulation within the nano-capsule formed by 44 affected the photochemistry of α-(n-alkyl) dibenzyl ketones 66 (R = Me through n-octyl).174 An extensive NMR study of these 2∶1 host–guest complexes revealed three types of guest packing within the capsule. For the smaller guests 66 (R = Me, Et and n-Pr) it was observed that each aromatic ring of the guest occupied a hemisphere of the host whilst the alkyl group was located at the equatorial region. For the mid-sized guests 66 (R = n-Bu, n-pentyl and n-hexyl), the size of the alkyl group meant that it could compete with the proximal aryl group for binding to a hemisphere. As a result, this aromatic ring was pushed into the equatorial region of the capsule with the alkyl group and the distal aromatic ring occupied the two hemispheres. Finally, with the large alkyl groups (66, R = n-heptyl and n-octyl) the proximal and distal rings exchanged positions. It was assumed that this change occurred because the bigger guests were filling the capsule to capacity and that this change in packing leads to a slight decrease in the length of the packed guest and hence a slight release of strain. These three different kinds of packing motif led to, in broad terms, three different photochemical outcomes. With the smallest of guests simple decarbonylation predominated, but as the alkyl group increased in size so rearrangement products analogous to those described in the aforementioned study became prominent. Decarbonylation again became prominent with the n-heptyl guest, but the rearrangement products were not observed in this case. Instead Type II Norrish photochemical products were observed. Finally, with the largest n-octyl guest Type II Norrish chemistry accounted for 90% of the products. Besides the fact that there was no evidence of products arising from radicals escaping the capsule and (statistically) undergoing recombination, the encapsulation reaction outcomes differed considerable from that seen in solution. For example, no rearrangement products were observed to occur when the photolysis of the free guest was carried out in hexanes or in buffered water. In addition, Type II Norrish products, if observed in free solution, account for only a few percent of the overall yield. In short, the capsule templated the formation of a range of products not normally seen in solution.
A paper on the effects of irradiating 2∶1 44–stilbene complexes and a 2∶2 44–styrene complex is another example of unusual photo-products from encapsulation.157 In this work the stoichiometry of the different complexes, as well as the orientation of the guests were determined by NMR. The stilbene guests, cis- and trans-4,4′-dimethylstilbene 67 (trans shown) and trans-4-methylstilbene 68 were observed to bind with one aromatic ring in each hemisphere. In the case of the trans isomer of 67, both methyl groups could anchor into the base of each hemisphere, whereas with the more rotund cis isomer this was not possible. That the trans-isomer formed a more stable complex was reflected in the products of irradiating the two capsular complexes. Thus both cis- and trans-isomers of 67 attained pseudo-stationary states consisting of an 85∶15 ratio of the trans and cis isomers (the former forming much faster than the latter), but in free solution the obtained ratio was 18% trans and 76% cis. In other words, stabilization from the anchoring of the methyl groups in the trans isomer complex changes the product outcome. This was also confirmed when trans-4-methylstilbene 68 was bound and encapsulated. For this guest the lack of a second anchor meant that the photochemical outcome of reaction on the encapsulated guest was identical to that when the reaction took place in hexane or water. Extending this work, the authors then examined the irradiation of the 2∶2 complexes with 4-Me-styrene. Here, NMR suggested that two subtly different packing motifs were present in the mixture of host and guest, and the ratio of these entities was identical to the ratio of the two products obtained after irradiation. Normally, in solution the products observed are 69 and 70; the former predominates by direct irradiation, the latter via triplet sensitization. In the capsule however the two observed products were 71 and 72. Precisely why the products are formed was not ascertained, but the authors suspected triplet sensitization by the host as one possibility.
The Gibb and Ramamurthy groups have also investigated highly selective photo-Fries reactions within the capsule formed by 44.175 These excited state reactions occur via singlet radical pairs and usually form a bewildering array of products. For example, ester 73 forms eight products upon irradiation in hexanes. In the confines of the capsule formed by host 44, only one product (74) was formed in 99% yield. Likewise, other esters gave exclusively (or mostly) the corresponding ortho-ketone rearranged product. The high selectivity was attributed to the cage effect of the capsule (radicals that form in a capsule can only react with the contents of that capsule), limited movement of the guest within the container, and rapid recombination of the radicals formed so that decarbonylation pathways did not manifest themselves.
Although host 44 is not chiral, the confining nature of the capsule cavity can be used to enhance stereoselective process carried out within it. For example, an examination of the photocyclization of tropolone ethers and the oxa-di-π-methane rearrangement of cyclohexadienones within the capsule revealed that the presence of distal chiral auxiliaries on each guest, whilst not leading to highly diastereoselective reaction in free solution, gave good diastereoselectivities in the capsule.176 To take one example, irradiation of cyclohexadienone 75 (R = (S) –OCH2CH(Me)CH2CH3) can lead to two different diastereomers 76 and 77 (Scheme 11). When this reaction was carried out in solution only 3% diastereomeric excess (de) was noted by gas chromatography. This result was obtained at both 278 K and 298 K. In contrast, NMR studies (COSY, NOESY, DOSY, and TOCSY) confirmed that cyclohexadienone 75 formed a stable 2∶1 host–guest complex with octaacid 44, and that irradiation of this complex gave 17% and 36% de at 298 K and 278 K respectively. Hence, in free solution the stereocenter on the sidechain is too remote to control the developing stereocenters in the ring system, but packing the guest in the confines of the host allows the sidechain stereocenter to influence the reaction to a much greater extent.
 | ||
| Scheme 11 The irradiation of cyclohexadienone 75 to give diastereomers 76 and 77. | ||
A related, and perhaps more illustrative example also reported by Gibb et al. concerns the photoelectrocyclization of pyridones within the same capsule.177 The series of guests examined, typified by 78 (Scheme 12), were all shown by NMR studies (COSY, NOESY, DOSY, and TOCSY) to bind in a 2∶1 host–guest complex. In the case of 78, a 4% de was observed in free acetonitrile, whereas de values of 73% and 92% were observed at 298 K and 278 K, respectively, when the guest was bound inside the host. Other guests examined, for example with different methyl substitutions on the pyridone ring, gave de values of between 21–70% at 278 K. Why these differences were observed was not automatically clear. The data from the NMR studies revealed that the chiral sidechain of the guest occupies one hemisphere of the capsule, and its packing motif does not change from complex to complex. In contrast, the position of the methyl group on the pyridone ring had a large influence on how this part of the guest bound to the other hemisphere of the host. However, how precisely these subtle changes in the constitution of the guest affect the diastereoselectivity of the reactions remains to be discovered.
Conclusions and outlook
Over the last five years there has been considerable activity regarding the dynamic assembly of large hosts in aqueous solution. Two-thirds to three quarters of this activity has involved metal coordination, a strategy that has proven to be of broad utility across all media types. Most recently however, the hydrophobic force has also proven to be a powerful tool for driving self-assembly in water. Together, these strategies have highlighted how the properties of encapsulated molecules can be dynamically altered, and as a result how many new chemical conversions, catalytic processes, and unusual phenomena can be engendered. Taking care not too tread on ice that is too thin, looking to the future it is likely that research groups will continue to make access to families of self-assembling systems easier, and that approaches that combine the metal-coordination and the hydrophobic effect strategies, or perhaps other strategies such as hydrogen bonding, will emerge. Regardless of the exact form of the field, it is apparent that self-assembly in water has many exciting, long-term possibilities.References
- B. C. Gibb, Nat. Chem., 2009, 1, 17–18 CrossRef CAS.
- B. C. Gibb, Nat. Chem., 2009, 1, 252–253 CrossRef CAS.
- S. E. Akhtar, J. Drug Targeting, 2008, 16, 517–637 CrossRef CAS.
- D. J. Cram, Science, 1983, 219, 1177–1183 CrossRef CAS.
- D. J. Cram and J. M. Cram, Container Molecules and Their Guests, Royal Society of Chemistry, Cambridge, 1st edn, 1994 Search PubMed.
- D. J. Cram, M. E. Tanner and R. Thomas, Angew. Chem., Int. Ed. Engl., 1991, 30, 1024–1027 CrossRef.
- R. Warmuth, Angew. Chem., Int. Ed. Engl., 1997, 36, 1347–1350 CrossRef CAS.
- D. A. Makeiff, K. Vishnumurthy and J. C. Sherman, J. Am. Chem. Soc., 2003, 125, 9558–9559 CrossRef CAS.
- R. Warmuth, Eur. J. Org. Chem., 2001, 423–437 CrossRef CAS.
- T. A. Robbins and D. J. Cram, J. Chem. Soc., Chem. Commun., 1995, 1515–1516 RSC.
- C. von dem Bussche-Hünnefeld, D. Bühring, C. B. Knobler and D. J. Cram, Chem. Commun., 1995, 1085–1087 RSC.
- J. Yoon, C. Sheu, K. N. Houk, C. B. Knobler and D. J. Cram, J. Org. Chem., 1996, 61, 9323–9339 CrossRef CAS.
- M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161–171 RSC.
- H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS.
- R. Wyler, J. De Mendoza and J. Rebek, Jr., Angew. Chem., Int. Ed. Engl., 1993, 32, 1699–1701 CrossRef.
- M. M. Conn and J. Rebek, Jr., Chem. Rev., 1997, 97, 1647–1668 CrossRef CAS.
- D. M. Vriezema, M. C. Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS.
- G. O. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS.
- S. M. Biros and J. Rebek, Jr., Chem. Soc. Rev., 2007, 36, 93–104 RSC.
- S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959–1976 CrossRef CAS.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1917 CrossRef CAS.
- K. Uekama, F. Hirayama and T. Irie, Chem. Rev., 1998, 98, 2045–2076 CrossRef CAS.
- E. Engeldinger, D. Armspach and D. Matt, Chem. Rev., 2003, 103, 4147–4586 CrossRef CAS.
- R. Villalonga, R. Cao and A. Fragoso, Chem. Rev., 2007, 107, 3088–3116 CrossRef CAS.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC.
- R. J. Hooley, S. M. Biros and J. Rebek, Jr., Chem. Commun., 2006, 509–510 RSC.
- R. J. Hooley, S. M. Biros and J. Rebek, Jr., Angew. Chem., Int. Ed., 2006, 45, 3517–3519 CrossRef CAS.
- R. J. Hooley, H. J. Van Anda and J. Rebek, Jr., J. Am. Chem. Soc., 2006, 128, 3894–3895 CrossRef CAS.
- R. J. Hooley, H. J. V. Anda and J. Rebek, Jr., J. Am. Chem. Soc., 2007, 129, 13464–13473 CrossRef CAS.
- S. M. Butterfield and J. Rebek, Jr., J. Am. Chem. Soc., 2006, 128, 15366–15367 CrossRef CAS.
- D. Podkoscielny, R. J. Hooley, J. Rebek, Jr. and A. E. Kaifer, Org. Lett., 2008, 10, 2865–2868 CrossRef.
- X. Liu, J. Sun and R. Warmuth, Tetrahedron, 2009, 65, 7303–7310 CrossRef CAS.
- D. L. Rousseau, Am. Sci., 1992, 80, 54–63.
- J. Maddox, J. Randi and W. W. Stewart, Nature, 1988, 334, 287–290 CrossRef CAS.
- K. Ovchinnikova and G. H. Pollack, Langmuir, 2009, 25, 542–547 CrossRef CAS.
- H. R. Corti and A. J. Colussi, Langmuir, 2009, 25, 6587–6589 CrossRef CAS.
- S. Granick and C. S. Bae, Science, 2008, 322, 1477–1478 CrossRef CAS.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS.
- Y. Levy and J. N. Onuchic, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 389–415 CrossRef CAS.
- R. Breslow, J. Phys. Org. Chem., 2006, 19, 813–822 CrossRef CAS.
- N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS.
- L. R. Pratt and A. Pohorille, Chem. Rev., 2002, 102, 2671–2692 CrossRef CAS.
- K. A. Dill, Biochemistry, 1990, 29, 7133–7155 CrossRef CAS.
- C. Tanford, The Hydrophobic Effect. Formation of Micelles & Biological Membranes, John Wiley & Sons, New York, 2nd edn, 1980 Search PubMed.
- J. Israelachvili, Intermolecular & Surface Forces, Academic Press, San Diego, 1991 Search PubMed.
- E. S. Stoyanov, I. V. Stoyanova and R. A. Reed, J. Am. Chem. Soc., 2010, 132, 1484–1485 CrossRef CAS.
- J.-W. Shin, N. I. Hammer, E. G. Diken, M. A. Johnson, R. S. Walters, T. D. Jaeger, M. A. Duncan, R. A. Christie and K. D. Jordan, Science, 2004, 304, 1137–1140 CrossRef CAS.
- M. Miyazaki, A. Fujii, T. Ebata and N. Mikami, Science, 2004, 304, 1134–1137 CrossRef CAS.
- A. Gutberlet, G. Schwaab, O. Birer, M. Masia, A. Kaczmarek, H. Forbert, M. Havenith and D. Marx, Science, 2009, 324, 1545–1548 CrossRef CAS.
- S. F. Dec, K. E. Bowler, L. L. Stadterman, C. A. Koh and E. D. J. Sloan, J. Am. Chem. Soc., 2006, 128, 414–415 CrossRef CAS.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS.
- Y. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658–663 CrossRef CAS.
- A. W. Omta, M. F. Kropman, S. Woutersen and H. J. Bakker, Science, 2003, 301, 347–349 CrossRef CAS.
- D. Harries, D. C. Rau and V. A. Parsegian, J. Am. Chem. Soc., 2005, 127, 2184–2190 CrossRef CAS.
- P. Setny and M. Geller, J. Chem. Phys., 2006, 125, 144717 CrossRef.
- J. Ewell, B. C. Gibb and S. W. Rick, J. Phys. Chem. B, 2008, 112, 10272–10279 CrossRef CAS.
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2004, 126, 3674–3675 CrossRef CAS.
- J. M. Rivera, T. Martín and J. Rebek, Jr., Science, 1998, 279, 1021–1023 CrossRef CAS.
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS.
- M. Yoshizawa, Y. Takeyama, T. Okano and M. Fujita, J. Am. Chem. Soc., 2003, 125, 3243–3247 CrossRef CAS.
- M. Yoshizawa, Y. Takeyama, T. Kusukawa and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 1347–1349 CrossRef CAS.
- M. Yoshizawa, S. Miyagi, M. Kawano, K. Ishiguro and M. Fujita, J. Am. Chem. Soc., 2004, 126, 9172–9173 CrossRef CAS.
- T. Yamaguchi and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 2067–2069 CrossRef CAS.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746–2747 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2007, 46, 8587–8589 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 11423–11429 CrossRef CAS.
- R. J. Hooley and J. Rebek, Jr., Org. Biomol. Chem., 2007, 5, 3631–3636 RSC.
- S. M. Butterfield and J. Rebek, Jr., Chem. Commun., 2007, 1605–1607 RSC.
- S. Richeter and J. Rebek, Jr., J. Am. Chem. Soc., 2004, 126, 16280–16281 CrossRef CAS.
- A. Gissot and J. Rebek, Jr., J. Am. Chem. Soc., 2004, 126, 7424–7425 CrossRef CAS.
- C. Gibson and J. Rebek, Jr., Org. Lett., 2002, 4, 1887–1890 CrossRef CAS.
- J. Kang, J. Santamaría, G. Hilmersson and J. Rebek, Jr., J. Am. Chem. Soc., 1998, 120, 7389–7390 CrossRef CAS.
- J. Chen, S. Körner, S. L. Craig, D. M. Rudkevich and J. Rebek, Jr., Nature, 2002, 415, 385–386 CrossRef CAS.
- F. E. Ziegler, A. Davis, D. W. Johnson and K. N. Raymond, Angew. Chem., Int. Ed., 2003, 42, 665–668 CrossRef CAS.
- S. Sato, J. Iida, K. Suzuki, M. Kawano, T. Ozeki and M. Fujita, Science, 2006, 313, 1273–1276 CrossRef CAS.
- T. Furusawa, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 5717–5719 CrossRef CAS.
- Y. Yamauchi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2008, 130, 5832–5833 CrossRef CAS.
- A. Shivanyuk and J. Rebek, Jr., J. Am. Chem. Soc., 2002, 124, 12074–12075 CrossRef CAS.
- A. Scarso, A. Shivanyuk and J. Rebek, Jr., J. Am. Chem. Soc., 2003, 125, 13981–13982 CrossRef CAS.
- A. Shivanyuk, A. Scarso and J. Rebek, Jr., Chem. Commun., 2003, 1230–1231 RSC.
- A. Scarso, H. Onagi and J. Rebek, Jr., J. Am. Chem. Soc., 2004, 126, 12728–12729 CrossRef CAS.
- A. Scarso, L. Trembleau and J. Rebek, Jr., J. Am. Chem. Soc., 2004, 126, 13512–13518 CrossRef CAS.
- M. Yamanaka and J. Rebek, Jr., Chem. Commun., 2004, 1690–1691 RSC.
- D. Ajami and J. Rebek, Jr., J. Am. Chem. Soc., 2006, 128, 15038–15039 CrossRef CAS.
- R. J. Hooley and J. Rebek, Jr., Org. Lett., 2007, 9, 1179–1182 CrossRef CAS.
- J. Rebek, Jr., Chem. Commun., 2007, 2777–2789 RSC.
- D. Ajami and J. Rebek, Jr., Angew. Chem., Int. Ed., 2008, 47, 6059–6061 CAS.
- J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296–313 RSC.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668–698 CrossRef CAS.
- M. G. Arriaga, G. Hobley and J. M. Rivera, J. Am. Chem. Soc., 2008, 130, 10492–10493 CrossRef CAS.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2004, 126, 11408–11409 CrossRef CAS.
- M. Fujita, D. Oguro, M. Mlyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469–471 CrossRef CAS.
- J. M. Davis, L. K. Tsou and A. D. Hamilton, Chem. Soc. Rev., 2007, 36, 326–334 RSC.
- S. Tashiro, M. Tominaga, M. Kawano, B. Therrien, T. Ozeki and K. Fujita, J. Am. Chem. Soc., 2005, 127, 4546–4547 CrossRef CAS.
- S. Tashiro and M. Fujita, Bull. Chem. Soc. Jpn., 2006, 79, 833–837 CrossRef CAS.
- K. Nakabayashi, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 5322–5325 CrossRef CAS.
- K. Nakabayashi, M. Kawano, T. Kato, K. Furukawa, S. I. Ohkoshi, T. Hozumi and M. Fujita, Chem.–Asian J., 2007, 2, 164–170 CrossRef CAS.
- K. Nakabayashi, Y. Ozaki, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 2046–2048 CrossRef CAS.
- Y. Nishioka, T. Yamaguchi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2007, 129, 7000–7001 CrossRef CAS.
- Y. Nishioka, T. Yamaguchi, M. Kawano and M. Fujita, J. Am. Chem. Soc., 2008, 130, 8160–8161 CrossRef.
- J. Clayden, A. Lund, L. Vallverdu and M. Helliwell, Nature, 2004, 431, 966–971 CrossRef CAS.
- S. Karthikeyan and V. Ramamurthy, J. Org. Chem., 2007, 72, 452–458 CrossRef CAS.
- S. Karthikeyan and V. Ramamurthy, J. Org. Chem., 2006, 71, 6409–6413 CrossRef CAS.
- S. Karthikeyan and V. Ramamurthy, Tetrahedron Lett., 2005, 46, 4495–4498 CrossRef CAS.
- M. Kawano, Y. Kobayashi, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2006, 128, 6558–6559 CrossRef CAS.
- K. Yamashita, M. Kawano and M. Fujita, Chem. Commun., 2007, 4102–4103 RSC.
- J. K. Klosterman, M. Iwamura, T. Tahara and M. Fujita, J. Am. Chem. Soc., 2009, 131, 9478–9479 CrossRef CAS.
- M. Yoshizawa, T. Kusukawa, M. Kawano, T. Ohhara, I. Tanaka, K. Kurihara, N. Niimura and M. Fujita, J. Am. Chem. Soc., 2005, 127, 2798–2799 CrossRef CAS.
- R. J. Mashl, S. Joseph, N. R. Aluru and E. Jakobsson, Nano Lett., 2003, 3, 589–592 CrossRef CAS.
- K. Koga, G. T. Gao, H. Tanaka and X. C. Zeng, Nature, 2001, 412, 802–805 CrossRef CAS.
- M. Yoshizawa, J. Nakagawa, K. Kumazawa, M. Nagao, M. Kawano, T. Ozeki and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 1810–1813 CrossRef CAS.
- K. Kumazawa, K. Biradha, T. Kusukawa, T. Okano and M. Fujita, Angew. Chem., Int. Ed., 2003, 42, 3909–3913 CrossRef CAS.
- M. Yoshizawa, K. Kumazawa and M. Fujita, J. Am. Chem. Soc., 2005, 127, 13456–13457 CrossRef CAS.
- K. Ono, J. K. Klosterman, M. Yoshizawa, K. Sekiguchi, T. Tahara and M. Fujita, J. Am. Chem. Soc., 2009, 131, 12526–12527 CrossRef CAS.
- Y. Yamauchi, M. Yoshizawa, M. Akita and M. Fujita, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10435–10437 CrossRef.
- Y. Ozaki, M. Kawano and M. Fujita, Chem. Commun., 2009, 4245–4247 RSC.
- K. Ono, M. Yoshizawa, T. Kato, K. Watanabe and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 1803–1806 CrossRef CAS.
- K. Ono, M. Yoshizawa, T. Kato and M. Fujita, Chem. Commun., 2008, 2328–2330 RSC.
- M. Yoshizawa, K. Ono, K. Kumazawa, T. Kato and M. Fujita, J. Am. Chem. Soc., 2005, 127, 10800–10801 CrossRef CAS.
- K. Ono, M. Yoshizawa, M. Akita, T. Kato, Y. Tsunobuchi, S. I. Ohkoshi and M. Fujita, J. Am. Chem. Soc., 2009, 131, 2782–2783 CrossRef CAS.
- T. Sawada, M. Yoshizawa, S. Sato and M. Fujita, Nat. Chem., 2009, 1, 53–56 CrossRef.
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS.
- S. Tashiro, M. Tominaga, Y. Yamaguchi, K. Kato and M. Fujita, Angew. Chem., Int. Ed., 2006, 45, 241–244 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 3874–3876 CrossRef CAS.
- S. Tashiro, M. Tominaga, Y. Yamaguchi, K. Kato and M. Fujita, Chem.–Eur. J., 2006, 12, 3211–3217 CrossRef CAS.
- A. V. Davis, D. Fiedler, F. E. Ziegler, A. Terpin and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 15354–15363 CrossRef CAS.
- S. M. Biros, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 12094–12095 CrossRef CAS.
- C. J. Hastings, M. D. Pluth, S. M. Biros, R. G. Bergman and K. N. Raymond, Tetrahedron, 2008, 64, 8362–8367 CrossRef CAS.
- A. V. Davis and K. N. Raymond, J. Am. Chem. Soc., 2005, 127, 7912–7919 CrossRef CAS.
- D. J. Cram, M. E. Tanner and C. B. Knobler, J. Am. Chem. Soc., 1991, 113, 7717–7727 CrossRef CAS.
- M. D. Pluth, B. E. F. Tiedemann, H. van Halbeek, R. Nunlist and K. N. Raymond, Inorg. Chem., 2008, 47, 1411–1413 CrossRef CAS.
- V. M. Dong, D. Fiedler, B. Carl, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 14464–14465 CrossRef CAS.
- D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745–748 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 11459–11467 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 6362–6366 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Org. Chem., 2008, 73, 7132–7136 CrossRef CAS.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 2798–2805 CrossRef CAS.
- B. E. F. Tiedemann and K. N. Raymond, Angew. Chem., Int. Ed., 2007, 46, 4976–4978 CrossRef CAS.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 9781–9797 CrossRef CAS.
- D. Fiedler, H. van Halbeek, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 10240–10252 CrossRef CAS.
- C. J. Hastings, D. Fiedler, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 10977–10983 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Org. Chem., 2009, 74, 58–63 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef.
- J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103–112 CrossRef CAS.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, Angew. Chem., Int. Ed., 2008, 47, 8297–8301 CrossRef CAS.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS.
- B. C. Gibb, in Organic Nano-Structures, ed. J. L. Atwood and J. W. Steed, John Wiley and Sons, 2007 Search PubMed.
- S. Liu and B. C. Gibb, Chem. Commun., 2008, 3709–3716 RSC.
- J. Y.-C. Chen, N. Jayaraj, S. Jo ckusch, M. F. Ottaviani, V. Ramamurthy and N. J. Turro, J. Am. Chem. Soc., 2008, 130, 7206–7207 CrossRef CAS.
- D. Podkoscielny, S. Gadde and A. E. Kaifer, J. Am. Chem. Soc., 2009, 131, 12876–12877 CrossRef CAS.
- M. Saitoha, T. Fukaminatoa and M. Irie, J. Photochem. Photobiol., A, 2009, 207, 28–31 CrossRef.
- A. Baldridge, S. R. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, J. Am. Chem. Soc., 2010, 132, 1498–1499 CrossRef CAS.
- N. Jayaraj, Y. Zhao, A. Parthasarathy, M. Porel, R. S. H. Liu and V. Ramamurthy, Langmuir, 2009, 25, 10575–10586 CrossRef CAS.
- A. Parthasarathy, L. S. Kaanumalle and V. Ramamurthy, Org. Lett., 2007, 9, 5059–5062 CrossRef CAS.
- M. Porel, N. Jayaraj, L. S. Kaanumalle, M. V. S. N. Maddipatla, A. Parthasarathy and V. Ramamurthy, Langmuir, 2009, 25, 3473–3481 CrossRef CAS.
- A. K. Sundaresan and V. Ramamurthy, Org. Lett., 2007, 9, 3575–3578 CrossRef CAS.
- A. K. Sundaresan and V. Ramamurthy, Photochem. Photobiol. Sci., 2008, 7, 1555–1564 RSC.
- M. D. Giles, S. Liu, R. L. Emanuel, B. C. Gibb and S. M. Grayson, J. Am. Chem. Soc., 2008, 130, 14430–14431 CrossRef CAS.
- M. D. Giles, S. Liu, R. L. Emanuel, B. C. Gibb and S. M. Grayson, Isr. J. Chem., 2009, 49, 31–40 CrossRef CAS.
- S. M. Grayson and B. C. Gibb, Soft Matter, 2010, 6, 1377–1382 RSC.
- C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2006, 128, 16498–16499 CrossRef CAS.
- C. L. D. Gibb and B. C. Gibb, Chem. Commun., 2007, 1635–1637 RSC.
- H. Sun, C. L. D. Gibb and B. C. Gibb, Supramol. Chem., 2008, 20, 141–147 CrossRef CAS.
- C. L. D. Gibb and B. C. Gibb, Tetrahedron, 2009, 65, 7240–7248 CrossRef CAS.
- D. Podkoscielny, C. L. D. Gibb, B. C. Gibb and A. E. Kaifer, Chem.–Eur. J., 2008, 14, 4704–4710 CrossRef CAS.
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2004, 126, 14366–14367 CrossRef CAS.
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674–3675 CrossRef CAS.
- A. Natarajan, L. S. Kaanumalle, S. Jockusch, C. L. D. Gibb, B. C. Gibb, N. J. Turro and V. Ramamurthy, J. Am. Chem. Soc., 2007, 129, 4132–4133 CrossRef CAS.
- A. Greer, Nature, 2007, 447, 273–274 CrossRef CAS.
- L. S. Kaanumalle and V. Ramamurthy, Chem. Commun., 2007, 1062–1064 RSC.
- C. L. D. Gibb, A. K. Sundaresan, V. Ramamurthy and B. C. Gibb, J. Am. Chem. Soc., 2008, 130, 4069–4080 CrossRef CAS.
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, Org. Biomol. Chem., 2007, 5, 236–238 RSC.
- A. K. Sundaresan, L. S. Kaanumalle, C. L. D. Gibb, B. G. Gibb and V. Ramamurthy, Dalton Trans., 2009, 4003–4011 RSC.
- A. K. Sundaresan, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, Tetrahedron, 2009, 65, 7277–7288 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |