Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy
Warwick B.
Dunn
*abc,
David I.
Broadhurst
d,
Helen J.
Atherton
ef,
Royston
Goodacre
ab and
Julian L.
Griffin
f
aManchester Centre for Integrative Systems Biology, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK. E-mail: warwick.dunn@manchester.ac.uk; Fax: +44 (0)161 3064556; Tel: +44 (0)161 3065197
bDepartment of Chemistry, Manchester Interdisciplinary Biocentre, University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK
cCentre for Advanced Discovery and Experimental Therapeutics, Manchester Biomedical Research Centre, Oxford Road, Manchester, M13 9WL, UK
dThe Anu Research Centre, Department of Obstetrics and Gynaecology, Cork University Maternity Hospital, University College Cork, Wilton, Cork, Ireland
eCardiac Metabolism Research Group, Department of Physiology, Anatomy and Genetics, University of Oxford, Oxford, UK
fDepartment of Biochemistry & Cambridge Systems Biology Centre, University of Cambridge, Cambridge, UK
First published on 17th August 2010
Abstract
The study of biological systems in a holistic manner (systems biology) is increasingly being viewed as a necessity to provide qualitative and quantitative descriptions of the emergent properties of the complete system. Systems biology performs studies focussed on the complex interactions of system components; emphasising the whole system rather than the individual parts. Many perturbations to mammalian systems (diet, disease, drugs) are multi-factorial and the study of small parts of the system is insufficient to understand the complete phenotypic changes induced. Metabolomics is one functional level tool being employed to investigate the complex interactions of metabolites with other metabolites (metabolism) but also the regulatory role metabolites provide through interaction with genes, transcripts and proteins (e.g. allosteric regulation). Technological developments are the driving force behind advances in scientific knowledge. Recent advances in the two analytical platforms of mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy have driven forward the discipline of metabolomics. In this critical review, an introduction to metabolites, metabolomes, metabolomics and the role of MS and NMR spectroscopy will be provided. The applications of metabolomics in mammalian systems biology for the study of the health–disease continuum, drug efficacy and toxicity and dietary effects on mammalian health will be reviewed. The current limitations and future goals of metabolomics in systems biology will also be discussed (374 references).
 Warwick B. Dunn | Warwick (Rick) Dunn is an Experimental Officer at The Manchester Centre for Integrative Systems Biology (http://www.mcisb.org/), specializing in the application of bio-analytical strategies in metabolomics and systems biology studies of microbial and mammalian systems. He is also significantly involved in the construction of a clinical systems biology centre in Manchester (CADET). BSc (Hons) and PhD degrees in Chemistry with Analytical Chemistry were obtained at The University of Hull in 1993 and 1997, respectively. He has applied metabolomic and systems biology strategies for eight years, six of these at The University of Manchester with Professors Kell and Goodacre. His interests include development and validation of bio-analytical methodologies, high-throughput metabolite identification, the study of yeast metabolism and the investigation of cardiovascular, bowel and kidney diseases. |
 David I. Broadhurst | David Broadhurst is a Postdoctoral Research Scientist, specializing in Experimental Design (DoE), Signal Processing, Statistics, Multivariate Data Analysis, Data Visualisation, and Bioinformatics. David has a BSc (Hons) degree in Electronic Engineering (Salford University), a MSc in Medical Informatics (City University & St. Thomas's Medical School), and a PhD in the “Application of Artificial Neural Networks and Evolutionary Algorithms to Chemometrics” (University of Wales, Aberystwth). He has spent the last 15 years working in the field of metabolomics. Over the past 5 years David has helped pioneer the use of Metabolomics in Human Pathology at The University of Manchester in Professor Douglas Kell's Bioanalytical Sciences Group. In 2009 he moved to the Anu Research Centre, University College Cork, where, in collaboration with Professor Louise Kenny, he is investigating presymptomatic metabolite biomarkers for major pregnancy diseases. |
 Helen J. Atherton | Helen Atherton received her BSc degree in Chemistry with Pharmacology from the University of Leeds, and her PhD in Biochemistry from the University of Cambridge. Her research, conducted under the supervision of Dr Julian Griffin focused on the application of metabolomics to characterise metabolic syndrome. Since early 2008 she has been a post-doctoral researcher at the University of Oxford where she uses hyperpolarized 13C-MRS to study in vivo cardiac metabolism. |
 Royston Goodacre | Roy Goodacre is Professor of Biological Chemistry at The University of Manchester. The research group's (http://www.biospec.net/) interests are broadly within bioanalytical chemistry, and in the application of a combination of a variety of modern analytical techniques (including MS, Raman, and IR) and advanced chemometrics and machine learning to the explanatory analysis of complex biological systems within a metabolomics context. |
 Julian L. Griffin | Julian Griffin received his DPhil from the University of Oxford in the laboratory of Prof. Sir George Radda, where he used 13C NMR spectroscopy to follow metabolism in cerebral tissue. He held a Fellowship in Radiology and Cardiology at Massachusetts General Hospital and Harvard Medical School, before returning to the UK to the lab of Prof. Jeremy Nicholson at Imperial College London. It was during his time at Imperial College London that he became involved in the use of metabolomics/metabonomics as a functional genomic tool. He was a recipient of a Royal Society University Fellowship, first held at Imperial College before setting up his own group at the University of Cambridge in 2003. His lab specialises in the use of a combination of NMR spectroscopy and mass spectrometry to phenotype mouse models of disease, and in particular in areas of type II diabetes/obesity, cancer and neuroscience. |
1. Introduction to metabolites, metabolomes and metabolomics
(i) Metabolites
The building blocks and information repositories of biological systems (organelles, cells, tissues, organs and organisms) can, in simplified terms, be divided into four main biochemical components; genes, transcripts, proteins and metabolites. Biological systems are constructed of and function through complex interactions of these components. Metabolites are in a unique position as they are the building blocks for all other biochemical species and structures including proteins (amino acids), genes and transcripts (nucleotides), and cell walls. In the post-genomics era metabolomics is a core scientific discipline, complementary to the study of other functional levels (genome, transcriptome and proteome).1–5 The study of the metabolome can be applied in isolation or in combination with other functional levels (systems biology).6–12 Metabolites and their relationship with other metabolites (defined as metabolism) and biochemical species are currently the major focus of metabolomic investigations to understand biological function/phenotype.Metabolites are defined as low molecular weight (in relation to proteins and nucleic acids) organic and inorganic chemicals which are the reactants, intermediates or products of enzyme-mediated biochemical reactions. The majority of metabolites are organic in class but the importance of inorganic metabolites including metals should be highlighted (for example, iron).13 Metallomics is the scientific study of the complement of metals in a biological system.14 Metabolites are functionally different to peptides, proteins, transcripts and genes though the exact divide is often blurred. For example, glutathione is a tripeptide composed of glutamate, cysteine and glycine monomers which is synthesised and functions metabolically, largely to protect the cell against reactive oxygen species. Similarly, DNA and RNA are synthesized from nucleotides, some of which also have important roles in cellular energy processes. The compositional diversity of metabolites provides wide ranges of physicochemical properties including molecular weight, hydrophobicity/hydrophilicity, acidity/basicity and boiling point. The range of molecular weight (from 1 amu (proton) to greater than 1500 amu e.g., gangliosides, lipids and small peptides) is significantly lower than observed for proteins, transcripts and genes. Hydrophobicity/hydrophilicity ranges from polar metabolites such as low molecular weight amino acids to high molecular weight non-polar lipids. Volatility ranges are from low boiling point metabolites present in breath including isoprene and carbon dioxide to high molecular weight lipids. This diversity ensures that investigation of the complete complement of metabolites is technically challenging and multiple strategies are commonly employed to provide a wide coverage. These include the use of different analytical techniques. Mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy, often coupled to chromatography, are the most prevalent and provide the emphasis of this review. This can be contrasted with the single analytical platforms which are generally applied for detection of proteins, transcripts and genes. General classification of metabolites can involve polarity (polar, non-polar), molecular weight and metabolite structure or reaction similarity. The most frequently applied method is similarity where metabolites are classified according to chemical core structure (e.g., fatty acids) or by presence in the same metabolic pathway or pathways (e.g., glycolysis). Multiple levels of complexity can be included in the classification, as has been shown in The Human Metabolome Database (HMDB).15
(ii) Metabolomes
The quantitative complement of metabolites in a biological system is defined as the metabolome.16,17 The complexity and size of the metabolome is dependent on the organism and sample type (blood, urine, CSF or tissue for example). Yeast has an estimated 1100 metabolites.18 The human metabolome is currently estimated to contain many thousands of metabolites as defined in metabolic reconstructions19,20 and HMDB.15 These are under-estimates of the actual number of metabolites expected to be defined in the future. Metabolic reconstructions and databases are compiled with bibliographic and experimental data21 but exhibit gaps in the present knowledge, commonly in areas of lipid metabolism (as shown for yeast)22 and human–gut microflora metabolism. The differences in the types of polar head group, fatty acid acyl chain length and the degree and position of unsaturation in lipids mean that the structural diversity of lipids is immense and the number of possible lipid species is >105.23 Furthermore, there are many xenobiotics that are commonly found in tissues, particularly humans who may be taking medications or eating a diverse diet. Other chemicals not classified as metabolites can also be present, for example persistent organic pollutants.Metabolomes can be classified according to their origin. Endometabolomes are related to intra-cellular metabolism, exometabolomes (alternatively referred to as the metabolic footprint or secretome) refer to extra-cellular metabolomes. In mammals the metabolome can be described by the sample type and include serum (or plasma), urine, cerebrospinal fluid (CSF), breath, tears, saliva, faecal and a variety of tissues. One metabolome can be interconnected with another metabolome. For example, serum and urine are biofluids integrating the metabolic composition of several tissue types and organs which are related to multiple biological and physiological processes. This is beneficial when investigating these biofluids as they are relatively easy to acquire and provide a metabolic snapshot on the mammalian system as a whole. Also, the interaction of human and gut microflora metabolomes play an important role in the health–disease status of mammals, including the cross-talk between these separate metabolomes.
Metabolomes are in essence a ‘parts list’ of metabolites combined with qualitative connectivity information (metabolic reactions). Informatics resources provide information on metabolites and qualitative information of the inter-relationship (connectivity) of metabolites in specific forms and details. The informatics resources available have been reviewed recently24 and include, among others, HMDB and the Small Molecule Pathway database (SMPDB)25 and KEGG.26 The inter-relationships within the metabolome, referred to as the metabolic network, are large, and can be inferred using bibliometrics and informatics.27,28 For example, the Nicholson metabolic maps are a visual guide to the complexity observed.29 For quantitative network descriptions further information are required (including metabolite and enzyme concentrations) and fall in the discipline of quantitative systems biology. A community consensus metabolic network for yeast has recently been described18 and a parallel effort relating to the human metabolic network is currently being performed. Experimental strategies to define metabolic networks are also being performed.30
The metabolome is composed of metabolites originating from a number of processes. Metabolism involves the catabolism (breakdown and energy producing) and anabolism (construction and molecule producing) of metabolites and other biochemicals. These involve endogenous metabolites synthesised and consumed within the biological system. Exogenous metabolites (drugs and nutrients from food as examples) are imported from outside the biological system and metabolised (exogenous metabolism). For example, drugs are metabolised in the body in phase I and phase II biotransformations to increase the reactivity (phase I) and hydrophilicity for excretion (phase II), which can also sometimes increase their toxicity. These phase II reactions include oxidation, hydrolysis, reduction or conjugation.31 There can be interactions between the metabolisms of two different organisms. Microflora in the mammalian gut provide a positive and essential symbiotic relationship with the mammal, and this system can be thought of as a superorganism.32 The microflora in and upon the mammal can provide a large impact on health and disease status.33,34
(iii) Metabolomics
The study of metabolites in biological systems, referred to as metabolomics, is primarily involved in the study of metabolism. Differential changes in the synthesis and consumption of metabolites are investigated. The phrase metabolism relates to the Ancient Greek metabolè, meaning change.35 Metabolism is the study of the chemical conversion of one metabolite to another metabolite through the interaction with an enzyme and in some cases co-factors (for example, ATP, NADH, co-enzyme A). Metabolism is regulated to ensure adequate biomass and energy production along with other requirements for growth and life. Central metabolism is those reactions and pathways required for energy, growth and nutrient supply and are conserved across many organisms (for example, the pathways of glycolysis and the citric acid cycle). Secondary metabolism is reactions or pathways associated with one or a limited number of organisms and are not required for survival (for example, antibiotic production in Streptomyces or alkaloid production in plants). The complexity of metabolic networks is exhibited by pleiotropy where a perturbation to a specific reaction (for example, gene knockout(s) resulting in the absence of a specific enzyme (isoforms)) may provide the consequential loss of direct production of a metabolite but can result in an indirect route of production via a series of metabolic reactions which may create a number of metabolic perturbations.36 This is a measure of the robustness of metabolic networks, often discussed in the evolution of metabolic networks.37Metabolites are involved in many other biochemical processes not directly (but often indirectly) related to their synthesis or consumption. These are also of scientific interest in metabolomics. Metabolites can act in the regulation of metabolism. Homeostasis provides a constant chemical environment within a biological system maintained by regulation of metabolism and other processes. This is particularly important for maintaining the osmotic potential of cells, with a number of high concentration metabolites also acting as osmolytes under various conditions. Increases or decreases in the concentration or availability of metabolic reactants in the environment can be self-regulated by a number of processes including the increase or decrease of the activity of enzymes responsible for the reactions through allosteric modification. Allosteric modifications involve the binding of given metabolites to a region of an enzyme which in turn either increases or decreases the rate of enzymatic action. This is often a rapid means of regulating metabolic flux within the cell. Covalent modification of proteins, such as phosphorylation, acetylation or ubiquitination, and transcriptional control through transcription factors provide regulation and the control over metabolism across multiple organs, such as processes like the Cori cycle. The timescale of protein modifications can be rapid when compared to transcriptional regulation. Dysregulation of these regulatory processes can result in disease onset or progression. For example, the hormone insulin regulates glucose and fat metabolism to increase storage as triglycerides or glycogen when the blood glucose concentration increases. A breakdown in this regulation is responsible for the onset of diabetes, either due to a failure to produce insulin in type I diabetes or insulin resistance in type II diabetes. This leads to decreased biological regulation of blood glucose concentration. Indeed the inappropriate storage of lipid is thought to be one of the causes of insulin resistance that predates full blown type II diabetes as part of lipotoxicity.38 Metabolites can also regulate other processes including gene transcription and recently, riboswitches (the interaction of RNA with metabolites) have been shown to modulate gene expression.39
A range of terminologies are applied in metabolomics and are described in Table 1. These can at times be perplexing with multiple terms defining the same process. Of greatest debate today is the scientific difference between metabolomics and metabonomics. Metabolomics is generally defined as the comprehensive study of all metabolites present in a biological system.1 Metabonomics is defined as “the quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification”.40 The differences are historical in origin. Metabolomics has its foundations in microbial and plant studies typically applying mass spectrometry. Metabonomics originated in the study of mammalian systems, particularly for toxicology, with NMR spectroscopy. Today the two terms are becoming synonymous and interconvertible as discussed recently.12
| Metabolomics |
| The study of the quantitative complement of metabolites in a biological system and changes in metabolite concentrations or fluxes related to genetic or environmental perturbations. Studies are typically holistic in nature though targeted studies are also encompassed in the term metabolomics. |
| Metabonomics |
| The quantitative measurement of the dynamic multi-parametric metabolic response of living systems to pathophysiological stimuli or genetic modification. Often, though not exclusively, focussed on biofluid analysis to follow systemic metabolism. |
| Endometabolome |
| The complement of metabolites located within a cell or tissue, often referred to as the intra-cellular metabolome. The intra-cellular contents are typically a composite of metabolites, enzymes and other biochemical species and are highly reactive and dynamic in nature. Sampling normally includes a metabolic quenching process to inhibit enzyme activity and halt metabolism. |
| Exometabolome |
| The metabolome present exterior to and in contact with cells and tissues and often referred to as the extra-cellular metabolome or metabolic footprint. Metabolic activity in the exometabolome is minimal as enzymes are typically not present or are at concentrations significantly lower than in endometabolomes. No metabolic quenching is often required and therefore the exometabolome provides a cumulative temporal picture of intra-cellular metabolism and metabolite uptake and secretion from a biological system. |
| Metabolic profiling |
| The holistic study of the metabolite complement of a biological system to define relative differences in the measured response or changes in the metabolite concentrations. Appropriate experimental design and analytical strategies are required to provide detection of 100–1000s of metabolites in a valid and robust manner. This term is often matched with metabolite profiling which originated and is applied in the pharmaceutical industry in the study of drug metabolism. |
| Metabolite fingerprinting |
| Global snapshot of the intra-cellular metabolome typically acquired with holistic and rapid acquisition analytical platforms. The complete sample or crude extract is analysed. Quantification and chemical identification is not typically available. Applied as a screening strategy for 100–1000s of samples before further targeted studies involving metabolic profiling. Provides a snapshot of metabolism at a single point in time. |
| Metabolic footprinting |
| Global snapshot of the extra-cellular metabolome, those metabolites secreted from a biological system (typically cells and tissues) or changes in metabolites consumed from the exometabolome. The metabolome measures the footprint of intra-cellular metabolism on the extra-cellular environment. Defines the inputs and outputs from biological systems and typically simpler to acquire and prepare samples than for cells and tissues. Provides a picture of metabolic changes occurring over time. Serum, urine, breath and CSF are defined as metabolic footprints of intra-cellular tissue and cell metabolism, although one could argue that there should be a distinction between fluids where homeostasis is necessary (e.g. blood plasma and CSF) and biofluids like urine and cell culture media where the environment is less rigorously controlled as a result of excretion, and thus may concentrate compounds that would be otherwise toxic inside the body. |
| Targeted analysis |
| The quantitative study of a small number of metabolites, typically related by chemical or biological similarity. Analytical methods include extensive separation of analytes and sample matrix and include the construction of calibration curves and quantification of metabolites. |
| Metabolic quenching |
| The process of inhibition of enzymes and halting of metabolic reactions. Normally performed by increasing or decreasing the temperature and/or by chemical degradation of protein structure. |
| Metabolite extraction |
| The process of separation of metabolites from the biological system and sample matrix. The level of complexity of separations is dependent on the experimental strategy applied. The complexity is greater for targeted analysis than for metabolic profiling. |
| Serum and plasma |
| Serum is the aqueous liquid fraction separated from clotted blood. Plasma is the aqueous liquid fraction of unclotted blood, and usually requires the addition of an anti-clotting factor (e.g., EDTA, citrate or heparin) which may interfere with subsequent analyses. They differ in composition by the presence (plasma) or absence (serum) of fibrogen. Serum and plasma are composed of water, metabolites, proteins, and salts, but not cells, and are sampled from the mammalian circulatory system. |
| Urine |
| An aqueous solution composed of waste products produced by filtration in the kidneys and stored in the bladder. Composed of water, urea, salts and metabolites, and may also contain significant amounts of protein in diseased individuals which can interfere with subsequent analyses. |
| Cerebrospinal fluid (CSF) |
| Aqueous fluid present in the spinal column, surrounding the brain and present in the intra-cerebral vesicles. Acts to protect the brain from mechanical and immunological damage and to provide the distribution of neuroendocrine factors. Composed of water, salts, metabolites and proteins, and is somewhat isolated from blood plasma from the semi-permeable blood–brain barrier. |
| Breath |
| Gas inhaled or expelled from the lungs during the process of breathing. Composed of volatile chemicals including oxygen, carbon dioxide, water, isoprene and other metabolites. Breath can be separated into condensable and non-condensable components. |
| Cell |
| A structure composed of a membrane or cell wall and containing an aqueous solution of biomolecules. Cells are sub-units of multi-cellular systems. Mammalian cells are eukaryotic and contain nuclei, unlike prokaryotic cells, and a range of sub-cellular compartments (e.g., mitochondria, Golgi and endoplasmic reticulum). |
| Tissue |
| An aggregate of cells of similar structure and which perform a similar function. Tissues can consist of a single cell type or more usually a conglomerate of multiple cells. |
| Descriptive statistics |
| Summarize a sample population by simply describing its observed characteristics numerically, or graphically. Numerical descriptors include mean, median, standard deviation, median absolute deviation, quartile ranges, and range for continuous data types (for example peak areas), while frequency and percentage are more useful in terms of describing categorical data (like detection of a metabolite over an experiment). |
| Inferential statistics |
| Use structure in the sample data to draw inferences about the population represented, whilst accounting for random, and systematic, error. These inferences may take the form of: asking yes/no questions about the data (hypothesis testing), describing associations within the data (correlation), modelling relationships within the data (regression), extrapolation, interpolation, or other modelling techniques like analysis of variance (ANOVA), time series and data mining. |
| Univariate statistical methods |
| Analysis methods accepting only one random variable at a time. Multivariate data can be analysed using univariate statistical methods by splitting up the data into a series of univariate vectors (in our case single metabolite vectors), which are each independently analysed. Any correlation between vectors is ignored; however distributions of univariate outcomes can be compiled, for example, a histogram of relative standard deviation across all detected metabolites. |
| Multivariate statistical method |
| Methods which take the form of statistical methods encompassing the simultaneous observation and analysis of more than one random variable. These may be descriptive (Principal Components Analysis), or inferential. Inferential multivariate methods can be further divided into unsupervised, where unbiased structural inference is made using algorithms that search for undefined structure in the data, and supervised, which is the multivariate equivalent of univariate hypothesis testing. |
Metabolomics is applied to fulfil a variety of objectives which will be described in greater depth later in this review. The study of the metabolome can offer a number of advantages, whether applied individually or in combination with other biochemical analyses.3,41 The metabolome is downstream of other biochemical species with biochemical information traditionally viewed as flowing from genome to transcriptome to proteome to metabolome. The metabolome is a sensitive measure of the biological phenotype, an indicator of both genetic and environmental (diet, drug, lifestyle) perturbations. These interactions are shown in Fig. 1. Changes in the metabolome (both metabolic flux and metabolite concentration) can be greater than observed in the proteome or transcriptome. It has been shown theoretically (with Metabolic Control Analysis)42 and experimentally36,43 that the change in enzyme concentration has a limited effect on metabolite flux but a greater effect on the concentrations of metabolites. The metabolome is highly dynamic in nature, the flux (rate of synthesis or consumption) of metabolites is measured in seconds compared to turnover in the proteome and transcriptome which are commonly measured in minutes to hours. This allows the metabolome to be a rapid indicator of environmental perturbations. Indeed, rapid metabolic changes within the cell are largely allosteric in nature relying on metabolites acting as inhibitors or activators, while changes in gene expression and covalent modification of enzymes can be slower, adaptive processes in mammals (e.g., as a result of hormonal action). Furthermore, many of the covalent modifications of proteins are mediated by metabolites such as ATP, acetyl-CoA, glucose and fats, and so metabolomics should be able to follow many (but not all) changes associated with both short and long term metabolic and physiological control. For these many reasons Van der Greef described the promise of applying metabolomics in clinical systems biology to detect early metabolic perturbations before disease symptoms are observed and more drastic measures are required.44
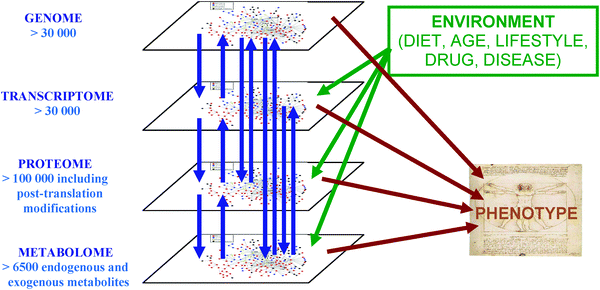 | ||
| Fig. 1 The complex interactions of functional levels (genome, transcriptome, proteome and metabolome) in biological systems. Bidirectional flows of biological information are observed between the genome, transcriptome, proteome and metabolome. The complex interaction of components from all the functional levels and the environment produces the phenotype, the output of the system measured in systems-level metabolomics and systems biology. | ||
Many offer the view that as the number of metabolites is lower than the number of genes, transcripts or proteins the metabolome is easier to investigate in a systems-wide study. This is now being realised as not to be the case! The wide ranges of physicochemical properties and metabolite concentrations ensure that the complexity and diversity is too great for fully comprehensive and holistic investigations. However, metabolomics does offer high-throughput applications where many hundreds of samples can be analysed every week. This reduces the financial costs per sample to acceptable levels and significantly lower than for proteome and transcriptome, though the purchasing costs for high-specification instruments are still high (typically greater than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 GB Pounds). However, many of these instruments are already found in the analytical groups of chemistry and biochemistry departments and in some ways the advent of metabolomics has given a new impetus to (bio)analytical chemistry. Finally, a metabolite present in multiple sample types can easily be detected with the same analytical platform with changes in sample preparation. This provides metabolomics laboratories the ability to investigate multiple biological systems and the development of centralised metabolomic facilities for regional use are being observed (for example, The Netherlands Metabolomics Centre).45
000 GB Pounds). However, many of these instruments are already found in the analytical groups of chemistry and biochemistry departments and in some ways the advent of metabolomics has given a new impetus to (bio)analytical chemistry. Finally, a metabolite present in multiple sample types can easily be detected with the same analytical platform with changes in sample preparation. This provides metabolomics laboratories the ability to investigate multiple biological systems and the development of centralised metabolomic facilities for regional use are being observed (for example, The Netherlands Metabolomics Centre).45
2. The development and growth of metabolomics
(i) The history of biochemistry
Metabolomics has a long history that significantly predates the coining of the word. Indeed, metabolism is the oldest branch of biochemistry, starting with pioneering studies by the likes of Buchner over a hundred years ago to understand the processes involved in glycolysis in so-called yeast juice. During the following 100 years a mass of research has increased our understanding of metabolism, and biochemistry in general, and thus the field of metabolomics stands on the shoulders of many biochemistry giants. These initial studies were largely reductionist in purpose and focused on small and specific areas of metabolism in a primarily qualitative manner. Today, these masses of data are being compiled into textbooks, encyclopedias and metabolic reconstructions to define the metabolic network in a holistic approach. These developments represent a shift in understanding and research; the focus of current studies is changing from reductionist to holistic and is increasingly providing a systems-wide understanding of biological function (systems biology). There is a shift in how scientists view metabolism. Traditionally metabolism has been viewed as a set of linear metabolic pathways which can be inter-related. Today metabolism is viewed as a network.18(ii) Early beginnings
The beginning of the global study of metabolites was observed in the 1960s and 1970s. Separately Horning and Pauling applied gas chromatography–mass spectrometry (GC-MS) to acquire metabolite profiles of human blood and urine vapour in 1968 and 1972, respectively.46,47 These studies were achieved because of preceding technological advances, here the development and interfacing of gas chromatographs and mass spectrometers. Similarly, the availability of NMR spectrometers in biological and medical departments also encouraged its use to profile metabolism in cells and biofluids.48,49 Brenner commented that the flow of new scientific discoveries originates from technical developments and this has been reviewed with a metabolomic and systems biology focus.50 Instrumental developments to provide greater sensitivity and separation resolution (e.g. UPLC, comprehensive GC × GC and 2D-NMR) and improvements in computational power and software needs have driven the ability to perform metabolomics research forward. The following twenty years provided few publications. One significant advance was the application of mass spectrometry for the diagnosis of inborn errors of metabolism.51 These are one of the first examples of comprehensive metabolic profiles being applied for clinical diagnosis and demonstrated the potential of metabolomics to the next generation of scientists.(iii) The emergence of metabolomics at the start of the 21st century
The sequencing of the first genomes in the late 1990s and early 21st century (including yeast52 and human53) welcomed in the post-genomic era and provided the real emphasis for metabolomics to develop and prosper. In 1997 and 1998, respectively, the research groups of Oliver and Ferenci were the first to define the metabolome.16,17 Two publications arrived within a twelve month period and are classified as the pioneering papers in metabonomics and metabolomics, respectively. In 1999, the Nicholson group at Imperial College in the UK published a paper defining metabonomics and the application of NMR to the study of human biofluids.40 In 2000, Fiehn and colleagues at the Max-Planck Institute of Plant Physiology published research defining the application of GC-MS to the study of plant metabolomes.54 From these roots has developed a flourishing scientific field. In 2009, 1503 papers were published (as defined in Web of Knowledge with the keywords [metabolom* OR metabonom*]) and the number of papers each year is increasing at an exponential rate as shown in Fig. 2. The majority of studies apply MS or NMR spectroscopy as the analytical instrument of choice. However, metabolomics is still a relatively small scientific field in comparison with proteomics and transcriptomics.![The growth in the number of publications described as [metabolomics OR metabonomics] in Web of Knowledge. The number of publications describing the application of NMR (black), MS (white) and others (shaded grey) is included to highlight their rate of application and influence on the development of metabolomics.](/image/article/2011/CS/b906712b/b906712b-f2.gif) | ||
| Fig. 2 The growth in the number of publications described as [metabolomics OR metabonomics] in Web of Knowledge. The number of publications describing the application of NMR (black), MS (white) and others (shaded grey) is included to highlight their rate of application and influence on the development of metabolomics. | ||
During the previous ten years metabolomics has advanced in stages. Many publications in the first 5 years described technological developments including the application of new analytical methods or instruments, as well as novel informatics approaches. Although these types of publications are still being observed, showing the growth of metabolomics, an increase in the number of biologically focused studies is being reported. There is a larger emphasis on standardisation, the importance of experimental design and quality assurance and the application of metabolomics to advance our understanding of biology. Metabolomics is now playing an important role in microbial, plant, environmental and mammalian studies although lessons are still being learnt from the complexity of data and the difficulties of quality and experimental robustness. These are being combined with systems biology studies as discussed below.
(iv) The role of metabolomics in systems biology
Systems biology is an emerging scientific discipline with the objective to study all (or a large proportion) of the biological components of a system, and more importantly, to study the complex interactions between these components. This is in contrast to traditional studies which are defined as reductionist and focussed on a small subset of the components and interactions.55 Biology in the previous 100 years has provided volumes of data regarding the components focussing on a given gene, protein or metabolite. However, in many cases this isolated knowledge of individual components has not provided accurate mechanistic understanding of complex phenotypes. These can include many mammalian diseases which can be described as multi-factorial, where there are multiple causes and multiple effects that interact with one another. Systems biology is increasingly being applied because it has been realised that the properties of a system are different to the properties of a single component. Sauer et al. discussed in 2007 that reductionist approaches have been hugely successful in separately identifying many of the components and single interactions in systems but have not provided quantitative information of the complete set of interactions that produce the function (or emergent properties) of a complete system. Systems biology has the objective to understand qualitatively and more importantly, quantitatively model and predict how genetic and environmental changes influence biological function at the systems level.56Fig. 1 describes the complex relationship of (bio)chemicals in mammalian systems and their interaction with other variables (including the environment) to produce the measured phenotype. The important transformation from reductionist to systems-wide studies in clinical applications has been previously reviewed.55,57,58Systems biology is an integrative science applying high-throughput experiments (for example, ‘omic measurements) along with theory and computational modelling to provide in silico (and predictive) models of components and their interactions. The strategies applied in systems biology are shown in Fig. 3. The main two types of studies performed are top-down and bottom-up. Top-down takes a holistic view of the system and aims to study the components and interactions of the complete system, generally on a semi-quantitative approach for example, metabolic profiling performs a holistic study of the metabolome and interactions of metabolites with other metabolites and biochemicals. Holistic studies of the proteome, transcriptome and epigenome can all be performed. By contrast bottom-up systems biology performs a quantitative study of specific components and interactions within the system, providing significantly greater accuracy and resolution compared to top-down approaches. For example, measurements of enzyme kinetics, protein concentrations and metabolite concentrations can be combined with metabolic reconstructions (for example see ref. 18) to construct in silico models of metabolism. One expects and hopes that these two approaches will meet in the middle. Alternatively one can adapt a ‘middle out’ strategy59 in which one starts at any level which contains sufficient data (e.g., on pathways) and reaches towards the other levels and components of the whole system. To fulfil these objectives systems biology is applied with a multi-disciplinary team performing genome-wide ‘omics’ measurements, biochemistry, biophysics, computational modelling, informatics and text mining among others. A number of excellent reviews describing the requirements and impact of systems biology are available.7,8,10–12,57,60–63
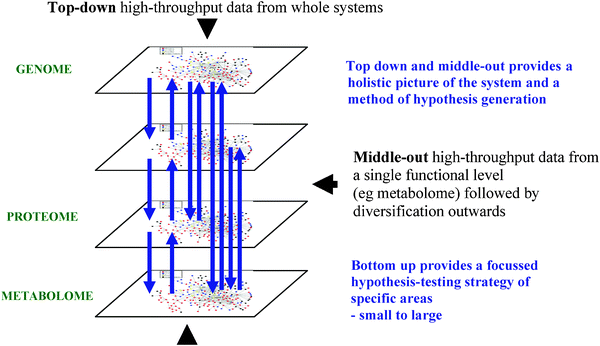 | ||
| Fig. 3 The experimental strategies applied in systems biology; bottom-up, top-down and middle-out. | ||
The role of metabolomics in systems biology is to define qualitatively or quantitatively the interactions of metabolites (and associated changes) in biological networks. Primarily, the components are metabolites and the interactions are metabolic reactions, on the holistic scale the metabolic network. However, in complex biological systems metabolites interact with other non-metabolite components in the regulation of biological processes (for example, metabolite interactions with mRNA riboswitches) and the study of metabolites provides indications of these processes. The development of holistic and inductive data acquisition strategies in the early years of the 21st century has advanced the role of metabolomics in systems biology. The application of metabolomics in the systems wide study of mammals is at the beginning of a long journey. A number of applications of metabolomics in top-down and middle-out strategies are described in Section 5.
3. Experimental strategies and experimental design
(i) Experimental workflows
The metabolomics experiment proceeds along a generic workflow which is specific to the experiment and sample type being studied. The workflow is shown in Fig. 4 and can be described as a metabolome pipeline.64 A combination of different expertises is required in multi-disciplinary teams including clinicians, analytical chemists, statisticians, epidemiologists, biologists, modellers and bioinformaticians. The components of the workflow begin with the design of the experiment, proceed through the biological and subsequent analytical experiment to data analysis and data storage. Each step in the workflow has multiple options and choosing the correct option for specific experiments is critical to ensure that robust and valid results are induced. Many scientists (including the authors) recommend and undertake development and validation of each step to ensure they are ‘fit-for-purpose’.65–69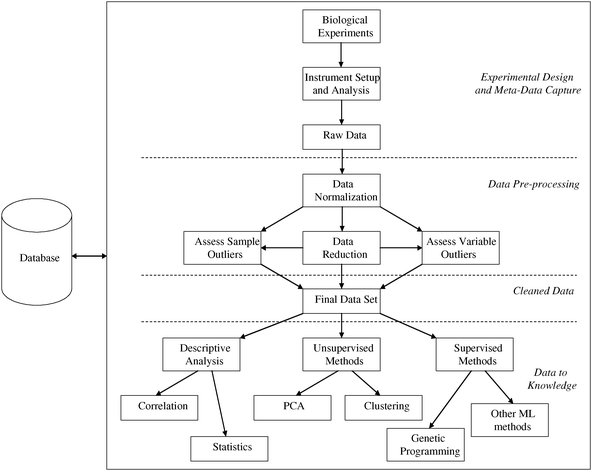 | ||
| Fig. 4 The metabolome pipeline. The integration of design, performance, storage and analysis of metabolomics experiments and their attendant data. Kindly reprinted from ref. 64 with permission from Springer. | ||
(ii) Metabolic profiling
In general terms, two types of workflows can be applied depending on the level of biological knowledge to be acquired; targeted studies and untargeted studies or metabolic profiling. Fig. 5 details the differences between the two workflows. Many metabolomic studies in the previous ten years have started with limited biological knowledge and for which a specific scientific hypothesis is not available. A general hypothesis is available (for example, there is a metabolic difference between humans diagnosed with cancer and healthy humans), but a specific hypothesis stating which metabolites are related to (patho)physiological changes is not available. In these studies the objective is to design an experiment to acquire valid data on a wide range of metabolites present in multiple metabolite classes or metabolic pathways and dispersed across the metabolic network. Subsequent analysis of the data can provide novel insights into changes in the metabolome related to the biological question being asked. These types of studies are inductive or hypothesis-generating.70 Traditionally, deductive or hypothesis testing studies were thought to be the only reliable method of scientific discovery. Many advances in biological understanding would not have been possible without inductive metabolomic studies (for example, ref. 71). Subsequent studies are hypothesis-testing or reductionist and aim to test a scientific hypothesis through the acquisition of data for a fewer number of metabolites, those metabolites highlighted in inductive experiments.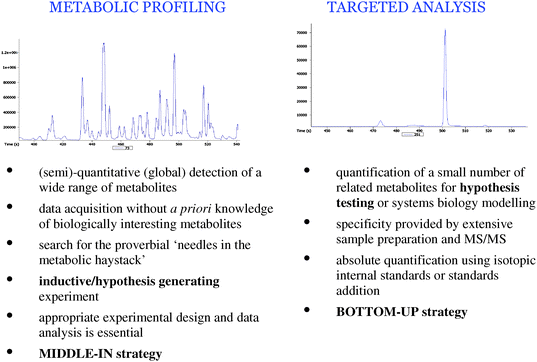 | ||
| Fig. 5 Comparison of metabolic profiling and targeted analysis strategies in metabolomics. | ||
The importance of appropriate experimental design in metabolomic studies is discussed in detail later. However, it is worth noting that many large-scale metabolomics studies are not financially feasible without convincing preliminary data. In studies such as those looking for risk-factors in the general population due to changes in lifestyle/diet (or similar epidemiological studies), or biomarker studies for diseases where patient numbers are statistically required to be in the 1000s the authors recommend three separate studies; (1) discovery study, (2) study validation and (3) cohort validation. Studies 1 and 2 use a highly constrained Design of Experiment (for example, a matched case–control design) where sample numbers range from 20–100s for each class and are sampled from two independent populations. These initial studies should be small enough to be financially viable as a pilot study but rigorously designed so that the resulting ‘biomarker’ metabolites are robust and independently validated. Study 3 expands the Design of Experiment to a cross-section of the complete ‘at-risk’ population employing larger sample numbers (n = 1000s). This final study defines the true utility of the ‘discovered’ markers in the target population. The journey through multiple studies is summarised in Fig. 6.
 | ||
| Fig. 6 The journey through multiple studies in epidemiological-type investigations. There are two highly constrained studies (discovery study and study validation) performed with tens or low hundreds of samples from two independent populations. A final cohort validation is performed on a cross-section of the complete ‘at-risk’ population employing thousands of samples so as to test the markers in the target population. | ||
Metabolic profiling, or untargeted analysis, is applied in inductive studies with an experimental objective to acquire analytical data relating to a wide range of metabolites in the metabolome. Sample collection, preparation and analysis are developed to provide detection of hundreds or thousands of metabolites in a single analysis. The obtained precision and accuracy is ‘fit-for purpose’ but lower than for targeted analysis and semi-quantitative data are acquired. Limited sample preparation is performed to ensure that metabolite loss is not present during processing steps. Relative changes in the measured responses (and not concentrations) of metabolites are calculated in most, but not all, applications. There is no construction of calibration curves for each metabolite because of the technical difficulty of preparing many hundreds of separate calibration curves, the availability of authentic chemical standards and most importantly the lack of metabolite information before analysis. These studies are performed with no or limited a priori information regarding the composition of the sample. The limitations of this strategy should be remembered in that no or limited absolute quantitative data are available, precision and accuracy are reduced to ensure detection of a large number of metabolites and chemical identification of all metabolites detected is currently not feasible on a routine and automated basis.
(iii) Targeted studies
At the opposite end of the spectrum are targeted studies, which are focused on a specific number of metabolites (typically less than 20) which are related in function or class and provide (absolute) quantitative metabolite concentrations with a high specificity, precision and accuracy. These are methods which traditional bio-analytical chemistry has applied for many decades and are applied in deductive or hypothesis-testing studies where the metabolites of biological interest are known. A greater level of sample preparation is used to separate the metabolites from all other metabolites and sample matrix components. Appropriate internal standards (commonly isotopic analogues of the metabolites to be quantified) should be applied to ensure accuracy. As these methods are well known in science and many of the developments discussed in this review will focus on the younger strategy of metabolic profiling.(iv) Semi-targeted studies
Recently, an intermediate strategy has been developed, sometimes described as semi-targeted analysis. Here experimental methodologies are developed to provide quantitative or semi-quantitative concentrations of metabolites with higher accuracy, precision and specificity than for metabolic profiling for up to 400 metabolites.72,73 These metabolites are chosen from a multitude of chemical classes and metabolic pathways to provide a wide coverage of metabolism, though are biased to those metabolites where authentic chemical standards are commercially available and relatively inexpensive to purchase. The strategy applies triple quadrupole mass spectrometers to provide a greater specificity compared to time-of-flight or Fourier transform instruments for metabolic profiling. This strategy assumes that metabolic changes will be reflected in the relative concentrations of these metabolites or is applied when a priori knowledge of the areas of metabolism of biological interest is known (e.g., TCA metabolites and heart disease).74 When biological knowledge is non-existent or limited there is the possibility that the metabolites of biological interest are not detected and metabolic profiling, where larger numbers of metabolites are detected, is more appropriate. However, it should be noted that metabolic profiling does not provide detection of the complete metabolome and therefore the possibility of not detecting the metabolite(s) of interest is still present but with a reduced probability. Metabolic profiling does not provide the automatic chemical identity of metabolites which this new strategy does and therefore provides a rapid and direct transfer of results to biological conclusions. Metabolite identification is one of the current areas requiring significant developments in metabolic profiling applications.75–77 Throughput is reduced because multiple injections for a single sample are required but accuracy and precision are greater than for metabolic profiling.(v) Design of metabolomic experiments
Metabolomic studies of mammalian systems generally adhere to one of two basic designs. Either: (A), they are studies of the metabolome in a highly controlled laboratory environment such as the perturbation of an in vitro tissue culture, or the effect of drug therapy in an animal model; or (B), they are epidemiological studies investigating metabolic factors affecting the health and disease of human populations (identification of biomarkers or risk indictors of diseases, drug efficacy and toxicity, and indicators of diet, lifestyle, age or particular time dependent conditions such as pregnancy).Studies of type A tend to be small (sometimes as low as 10 samples) as experimental conditions can be highly controlled, such that the treatment, or exposure, under examination is the only random variable. The treatment/exposure can often be quite extreme, compared to a human study, thus the expected change in the metabolome is much greater allowing suitable statistical power to be achieved with lower sample numbers. These studies can also be constrained by external factors such as the availability/cost of collecting samples or breeding animals. Studies of type B, until very recently, have also been small. However, as discussed by Broadhurst and Kell,78 to enable a greater understanding of the metabolic status of humans, medium to large-scale epidemiological studies are required in order to take account of the substantial diversity observed in physiology, metabolic status, and lifestyle in the general human population. Large-scale studies are required also to boost the power of any subsequent statistical analysis, so that subtle differences within the subject cohort can be detected. For example, given an identical change in metabolite response the statistical confidence interval for a biomarker will decrease as the sample size increases, thus reducing the probability of false discovery.
Fortunately, through recent advances in analytical equipment and methodology, it is now economically viable to analyse the metabolic profile of many hundreds of samples in a single week, and therefore thousands over several months. This scaling-up of metabolomic studies from small laboratory based proof of principle to full blown epidemiological studies requires that great care be taken in the selection of participants (Study Design), the collection of the biological samples, and the design of the analytical experiment (Design of Experiment), in order to make subsequent data analysis unbiased and fit-for-purpose.
(vi) Study design
In epidemiology, a study design can either be controlled (i.e., experimental) or observational. Controlled studies will generally be a comparison between two or more treatments, where the experimentalist controls the treatment (or exposure). Often one compares against a standard vehicle, placebo, or traditional treatment. Experiments can also often be multi-factorial, comparing multiple factors at once (e.g. the comparison of two treatments at multiple time-points). Observational studies involve the analysis of a population in which the ‘observer’ has no direct control over the assignment of subjects into treated and untreated populations (or exposed and not exposed). Observational studies break down into four types: case–control, where factors that may contribute to a medical condition are assessed by comparing subjects who have that condition (the ‘cases’) with patients who do not have the condition but are otherwise similar (the ‘controls’); cross-sectional, where a cross-section of a given population is compared at a given time-point irrespective of disease outcome, or exposure; cohort, where two groups of people are established as exposed versus non-exposed, and these groups are followed over time for occurrence of disease; and longitudinal, where a cohort is followed over a long period of time in order to study developmental trends.Two special cases of these general classes that are of particular interest to metabolomics are: the nested case–control study, where the case–control sub-populations are taken, and matched, from a single cross-sectional population; and the crossover study. A longitudinal study where subjects receive a sequence of different treatments (or exposures) and thus each subject acts as his/her own control. The prominent characteristic linking these two types of design is the highly constrained matching of comparison groups. Optimal matching occurs when each exposed subject is matched to a comparable unexposed subject to whom all the measurable parameters are equal in every aspect except the exposure of interest. This of course happens automatically in a crossover study. A slightly less constrained, but still robust, matching process would be to perform matching on a population basis. That is, each comparison group is matched by all measurable parameters such that both groups can be considered statistically as being drawn from the same population, except on the basis of the exposure of interest.
By strongly matching comparison groups any difference in metabolome can be more closely associated with the exposure of interest (i.e. the analysis is not biased). This is particularly important in metabolic profiling studies due to their holistic, ‘measure everything’ nature.
(vii) Design of experiment
When the number of samples in a given metabolic profiling experiment is small, and the study design is highly constrained, the design of experiment (DoE) is relatively straight-forward. All the samples can be analysed in a single analytical batch in a relatively short period. The only recommended action is that the sample preparation order and injection order be randomised so that no run-order bias is introduced into subsequent statistical analysis.In medium to large-scale epidemiological metabolomic studies far more care in the DoE is necessary. By far the biggest constraint on a large-scale metabolomic experiment is that all the samples cannot be run in a single analytical batch. Obvious issues of instrument reproducibility in the medium to long-term and necessary periodic maintenance come into play. The issue of reproducibility is very much instrument-dependent. In NMR spectroscopy, instrument reproducibility is very good, as the sample does not physically interact with the operating parts of the instrument and therefore changes in sensitivity from instrument contamination are not observed. However, this is not the case with LC- or GC-MS. In any chromatography–mass spectrometry system the sample unavoidably interacts directly with the instrument. This inevitability leads to changes in measured analyte response over time both in terms of chromatography and mass spectrometry. The degree, and timing, of signal attenuation is not consistent across all measured analytes and it is also dependent on the type of biofluid measured. It is advised that Quality Control samples (QCs) are periodically analysed throughout an analytical run in order to provide robust Quality Assurance for each chemical feature detected. The QC samples should be identical (drawn from a pool) for the whole Analytical Experiment. It has been shown that for human serum, changes in response due to sample–instrument interaction requires that a single metabolomic experiment should be broken up into batches of approximately 90 injections (60 samples and 30 QCs—a QC analysed every fourth sample), followed by an instrument cleaning step.68 Later, data conditioning algorithms can use the QC responses as the basis to assess the quality of the data, remove peaks with poor repeatability, correct the signal attenuation, and concatenate batch data together post chemical analysis and prior to statistical analysis.11,68,79,80 After signal correction and batch-integration each detected peak should be required to pass strict Quality Assurance criteria. While there are no generally accepted criteria for the assessment of repeatability in metabolomic data sets, the Food and Drug Administration (FDA) in the USA suggests a range of criteria that should be applied. In the guidance for bioanalytical method validation in industry81 the FDA recommends for single analyte tests that tolerance limits are set such that the measured response detected in two-thirds of QC samples is within 15% of the QC mean, except for compounds with concentrations at or near the limit of quantification (LOQ), in these cases a tolerance of 20% is acceptable. In the case of metabolic profiling applying LC-MS, the methods are not specific for one analyte of interest, but instead the aim is to detect thousands of analytes, therefore an acceptance tolerance of 20% would seem to be appropriate. Any peak that did not pass the QA criteria should be removed from the dataset and thus ignored in any subsequent data analysis.
Signal correction and batch-integration can never be perfect so it is important not to introduce any systemic bias into a study when choosing the order of injection and batch membership. It is recommended that within-batch run-order is assigned stochastically to each sample, such that the sample order is random but stratified by exposure group. Also it is recommended that each batch is stratified comparably to the total experiment population. That is, each batch contains a representative cross-section of the total study. Again this will reduce bias in the data analysis.
Bias is another important consideration. The problem is often referred to as a problem of ‘confounding variables or confounding factors’, although the latter phrase has a slightly different emphasis and meaning in the epidemiological literature (‘‘confounding is a distortion in the estimated exposure effects that result from differences in risk between the exposed and unexposed that are not due to exposure’’).82 Imagine a study in which we wished to measure biomarkers for ethnicity, and compared the serum or urine metabolome of samples taken from Japanese and Russian people. No doubt we would find differences, but it would be quite wrong to ascribe these to ethnicity as the differences are just as likely to be due to something else that co-varies with ethnicity. Diet is likely the most important co-varying difference here.
Reproducible standard operating procedures (SOPs) are essential to ensure that samples are collected, stored and transported in an identical manner in all countries. Ransohoff83 refers to bias as ‘‘the most important ‘threat to validity’ that must be addressed in the design, conduct and interpretation of such (i.e. biomarker) research’’, and he comments that ‘‘Bias can be so powerful in non-experimental observational research that a study should be presumed ‘guilty’—or biased—until proven innocent’’. Bias cannot be compensated for by large sample numbers—in fact this can even make things worse by persuading readers of the validity of spurious differences that are actually due simply to confounding factors that happen to correlate with the class discrimination of interest. Naturally the correlation improves with sample size, as does the statistical confidence in the defined difference.
Bias can be exceptionally difficult to remove, although careful age and gender matching is a good start. Having a gender bias (in which say males are more common in the case than in the control cohort) means that there is a danger of creating a model that is actually discriminating on gender. It has been highlighted that gender and drug intake can be observed in disease biomarker studies.84 Bias can be introduced at every stage of the metabolomic workflow as well as the study design. It is important that samples from each comparison groups are collected, transported, stored, analytically prepared and injected into the analytical instrument in a standard and, as far as possible, identical way. If in a case–control study cases are collected at one study centre and controls are collected at a different study centre then, again, no doubt we would find differences, but it would be wrong to ascribe these to disease exposure as the differences in the metabolic profiles are just as likely to be due to some factor regarding the collection and storage procedures.
(viii) Sample collection and preparation
The objective of sample collection and extraction is to ensure that a sample is acquired and analysed which is representative of the metabolome in the sample before collection. In targeted studies the limited number of metabolites of interest is known and highly-specific analytical methods can be developed and validated to ensure that specificity, accuracy and precision are appropriate. In metabolic profiling studies, methods are developed to provide a holistic profile of metabolites with a wide range of physicochemical properties. The accuracy and precision are inherently reduced as a consequence of the comprehensive nature of the study. There are many different methods to achieve the same experimental goals. Those commonly used are discussed below.The methods of sample collection are technically different to those applied in proteomics, transcriptomics or genomics. Many metabolomes are highly dynamic and operate with high metabolic fluxes compared to the other functional levels. The flux of metabolites is measured in units of seconds for many metabolites compared to minutes and hours for proteins and transcripts and is highly dependent on the metabolite, enzyme and environmental conditions. The process of sample collection and preparation is typically separated in to two steps: (a) quenching of metabolic activity and (b) extraction of metabolites into an appropriate solvent for analysis.
Quenching is a process where metabolism, or more specifically enzymatic activity, is decreased or stopped so as to obtain a sample where metabolic flux is eliminated. This is typically performed by increasing or decreasing the temperature of the sample and/or providing chemical inactivation of enzymes, specifically alterations in the 3-D protein structure by addition of organic solvents and/or heat. Quenching is more technically demanding for tissues and cells compared to biofluids because of the risk of cell membrane permeability being increased resulting in leakage of metabolites from the cell or tissue. The complexity of sample preparation is dependent on the experimental strategy to be applied. Greater levels of metabolite separation from matrix are observed for targeted analysis (for example, solid phase extraction or liquid–liquid extraction) compared to metabolic profiling where extractions are optimised to detect as many metabolites as possible.
These processes of sample collection and preparation inhibit metabolic flux and in most studies disrupt the spatial distribution of metabolites in extraction processes. In metabolomics, data will show a representative snapshot of the metabolome of a sample. Temporal changes are typically investigated by multiple sampling of the system though recent developments have allowed in vivo temporal changes to be studied. Spatial mapping can also be performed by the use of NMR in the form of magnetic resonance imaging or spatial imaging with mass spectrometry, both of which are discussed later in this review.
Tissues, cells, urine and cerebrospinal fluid (CSF) are collected and the temperature immediately reduced to sub-zero temperatures and samples are stored at −80 °C.35 Blood requires an extra step of preparation to allow separation of serum or plasma and these are performed at temperatures of 4 °C for up to 12 hours before freezing and storage. For blood sera, blood is allowed to clot before centrifugation and storage of the liquid phase (serum). For plasma, blood is collected into tubes containing anti-coagulants (citrate, EDTA, heparin) to stop clotting and the liquid plasma phase is collected.35 Even with precautions of reduced temperatures there is still the possibility, though significantly reduced, of enzymatic activity in these blood samples. The collection of samples should be performed with high-quality plastics and specific types of collection tubes are not recommended, including gel-based serum collection tubes. The validation of methods for sample collection of human biofluids and tissues is essential as samples are not collected in the confines of a well-regulated academic laboratory but typically in clinics. Validated standard operating procedures (SOP) are now available and significant research has been performed to assess sources of variability and fitness for purpose.66,85 Biological samples acquired from mammals are complex and contain metabolites as well as low and high concentration matrix components (polymers including cell walls and proteins, inorganic salts, lipids). Typically there is a process to separate matrix species from the metabolites of interest while ensuring maximum recovery of metabolites. This is an extraction step and the process is dependent on sample type, experimental strategy (targeted analysis or metabolic profiling) and analytical instrument to be employed.
The most complex and experimentally difficult system to extract is tissue. The release of intra-cellular metabolites into the extraction solvent typically requires homogenisation and mechanical or chemical lysation of cell walls to release the metabolites.35,86 Other methods employ freeze clamping and it should be emphasised that no single method for quenching and extraction is applicable to all sample types and metabolites. The ruggedness of tissue structure and ease of homogenisation and lysation are dependent on the type of tissue, for example muscle tissue is significantly more rugged than liver or kidney tissue. Typically, greater than 30 mg of tissue is required. A range of methods have been developed for extraction of tissues and include tissue homogenisation and chemical or physical methods for cell lysation.35,86 It should always be remembered that tissues will contain blood and separation of blood and tissue metabolomes is technically demanding. The best approach for rapid tissue collection is to wash the tissue at a reduced temperature before freezing.
Serum and plasma obtained from blood are one of the most complex biofluids. They contain high concentrations of proteins which are removed by deproteinisation during extraction processes. The type of extraction performed is dependent on the metabolites of interest and a number of studies have been performed to investigate the most appropriate strategies.87,88 None of these studies have applied a multi-platform approach though and this is still required. Extraction into an organic solvent in excess (ethanol, methanol, acetonitrile or acetone) is performed. Metabolites in serum and plasma are both freely available in the liquid fraction and are bound to proteins. It is assumed that extraction processes degrade metabolite–protein complexes but limited research has been performed in metabolic profiling. Research elsewhere has applied proteolysis to release bound metabolites. The lipid content of serum and plasma can be significantly greater than many other metabolites and can mask metabolite detection. Want et al. and Wilson et al. have separately developed methods to remove abundant lipids, specifically phospholipids.89,90
Urine acquired from healthy mammals has a very low protein content and preparation steps are simple and normally involve dilution and analysis.91 However, high concentrations of urea are present (up to 2%) which are detrimental to GC-MS instrumentation and data quality. Traditional urine analysis applying GC-MS is performed after urease treatment (for example diagnosis of inborn errors of metabolism) to remove the high concentration of urea.51 However, one study has shown the negative effect this process can have on the concentration of other metabolites.92 CSF is protein and urea free and limited sample preparation is also required for this biofluid.
Sample throughput is dependent on the type of sample, the experimental strategy applied and the availability of automation. Sample preparation is composed of a limited number of processes in metabolic profiling and many steps (liquid handling and extraction) can be automated. Analytical instrument throughput is typically tens or hundreds of samples a day and automation of sample preparation allows a similar throughput for samples in a controlled process which can operate 24 hours a day and seven days a week if necessary.
(ix) Analytical instrumentation
A large range of analytical platforms have been applied in metabolomic investigations. MS and NMR spectroscopy are the two techniques applied most frequently in metabolic profiling and will be discussed in more detail in this review. However, many other techniques are applied and include Fourier transform infrared and Raman spectroscopy93 and chromatography with detectors other than mass spectrometry or NMR spectroscopy (for example flame ionisation detectors).94 Although outside the scope of this review the multitude of technologies available should always be considered as one platform typically offers specific advantages dependent on the application required. For example, electrochemical detection provides a level of specificity in the detector to allow the study of electrochemically active metabolites, particularly for redox active metabolites.95 However, the choice of an appropriate analytical strategy is difficult compared to traditional analytical chemistry. Universal detection is essential in holistic methods. The wide diversity of metabolites (physicochemical properties and concentration) ensures that no one single analytical platform is appropriate for all investigations.The platforms of mass spectrometry and NMR spectroscopy provide the greatest frequencies of applications in metabolomics today. The techniques and their applications in metabolomics will now be discussed.
(x) Mass spectrometry
Early developments of mass spectrometry occurred more than a century ago with the pioneering work of Thomson and Aston, which has been reviewed recently.96 In the period since, great advances have been observed and the instruments of today provide many advantages in their application in metabolomics.77,97,98 Although this review is not a tutorial a concise introduction to the operation of mass spectrometers is required. For more detailed descriptions a number of books and reviews77,97,98 are available.Mass spectrometers operate by the formation of positively or negatively charged species (ions) from analytes of interest, separation of ions according to their mass-to-charge ratio (m/z) and detection of ions. Separation and detection is performed under high vacuum pressures to reduce the number of ion–ion or ion–molecule collisions which can influence the mass resolution, mass accuracy and sensitivity of instruments. Ion formation in ionisation (or ion) sources can be performed at high vacuum pressures (for example, MALDI or electron impact) or at atmospheric pressure (for example, electrospray (ESI) and Atmospheric Pressure Chemical Ionisation (APCI)). The m/z is the measured parameter in MS with the majority of ionised metabolites being singularly charged because of their low molecular weight which is capable of carrying single charges only, compared to proteomics where analytes are of high molecular weight and multiply-charged species are detected. Mass spectrometers can scan the mass ranges of interest, which for metabolomics is typically from 20 amu to 1500 amu. Scan times are typically rapid because of fast electronics and allow multiple mass spectra to be acquired every second, aiding both metabolite detection and structural elucidation by MSn scans. The advances in electronics and manufacturing precision have provided a suite of high-specificity platforms for metabolomic investigations. Time-of-flight, quadrupole, Fourier transform (FT) and hybrid (Q-TOF, ion trap–Orbitrap, triple quadrupole) instruments are applied in the majority of applications because of the advantages they provide for a given application. The generic advantages include high sensitivity (typical limits of detection of low micromoles per litre), fast scan or acquisition rates applicable for detection of narrow (less than 3 s) chromatographic peaks, the ability to provide high mass resolutions and mass accuracy and they allow chemical identification of metabolites. Most instruments in metabolomics studies provide one or more of these advantages.
A range of ion sources are employed though two are used with the highest frequency. Electron impact ionisation is a technique applied with gas chromatography where the column eluant is introduced to the source operating under a vacuum. An electron current emitted from a filament is accelerated through the sample region. Quantum mechanical interactions between electrons and gas molecules provide the ejection of an electron as the most probable mechanism of ion formation, though negatively charged ions from electron capture can also be formed at a significantly lower rate than electron loss. The ionisation process is applicable to all metabolites entering the source. The energy of ions required for ionisation is typically set at 70 eV and this imparts a high level of energy to the ionised molecule. As the system is under vacuum and energy cannot be lost through ion–molecule collisions the energy is lost through covalent bond fission. This produces a fragmentation pattern and a mass spectrum highly characteristic of the molecule. This can be applied for chemical identification.
The second commonly applied ionisation technique is electrospray, used with liquid introduction systems including liquid chromatography and capillary electrophoresis. These operate at atmospheric pressure and allow the coupling of liquid systems to mass spectrometry. During ionisation from liquid samples evaporation of the solvent is required. If this was performed under vacuum the vacuum pressure would be quickly lost. The introduction of atmospheric pressure ion sources allowed ion formation at atmospheric pressure and subsequent extraction of ions only into the vacuum region of the mass spectrometer. This was a significant technological advance and allowed the routine and robust interfacing of liquid chromatography platforms with mass spectrometers. Molecules in the liquid phase are charged by the non-covalent addition or loss of chemical species (for example, H+, NH4+, Na+ or K+). The liquid flow is then nebulised into a droplet spray and continued fission and solvent evaporation provides desolvated charged ions which are accelerated from the atmospheric region to the vacuum region of the mass spectrometer. Positive and negative charged ions are formed depending on the electrical potentials within the source and physicochemical properties of the metabolite (for example, organic acids are thermodynamically more probable to lose a proton than gain a proton and so are typically detected in negative ion mode). Generally samples are analysed twice, once in positive and then again in negative ion mode. Other ion sources are applied less frequently including chemical ionisation (GC-MS) and APCI (LC-MS).
Mass spectrometry is typically applied to the analysis of gaseous or liquid sample, though solid samples such as tissues can be analysed either directly or after extraction processes. Mass spectrometry offers a number of advantages over other analytical techniques including sensitivity, chemical identification capabilities and when combined with chromatography the ability to detect hundreds or thousands of metabolites in a given sample. Mass spectrometry is the tool of choice if a wide ranging metabolic profile or quantitative analysis of a few metabolites is required. However, these systems provide disadvantages also. The samples physically interact with the instrument and this can cause changes in response over short or medium periods of time. The application of quality assurance through the periodic analysis of QC samples is important in mass spectrometric studies.11,68,79,80 Although, chemical identification is possible, automated and high-throughput approaches for identification in metabolic profiling studies are lacking at present and identification of all detected features is currently not possible.75–77 Although quantification is achievable, the response factor for a metabolite is dependent on the sample matrix which can change between samples creating differences in measured responses for identical metabolite concentrations. This is particularly true for ESI in LC-MS and CE-MS. The inclusion of a chemical analogue of the metabolites of interest (an internal standard, for example 13C-glucose for the quantification of glucose) is applied for targeted analysis to compensate for these differences, though is not applicable for metabolic profiling where the metabolites of interest are not known a priori and the inclusion of hundreds of internal standards is not experimentally or financially achievable.
(xi) Direct infusion mass spectrometry
Mass spectrometry can be applied with or without chromatographic or electrophoretic separation before detection. Direct infusion (or injection) mass spectrometry (DIMS) is applied with ESI-mass spectrometers where the sample is directly introduced into the mass spectrometer and this can be performed in an automated flow injection mode.99 A single summed or averaged mass spectrum is acquired for each sample as shown in Fig. 7. As metabolome samples are highly complex an instrument with high mass resolution and hence mass accuracy is required to ensure fit-for-purpose mass separation of the majority of metabolites detected. Mass resolution defines the mass peak width (for Full Width Height Maximum (FWHM) calculations), higher mass resolutions provide narrower peak widths and the ability to separately detect metabolites of similar but not identical accurate mass. Mass accuracy defines the error of the determined mass of a metabolite with the theoretical mass. High mass resolution and accuracy instruments provide the ability to separately detect ions of similar accurate mass and allow accurate mass determination for putative metabolite identification. Definitive metabolite identification is limited as metabolites with the same accurate mass but different chemical structures (for example, stereoisomers such as glucose and fructose) will be detected as a single m/z. These high-specification instruments include TOF and FT instruments. DIMS provides a high-throughput system where up to 60 samples per hour can be analysed though with a reduced capacity for definitive metabolite identification and an increased level of ionisation suppression as the complete sample and matrix are ionised at the same time and competition for charge is high. Ionisation suppression is observed in ESI when multiple species are competing for the available charge, common in complex metabolome samples. The frequency of DIMS applications is relatively low though. Recent advances have shown improvements in both the mass accuracy and number of metabolites detected. Southam and colleagues have presented Single Ion Monitoring (SIM)-stitching experiments applying multiple and adjacent SIM windows in a FT-MS instrument.100 Space-charging effects observed in trap-based instruments can reduce the instrument sensitivity and mass accuracy through interactions of different ‘packets’ of ions in an orbital motion. To solve this problem a reduced ion current was required and therefore smaller SIM mass windows (30 amu in the published example) across the mass range were acquired with lower total ion currents in each SIM window followed by the stitching together of all data to produce a single mass spectrum for each sample. This provided an improved mass accuracy and increased number of detected features, a number similar to that detected using LC-MS. This strategy can be employed for profiling of metabolomes with short analysis times (5.6 minutes per sample in the quoted example, quicker than typical LC-MS analysis times). The authors (WD, DB, RG) have applied this to the characterisation of metabolomes using UPLC-MS and requiring 2–3 days of instrument time per sample (unpublished data).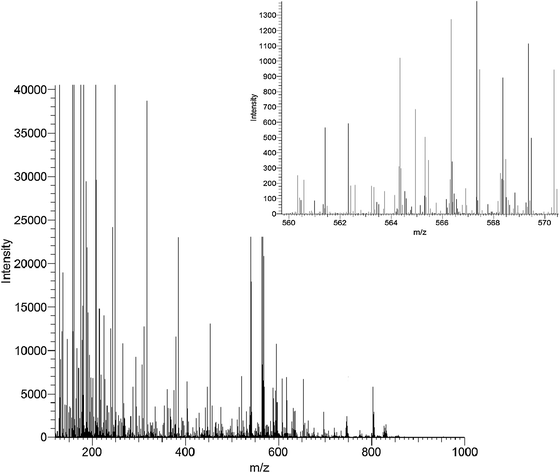 | ||
| Fig. 7 A typical mass spectrum acquired from Direct Infusion Mass Spectrometry of human serum. | ||
(xii) Gas chromatography–mass spectrometry
Chromatographic separation can be divided into three classes; gas chromatography, liquid chromatography and capillary electrophoresis. Gas chromatography is the oldest hyphenated technique being applied for 50 years in combination with MS. GC provides high chromatographic resolving power with peak widths typically of less than 3 seconds. Separations are today performed with capillary columns onto which a stationary phase is coated on the inner surface and through which a carrier gas flows at 1–2 mL min−1. This flow rate allows direct introduction of the complete eluant into an electron impact ion source. Chromatographic separation of complex samples are optimised generally with different stationary phases and the ramping of the oven temperature from low to high temperatures, though other factors including stationary phase thickness, column i.d. and carrier gas flow rate are also varied. Metabolomic samples are complex and ‘dirty’. In GC-MS the non-volatile components of the sample are introduced into the heated injection inlet and may pass to the start of a GC column but rarely are introduced into the source. This allows robustness in instrument operation where columns can be applied for many months with routine maintenance involving removal of small sections of the inlet end of the column and replacement of the GC injection liner. The frequency of replacement of the injection liner is defined by the researcher and automated replacement after every 1–10th injection is achievable. A guard column can also be applied to inhibit sample components passing on to the analytical column. A column can be employed for hundreds or thousands of injections, much higher than for LC-MS where columns are typically changed every 100–300 injections.GC-MS is applied to the analysis of metabolites of low boiling points to enable vaporisation and travel through a column at temperatures less than 350 °C. The majority of endogenous metabolites do not have sufficient volatility. Chemical derivatisation is typically applied to increase the range of metabolites detectable by GC-MS. Here oximation followed by trimethylsilylation (TMS) to remove intra- and inter-molecular hydrogen bonding is the most common due to its holistic applicability for metabolites of different functionality (CO2H, NH2, OH, SH).54,101 A typical m/z 73 single ion chromatogram of serum is shown in Fig. 8. Other methods have been applied and provide higher levels of specificity or faster completion times including chloroformate derivatives.102 Oximation and TMS reaction times range from 15 min to overnight, chloroformate reactions are less than 2 min. The stability of derivatisated metabolites is also different; the presence of water in TMS derivatives is detrimental as it produces hydrolysis of TMS ester. This is not the case for chloroformate derivatives. The derivatisation process can be automated and placed in-line with derivatisation completion just before injection to ensure that sample stability is not compromised. Typically, 10–100 injections per day can be performed.101 However, results have shown that increased numbers of metabolites are detected when longer analysis times are employed.65,103
 | ||
| Fig. 8 A typical m/z 73 single ion chromatogram of urine acquired using GC-MS. | ||
(xiii) Comprehensive GC × GC-MS
More recently a technique with greater chromatographic resolving power than conventional GC has been introduced and applied with some success in metabolomics.104–106 So called ‘Comprehensive’ GC × GC-ToF-MS employs two chromatographic columns of differing column chemistry to provide separations in two dimensions. The first column is a 30–60 m column and the second column is a shorter (typically 1–3 m) column of different stationary phase chemistry with a modulator located between the columns to focus the column eluant from column 1 and introduce this as a focussed plug on to column 2. Retention times are typically minutes and seconds for columns 1 and 2, respectively. Sample focussing and transfer from column 1 to 2 are typically temperature based (cold nitrogen jets for focussing and hot nitrogen jets for release) though pressure based systems are also available. Comprehensive GC × GC-MS can provide increased sensitivity caused by the focussing effect and narrower peak widths associated with the system, providing the detection of lower concentration metabolites not detected by conventional GC-MS. However, initial problems with the systems have been observed particularly with the accuracy and reproducibility of raw data processing. The use of second columns with narrow internal diameters and thin stationary phase thickness is improving the chromatography and therefore accuracy of data processing,105 further steps are still necessary to provide fully-automated operation.(xiv) Liquid chromatography–mass spectrometry
The routine application of LC-MS is a more recent observation, particularly after the commercial introduction of atmospheric pressure ionisation sources in the 1990s. Before ESI, there were other less reproducible or robust techniques of sample introduction and ionisation. However, the application of LC-MS has increased during the previous ten years.107 Liquid chromatography provides separations as a result of metabolite equilibration between a liquid mobile phase and a solid (or liquid) stationary phase. A mobile phase traverses a LC column (at flow rates of 0.1–2.0 mL min−1) packed with particles on which stationary phase is present. In traditional LC, chromatographic resolving power and peak widths are dependent on the column dimensions (i.d. and length), stationary phase, mobile phase flow rate and temperature. Peak widths are typically wider than for GC, and LC is not thought of as providing high chromatographic resolution. However, in 2004 a new instrument for LC was introduced by Waters and subsequently by other companies. Waters termed this Ultra Performance Liquid Chromatography (UPLC) and employed the capabilities of narrow peak widths provided by higher flow rates, increased pressures and smaller diameter column packings.108,109 For the first time sub-2 μm stationary phase particles were applied and this was only possible because of advances in instrument and column chemistry design which allowed the 3-fold increase in pressure to be maintained without detriment to instrument or column performance and lifetime. UPLC can provide chromatographic resolution equivalent to GC and also provides a higher sensitivity than conventional LC. Wilson and colleagues have reviewed the impact this technological advance has provided in metabolomics.110 A typical base peak ion (BPI) chromatogram is shown in Fig. 9. UPLC-MS can provide the detection of thousands of features in a given sample and different column chemistries can be applied, the most commonly applied are reversed-phase C8 or C18 bonded stationary phases. These reversed phase separations employ a solvent system which starts with a high water content and a gradient elution increases the organic solvent (methanol or acetonitrile) to provide chromatographic separation.68,111,112 This is ideal for relatively non-polar metabolites, including lipids, though is not applicable for polar metabolites including sugars and some amino acids. Here, Hydrophilic Interaction Chromatography (HILIC) is starting to be investigated where separations are performed with a hydrated silica column and with gradient elutions running from high organic to high aqueous.113,114 This allows separation of more polar compounds compared to non-polar lipids which are poorly retained. Serum and plasma are deproteinised in methanol or acetonitrile solvents and therefore lyophilisation followed by reconstitution in water is not required as is observed for reversed phase systems. Combinations of both types of separations are feasible.115 Generally, no derivatisation is performed in LC-MS metabolic profiling but this can be applied for more targeted analyses or to increase selectivity or sensitivity.116 UPLC provides rapid analysis times if required and optimised appropriately though as for GC, the number of metabolites can be shown to increase as analysis time increases.68 | ||
| Fig. 9 A typical base peak ion (BPI) chromatogram of plasma acquired using UPLC-MS. | ||
(xv) Capillary electrophoresis–mass spectrometry
Capillary electrophoresis (CE) is the third platform applied for metabolite separation before MS detection in metabolomics.117–119 Here, electrically charged species (LC and GC apply neutral charged species) are separated in an electrically conductive liquid phase under an externally applied electrical field and resulting in electro-osmotic flow. The electrophoretic migration velocity is dependent on the electrical field strength, the ionic charge and the metabolite cross-sectional diameter. Columns are normally narrow i.d. capillary columns, typically silica. CE provides separation efficiency equivalent to or better than UPLC and GC-MS and smaller sample volumes are required, as are volumes of organic solvents or high-purity gases. CE-MS is less frequently applied than GC-MS and LC-MS, with specific centres of excellence observed in Japan and the Americas. Typically, samples are analysed in duplicate or triplicate in different modes for the analysis of cationic and anionic polar metabolites separately. The analysis of non-polar metabolites is technically limiting. The technique was initially introduced in 2003 and due to technical challenges limited applications are still observed.(xvi) Nuclear magnetic resonance spectroscopy
NMR has become an invaluable tool for chemists and structural biologists, and for more than 20 years has also been used extensively in metabolic profiling research. The ubiquity of protons in cellular metabolites and the fact that other nuclei are observable by NMR spectroscopy (e.g.31P and 13C) mean that a relatively large number of different metabolites can be detected simultaneously. NMR spectroscopy benefits from being quantitative, highly reproducible and, unlike other profiling modalities, non-selective; that is to say, the sensitivity of this technique is independent of the hydrophobicity or the pKa of the compounds being analysed. Furthermore, the resonances present in an NMR spectrum provide large amounts of structural information, and enable the identification of individual constituents within a sample through the interpretation of, amongst other features, chemical shifts and coupling constants. However, because of the small energy differences between ground and excited energy levels relative to thermal energy, and hence small population differences, the technique does suffer from relatively low sensitivity, particularly when compared with mass spectrometry. In this respect there is a drive to ever higher magnetic fields to improve the sensitivity of the experiment.The majority of metabolomic samples analysed by NMR spectroscopy are in solution state, although it is possible to analyse intact tissue samples using high resolution magic angle spinning (MAS) NMR.120,121 Samples typically are either biofluids, such as urine or plasma, or metabolites extracted from tissue samples and subsequently re-dissolved in solvent. NMR is a non-destructive technique thereby allowing several analyses to be conducted on the same sample. In contrast to MS-based methods, sample preparation for NMR-based metabolomic experiments is relatively minimal. A small amount of deuterated solvent such as D2O or chloroform (CDCl3) is added in order to provide a frequency lock signal which is used to control for drifting of the magnetic field. A chemical shift reference compound such as TSP may also be added. Additionally, depending upon the type of sample, it may be necessary to buffer the pH using a phosphate based buffer; a number of metabolites such as citrate and histidine show significant pH dependent chemical shift variation. All ionisable metabolites can show some pH-dependent chemical shift. The addition of a pH buffer minimises this effect, although there may still be some differences between samples which have to be considered during data interpretation.122 In general, 3 mm or 5 mm NMR tubes are used for analyses, and require approximately 200 and 600 microlitres of sample, respectively. Such volumes completely fill the observe volume of the coil, thus maximising sensitivity and allowing an easier shim (the process whereby the magnetic field is made more homogeneous to ensure narrow line widths in the subsequently acquired NMR spectra). Alternatively, samples can be analysed via flow injection NMR to increase the rate of sample throughout.123 This technique involves the sequential direct loading of samples into the magnet from a 96-well plate. Post acquisition, the sample is directly transferred out of the magnet to be retained or disposed of, and the transfer capillary is washed to avoid sample contamination or spill-over.
The majority of NMR-based metabolomic studies use a simple one-dimensional solvent suppressed 1H NMR pulse sequence to acquire the data. The 1D NOESYPR1D is particularly popular as it provides good solvent suppression while maintaining a flat baseline. Signal attenuation is an important consideration when comparing NMR data, as it is essential that the same technique of water suppression is applied in all experiments to prevent attenuation differences of off-resonant peaks being mistakenly interpreted as biological variation. A 1H NMR spectrum of a liver tissue extract is shown in Fig. 10.
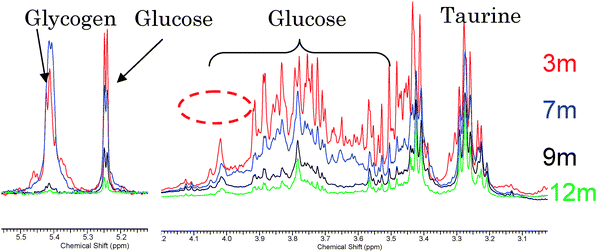 | ||
| Fig. 10 A 1D 1H NMR spectrum of extracts of liver tissue across an ageing time course from 3 months (3 m) to 11 months (11 m). | ||
Another consideration when acquiring metabolomic data is that many biological samples, particularly biofluids such as plasma which may not have been pre-treated or extracted, often contain large molecular weight molecules such as phospholipids, triglycerides and lipoproteins which give rise to broad signals in the resultant spectra. These may obscure the narrow resonances arising from lower molecular weight molecules such as sugars and amino acids, yet these smaller molecules are often of greater biological interest. To facilitate the observation of narrower resonances, the 1D-1H Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence can be applied. This produces T2 spectral editing, thus attenuating the contribution that large, motionally restrained metabolites such as lipids make to the resultant spectrum. Similarly diffusion ordered spectroscopy (DOSY) has been used to attenuate small molecules, and selectively examine large molecules.124–126
Undoubtedly, the largest disadvantage of NMR spectroscopy relative to other analytical modalities is its inherent insensitivity. Therefore, NMR spectroscopy can only reliably detect and quantify metabolites present in relatively high concentrations. Using a simple one-dimensional pulse sequence typically 20–40 metabolites can be detected in tissue extracts,125,127 30–100 metabolites in urine,122,128 and 20–30 metabolites in blood plasma or serum.84,129 2D-NMR has recently shown improvements in the number of metabolites detected and identified through the use of cryoprobes and larger field strengths. Despite this 1H-NMR spectroscopy has proved to be highly discriminatory in the classification of certain phenotypes, toxicological insults and disease processes. For example, Raamsdonk and colleagues used metabolomics as part of a preliminary study of functional genomics in Saccharomyces cerevisiae. The aim of the work was to use genes of known function to elucidate the role of unstudied genes in an approach they termed functional analysis of co-responses in yeast (FANCY)43 which could be expanded to the entire genome of yeast. This approach allowed the co-clustering of genes of a similar function (e.g., glycolytic, oxidative phosphorylation) demonstrating that genes of unknown function could be examined by this approach. Since then, similar NMR based profiling methods have been applied to elucidate key regulatory points on metabolic pathways,130 and to metabolically profile cell culture media as part of metabolic footprinting.131
The insensitivity of NMR and its ability to classify phenotypes and/or disease processes may seem somewhat contradictory. However the success of this technique appears to be attributable to the high concentration metabolites it detects. Many of these metabolites, such as ATP and glutamate, are found in several metabolic pathways, and in terms of the metabolic network of the cell, these metabolites represent points which can be perturbed by a number of stimulations. However, restricting the coverage of the metabolome to such a small number of metabolites may hinder the isolation of metabolites as unique biomarkers for disease processes and confound the deduction of which pathways are perturbed during a given modification. It is also possible that the effects measured are non-specific to the disease being studied (biases or confounders). This problem has been highlighted by a number of studies, for instance Connor et al. observed that a number of metabolic alterations previously described as biomarkers of liver and kidney toxicity were actually effects of food restriction in sick animals post-toxic insult.128 In another example conducted at Papworth Hospital (Cambridgeshire, UK) the potential of an NMR based metabolomic approach in the prediction of various stages of occlusion of coronary arteries was demonstrated.132 Blood samples from patients with severe atherosclerotic disease could be differentiated from blood samples taken from patients with normal coronary arteries, as determined by angiography, using 1H NMR spectroscopy with greater than 90% specificity. The difference between the sample groups could be attributed largely to subtle changes in lipoprotein composition. However, Kirschenlohr and colleagues have since identified a number of confounders for a diagnosis based primarily on lipid composition, in particular gender and statin treatment (a common therapy for coronary artery disease) which may have biased the results of the original study.84 When data were re-modelled, confining them to only one gender and treatment, the predictive power of the generated models to predict coronary artery disease was reduced by approximately 30% depending on the patient population being compared (i.e., gender, statin treatment, severity of disease).
In an attempt to overcome some of these issues associated with sensitivity in NMR based metabolomics, a number of strategies are being developed to increase the sensitivity of NMR. For instance, cryoprobes have proved to be particularly useful in improving signal to noise for 13C NMR based metabolomics. Cryoprobes have the electronic circuitry of the probe and amplifier chilled to reduce electronic noise133,134 and can provide improvements of the order of 4-fold for 13C NMR spectroscopy. Another physical method to improve sensitivity is to move to smaller coils, which not only require less material, but are also intrinsically more sensitive.135,136 Furthermore hyphenated approaches such as liquid chromatography can selectively concentrate metabolites during the chromatographic run and be analyzed either in real-time or using stop-flow techniques.137,138 Finally, one recent area of much interest is the possibility to use hyperpolarised substrates to selectively enhance the resonances of key metabolites. In this approach magnetisation is transferred from a free radical to the substrate of interest (often 13C labelled metabolites) in a solid, usually in the form of freezing the sample using liquid nitrogen within a magnetic field, and irradiating the sample with microwaves to transfer polarisation to the free electron in the free radical. Magnetisation is then built up on the labelled substrate by the nuclear Overhauser effect. The sample is then defrosted rapidly and injected into the biological system. This has been used to follow metabolism in real time in tumours, the heart and the brain.139–141 However, because there is a time delay between creating the magnetisation and delivering the sample to the region of interest, most studies have focussed on resonances with long T1 relaxation times. This has prohibited the use of many metabolites. While this is a major current limitation, this is also an area of active research and so may be circumvented in the future and provide a revolution in spectroscopy in vivo.
In addition to limitations associated with detection limits 1H NMR spectroscopy also suffers from a large number of co-resonances, whereby different metabolites are found to have resonances in the same region of the NMR spectrum. This can be solved to a degree by the use of two-dimensional NMR techniques123 or the use of nuclei with more dispersion, such as 13C.129
(xvii) Processing of raw analytical data
Data acquired on analytical instrumentation are complex and can be exported in multiple different computer-readable formats depending on the type of instrument and the instrument manufacturer’s preferences. These data are defined as raw data and only occasionally are these data passed forward for data analysis. Commonly, raw data are converted and exported in a specific format before a pre-processing step is performed. These processes are performed with two objectives. The first is to reduce the file size through a reduction of data complexity and provide data in a format suitable for import into a range of software packages. Raw data files for MS can be large (10–1000 MB), while those for most one-dimensional NMR experiments are more modest (∼200 KB per spectrum). A second reason for a pre-processing step is to provide alignment of data to ensure that metabolites or features are identified as the same metabolite or feature for all samples analysed. Inaccuracies in this process will provide multiple reports of a single feature (e.g., a metabolite feature could be reported as metabolite 10 in one sample and metabolite 15 in a second sample). This is highly detrimental to subsequent data analysis processes. ‘Drift’ in the parameters applied to identify specific features or metabolites is commonly observed for mass spectrometry (retention time, migration time, accurate mass, response) and NMR spectroscopy (chemical shift associated with changes in pH or osmolarity). Raw data processing typically converts continuous data to segmented data. For example in chromatography–mass spectrometry the continuous 3D data (retention time vs. response vs. mass) is converted to a 2D matrix of chromatographic peak vs. peak area.The processing of NMR spectra originally involved an approach referred to as ‘bucketing’ which is a simple automated manner of integration of the spectra into buckets of for example ∼0.04 ppm which also reduces the impact of small changes in chemical shift.122 One problem with this approach is that the integral regions increase the number of co-resonant peaks in the spectrum, confounding the discrimination power of key metabolic changes. To circumvent this software packages have been produced that allow peak fitting of standard spectra.142 Some researchers have decided to live with the effects of chemical shift variations and use the total NMR spectrum, benefiting from recent improvements in computational power.143 Finally, others have approached the problem by making use of the mathematical structure of the free induction decay acquired during the NMR spectrum to allow automatic peak picking.144 Finally, it should be noted that while the vast majority of spectra involve 1D techniques, with improvements in probe sensitivity and the movement to higher field strengths some have opted to use multidimensional NMR spectra, thereby reducing the effects of co-resonances and also aiding chemical assignment.127,145
In mass spectrometry-based metabolomics, files are typically converted from the proprietary instrument manufacturer raw data format to a text-based file format known as NetCDF (network common data format).146 This is a common format which is compatible with many other software packages and is available as an open source program. However, this format is not defined as a standard format in MS. Three other open source, XML-based data formats are available: mzXML,147 mzData148 and a third format mzML which is a fusion of the other two formats.148 XML (eXtensible Markup Language) is a methodology where rules for encoding electronic documents to be applied in systems biology and from many different sources are defined.149 This allows the fusion of data from multiple sources including genomics, proteomics and computational models to be applied in systems biology. These formats for MS data have been developed within the proteomics and systems biology communities though are infrequently applied in metabolomics, for two reasons. These formats are not currently supported by many of the available software programs applied for the conversion of raw instrumental data and for pre-processing of metabolomics data. The second reason is the lack of knowledge by the users in the availability of different formats and therefore the ability to convert from the traditional formats (including netCDF) to new standardised formats. However, assistance is also required from the systems biology community to ensure that these formats are appropriate for metabolomics data. The same problems are observed with NMR data also. Here while there is an agreed cross-platform data format of JCAMP, the majority of users prefer to use the vendors own format, although there are a number of software packages which can readily convert between formats.
Data pre-processing is performed using files encoded in a common format. Data are commonly binned in DIMS applications to provide alignment for small levels of mass drift observed. Binning of data is provided where the responses for all ions within a defined mass range (‘bin’) are summed and reported as a single response. The mass bin width is dependent on the mass resolution of the instrument used, 1 or 0.1 amu mass windows are commonly applied.100,150–152 Data analysis is performed and mass bins of statistical significance can be interrogated to define the specific accurate masses which drive the observed statistical significance. However, alignment of DIMS data can also be performed without the requirement for binning.153
For chromatography-MS and CE-MS, alignment of the retention time or migration time is required and a collection of software packages are now available to convert the raw data (a 3D matrix of time vs. mass vs. intensity) to a matrix of chromatographic peaks (with associated retention time and accurate mass and/or fragmentation mass spectrum) and peak area or height. This process is sometimes referred to as ‘deconvolution’ and provides alignment of retention time and accurate mass. Software packages applied include those available as open-source (XCMS, Metalign, MZmine, MathDAMP154–157) and others which are instrument company specific (e.g., SIEVE supplied by ThermoFisher Scientific and MarkerLynx supplied by Waters). The software listed is a range but the list is not exhaustive and new or revised programs are becoming available. A review of data pre-processing of LC-MS data has been published.158
Pre-processing of chromatographic data can be inaccurate, caused by the complexity of the data and sub-optimal chromatographic separation when compared to traditional Analytical Chemistry where samples are less complex and variations in peak shapes are not observed. A reduction in the complexity of the chromatogram provided through longer analysis times or more dilute samples can provide improvements in accuracy with a loss of the number of metabolites detected.65,103 Metabolomics is often referred to as a high-throughput strategy. However, there is a compromise between accuracy, metabolome coverage and throughput which should always be considered. Improvements in the accuracy of data pre-processing would undoubtedly increase throughput. One of the main problems is that peaks detected by the instrumental platform are not reported by the pre-processing software and provide a data matrix for analysis with intermittent missing values. Some software packages return to the data to integrate retention time windows where a missing value is observed.156
(xviii) Data analysis
The fundamental goal of any metabolomics experiment is to convert raw data into biological knowledge. At a most basic level this will be the knowledge that there is a significant change in the metabolome which directly reflects a change in an experimental condition or observed exposure. However, in a mammalian study the goal is more likely to uncover a phenotypic signature of disease etiology and pathophysiology, to pinpoint diagnostic biomarkers of disease or to determine biomarkers of drug efficacy/toxicity.The type of question that one wants to answer generally drives the selection of analytical workflow. Fig. 11 shows a simplified view of a metabolomics workflow from the perspective of data analysis. The Study Design, as discussed previously, involves collecting all possible clinical information such as gender, age, physiological traits, disease status, drug use, and so on (so called clinical metadata) so that this can be used to statistically assess the study for bias and confounding factors. Similarly, the Design of Experiment will produce a database of experimental metadata such as a time-stamp for sample preparation and sample injection, the analytical batch number, and any other such data that seem relevant. These data are used statistically to assess sources of experimental bias.
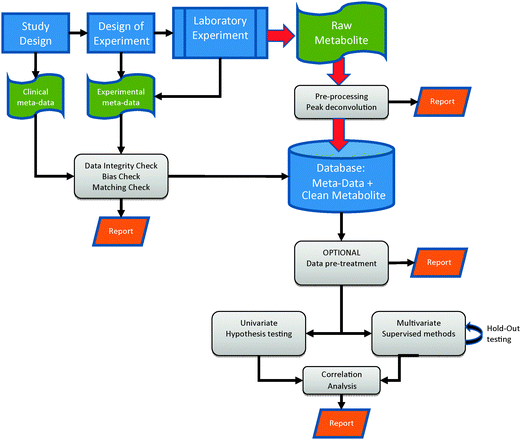 | ||
| Fig. 11 The workflow for data analysis in a holistic metabolomics experiment. | ||
Once Raw Instrument Data are obtained, they need to be converted into a matrix size of N × M where M is the number of metabolites (or metabolite features) and N is the number of biological (and technical replicate if appropriate) samples. This process, known as pre-processing or peak-deconvolution, is discussed in the previous section. The resulting data are now considered ‘clean’ and in a form suitable for statistical analysis.
Before statistical analysis is performed it is often essential to pre-treat the data such that data are normalized, scaled, or transformed;159 missing values are imputed;160 and outliers detected and removed.161 It may also be advantageous to subject the raw data matrix to some sort of data reduction, or clustering, algorithm.162–165 These algorithms, often called unsupervised learning methods, project the ‘raw’ extremely high-dimensional data (M) onto a lower dimensional basis function (P), such that the maximal amount of experimental information is conserved. Thus the low dimension projection describes the generalised, or latent, structure of the experimental data. For example, using Principal Components Analysis (PCA)166 data can be projected onto a number (P ≪ M) of Principal Components each describing, by descending degree, the directions of maximal multivariate variance in the data. Usually all the major causes of variance can be described in the first few principal components. The process of data-reduction (or dimensionality reduction) can either be used as a means of visualising the global change in the metabolome (e.g., in the form of a PCA scores plot) or as a pre-treatment step for hypothesis-based multivariate statistical/classification models, known as supervised learning methods.162,165,167–170
Another common form of pre-treatment is signal correction. Signal correction is performed to try and reduce the effects of either known or unknown bias in the data set. As discussed earlier, if QC samples have been periodically analysed throughout a run, then the effect of instrument drift can be effectively subtracted from the data set. If the causes of bias are not known then a multivariate technique referred to as Orthogonal Signal Correction (OSC) can be implemented.171 There are several flavours of OSC,172–177 but in principle they are similar. As with the unsupervised learning methods the aim here is to project the multivariate data onto a basis function of lower dimensionality. However, the basis function is not optimised by maximising all experimental variance but by maximising any variance which is orthogonal to the direction of maximum discrimination based on the treatment class. The projection of this basis function is then reverse-engineered and subtracted from the original data set. In more simple terms the algorithms remove (or correct for) any latent multivariate effects in the data that are completely uncorrelated with the treatment. OSC methods are very powerful and it is easy to ‘over-train’ the model such that the final data set no longer accurately represents the underlying measured biology, resulting in inaccurate experimental conclusions.178
The Statistical Analysis performed in a metabolomic workflow usually takes the form of hypothesis generation. Starting with a base-hypothesis (for example, “Is there a difference in the metabolome between exposed and non-exposed subjects?”) the statistical analysis goes on to suggest possible metabolite features that provisionally prove that hypothesis to be correct. These hypotheses should then be validated using classical biochemistry or targeted analyses. Using univariate statistical tests such as Student's t-test, ANOVA and non-parametric Kruskal–Wallis, isolated metabolite markers can be investigated in turn. See below for a discussion of Receiver–Operator Characteristic (ROC). Alternatively patterns of correlated biomarkers can be investigated using supervised multivariate statistical methods, where knowledge about class membership is used to help find discriminatory groups of metabolites that are significant in combination (biomarker signature), when they may not be significant individually. This is of particular interest in diseases which are considered to have a multi-factorial aetiology, or if the power of the study is insufficient for single biomarker discovery, such that the combination of metabolites in a given metabolic pathway is significant when combined. By far the most popular multi-purpose supervised algorithm in the metabolomics community is PLS-DA (Partial Least Squares Discriminant Analysis).179–181 However note that “A necessary condition for PLS-DA to work reliably is that each class is tight and occupies a small and separate volume in X-space. Moreover, when some of the classes are not homogeneous and spread significantly in X-space, the discriminant analysis does not work”.181 In clinical, and especially epidemiological, data the boundaries between treatment groups are often overlapping, or ‘fuzzy’. Also the phenotype of the condition under study may only be evident in a very small percentage of the measured metabolome. These factors often make PLS models of the whole metabolome ineffectual. Fortunately, there are many other algorithms whose effectiveness is dependent on the choice of workflow (e.g. Canonical Variate Analysis (CVA);168 Artificial Neural Networks;170 Rule Induction;182 Inductive Logic Programming;183 Random Forests;184,185 Evolutionary Computation;186–189 Radial Basis Function Networks190 which allow disjoint relationships to be revealed which may be useful in understanding multi-factorial processes). Several specific reviews on this subject are available.3,7,64,191–194 Alternatively variable selection strategies may be combined with existing modelling methods, to search for the regions of the metabolome which most accurately model the phenotype in question.195,196 For example, Broadhurst et al.197 combined an evolutionary computation based search algorithm (Genetic Algorithm) together with a PLS regression model, to form a GA-PLS ‘data-mining’ tool; alternatively this GA ‘wrapper’ can be used prior to CVA.198 In addition, for identifying the stage of disease (e.g., Gleason staging for prostate cancer) one may seek to correlate metabolites with the quantitative progress (stage) of disease. This can be performed by univariate correlation analysis such as Pearson's product moment correlation, or in a multivariate manner using PLS regression.169 As with all supervised modelling methods these algorithms are very powerful and can easily find random associations, unless very rigorous model validation is performed.78,194,199,200
(xix) Data visualization
Data visualization is an important issue in metabolomics experiments due to the vast quantities of data collected and the complexity of the modelling methodologies. As described above multivariate projection methods can be used to visualise any general structure in the data. However, directly interpreting the scores plots and the associated loadings plots can be difficult. Equally, graphically comparing multiple univariate results can be challenging. A full discussion of this subject is beyond the scope of this paper but is reviewed here.67 One particularly useful visualization method which thoroughly illustrates the biomarker utility of either a single metabolite or multivariate predictive model is the Receiver–Operator Characteristic, or ROC, curve.201,202 ROC curves are limited to two-state experimental designs (e.g., case–control), and are constructed by plotting the sensitivity versus 1-specificity of a hypothetical decision boundary moving across the total range of the predictive score. This plot will necessarily include the points (0,0) and (1,1). If the area under the ROC curve (the AuROC) is 0.5 (the lower limit) the variable is distributed similarly between cases and controls, such that any diagnostic test based on it is valueless for discrimination. If the area under the ROC curve is 1, there is complete separation of the two populations and therefore samples can be classified with 100% sensitivity (no false negatives) and 100% specificity (no false positives). Fig. 12 shows a comparison of 5 potential metabolite biomarkers with a known ‘gold standard’ using ROC curves. In this example the metabolite pseudouridine has an AuROC of 0.96 and is therefore considered to be an effective biomarker of heart failure.203 Multiple ROC curves on a single axis can soon become extremely cluttered, as an alternative, when comparing multiple univariate biomarkers, or multiple model predictions, a plot of p-value versus AuROC can be constructed. In such a plot (Fig. 13) the more effective biomarkers approach the top left hand corner of the plot (i.e., low p-value and high AuROC).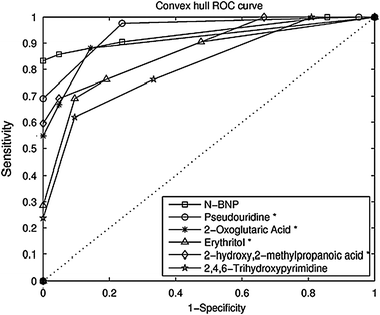 | ||
| Fig. 12 An example of receiver–operator characteristic (ROC) plots for five metabolite peaks including pseudouridine and 2-oxoglutarate and the current gold standard of N-BNP. If the area under the ROC curve is 0.5 (the lower limit) the variable is distributed similarly between cases and controls, such that any diagnostic test based on it is valueless for discrimination. If the area under the ROC curve (the AuROC) is 1, there is complete separation of the two populations and therefore samples can be classified with 100% sensitivity (no false negatives) and 100% specificity (no false positives). Kindly reprinted from a study related to heart failure203 with permission from Springer. | ||
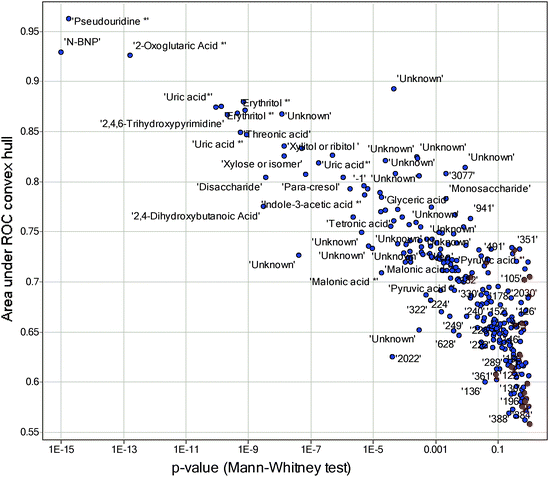 | ||
| Fig. 13 An example of plots describing the relationship between area under ROC curve and p-values for various metabolites. These plots are applicable when comparing univariate biomarkers or multiple model predictions. The more effective biomarkers approach the top left hand corner of the plot (i.e., low p-value and high AuROC). Kindly reprinted from a study related to heart failure203 with permission from Springer. | ||
(xx) Model validation and multiple testing
The types of multivariate modelling methods used in metabolomics (and indeed in other ‘omics studies) are known as data driven55,204–206 rather than knowledge driven (physically-based modelling). That is, no assumptions about underlying causality, or structure, in the metabolomic data are assumed. In such methods, often known as machine learning methods, both model parameters, model structure, and included variables are estimated. This massive amount of flexibility makes these machine learning algorithms incredibly powerful. With great power comes great responsibility;207 as pointed out by Efron and Tibshirani “Left to our own devices …we are all too good at picking out non-existent patterns that happen to suit our purposes”.208There are many publications, across all the biological sciences, pointing out the potential folly of using profiling techniques such as metabolomics, proteomics, transcriptomics, and genomics in order to discover clinically significant biomarkers.78,209–212 This criticism focuses mainly on the idea that these methods are just ‘fishing expeditions’ and you are just as likely to discover biomarkers that are randomly correlated to the effect of interest, due to the massively parallel significance testing that is performed. For example, if a hypothesis is tested using a univariate significance test and a calculated p-value of 0.05 is produced, this means that there is a one in twenty chance that the biomarker is really a false positive (false discovery). This is fine if there is only one test. However, if you perform 1000 tests you would expect to see 50 false positives—i.e. 50 random findings. So the more tests you do the more chance there is of finding a biomarker which is not biologically positive or relevant. The difficulty is checking whether the biomarker is valid, or not. P-Values can be corrected for multiple testing (Bonferonni correction; Benjamini and Hochberg; False Discovery Rate); however, the validity of these methods in ‘omic type studies has been questioned.213,214
When one uses multivariate statistics the multiple testing effects are amplified, as the significance of combinations of metabolites is being investigated. The more metabolites measured the more combinations possible. The combinatorial effects are further amplified by machine learning methods, as a number of model structures will be tested in parallel. The answer to this question of scientific robustness which has been adopted by the machine learning community is to use a subset of the complete data—the hold-out set—that is not used in the generation of the model in any way at all.215 The set used in producing the model is called the training set. Models built using the training data can then be independently validated using the hold-out set. The obvious difficulty in this design is making sure that the hold-out set is suitably representative of the training set, both in terms of clinical/experimental metadata and in terms of the metabolite profiles themselves. This is not a simple task. An alternative method of independent model validation is to use permutation testing. Here a reference distribution of model effectiveness (Q2 or Area under ROC curve) is obtained by training the chosen model type/structure to multiple random rearrangements of the labels on the observed data points. The ‘true’ model score can then be compared to this distribution of all possible models. For a more comprehensive discussion see Westerhuis et al.200 and Bijlsma et al.194
The most clinically robust method of validating biomarkers (or biomarker patterns) is to repeat the experiment with an independent sample set.74,216 If the same biomarkers appear in a completely independent study then they are much more likely to be true. Counter intuitively, the strength of validity increases for patterns of metabolites. Without going into the probability theory, it is easy to appreciate that if a combination of 5 metabolites {p,q,r,s,t} out of 1000 measured metabolites reflects a given disease phenotype for experiment 1 and the same 5 metabolite ‘rule’ is also effective in experiment 2 then the probability of these two consecutive findings being random is minuscule, much like the same winning lottery ticket being picked two weeks in a row.
A comprehensive discussion of strategies for avoiding false discoveries and good model validation practice are beyond the scope of this paper. The authors suggest the following reviews.78,200
Following the development of rules or models which are predictive of disease or drug toxicity/efficacy further knowledge concerning the pathophysiological processes may be essential. Here structures in the combination of metabolites defined as ‘biologically interesting’ are interrogated. Typically, these metabolites are classified, for example, by metabolite class or metabolic pathway as defined in databases such as KEGG and HMDB.217
5. Applications of metabolomics in mammalian studies
The growth of metabolomics as a scientific discipline has been exponential in the last ten years. 1503 papers are listed in Web of Knowledge in 2009, compared to 20 in the year 2000. The discipline has shown great promise in advancing our knowledge of mammalian systems, though significantly more work is required to demonstrate its applicability to a wider audience of scientific researchers. Success stories are being observed71 and some applications originating from industrial sources (for example, pharmaceutical companies) are never communicated to the scientific community.(i) Sample types
A wide array of mammalian biofluids, cells and tissues have been investigated in metabolomic studies. Biofluids including serum and plasma,68,218 urine,219 bile,220 faecal water,221 cerebrospinal fluid (CSF),222 saliva223 and embryo cell media224 have all been studied. Many tissue types have also been investigated including liver,225 kidney,226 cancerous tumours,227 gastrointestinal,228 placental,216 brain229 and adipose.6 Eukaryotic cells studied include Chinese Hamster Ovarian,230 human lung epithelial,231 human glioblastoma,232 rat basophil leukemia,233 cancer234 and stem.235The choice of the sample type to investigate is dependent on the experimental objective and sample availability. Logical reasoning defines that the sample type closest to the physiological area of interest would provide the greater probability of detection of the greatest number and magnitude of metabolic differences. As one moves away from the physiological area other biological processes dilute or complicate the metabolic profile. For example, study of drug toxicity of the kidney suggests that investigating kidney tissue would be appropriate and this is routinely employed. However, the acquisition of suitable numbers of tissue samples can be difficult. Biopsies are clinically difficult to acquire, painful (so longitudinal studies are limited), and tissues are recognised to be heterogeneous. Collection of complete tissues is generally only possible after death and so animal models are commonly applied. However, the three Rs are guiding principles for the use of animal testing and recommend reduction, replacement or refinement wherever possible. Placental tissue and skin can be obtained without the requirement for invasive sampling and are alternatives to animal models.236 The process of sample collection and preparation of tissue is time consuming and expensive. In human studies for health and safety reasons of the clinic (compared to laboratory) freezing of tissue is often performed in a separate location to the operating room and this temporal difference can provide changes in the tissue metabolome.
The collection of biofluids can be less evasive than tissues. Urine and faecal water collection are non-invasive. Blood collection is minimally invasive and routine with limited complications. However, the collection of CSF requires a lumbar puncture procedure which is technically demanding and can result in clinical complications. To illustrate the power of biofluid based analyses, regarding the study of drug toxicity of the kidney, if biopsies are not available urine is an appropriate biofluid to study as urine is a by-product of kidney function. Serum and plasma could also be described as an integrative biofluid as its passage around the body and physical contact with several organs of centralised function provides a suitable biofluid for an integrative phenotypic assessment of mammals, a metabolic footprint of biological function. However, in many cases the hard work of the kidneys and liver can maintain the composition of blood within very narrow limits. To circumvent this homeostatic regulation the collection of blood from specific areas of the body can be highly discriminatory and provide additional information (for example, collection from the coronary sinus artery in the study of the heart).74 Although CSF requires a highly-invasive sampling procedure this fluid provides highly selective information on the central nervous system, especially in view of the blood–brain barrier and limited transfer of metabolites across this barrier.
(ii) Biomarkers and risk factors/assessments of diseases and disease pathophysiology
A health–disease continuum exists for all mammals. As humans we are defined as healthy or ill, though in reality we exist at a point between the two extremes of health and illness. Metabolomics is playing a large role in the discovery of ‘biomarkers’ or risk factors associated with specific diseases and also in acquiring greater pathophysiological understanding of the onset and progression of diseases. Many of these studies are based around animal models, where a low level of inter-animal variability is acquired from the careful control of genetic and environmental factors in a laboratory. Alternatively, the general population is studied where inter-human metabolic variability is high caused by the large variations observed in genome, lifestyle, diet, age and body mass index (BMI) for example.237,238 While it is not possible to include a complete review of metabolomics in disease models and human patients, we hope the selective description of three large disease areas will give the reader a flavour of the approaches currently being used both at the bench and the bed side. It is hugely important to provide the translation of these advances from the bench to the bed side to allow the human population worldwide to benefit from these developments, either through new biomarkers of disease or the development of new interventions (e.g. drugs) by producing markers of efficacy.Since the completion of the human genome, focus has switched to understanding gene function in situ. Metabolomic-based approaches to functional genomics are relatively rapid, and cheap on a per-sample basis when compared with other common -omic approaches such as transcriptomics. They often prove to be significantly less labour intensive than conducting transcriptomic or proteomic based phenotyping analyses and yet still provide a comprehensive global systems description of biological effects. This makes metabolomics an ideal profiling tool for the exploration of naturally occurring and transgenic disease models. Many metabolomic studies to date have focussed on investigating disease in model organisms. The refinement of knock-out and knock-in strategies combined with accumulating sequence data has accelerated the generation of accurate disease models. The mouse is currently the most widely used tool in studies of mammalian genomics. Metabolic profiling techniques have been successfully used to characterise metabolic pathways disrupted in mouse models of human diseases including cardiac disease,239 type 2 diabetes mellitus240 and atherosclerosis.241 Additionally, the implementation of metabolomics as a screen in large scale mutagenesis programs has proven successful in identifying those mutants which possess clinically relevant phenotypes. Using this approach, models of various human metabolic diseases have been identified, including a model of maple syrup urine disease (branched chain ketoaciduria), and a model of lipotoxic cardiomyopathy which could be used to investigate the mechanisms of cardiac fibrosis and hepatic steatosis.242,243
Cardiovascular disease has been extensively profiled using metabolomics with the primary aim of improving diagnosis. In particular, the use of 1H NMR spectroscopy is well documented and its application has been used to monitor atherosclerotic disease progression,244 to differentiate underlying causes of heart disease,239 and to monitor the effects of genetic modification on cardiac metabolism.245
Due to the multi-factorial nature of cardiovascular disease, many of the available mouse models only recapitulate a fraction of the symptoms associated with this disorder. For example, most mouse strains are naturally resistant to atherosclerosis even when on a high fat and calorie rich diet. However, the ApoE knock-out mouse is a model of human atherosclerosis.246 The high circulating lipid levels in the mutant are due to a reduced capacity to clear fatty acids from the plasma, resulting in the development of atherosclerotic plaques at approximately 25 weeks and this has been the subject of metabolomic studies.241 The inability to recapitulate all features of human cardiovascular disease fully in animal models has resulted in an increasing number of human metabolomic experiments being conducted. Such studies are complicated by factors including uncertainty in the timing of disease onset and profound inter-patient variability. Nevertheless, a study conducted at Papworth Hospital (Cambridgeshire, UK) by Brindle and colleagues and discussed earlier highlighted the potential of metabolomic based approaches in the prediction of various stages of occlusion of coronary arteries.132 However, Kirschenlohr and colleagues have since identified a number of confounders for a diagnosis based primarily on lipid composition, in particular gender and statin treatment (a common therapy for coronary artery disease)84 which may have biased the results of the original study. Therefore, large patient cohorts and classification of patients according to risk factors or drug exposure is advocated to minimise contributions from such confounding clinical effects.84 However, large cohorts will not necessarily remove or highlight confounders or biases and can magnify the effects of instrument drift as samples are run across multiple batches. Mass spectrometry has also been applied to the study of cardiovascular disease including the identification of serum metabolic biomarkers of heart failure,203 where pseudouridine and 2-oxoglutaric acid were defined as potential markers and which are being assessed in further targeted work to define whether these differences are the cause or effect of the pathophysiology of heart failure. Gerszten and colleagues have applied targeted analysis of up to 250 metabolites to study heart-related diseases including myocardial ischemia73 and planned myocardial infarction.74 Interestingly, the role of TCA metabolites has been highlighted in many of these studies, demonstrating that cellular damage can be detected directly.
The development of the db/db and the ob/ob mouse models, with deficiencies in leptin signalling and leptin production, respectively, has significantly aided research into the mechanistic causes of insulin resistance.247,248 These mice were observed to be obese, hyperphagic, hyperinsulinaemic and dyslipidaemic, and they developed severe hyperglycaemia under fasting conditions.249 Metabolomic analysis of urine from the db/db mouse identified profound perturbations in nucleotide metabolism, including that of N-methylnicotinamide and N-methyl-2-pyridone-5-carboxamide, which were suggested to represent novel biomarkers for following the progression of type 2 diabetes mellitus.240 Dumas and co-workers have similarly used NMR-based urinary metabolic profiles to examine correlations between the metabolome and Quantitative Trait Loci (QTL) to understand mechanisms that pre-dispose or protect strains of mice from the development of insulin resistance and type II diabetes.250 Furthermore, the metabolic perturbations of metabolic syndrome (combination of medical problems which increase the risk of cardiovascular and heart diseases) have also been investigated using the PPAR-α null mouse. The PPARs comprise a family of nuclear hormone receptors involved in lipid metabolism. Hypoglycaemia, a consequence of impaired liver fatty acid β-oxidation and reduced gluconeogenesis, was monitored in this model using stable isotope techniques.251 The results implicated PPAR-α in the regulation of substrate utilisation for hepatic glucose production in the fasted and fed states. Following on from this study, the systemic effects of the PPAR-α mutation have been defined. Using a combination of 1H NMR and GC-MS metabolic changes have been followed in the heart, liver, skeletal muscle and adipose tissue of the PPAR-α null mouse,252 a true systems-wide study.
As insulin resistance is thought to be closely linked with so-called lipotoxicity, the accumulation of fat in tissues other than adipose resulting in metabolic impairment, it has also proved profitable to study the changes in the lipidome directly using LC-MS. Using such an approach Medina-Gomez and colleagues demonstrated the importance of PPARγ2 in controlling adipose tissue expandability and preventing the accumulation of fat in peripheral tissues.253 This approach has also been used to monitor the influence of the altered lipidome in mouse models of β-pancreatic cell failure which proved to be more predictive of the ultimate disease compared with many traditional markers of metabolic stress in these mice.254
Despite the huge challenges associated with studying disease in humans this has not deterred researchers in the hope of finding predictive markers of disease or defining the mechanisms of pathology using metabolomics. Much work has focussed on understanding the role of lipotoxicity and its role in insulin resistance in humans. Kolak and colleagues have used LC-MS lipidomics to examine inflammation in adipose tissue in obese women, demonstrating that the content of ceramides and long chain fatty acids in triglycerides in this tissue correlated with the degree of fatty liver when comparing women with similar body mass index but a range of hepatic steatosis.255 Examining why some people develop obesity and others show marked resistance, Pietiläinen and co-workers have examined adipose tissue in weight discordant monozygotic twins. At the transcriptional level there was evidence of a decrease in branch chain amino acids (BCAA) in the siblings with obesity. This was correlated with an increase in serum concentrations of these amino acids, suggesting that BCAA have a role in weight regulation.256 Newgard and co-workers have similarly followed the effects of BCAA and high fat feeding in rats, demonstrating that BCAA influenced TOR signalling and the development of insulin resistance.257 Recently, there have been discussions on whether specific and predictive biomarkers are appropriate or whether instead metabolic profile changes should be employed to define or undertake risk assessments.258
Other cardiovascular diseases have been studied. Kenny and colleagues have identified small molecular markers of preeclampsia in blood plasma demonstrating the potential impact metabolomic studies will have in the clinic in terms of biomarker discovery.259,260 Studies employing placental tissue cultures have provided pathophysiological links between hypoxia and pre-eclampsia.216,236 Specific and identical metabolic changes have been observed in plasma and tissue (for example, glutamate), showing the importance to integrate data from multiple sample types including biofluids and tissues so as to provide greater confidence to new discoveries.
Recently, studies have been designed to incorporate serial sampling before and after a controlled intervention thereby enabling patients to act as their own control thus reducing the influence of the aforementioned confounders. For example, in a study by Lewis and co-workers, serial blood samples were taken from patients undergoing alcohol septal ablation treatment for hypertrophic obstructive cardiomyopathy, before and after a planned myocardial infarction (MI).74 Using a targeted MS-based approach, perturbations in pyrimidine, tricarboxylic acid cycle and pentose phosphate pathway metabolism were identified through changes in the concentration of aconitic acid, hypoxanthine, trimethylamine-N-oxide and threonine. These findings were subsequently validated in plasma from patients of spontaneous MI. The authors of this review highly recommend the validation of results as described in the paper by Lewis and colleagues and discussed earlier in this review. The authors conclude that the study design enhanced their power to identify statistically meaningful changes associated with MI which in turn enabled the detection of very early myocardial injury. In another similar study, myocardial substrate utilisation in humans with coronary artery disease or left ventricular dysfunction was investigated during surgical ischaemia/reperfusion (I/R). This study revealed a number of pertinent metabolic alterations associated with I/R, including increased circulating concentrations of acetylcarnitine and impaired cardiac tricarboxylic acid cycle flux.261
The investigation of brain metabolism using 1H NMR based metabolomics is also well established, with a diverse array of applications including the characterisation of regional variation, brain tumours and neurological disorders.262–264 Since the brain is heavily compartmentalised, a study by Tsang and co-workers used metabolic profiling to characterise distinct neuroanatomical regions in rats ex vivo by high resolution magic angle spinning (HRMAS) 1H NMR.264 Clear biochemical differences were defined between the brain stem, frontal cortex, cerebellum and hippocampus. This provides an invaluable baseline reference for further HRMAS 1H NMR spectroscopic studies to monitor disease and specific pharmacological insults within the brain. Furthermore, using HRMAS 1H NMR spectroscopy, it was possible to characterise an accumulation of polyunsaturated fatty acids in BT4C gliomas in rats during gene-therapy-induced apoptosis.265 Such lipids are easily detectable in vivo by magnetic resonance spectroscopy (MRS) and could be used to monitor the efficacy of gene therapy in patients with glioma.263 As a complement to this study, the low molecular weight intermediate composition of the same rat gliomas was subsequently quantified and it was demonstrated that myo-inositol, glycine and taurine concentrations correlated with tumour cell density, whereas the overall concentration of choline-containing compounds was unaffected by cell loss.266 Another study has combined MRS with automated pattern recognition techniques to help radiologists categorise brain tumours according to histological type and grade.267 Using metabolic profiling, it was possible to discriminate between meningiomas, low grade astrocytomas and aggressive tumours such as glioblastomas and metastases. This highlights the ability to transfer knowledge from the laboratory to the bedside to assist in healthcare and potentially provide better outcomes by earlier diagnosis or improved interventions. Spectral profiles prepared from intact tissue, tissue extracts and biofluids have also proven to be highly discriminatory for a number of neurological diseases, including spinocerebellar ataxias, Huntington's disease, schizophrenia, Lesch–Nyhan syndrome and Duchenne muscular dystrophy.262,268–272 For example, metabolic profiles derived from cerebral tissue of a mouse model of spinocerebellar ataxia-3 demonstrated metabolic abnormalities in the cerebellum and also in the cerebrum, which has not previously been implicated in the disease.262 Similarly, metabolic deficits in a mouse model of Huntington's disease have been characterised, suggestive of a redistribution of neural osmolytes and an alteration in glutamate–glutamine cycling.272
Metabolic profiling of cerebral spinal fluid (CSF) has also been conducted with the aim of establishing biomarkers of diseases affecting the central nervous system. Using an NMR spectroscopy based approach, it has been possible to differentiate CSF samples of first-onset schizophrenia patients from healthy controls.273 CSF has been used to diagnose differentially viral, tubercular and bacterial meningitis in children.274 Another recent study used NMR spectroscopy to identify CSF biomarkers of the neurological disorders idiopathic intracranial hypertension (IIH) and multiple sclerosis. The metabolic profiles obtained could predict disease diagnosis in a second cohort of patients with 80% specificity.275 Schizophrenia has been studied with metabolomics276 and systems biology showing the significant changes in energy metabolism in the mitochondria and oxidative stress.277 The role of hypoxia and/or oxidative stress is increasingly being implemented in a number of diseases including pre-eclampsia, Parkinson's disease, Alzheimer's disease, heart failure, atherosclerosis and tissue inflammation.
In addition to cardiovascular disease and neurodegeneration, the other major research area that has benefitted from the application of metabolomic tools is cancer. The first applications focussed on the discrimination of tumour types in brain tissue using in vivo NMR spectroscopy, solution state extracts and even intact tissues.278–280 While NMR spectroscopy based approaches have dominated metabolomics in cancer research to date, in part because of the potential of moving from tissue extracts to carrying out NMR either in situ or in vivo, there has been a recent increase in MS-based studies. GC-MS methods have been used to characterise ovarian tumours,281 kidney cancer92 and colon cancer.217 Similar progress has been made in understanding the progression of prostate cancer, with spermine and sarcosine concentrations having a prominent role in discriminating tumours according to aggressiveness.71,282
(iii) Drug discovery, toxicity, and efficacy
Metabolomics has been widely used in the field of drug toxicology as it offers the potential for identifying and assessing toxic effects during the early stages of compound development, saving money, time and resources for other drugs in the pipeline.283–285 Many published examples are available, of which only a few will be discussed here. Metabolomics can be used to search for biomarkers which are characteristic of a particular type of toxicity. Alternatively, it can be used to construct databases from which models can be built to try to predict the toxicity of unknown compounds without detailed analysis of the changes occurring due to each compound. The putative biomarkers are more acceptable if they can be linked to a mechanism as many of the changes commonly detected can be the result of non-specific toxicity, often due to loss of body weight or general stress.128,286The Consortium for Metabonomic Toxicology (COMET), a collaboration between Imperial College London and six pharmaceutical companies, is an example of the creation of a large database of metabolomic data for the prediction of toxic effects. It was set up to investigate the use of metabolomics/metabonomics in preclinical toxicological screening of drug candidates, with a focus on biofluids.287 A database of 147 compounds selected as being model toxins, mainly targeting the liver or kidney, was compiled along with associated meta-data including histopathology and clinical chemistry.288 Using a subset of these compounds a model was developed to distinguish liver and kidney toxicity. When the model could make predictions the error rate was 8%. However, in 39% of cases, a prediction could not be made.289 More work will be required to increase the success rate of the predictions for this ambitious but essential program.
An example of detection of biomarkers which have a mechanistic explanation is found in a study of urine from rats exposed to peroxisomal proliferation. Normally to determine peroxisomal proliferation a liver sample is required for electron microscopy to directly visualize the changes. Urinary N-methylnicotinamide (NMN), which is formed from nicotinamide and is one of the end points of the tryptophan–NAD+ pathway, was found to correlate with the density of peroxisomes in liver. It was proposed that increased flux through the tryptophan–NAD+ pathway is the cause of the increase in urinary NMN and gene expression data were used to support this hypothesis.290–292
Urinary metabolomics often identifies changes in the same subset of high concentration metabolites, many of which are involved in the citric acid cycle and energy homeostasis.283 The levels of urinary creatine and taurine are commonly perturbed in response to hepatotoxins, but often the direction of the change varies. However, Clayton et al. studied three model hepatotoxins which caused necrosis, steatosis and cholestasis and suggested a hypothesis for the different changes in levels of creatine and taurine in terms of cysteine synthesis in the liver.293 In a similar experiment Mortishire-Smith and colleagues used metabolomics to elucidate the mechanism of toxicity in a candidate drug. Medium chain dicarboxylic acids were identified in urine and triglycerides increased in the liver leading to the hypothesis that the compound disrupted fatty acid metabolism and inhibited β-oxidation. This was then confirmed using in vitro assays.294
(iv) Lipidomics
The full complement of lipids present in a biological sample is defined as the lipidome and can be viewed as a sub-category of the metabolome. However, the most comprehensive database of lipids (Lipid Maps295) describes 21715 separate lipids compared to the 7800 metabolites defined in the Human Metabolome Database.15 Lipids constitute a large proportion of the mammalian metabolome and are employed in diverse roles including energy storage, cell membranes and signalling. Lipidomics has been defined as “the full characterisation of lipid molecular species and of their biological roles with respect to expression of proteins involved in lipid metabolism and function, including gene regulation”.296 The importance of lipids in disease pathophysiology and as biomarkers297,298 and their role in signalling processes299 is increasing rapidly and their importance in structural roles and energy storage is essential. A number of reviews are available which detail the application of lipidomics.298,300,301
Specific experimental systems are employed for lipidomics which often differ when compared with metabolomic analyses, to reflect the great diversity of lipids found inside the cell and the similar chemical properties they possess. In addition to using specific assays based on MS and NMR, thin layer chromatography and solid phase extraction have been widely used. Shotgun lipidomics employs the direct infusion of samples without chromatographic separation and although offers disadvantages as described earlier for DIMS (i.e., ionisation suppression and separation of stereoisomers), the technique has been applied routinely in a high throughput manner. It is recommended that samples are analysed three times to expand the range of detectable lipids: (i) negative ion mode with no modifier for anionic lipid species, (ii) addition of lithium hydroxide in negative ion mode detects the weak anionic species and (iii) positive ion mode with a weak acid such as formate to detect neutral and polar lipids.23 The double-bond position in unsaturated lipids can now be determined with ozone-based reactions.302 Extraction methods apply a non-polar solvent system, typically chloroform, with a range of different physical methods.303 Currently, the focus of informatics and analytical excellence in lipidomics is Lipidmaps295,304 where specific methods for lipid class study and a database of all known lipids are available. Seven specific classes of lipids have been defined. These are fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, sterol lipids, prenol lipids and saccharolipids. The abilities to derive knowledge from data are dependent on the informatics applied to integrate large data sets from different sources. Oresic and colleagues recently reviewed the current expertise and limitations.305
A range of diseases and physiological dysregulation have implicated the role of lipids including diabetes,306 heart disease,307,308 mitochondria,309 traumatic and ischemic brain injury,310,311 mediators in diseases312 including the regulation of pain sensitivity313 and lipid involvement in cell death.314 The role of lipidomics in metabolomics is expected to increase in the coming years.
(v) Nutrigenomics and the role of metabolomics
The impact of food and diet on metabolism in mammals is poorly understood. The body consumes many different dietary metabolites, including nutrients, but little is known on how these influence physiology and metabolism. Optimal nutrition is known to benefit the health of humans and has the potential to eliminate specific diseases. The influence of diet on the progression of diseases is becoming clear (for example, diabetes). Nutritional assessments are an essential part of the toolbox to personalised assessments of the interaction between diet and health. Recently, reduced calorific intake has been shown to be important to health and to improve outcome after disease interventions.315,316A pioneering paper in 2002 highlighted the role metabolomics will play in providing individual metabolic assessments and many positive results have been observed so far. However, there is still a long road to follow and the development of metabolomics and nutritional assessments will walk hand-in-hand as the difficulties are observed and overcome in both.317
However, in the human population where inter-subject variation is high there are many confounders associated with these studies. These include metabolic differences of individuals which provide different metabolic signatures inter-dispersed with nutrient signals. The gut microflora is known to have beneficial influences on human health and the ability to accurately map diet records of food components with metabolic profiles is required. Before the introduction of metabolomics a limited and specific number of nutrients and metabolites were studied. The majority of the 20th century has focussed on the discovery of vitamins and nutrients which provide prevention of deficiency diseases. Separately, a discovery of polyphenols in red wine and their beneficial protection against oxidative stress in the body has been observed. Metabolomics has provided the holistic study of the interaction between diet and health. The number of metabolites in food is significantly greater than the number of nutrients and the goal is to determine how the interactions between all of these influence health. In order to understand how diet interacts with health there is a requirement to determine specific markers of nutrient or food intake318,319 and to measure the chronic and acute effect of diet on metabolism and physiology.
(vi) The application of stable isotopes in metabolomics
Stable isotopes are defined as entities of an element which differ in mass, a result of differing numbers of neutrons and the same number of electrons and protons. Stable isotopes are not radioactive, the relative abundance of isotopes remains constant. For example carbon-12 and carbon-13 (12C and 13C) are isotopes of the same element. The abundance of each isotope is element specific and typically the isotope of lowest mass is the most abundant. For example, the ratio of abundances for 12C and 13C is 98.9∶1.1. A number of common elements have two stable (i.e. not radioactive) isotopes including carbon, nitrogen, sulfur, chlorine and bromine. The introduction of an unnaturally high ratio of an isotope can be employed in metabolomics for two types of studies: tracer or flux distribution studies and flux analysis studies.Tracer studies are applied to define the path of an element (and related to the source metabolite) through a metabolic network. The metabolites enriched above the natural level of 13C when a 13C carbon source is introduced can be expected to be linked metabolically to the source of 13C. Glucose is typically, but not exclusively, the carbon source. The route through the metabolic network can also be defined by the carbon atom(s) of a metabolite enriched in 13C (positional isotopomer distribution). These ‘tracer’ studies are reviewed excellently in a recent paper where MS and NMR were applied to these types of studies in mammalian systems.320 These studies can be extended to define the flux distribution. For example, the distribution of an isotope to specific metabolites in the metabolic network can define the relative fluxes through specific metabolic pathways which lead to specific metabolites. For example, the determination of the flux distribution in proteinogenic amino acids is employed to define flux to pathways involved in amino acid metabolism.321 One human-based study has provided stable isotope resolved metabolomic (SIRM) analysis following 13C glucose infusion into humans diagnosed with lung cancer. This provided in vivo, rather than in vitro, insights into metabolism of tumours and showed increased flux through the glycolysis and TCA cycle pathways.322
Metabolomics typically studies the metabolite concentration in a pool and provides a snapshot of metabolism. However, metabolite concentrations are influenced by the metabolic flux of reactions and the determination of concentration and flux is important to define temporal changes. Here, an isotopically enriched metabolite is added to the system and the changes in the 12C and 13C abundances of metabolites downstream at multiple time points (optimised to define the increase and decrease in the abundance appropriately) are measured. These applications have been reviewed by Sauer and Zamboni in microbial systems.323,324 For pathways where flux is high (for example, glycolysis), rapid sampling systems have been developed for microbial systems. Here, twenty samples were collected over a sixteen second period.325 These types of studies are performed in cell-based rather than tissue-based systems as rapid sampling and quenching is required and therefore examples in whole mammalian systems are rare although perfused organs have commonly been investigated.326,327 Such approaches allow the measurement of fluxes, particularly of the TCA cycle, in functioning organs. Also, benefiting from the ready uptake of glucose by the brain 13C MRS has been applied to follow brain metabolism, including estimating TCA flux rates, in animals and humans in vivo.328,329
Applications of isotopes in mammalian systems are typically tracer studies or flux distribution studies rather than flux measurements because of the technical demands of rapid sampling of mammalian systems. Rabinowitz and colleagues have applied systems-wide metabolic flux profiling to determine that metabolic flux in many central metabolic pathways present in mammalian cells is upregulated following induction by human cytomegalovirus, including TCA cycle and fatty acid biosynthesis. Pharmacological inhibition of fatty acid biosynthesis showed reduced replication of the virus.330
(vii) Spatial mapping of metabolite distributions in tissues and cells
The majority of metabolomic experiments ignore the spatial information of the metabolome, extracting metabolites from relatively large tissue areas. For example, tissue studies perform the extraction of intracellular metabolites into an appropriate extraction solvent for further analysis. Although this determines global changes, specific information on the spatial distribution of metabolites is lost. A migration to spatial mapping of metabolites is appropriate where MS and NMR can be applied.Mass spectrometry imaging employs a range of ionisation techniques including matrix assisted laser desorption ionisation (MALDI331), secondary ion mass spectrometry (SIMS332,333) and desorption electrospray ionization (DESI).334 Here, a focussed laser or ion beam or solvent spray results in the ionisation of metabolites and their fragments from the surface of a sample prior to mass analysis. The level of sputtering and sample removal can be controlled and depth profiling can be performed, as shown for SIMS using a C60+ ion beam on frog oocytes.332 The resolution of imaging is highly dependent on the diameter of the ion or laser beam; typically with SIMS having better resolution (μm scale) than MALDI, and DESI currently having very poor resolution. More recently, nanostructure-initiator mass spectrometry (NIMS) has been investigated for spatial profiling of metabolites without the need for matrix (as is observed in MALDI) and with reduced fragmentation (as is observed in MALDI and SIMS).335
Magnetic resonance imaging (MRI), or in vivo chemical shift imaging (a spectroscopic variant of MRI) has long been used to follow a host of diseases in animal models and humans in vivo, and this is an expanding field in drug discovery.336,337 This provides advantages in human physiology as in vivo studies are closer to the observed phenotype than animal or tissue models. It also circumvents the need for quenching metabolism, and indeed from in vivo spectroscopic studies of brain metabolism one can determine that the intracellular concentration of lactate is ∼1 mM, compared with the >10 mM concentration detected in tissue extracts as a result of post mortem metabolism of glucose and glycogen. Despite over 20 years of activity this is an expanding field and two of its pioneers, Lauterbur and Mansfield, received the Nobel prize in 2003 in recognition of this. Activatable molecular probes which provide an increase in detectable signal following interaction with an enzyme during metabolism has been shown to provide advantages in cancer metabolomics.338
(viii) Metabolomics role in systems biology
Three specific publications, which highlight the growing potential of metabolomics in combination with systems biology, will be discussed further here.Sreekumar et al. have applied metabolomics to decipher metabolic alterations observed in tissue and biofluids (urine and plasma) associated with prostate cancer.71 A combination of GC-MS and LC-MS provided the detection of 1126 unique metabolites. The metabolic profiles were able to distinguish between benign, clinically localised and metastatic prostate cancer and provided evidence of the role of sarcosine in cancer cell invasion and its predictive ability when measured in biofluids. This study was one of the first to highlight the role of inductive metabolomics in the discovery of metabolic disease biomarkers and provide hypotheses which could be tested relating to the pathophysiology of disease in a targeted systems biology study.
Gieger and colleagues have undertaken a genome-wide association study with metabolomics data.339 Quantitative data for 363 metabolites in 284 male participants were acquired. Associations between single nucleotide polymorphisms (SNPs) and metabolism were observed and accounted for 12% of the total variation measured in the metabolic profiles. The results showed that holistic data from different functional levels (genome and metabolome) can be acquired, integrated and analysed to show that common genetic polymorphisms can induce major differences in the metabolic network. These types of studies provide the appropriate tools and data to enable personalised medicine to become a reality.
Shlomi and co-workers have described how model-based, and not experimentally derived, data can be applied to predict human inborn errors of metabolism.340 Diagnosis of inborn errors of metabolism and disease phenotypes is typically performed by the holistic acquisition of data from healthy and diseased subjects followed by data analysis to determine metabolic differences. This process is time-consuming and relatively expensive. This publication described a computational approach to systematically predict metabolic biomarkers from stoichiometric metabolic models. The results showed that genome-scale metabolic models can be applied to predict errors in metabolism. The concentrations of 233 metabolites were predicted to be up or down regulated as a result of 176 dysfunctional enzymes. This approach is attractive as it focuses the metabolomic experiment to a specific set of metabolites for further targeted studies without the requirement for metabolic profiling to generate hypotheses. However, the method is limited by the knowledge gaps present in current genome-scale metabolic reconstructions.
The role of metabolomics in the systems-wide study of mammalian systems is at its infancy and suggests many potential advantages and applications. The study of disease pathophysiology, identification of metabolic biomarkers and the study of drug toxicity and efficacy have shown interesting advances in recent years and further advances in the years to come are expected. The role of systems biology in personalised medicine, where nutrition and drug treatment are tailor made to the individual (rather than the population as is currently observed) or the risk assessed depending on the measured response of metabolites, proteins and genes in an individual, is exciting but at a very early stage of development. Most studies currently perform population-based research where the ‘average’ response and associated variation to diet or drugs are measured. However, people are individuals and each person's metabolism reacts differently to food and drug intake which can, for example, determine the dosage of drug which will be effective or the drug concentration at which toxicity is observed. Personalised medicine can, for example, provide information to determine the current drug (from a library of many) and dosage to apply. Genetics has already provided levels of personalised risk assessment and treatment. For example, the BRCA1 and BRCA2 genes are implicated in the development of breast and ovarian cancer and detection can allow specific treatment to be chosen after counselling (removal of the breast and ovaries).341
6. Growing pains
(i) Chemical identification of metabolites
In the majority of metabolomic investigations there is the requirement to convert the unidentified feature of biological interest to a known chemical entity, a metabolite. The use of MS and NMR spectroscopy, which are respected as powerful tools for chemical characterisation in traditional analytical chemistry, should provide simple and automated methods to perform this. However, these automated processes have not been developed to provide high-throughput and automatic identification of many hundreds or thousands of metabolites in a single sample. Chemical identification in metabolomics is still a manual or semi-automated process, typically applied only to metabolites of biological interest rather than all metabolites detected. The process of automation is difficult as it requires the transfer of the logical knowledge of chemists to software programs while ensuring accuracy in results, especially the absence of false positives. Research has been performed to provide automation which is available to a limited extent in a range of commercially available software, though is currently lacking in open source software.NMR is commonly applied in laboratories across the world for structural interpretation of chemicals, proteins and protein–ligand complexes. However, metabolomic experiments are particularly challenging as identification has to be performed in a complex mixture of metabolites, where there may be significant peak overlap. However, moving to higher dimensions is advantageous by providing a reduction in the spectral complexity. This reduction provides an increase in the number of metabolites detected and identified. The application of homonuclear techniques like COSY (COrrelation SpectroscopY) and TOCSY (TOtal Correlation SpectroscopY) investigates the coupling between protons. Heteronuclear approaches like HSQC (Heteronuclear Single Quantum Coherence spectroscopy) or HMBC (Heteronuclear Multiple Bond Correlation) investigate coupling between protons and another nuclei (typically 13C). A typical 2D NMR spectrum of a yeast extract is shown in Fig. 14. These spectra can then be used to search through a variety of on-line databases such as the HMDB,17 the BioMagResBank (BMRB)342 and the Madison Metabolomics Consortium Database (MMCD).343 Finally, some are developing automated tools for spectral assignments, using two and three dimensional techniques for assignments through on-line databases.344
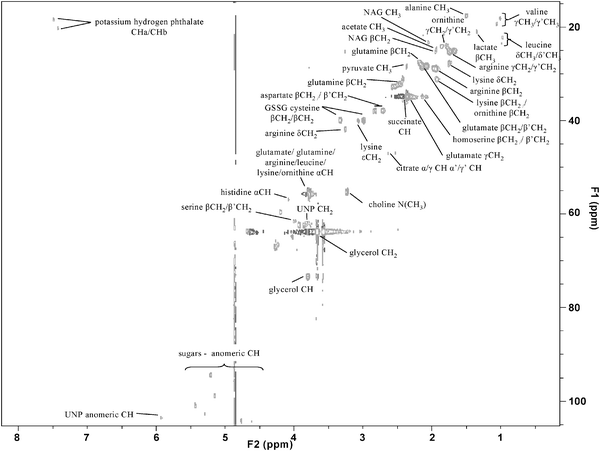 | ||
| Fig. 14 A 2D NMR spectrum acquired from a yeast cell extract applied in a model of Batten disease. | ||
The complexity of mass spectrometric data is high. Many hundreds of metabolites are detected and the process of chemical derivatisation (in GC-MS) and electrospray ionisation (in DIMS, LC-MS and CE-MS) can increase the complexity. The production of multiple derivatisation products following trimethylsilylation is well-known and can increase the complexity of GC-MS chromatograms. Other methods of derivatisation are more specific and can provide single products for each metabolite.345 Recent studies have described the wide range of ions detected in ESI-based studies.75,76 These include adducts, fragments, isotope and multiply-charged peaks common to all instrument types and instrument-specific peaks observed only with a limited number of instruments (for example, Fourier Artefact peaks have been observed with the Orbitrap mass analyser for metabolites present at a high concentration75). Recent research in Manchester has shown that the single metabolite tryptophan is detected as 11 different features in ESI-MS using specific analytical methods and platforms (unpublished data).
The introduction of more powerful mass spectrometric tools for identification of metabolites and valid workflows which should be employed have been observed.75,76,346–349,350 Fiehn and Kind should be congratulated on the early work in this field including the seven golden rules which all metabolomic researchers using mass spectrometry should apply.351 These describe the requirements for correct identification of elemental (or molecular) formulae and the appropriate rules to apply to constrain the number of possible elemental formulae. However, limited advances have been observed in the previous three years since these pioneering publications. Two classifications of identification are applied in metabolomics, putative and definitive.352 Putative annotation or characterisation employs typically one experimentally-defined parameter (e.g., accurate mass), though combinations can be applied, to identify a metabolite. The parameter or parameters applied are not matched to those of an authentic chemical standard. In GC-MS the electron impact fragmentation mass spectrum is applied, which can be a highly specific method for metabolite identification because of the complexity of molecular and fragment ions present in the mass spectrum. In LC-MS, CE-MS and DIMS the accurate mass of an analyte is typically applied which is matched to a metabolite in specific databases either directly or via an intermediate step of matching accurate mass to molecular formulae before conversion of this to a metabolite. It is highly recommended to apply the two-step process as databases are not fully comprehensive and currently do not contain information on all metabolites present in biological systems. There is a high probability of false positives in the single step process. The two-step process should provide matching of accurate mass to the molecular formulae of chemicals present in metabolomic and chemical-focussed databases (for example PubChem353 or ChemSpider354). Detected features may be chemicals introduced during sample collection, preparation and analysis and metabolomic databases are not comprehensive. Inclusion of the seven golden rules can subsequently be applied with other methods to provide increased specificity and confidence while reducing the number of possible molecular formulae. The measured accurate mass can be matched to multiple metabolites with the same molecular formula but different structural arrangement (stereoisomers; for example, glucose or fructose) or matched to metabolites with different molecular formula and similar or identical molecular mass.
The application of fragmentation mass spectra is achievable with many LC-MS instruments applied in metabolomics (triple quadrupole, Q-TOF and trap-based instruments) and can be highly specific. The mass spectra acquired from the collision induced dissociation (CID) of the isomers glucose-1-phosphate and glucose-6-phosphate are different, showing the ability to distinguish between metabolites of similar molecular structures. MSn where n > 2 combined with spectral trees can also be applied in specific trap-based instruments to increase the accuracy of identification and reduce the possibility of a false positive/misassignment. The adduct pattern can be applied to reduce the number of molecular formulae matches in electrospray data. For increased confidence and where definitive identification is not possible isolation of the metabolite by fractionation and chemical characterisation using MS, NMR, elemental analysis and UV/IR spectroscopy should be performed.355 This is labour-intensive, not high-throughput, requires sufficient material and sometimes is beyond the capabilities of current analytical tools. Recently, published research has defined metabolites with a link to an electronic source and this is commended to provide a direct link between results and further information.
However, without the comparison of multiple parameters acquired for a metabolite detected in a sample with an authentic chemical standard no level of high confidence can be achieved. Matching of data to those acquired for authentic chemical standards is classified as definitive identification. Typically, two orthogonal properties are applied: retention time or migration time as a chromatographic property (associated with boiling point or hydrophobicity/hydrophilicity) and accurate mass and/or fragmentation mass spectrum and/or NMR spectrum (associated with chemical structure). For this reason DIMS can typically only provide putative identification of metabolites. Definitive identification can be performed for a limited number of metabolites after putative identification and the purchase of the relevant authentic standards.
A singe-stage process for definitive identification is achievable with the use of mass spectral libraries, though this can be limited and provide false positives for structurally similar metabolites. Mass spectral libraries are constructed by the analysis of authentic chemical standards applying specific analytical instruments and methods. In metabolomics, all possible metabolites are not commercially available or the purchasing costs are high.75,77 Therefore a comprehensive library is highly unlikely. However, libraries have been constructed which are either highly specific to metabolomics (i.e., only contain metabolites as entries) or are less specific and provide data on a wide range of chemicals. This has especially been observed for GC-MS where NIST/EPA/NIH libraries are commercially available and provide electron impact fragmentation spectra on greater than 191000 entities and provide other data including MS/MS mass spectra and Kovats retention indices (RI) values for greater than 44000 chemicals. Metabolomic-specific libraries have been constructed and report retention index (a normalised retention time parameter) and fragmentation mass spectrum.75,356–358 The transferability of these libraries between different instruments and laboratories is relatively high though systematic errors can be introduced in the reported retention index with different instrumental methods. However, a limited number of column chemistries are applied (95% methyl–5% phenyl is the most common in metabolomics) which limits the impact of this technical difficulty. The reproducibility of the electron energy and fragmentation process across all instruments is high and provides good matching of mass spectra between metabolomic samples and libraries.
The availability and transferability of LC-MS mass spectral libraries is limited in metabolomics. Technical issues have limited construction. Retention times vary greatly between different LC columns and chromatographs and do not allow retention times to be transferred accurately between different methods as is possible for GC-MS. The fragmentation process is also highly variable depending on the instrument applied as has been shown previously.359 The application of a calibration point for instrument tuning before analysis can provide mass spectra acquired on different instrument types which are comparable.360 The construction and development of libraries based on LC-MS data which are reproducible and transferable is of high importance in metabolomics but has currently not been fulfilled and there are no indications that this will be performed in the next 5–10 years.
(ii) Standardisation
Greater than 200 laboratories worldwide are estimated to perform metabolomics research, a field that is undergoing analytical evolution. Each laboratory operates with different viewpoints regarding the optimal experimental design, analytical experiment and data analysis tools. The ability to adhere to standardised methods and tools for the foreseeable future is unlikely to be acceptable in metabolomics. However, the ability to share and disseminate methods and results is essential and appropriate reporting standards are necessary for successful data dissemination. Details of the experimental methods are required to provide comparability between different experiments and the possibility of meta-analyses of data from different studies, as is performed in clinical studies. Data reporting standards should describe the minimal information content required for unambiguous interpretation of experimental methods and biological data, the common language (through the use of ontologies) and the appropriate data formats for exchange. Reporting standards provide the ability for information to be accessible, comparable and interpretable for the complete scientific community.In 2005 the Metabolomics Standards Initiative (MSI), in cooperation with The Metabolomics Society, was appointed the role of developing and communicating standards for the metabolomics community and originated from significant work provided by two separate groups: Lindon and colleagues provided standards for data exchange and the communication of results between academia and pharmaceutical companies, largely focussed on NMR spectroscopy;287 while Jenkins and colleagues constructed a generic data model for data storage and exchange in the plant community (ArMET) largely focussed on mass spectrometry.361 The MSI subsequently emerged, and is a group of international and eminent volunteers from the metabolomics community who are developing community-consensus standards. The MSI is separated into working groups, each concentrating on a specific area. In 2007, the MSI published a set of papers to provide communication of preliminary research, highlight the necessity for these standards and raise community awareness.362 The papers described requirements (rather than finalised standards) developed by each of the working groups and include reporting requirements for biological samples (mammalian,363 microbial,364 plant365 and environmental366), chemical analysis,352 NMR experiments,367 data analysis,368 data exchange369 and ontologies.370
Currently, limited numbers of research groups freely provide their data to the scientific community, though recently the provision of data as supplementary with published manuscripts is being observed. There is the requirement for a greater number of research groups to allow their data to be freely available and funding organisations are including this as a necessity for funding. Decisions on whether raw data or pre-processed data will be made available and the restraints of file sizes of raw data have to be made. The complexity and inter-operability of different sources of data (biological, clinical) provide extra complexity to these databases. For example, clinical-based metabolomics require not only storage of analytical data but also clinical metadata specific to the subjects from which samples are required. The Husermet and COMET projects have shown that this complexity can be present and still integratable.
Two specific areas of importance is the requirement for standardisation of controlled vocabularies (or ontologies) and data exchange. Ontologies are defined as formal representations of a set of concepts within a domain and the relationships between the concepts. One example is the naming of metabolites where multiple synonyms are available. To many scientists glucose and β-D-glucose are recognised as the same entity. To a logical computer program these are two separate entities as the names (annotations) do not match (for glucose there are 79 synonyms in PubChem (CID 5793); the chances of confusion are clear). Standardisation is essential in this area and recent work in the yeast metabolomics and systems biology community has provided recommendations to how metabolites should be named.18 Metabolites must be annotated with external references available to the scientific community and it is recommended to apply ChEBI (CHemical Entities of Biological Interest) as the primary source of annotation. If the metabolite is not present in ChEBI then KEGG followed by HMDB followed by PubChem is recommended. Each metabolite is annotated with a name and a database independent representation for small molecules, specifically InChI (INternational CHemical Identifier) or SMILES (Simplified Molecular Input Line Entry System). The charge state of the metabolite, dependent on the environmental pH, should also be considered and be accurately reported. For example, malonic acid (neutral species) or malonate (negatively charged species). ChEBI reports multiple entries for a single metabolite specific to charge state.
The appropriate standards for ontologies and data exchange allow the exchange (usually via web services) and seamless integration of data from multiple sources to be applied in systems biology. Here data from genomic, transcriptomic, proteomic and metabolomic experiments may be combined in the construction of quantitative network models, including models of metabolism. This is essential for systems biology to be successful. The automation, accuracy and rapid performance are only possible when standards for ontologies and data exchange are available. Recent advances in automation to provide efficient retrieval of scientific terms to provide the construction of ontologies have been developed with the application of text-mining, an automated informatics process to acquire high-quality data from text.371
(iii) Integration of datasets from multiple sources
The success of systems biology will depend on the integration and analysis of data from different sources including high-throughput ‘omics data and clinical data. For this to be successful databases to store and disseminate data (for example, MeMo372) are required and these have been reviewed recently.24 In metabolomics, early research has focussed on the study of correlations between components of different data sets (for example, metabolite–metabolite, metabolite–transcript) using methods including pairwise metabolite–transcript comparison373 and Bayesian methods to combine correlation and meta-data to provide greater understanding of biological changes.374 Significant impetus is required to provide the routine study of interactions between different functional levels with data acquired in holistic approaches.Concluding comments
The role of metabolomics in the systems-wide study of mammals is rapidly increasing and evolving. The importance of metabolites in metabolism and regulation of physiological processes is increasingly being highlighted in disease studies to identify biomarkers, to define disease pathophysiology and in drug studies to define efficacy and toxicity. We are aware that the previous 100 years have provided significant advances in qualitative knowledge of the metabolites and interactions (metabolism) from many reductionist-type studies. However, these have not studied the systems as a whole to define emergent properties which are increasingly becoming apparent as essential to understand multi-factorial interactions of causes and effects of disease, diet and drugs. Only now are these avalanches of data from the previous 100 years being combined to allow systems-wide studies to be performed. The rapid advance in metabolomics has been created by the technological advances to allow high-throughput holistic investigations of metabolomes (for example, advances in analytical platforms and informatics) and to provide computational power and technologies to allow the analysis and modelling of the large volumes of data provided. Only now are we starting to see the advantages that systems wide studies will provide and the study of the metabolome to define system-wide properties and phenotypes is at the start of a long and prosperous path in the next 50 years. However, we should always remember that the goal of these studies is to drive forward knowledge of the understanding of us as humans and to enable improved health status, including healthy ageing and better interventions in diseases. The economic impact of these advances will be large.Acknowledgements
WD and RG wish to thank the BBSRC and EPSRC for financial support of The Manchester Centre for Integrative Systems Biology (BB/C008219/1). RG also thanks the EU Framework VI initiative for funding the metabolomics project META-PHOR (FOOD-CT-2006-036220). DB wishes to thank the Wellcome Trust and Science Foundation Ireland for financial support. Work in JLG's laboratory is funded by the EU (MetaCancer), the Medical Research Council, the BBSRC, the Wellcome Trust, GlaxoSmithKline and Syngenta. WD and DB wish to thank members of the Manchester Biomedical Research Centre for many thought-provoking discussions.References
- O. Fiehn, Plant Mol. Biol., 2002, 48, 155 CrossRef CAS.
- W. B. Dunn and D. I. Ellis, TrAC, Trends Anal. Chem., 2005, 24, 285 CrossRef CAS.
- R. Goodacre, S. Vaidyanathan, W. B. Dunn, G. G. Harrigan and D. B. Kell, Trends Biotechnol., 2004, 22, 245 CrossRef CAS.
- M. J. Gibney, M. Walsh, L. Brennan, H. M. Roche, B. German and B. van Ommen, Am. J. Clin. Nutr., 2005, 82, 497 CAS.
- J. L. Griffin, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 147 CrossRef.
- H. J. Atherton, M. K. Gulston, N. J. Bailey, K. K. Cheng, W. Zhang, K. Clarke and J. L. Griffin, Mol. Syst. Biol., 2009, 5, 259.
- D. B. Kell, FEBS J., 2006, 273, 873 CrossRef CAS.
- F. J. Bruggeman and H. V. Westerhoff, Trends Microbiol., 2007, 15, 45 CrossRef CAS.
- F. P. J. Martin, Y. Wang, N. Sprenger, I. K. S. Yap, T. Lundstedt, P. Lek, S. Rezzi, Z. Ramadan, P. van Bladeren, L. B. Fay, S. Kochhar, J. C. Lindon, E. Holmes and J. K. Nicholson, Mol. Syst. Biol., 2008, 4, 157.
- L. K. Schnackenberg, Expert Rev. Mol. Diagn., 2007, 7, 247 CrossRef CAS.
- J. van der Greef, S. Martin, P. Juhasz, A. Adourian, T. Plasterer, E. R. Verheij and R. N. McBurney, J. Proteome Res., 2007, 6, 1540 CrossRef CAS.
- J. K. Nicholson and J. C. Lindon, Nature, 2008, 455, 1054 CrossRef CAS.
- D. B. Kell, BMC Med. Genomics, 2009, 2, 2 Search PubMed.
- S. Mounicou, J. Szpunar and R. Lobinski, Chem. Soc. Rev., 2009, 38, 1119 RSC.
- D. S. Wishart, C. Knox, A. C. Guo, R. Eisner, N. Young, B. Gautam, D. D. Hau, N. Psychogios, E. Dong, S. Bouatra, R. Mandal, I. Sinelnikov, J. G. Xia, L. Jia, J. A. Cruz, E. Lim, C. A. Sobsey, S. Shrivastava, P. Huang, P. Liu, L. Fang, J. Peng, R. Fradette, D. Cheng, D. Tzur, M. Clements, A. Lewis, A. De Souza, A. Zuniga, M. Dawe, Y. P. Xiong, D. Clive, R. Greiner, A. Nazyrova, R. Shaykhutdinov, L. Li, H. J. Vogel and I. Forsythe, Nucleic Acids Res., 2009, 37, D603 CrossRef CAS.
- S. G. Oliver, M. K. Winson, D. B. Kell and F. Baganz, Trends Biotechnol., 1998, 16, 373 CrossRef CAS.
- H. Tweeddale, L. Notley-McRobb and T. Ferenci, J. Bacteriol., 1998, 180, 5109 CAS.
- M. J. Herrgard, N. Swainston, P. Dobson, W. B. Dunn, K. Y. Arga, M. Arvas, N. Bluthgen, S. Borger, R. Costenoble, M. Heinemann, M. Hucka, N. Le Novere, P. Li, W. Liebermeister, M. L. Mo, A. P. Oliveira, D. Petranovic, S. Pettifer, E. Simeonidis, K. Smallbone, I. Spasic, D. Weichart, R. Brent, D. S. Broomhead, H. V. Westerhoff, B. Kirdar, M. Penttila, E. Klipp, B. O. Palsson, U. Sauer, S. G. Oliver, P. Mendes, J. Nielsen and D. B. Kell, Nat. Biotechnol., 2008, 26, 1155 CrossRef CAS.
- H. W. Ma, A. Sorokin, A. Mazein, A. Selkov, E. Selkov, O. Demin and I. Goryanin, Mol. Syst. Biol., 2007, 3, 135.
- N. C. Duarte, S. A. Becker, N. Jamshidi, I. Thiele, M. L. Mo, T. D. Vo, R. Srivas and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1777 CrossRef CAS.
- M. L. Mo, N. Jamshidi and B. O. Palsson, Mol. BioSyst., 2007, 3, 598 RSC.
- I. Nookaew, M. C. Jewett, A. Meechai, C. Thammarongtham, K. Laoteng, S. Cheevadhanarak, J. Nielsen and S. Bhumiratana, BMC Syst. Biol., 2008, 2, 71 CrossRef.
- X. L. Han and R. W. Gross, Mass Spectrom. Rev., 2005, 24, 367 CrossRef CAS.
- E. P. Go, J. Neuroimmune Pharmacol. Ther., 2010, 5, 18 Search PubMed.
- A. Frolkis, C. Knox, E. Lim, T. Jewison, V. Law, D. D. Hau, P. Liu, B. Gautam, S. Ly, A. C. Guo, J. Xia, Y. Liang, S. Shrivastava and D. S. Wishart, Nucleic Acids Res., 2010, 38, D480 CrossRef CAS.
- http://www.genome.jp/kegg/ .
- H. Jeong, B. Tombor, R. Albert, Z. N. Oltvai and A. L. Barabasi, Nature, 2000, 407, 651 CrossRef CAS.
- E. Ravasz, A. L. Somera, D. A. Mongru, Z. N. Oltvai and A. L. Barabasi, Science, 2002, 297, 1551 CrossRef CAS.
- http://www.iubmb-nicholson.org/chart.html .
- R. Breitling, S. Ritchie, D. Goodenowe, M. L. Stewart and M. P. Barrett, Metabolomics, 2006, 2, 155 CrossRef CAS.
- J. Timbrell, Principles of Biochemical Toxicology, Taylor and Francis, 2001 Search PubMed.
- R. Goodacre, J. Nutr., 2007, 137, 259S CAS.
- F. Guarner and J. R. Malagelada, Lancet, 2003, 361, 512 CrossRef.
- J. K. Nicholson, E. Holmes and I. D. Wilson, Nat. Rev. Microbiol., 2005, 3, 431 Search PubMed.
- S. G. VillasBoas, J. Nielsen, J. Smedsgaard, M. A. E. Hansen and U. RoessnerTunali, Metabolome Analysis: An Introduction, John Wiley and Sons, 2007 Search PubMed.
- N. Ishii, K. Nakahigashi, T. Baba, M. Robert, T. Soga, A. Kanai, T. Hirasawa, M. Naba, K. Hirai, A. Hoque, P. Y. Ho, Y. Kakazu, K. Sugawara, S. Igarashi, S. Harada, T. Masuda, N. Sugiyama, T. Togashi, M. Hasegawa, Y. Takai, K. Yugi, K. Arakawa, N. Iwata, Y. Toya, Y. Nakayama, T. Nishioka, K. Shimizu, H. Mori and M. Tomita, Science, 2007, 316, 593 CrossRef CAS.
- T. Handorf, O. Ebenhoh and R. Heinrich, J. Mol. Evol., 2005, 61, 498 CrossRef CAS.
- D. M. Muoio and C. B. Newgard, Nat. Rev. Mol. Cell Biol., 2008, 9, 193 CrossRef CAS.
- T. M. Henkin, Genes Dev., 2008, 22, 3383 CrossRef CAS.
- J. K. Nicholson, J. C. Lindon and E. Holmes, Xenobiotica, 1999, 29, 1181 CrossRef CAS.
- W. B. Dunn, N. J. C. Bailey and H. E. Johnson, Analyst, 2005, 130, 606 RSC.
- D. B. Kell and P. Mendes, in Technological and Medical Implications of Metabolic Control Analysis, ed. A. Cornish-Bowden and M. L. Cardenas, Kluwer Academic Publishers, Dordrecht, 1st edn., 1999, pp. 3–25 Search PubMed.
- L. M. Raamsdonk, B. Teusink, D. Broadhurst, N. S. Zhang, A. Hayes, M. C. Walsh, J. A. Berden, K. M. Brindle, D. B. Kell, J. J. Rowland, H. V. Westerhoff, K. van Dam and S. G. Oliver, Nat. Biotechnol., 2001, 19, 45 CrossRef CAS.
- J. van der Greef, P. Stroobant and R. van der Heijden, Curr. Opin. Chem. Biol., 2004, 8, 559 CrossRef CAS.
- http://www.metabolomicscentre.nl/ .
- E. C. Horning, Clin. Chem., 1968, 14, 777.
- L. Pauling, A. B. Robinson, R. Teranish and P. Cary, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 2374 CrossRef CAS.
- S. L. Howells, R. J. Maxwell, A. C. Peet and J. R. Griffiths, Magn. Reson. Med., 1992, 28, 214 CAS.
- K. L. Behar, J. A. Denhollander, M. E. Stromski, T. Ogino, R. G. Shulman, O. A. C. Petroff and J. W. Prichard, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 4945 CrossRef CAS.
- D. B. Kell, Biochem. Soc. Trans., 2005, 33, 520 CrossRef CAS.
- I. Matsumoto and T. Kuhara, Mass Spectrom. Rev., 1996, 15, 43 CAS.
- A. Goffeau, B. G. Barrell, H. Bussey, R. W. Davis, B. Dujon, H. Feldmann, F. Galibert, J. D. Hoheisel, C. Jacq, M. Johnston, E. J. Louis, H. W. Mewes, Y. Murakami, P. Philippsen, H. Tettelin and S. G. Oliver, Science, 1996, 274, 546 CrossRef CAS.
- J. C. Venter, M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanatides, R. M. Ballew, D. H. Huson, J. R. Wortman, Q. Zhang, C. D. Kodira, X. Q. H. Zheng, L. Chen, M. Skupski, G. Subramanian, P. D. Thomas, J. H. Zhang, G. L. G. Miklos, C. Nelson, S. Broder, A. G. Clark, C. Nadeau, V. A. McKusick, N. Zinder, A. J. Levine, R. J. Roberts, M. Simon, C. Slayman, M. Hunkapiller, R. Bolanos, A. Delcher, I. Dew, D. Fasulo, M. Flanigan, L. Florea, A. Halpern, S. Hannenhalli, S. Kravitz, S. Levy, C. Mobarry, K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley, K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramouliswaran, R. Charlab, K. Chaturvedi, Z. M. Deng, V. Di Francesco, P. Dunn, K. Eilbeck, C. Evangelista, A. E. Gabrielian, W. Gan, W. M. Ge, F. C. Gong, Z. P. Gu, P. Guan, T. J. Heiman, M. E. Higgins, R. R. Ji, Z. X. Ke, K. A. Ketchum, Z. W. Lai, Y. D. Lei, Z. Y. Li, J. Y. Li, Y. Liang, X. Y. Lin, F. Lu, G. V. Merkulov, N. Milshina, H. M. Moore, A. K. Naik, V. A. Narayan, B. Neelam, D. Nusskern, D. B. Rusch, S. Salzberg, W. Shao, B. X. Shue, J. T. Sun, Z. Y. Wang, A. H. Wang, X. Wang, J. Wang, M. H. Wei, R. Wides, C. L. Xiao and C. H. Yan, et al. , Science, 2001, 291, 1304 CrossRef CAS.
- O. Fiehn, J. Kopka, P. Dormann, T. Altmann, R. N. Trethewey and L. Willmitzer, Nat. Biotechnol., 2000, 18, 1157 CrossRef CAS.
- D. B. Kell and S. G. Oliver, Bioessays, 2004, 26, 99 CrossRef.
- U. Sauer, M. Heinemann and N. Zamboni, Science, 2007, 316, 550 CrossRef CAS.
- A. C. Ahn, M. Tewari, C. S. Poon and R. S. Phillips, PLoS Med., 2006, 3, 709.
- C. Auffray, G. Clermont, Y. Moreau, D. M. Rocke, D. Dalevi, D. Dubhashi, D. R. Marshall, P. Raasch, F. Dehne, P. Provero, J. Tegner, B. J. Aronow, M. A. Langston and M. Benson, Genome Medicine, 2009, 1, 88 Search PubMed.
- D. Noble, Science, 2002, 295, 1678 CrossRef CAS.
- S. Van Dien and C. H. Schilling, Mol. Syst. Biol., 2006, 2, 35.
- D. B. Kell, Drug Discovery Today, 2006, 11, 1085 CrossRef CAS.
- J. Nicholson, Drug Metab. Rev., 2005, 37, 21.
- A. C. Ahn, M. Tewari, C. S. Poon and R. S. Phillips, PLoS Med., 2006, 3, 956.
- M. Brown, W. B. Dunn, D. I. Ellis, R. Goodacre, J. Handl, J. D. Knowles, S. O'Hagan, I. Spasic and D. B. Kell, Metabolomics, 2005, 1, 39 CrossRef CAS.
- P. A. Guy, I. Tavazzi, S. J. Bruce, Z. Ramadan and S. Kochhar, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 871, 253 CrossRef CAS.
- W. B. Dunn, D. Broadhurst, D. I. Ellis, M. Brown, A. Halsall, S. O'Hagan, I. Spasic, A. Tseng and D. B. Kell, Int. J. Epidemiol., 2008, 37, i23 Search PubMed.
- C. L. Winder, W. B. Dunn, S. Schuler, D. Broadhurst, R. Jarvis, G. M. Stephens and R. Goodacre, Anal. Chem., 2008, 80, 2939 CrossRef CAS.
- E. Zelena, W. B. Dunn, D. Broadhurst, S. Francis-McIntyre, K. M. Carroll, P. Begley, S. O'Hagan, J. D. Knowles, A. Halsall, I. D. Wilson and D. B. Kell, Anal. Chem., 2009, 81, 1357 CrossRef CAS.
- P. Jonsson, S. J. Bruce, T. Moritz, J. Trygg, M. Sjostrom, R. Plumb, J. Granger, E. Maibaum, J. K. Nicholson, E. Holmes and H. Antti, Analyst, 2005, 130, 701 RSC.
- D. B. Kell and S. G. Oliver, BioEssays, 2003, 26, 99.
- A. Sreekumar, L. M. Poisson, T. M. Rajendiran, A. P. Khan, Q. Cao, J. D. Yu, B. Laxman, R. Mehra, R. J. Lonigro, Y. Li, M. K. Nyati, A. Ahsan, S. Kalyana-Sundaram, B. Han, X. H. Cao, J. Byun, G. S. Omenn, D. Ghosh, S. Pennathur, D. C. Alexander, A. Berger, J. R. Shuster, J. T. Wei, S. Varambally, C. Beecher and A. M. Chinnaiyan, Nature, 2009, 457, 910 CrossRef CAS.
- W. Lu, B. D. Bennett and J. D. Rabinowitz, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 871, 236 CrossRef CAS.
- M. S. Sabatine, E. Liu, D. A. Morrow, E. Heller, R. McCarroll, R. Wiegand, G. F. Berriz, F. P. Roth and R. E. Gerszten, Circulation, 2005, 112, 3868 CrossRef CAS.
- G. D. Lewis, R. Wei, E. Liu, E. Yang, X. Shi, M. Martinovic, L. Farrell, A. Asnani, M. Cyrille, A. Ramanathan, O. Shaham, G. Berriz, P. A. Lowry, I. F. Palacios, M. Tasan, F. P. Roth, J. Y. Min, C. Baumgartner, H. Keshishian, T. Addona, V. K. Mootha, A. Rosenzweig, S. A. Carr, M. A. Fifer, M. S. Sabatine and R. E. Gerszten, J. Clin. Invest., 2008, 118, 3503 CrossRef CAS.
- M. Brown, W. B. Dunn, P. Dobson, Y. Patel, C. L. Winder, S. Francis-McIntyre, P. Begley, K. Carroll, D. Broadhurst, A. Tseng, N. Swainston, I. Spasic, R. Goodacre and D. B. Kell, Analyst, 2009, 134, 1322 RSC.
- J. Draper, D. P. Enot, D. Parker, M. Beckmann, S. Snowdon, W. Lin and H. Zubair, BMC Bioinformatics, 2009, 10, 227 CrossRef.
- W. B. Dunn, Phys. Biol., 2008, 5, 011001 Search PubMed.
- D. I. Broadhurst and D. B. Kell, Metabolomics, 2006, 2, 171 CAS.
- T. Sangster, H. Major, R. Plumb, A. J. Wilson and I. D. Wilson, Analyst, 2006, 131, 1075 RSC.
- F. M. van der Kloet, I. Bobeldijk, E. R. Verheij and R. H. Jellema, J. Proteome Res., 2009, 8, 5132 CrossRef CAS.
- CDER, in Guidance for Industry, Bioanalytical Method Validation, FDA, Centre for Drug Valuation and Research, 2001 Search PubMed.
- K. J. Rothman and S. Greenland, Modern epidemiology, Lippincott, Williams & Wilkins, 2nd edn, 1998 Search PubMed.
- D. F. Ransohoff, Nat. Rev. Cancer, 2005, 5, 142 CrossRef CAS.
- H. L. Kirschenlohr, J. L. Griffin, S. C. Clarke, R. Rhydwen, A. A. Grace, P. M. Schofield, K. M. Brindle and J. C. Metcalfe, Nat. Med. (N. Y.), 2006, 12, 705 CrossRef CAS.
- O. Teahan, S. Gamble, E. Holmes, J. Waxman, J. K. Nicholson, C. Bevan and H. C. Keun, Anal. Chem., 2006, 78, 4307 CrossRef CAS.
- H. F. Wu, A. D. Southam, A. Hines and M. R. Viant, Anal. Biochem., 2008, 372, 204 CrossRef CAS.
- E. J. Want, G. O'Maille, C. A. Smith, T. R. Brandon, W. Uritboonthai, C. Qin, S. A. Trauger and G. Siuzdak, Anal. Chem., 2006, 78, 743 CrossRef CAS.
- S. J. Bruce, I. Tavazzi, V. Parisod, S. Rezzi, S. Kochhar and P. A. Guy, Anal. Chem., 2009, 81, 3285 CrossRef CAS.
- F. Michopoulos, L. Lai, H. Gika, G. Theodoridis and I. Wilson, J. Proteome Res., 2009, 8, 2114 CrossRef CAS.
- E. J. Want, C. A. Smith, C. A. Qin, K. C. VanHorne and G. Siuzdak, Metabolomics, 2006, 2, 145 CrossRef CAS.
- H. G. Gika, G. Theodoridis, J. Extance, A. M. Edge and I. D. Wilson, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 871, 279 CrossRef CAS.
- T. Kind, V. Tolstikov, O. Fiehn and R. H. Weiss, Anal. Biochem., 2007, 363, 185 CrossRef CAS.
- D. I. Ellis and R. Goodacre, Analyst, 2006, 131, 875 RSC.
- S. A. Fancy, O. Beckonert, G. Darbon, W. Yabsley, R. Walley, D. Baker, G. L. Perkins, F. S. Pullen and K. Rumpel, Rapid Commun. Mass Spectrom., 2006, 20, 2271 CrossRef CAS.
- M. Bogdanov, W. R. Matson, L. Wang, T. Matson, R. Saunders-Pullman, S. S. Bressman and M. F. Beal, Brain, 2008, 131, 389 CrossRef.
- I. W. Griffiths, Rapid Commun. Mass Spectrom., 1997, 11, 3.
- K. Dettmer, P. A. Aronov and B. D. Hammock, Mass Spectrom. Rev., 2007, 26, 51 CrossRef CAS.
- S. G. Villas-Boas, S. Mas, M. Akesson, J. Smedsgaard and J. Nielsen, Mass Spectrom. Rev., 2005, 24, 613 CrossRef CAS.
- S. Vaidyanathan, D. B. Kell and R. Goodacre, J. Am. Soc. Mass Spectrom., 2002, 13, 118 CrossRef CAS.
- A. D. Southam, T. G. Payne, H. J. Cooper, T. N. Arvanitis and M. R. Viant, Anal. Chem., 2007, 79, 4595 CrossRef CAS.
- P. Begley, S. Francis-McIntyre, W. B. Dunn, D. I. Broadhurst, A. Halsall, A. Tseng, J. Knowles, R. Goodacre, D. B. Kell and H. Consortium, Anal. Chem., 2009, 81, 7038 CrossRef CAS.
- X. M. Tao, Y. M. Liu, Y. H. Wang, Y. P. Qiu, J. C. Lin, A. H. Zhao, M. M. Su and W. Jia, Anal. Bioanal. Chem., 2008, 391, 2881 CrossRef CAS.
- S. O'Hagan, W. B. Dunn, M. Brown, J. D. Knowles and D. B. Kell, Anal. Chem., 2005, 77, 290 CrossRef CAS.
- W. Welthagen, R. A. Shellie, J. Spranger, M. Ristow, R. Zimmermann and O. Fiehn, Metabolomics, 2005, 1, 65 CrossRef CAS.
- M. M. Koek, B. Muilwijk, L. L. P. van Stee and T. Hankemeier, J. Chromatogr., A, 2008, 1186, 420 CAS.
- K. M. Pierce, J. C. Hoggard, R. E. Mohler and R. E. Synovec, J. Chromatogr., A, 2008, 1184, 341 CrossRef CAS.
- J. W. Allwood and R. Goodacre, Phytochem. Anal., 2010, 21, 33 CrossRef CAS.
- M. E. Swartz, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 1253 CrossRef CAS.
- J. H. Granger, A. Baker, R. S. Plumb, J. C. Perez and I. D. Wilson, Drug Metab. Rev., 2004, 36, 504.
- I. D. Wilson, J. K. Nicholson, J. Castro-Perez, J. H. Granger, K. A. Johnson, B. W. Smith and R. S. Plumb, J. Proteome Res., 2005, 4, 591 CrossRef CAS.
- S. J. Bruce, P. Jonsson, H. Antti, O. Cloarec, J. Trygg, S. L. Marklund and T. Moritz, Anal. Biochem., 2008, 372, 237 CrossRef CAS.
- D. J. Crockford, J. C. Lindon, O. Cloarec, R. S. Plumb, S. J. Bruce, S. Zirah, P. Rainville, C. L. Stumpf, K. Johnson, E. Holmes and J. K. Nicholson, Anal. Chem., 2006, 78, 4398 CrossRef CAS.
- A. Kamleh, M. P. Barrett, D. Wildridge, R. J. S. Burchmore, R. A. Scheltema and D. G. Watson, Rapid Commun. Mass Spectrom., 2008, 22, 1912 CrossRef CAS.
- H. G. Gika, G. A. Theodoridis and I. D. Wilson, J. Sep. Sci., 2008, 31, 1598 CrossRef CAS.
- Y. Wang, R. Lehmann, X. Lu, X. J. Zhao and G. W. Xu, J. Chromatogr., A, 2008, 1204, 28 CrossRef CAS.
- S. J. Barry, R. M. Carr, S. J. Lane, W. J. Leavens, S. Monte and I. Waterhouse, Rapid Commun. Mass Spectrom., 2003, 17, 603 CrossRef CAS.
- K. Urano, K. Maruyama, Y. Ogata, Y. Morishita, M. Takeda, N. Sakurai, H. Suzuki, K. Saito, D. Shibata, M. Kobayashi, K. Yamaguchi-Shinozaki and K. Shinozaki, Plant J., 2009, 57, 1065 CrossRef CAS.
- E. E. K. Baidoo, P. I. Benket, C. Neususs, M. Pelzing, G. Kruppa, J. A. Leary and J. D. Keasling, Anal. Chem., 2008, 80, 3112 CrossRef CAS.
- T. Soga, Y. Ohashi, Y. Ueno, H. Naraoka, M. Tomita and T. Nishioka, J. Proteome Res., 2003, 2, 488 CrossRef CAS.
- B. Sitter, T. F. Bathen, M. B. Tessem and I. S. Gribbestad, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 54, 239 CrossRef CAS.
- T. F. Bathen, L. R. Jensen, B. Sitter, H. E. Fjoesne, J. Halgunset, D. E. Axelson, I. S. Gribbestad and S. Lundgren, Breast Cancer Res. Treat., 2007, 104, 181 CrossRef.
- B. M. Beckwith-Hall, J. K. Nicholson, A. W. Nicholls, P. J. D. Foxall, J. C. Lindon, S. C. Connor, M. Abdi, J. Connelly and E. Holmes, Chem. Res. Toxicol., 1998, 11, 260 CrossRef CAS.
- M. Spraul, M. Hofmann, P. Dvortsak, J. K. Nicholson and I. D. Wilson, Anal. Chem., 1993, 65, 327 CrossRef CAS.
- J. L. Griffin, J. Troke, L. A. Walker, R. F. Shore, J. C. Lindon and J. K. Nicholson, FEBS Lett., 2000, 486, 225 CrossRef CAS.
- O. M. Rooney, J. Troke, J. K. Nicholson and J. L. Griffin, Magn. Reson. Med., 2003, 50, 925 CrossRef CAS.
- L. M. Smith, A. D. Maher, O. Cloarec, M. Rantalainen, H. R. Tang, P. Elliott, J. Stamler, J. C. Lindon, E. Holmes and J. K. Nicholson, Anal. Chem., 2007, 79, 5682 CrossRef CAS.
- J. L. Griffin, H. J. Williams, E. Sang and J. K. Nicholson, Magn. Reson. Med., 2001, 46, 249 CrossRef CAS.
- S. C. Connor, W. Wu, B. C. Sweatman, J. Manini, J. N. Haselden, D. J. Crowther and C. J. Waterfield, Biomarkers, 2004, 9, 156 CAS.
- J. T. Brindle, H. Antti, E. Holmes, G. Tranter, J. K. Nicholson, H. W. L. Bethell, S. Clarke, P. M. Schofield, E. McKilligin, D. E. Mosedale and D. J. Grainger, Nat. Med. (N. Y.), 2002, 8, 1439 CrossRef CAS.
- J. G. Bundy, H. C. Keun, J. K. Sidhu, D. J. Spurgeon, C. Svendsen, P. Kille and A. J. Morgan, Environ. Sci. Technol., 2007, 41, 4458 CrossRef CAS.
- J. G. Bundy, B. Papp, R. Harmston, R. A. Browne, E. M. Clayson, N. Burton, R. J. Reece, S. G. Oliver and K. M. Brindle, Genome Res., 2007, 17, 510 CrossRef CAS.
- J. T. Brindle, H. Antti, E. Holmes, G. Tranter, J. K. Nicholson, H. W. Bethell, S. Clarke, P. M. Schofield, E. McKilligin, D. E. Mosedale and D. J. Grainger, Nat. Med. (N. Y.), 2002, 8, 1439 CrossRef CAS.
- H. C. Keun, O. Beckonert, J. L. Griffin, C. Richter, D. Moskau, J. C. Lindon and J. K. Nicholson, Anal. Chem., 2002, 74, 4588 CrossRef CAS.
- P. Styles, N. F. Soffe, C. A. Scott, D. A. Cragg, F. Row, D. J. White and P. C. J. White, J. Magn. Reson., 1984, 60, 397 CAS.
- J. L. Griffin, A. W. Nicholls, H. C. Keun, R. J. Mortishire-Smith, J. K. Nicholson and T. Kuehn, Analyst, 2002, 127, 582 RSC.
- G. Schlotterbeck, A. Ross, R. Hochstrasser, H. Senn, T. Kuhn, D. Marek and O. Schett, Anal. Chem., 2002, 74, 4464 CrossRef CAS.
- N. J. C. Bailey, P. D. Stanley, S. T. Hadfield, J. C. Lindon and J. K. Nicholson, Rapid Commun. Mass Spectrom., 2000, 14, 679 CrossRef CAS.
- A. J. Simpson, L. H. Tseng, M. J. Simpson, M. Spraul, U. Braumann, W. L. Kingery, B. P. Kelleher and M. H. B. Hayes, Analyst, 2004, 129, 1216 RSC.
- K. Golman, R. in't Zandt, M. Lerche, R. Pehrson and J. H. Ardenkjaer-Larsen, Cancer Res., 2006, 66, 10855 CrossRef CAS.
- K. Golman, R. in't Zandt and M. Thaning, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11270 CrossRef CAS.
- M. A. Schroeder, H. J. Atherton, D. R. Ball, M. A. Cole, L. C. Heather, J. L. Griffin, K. Clarke, G. K. Radda and D. J. Tyler, FASEB J., 2009, 23, 2529 CrossRef CAS.
- A. M. Weljie, J. Newton, P. Mercier, E. Carlson and C. M. Slupsky, Anal. Chem., 2006, 78, 4430 CrossRef CAS.
- O. Cloarec, M. E. Dumas, A. Craig, R. H. Barton, J. Trygg, J. Hudson, C. Blancher, D. Gauguier, J. C. Lindon, E. Holmes and J. Nicholson, Anal. Chem., 2005, 77, 1282 CrossRef CAS.
- D. V. Rubtsov and J. L. Griffin, J. Magn. Reson., 2007, 188, 367 CrossRef CAS.
- M. Rantalainen, O. Cloarec, O. Beckonert, I. D. Wilson, D. Jackson, R. Tonge, R. Rowlinson, S. Rayner, J. Nickson, R. W. Wilkinson, J. D. Mills, J. Trygg, J. K. Nicholson and E. Holmes, J. Proteome Res., 2006, 5, 2642 CrossRef CAS.
- R. Rew and G. Davis, IEEE Computer Graphics and Applications, 1990, 10, 76 Search PubMed.
- P. G. A. Pedrioli, J. K. Eng, R. Hubley, M. Vogelzang, E. W. Deutsch, B. Raught, B. Pratt, E. Nilsson, R. H. Angeletti, R. Apweiler, K. Cheung, C. E. Costello, H. Hermjakob, S. Huang, R. K. Julian, E. Kapp, M. E. McComb, S. G. Oliver, G. Omenn, N. W. Paton, R. Simpson, R. Smith, C. F. Taylor, W. M. Zhu and R. Aebersold, Nat. Biotechnol., 2004, 22, 1459 CrossRef CAS.
- S. Orchard, L. Montechi-Palazzi, E. W. Deutsch, P. A. Binz, A. R. Jones, N. Paton, A. Pizarro, D. M. Creasy, J. Wojcik and H. Hermjakob, Proteomics, 2007, 19, 3436 CrossRef.
- http://www.w3.org/XML/ .
- R. Goodacre, S. Vaidyanathan, G. Bianchi and D. B. Kell, Analyst, 2002, 127, 1457 RSC.
- W. B. Dunn, S. Overy and W. P. Quick, Metabolomics, 2005, 1, 137 CrossRef CAS.
- H. M. Parsons, D. R. Ekman, T. W. Collette and M. R. Viant, Analyst, 2009, 134, 478 RSC.
- M. A. E. Hansen and J. Smedsgaard, Metabolomics, 2007, 3, 41 CrossRef CAS.
- A. Nordstrom, G. O'Maille, C. Qin and G. Siuzdak, Anal. Chem., 2006, 78, 3289 CrossRef.
- A. Lommen, Anal. Chem., 2009, 81, 3079 CrossRef CAS.
- M. Katajamaa and M. Oresic, BMC Bioinformatics, 2005, 6, 179 CrossRef.
- R. Baran, H. Kochi, N. Saito, M. Suematsu, T. Soga, T. Nishioka, M. Robert and M. Tomita, BMC Bioinformatics, 2006, 7, 530 CrossRef.
- M. Katajamaa and M. Oresic, J. Chromatogr., A, 2007, 1158, 318 CrossRef CAS.
- R. A. van den Berg, H. C. Hoefsloot, J. A. Westerhuis, A. K. Smilde and M. J. van der Werf, BMC Genomics, 2006, 7, 142 CrossRef.
- D. B. Rubbin and R. J. A. Little, Statistical Analysis with Missing Data, John Wiley & Sons Inc, 2002 Search PubMed.
- J. C. Lindon, E. Holmes and J. K. Nicholson, Pharm. Res., 2006, 23, 1075 CrossRef CAS.
- R. O. Duda, P. E. Hart and D. E. Stork, Pattern classification, John Wiley, 2nd edn, 2001 Search PubMed.
- J. B. Kruskal and M. Wish, Multidimensional scaling, Sage, 1978 Search PubMed.
- B. S. Everitt, Cluster Analysis, Edward Arnold, 1993 Search PubMed.
- T. Hastie, R. Tibshirani and J. Friedman, The elements of statistical learning: data mining, inference and prediction, Springer-Verlag, 2001 Search PubMed.
- I. T. Jolliffe, Principal Component Analysis, Springer-Verlag, 1986 Search PubMed.
- R. A. Fisher, The design of experiments, Oliver & Boyd, 6th edn, 1951 Search PubMed.
- W. J. Krzanowski, Principles of Multivariate Analysis: A User's Perspective, Oxford University Press, 1988 Search PubMed.
- H. Martens and T. Næs, Multivariate calibration, John Wiley, 1989 Search PubMed.
- B. D. Ripley, Pattern recognition and neural networks, Cambridge University Press, 1996 Search PubMed.
- S. Wold, H. Antti, F. Lindgren and J. Ohman, Chemom. Intell. Lab. Syst., 1998, 44, 175 CrossRef CAS.
- J. Sjoblom, O. Svensson, M. Josefson, H. Kullberg and S. Wold, Chemom. Intell. Lab. Syst., 1998, 44, 229 CrossRef CAS.
- C. A. Andersson, Chemom. Intell. Lab. Syst., 1999, 47, 51 CrossRef CAS.
- J. A. Westerhuis, S. de Jong and A. K. Smilde, Chemom. Intell. Lab. Syst., 2001, 56, 13 CrossRef CAS.
- L. Eriksson, J. Trygg, E. Johansson, R. Bro and S. Wold, Anal. Chim. Acta, 2000, 420, 181 CrossRef CAS.
- P. D. Harrington, J. Kister, J. Artaud and N. Dupuy, Anal. Chem., 2009, 81, 7160 CrossRef CAS.
- J. Trygg and S. Wold, J. Chemom., 2002, 16, 119 CrossRef CAS.
- I. Esteban-Diez, J. M. Gonzalez-Saiz and C. Pizarro, Anal. Chim. Acta, 2004, 514, 57 CAS.
- H. Wold, in Perspective in probability and statistics: Papers in honour of M.S. Bartlett, ed. J. Gani, Academic Press, London, 1975, pp. 117–142 Search PubMed.
- S. Wold, J. Trygg, A. Berglund and H. Antti, Chemom. Intell. Lab. Syst., 2001, 58, 131 CrossRef CAS.
- L. Eriksson, E. Johansson, N. Kettaneh-Wold and S. Wold, Multi- and megavariate data analysis: principles and applications, Umetrics Academy, 2001 Search PubMed.
- B. K. Alsberg, R. Goodacre, J. J. Rowland and D. B. Kell, Anal. Chim. Acta, 1997, 348, 389 CrossRef CAS.
- R. D. King, A. Srinivasan and L. Dehaspe, J. Comput.-Aided Mol. Des., 2001, 15, 173 CrossRef CAS.
- L. Breiman, Mach. Learn., 2001, 45, 5 CrossRef.
- D. P. Enot, M. Beckmann and J. Draper, Computational Life Sciences II Second International Symposium, ed. S. Istrail, P. Pevzner, and M.Waterman, Springer, Berlin, 1st edn., 2006, pp. 226–235 Search PubMed.
- R. Goodacre and D. B. Kell, in In Metabolic profiling: its role in biomarker discovery and gene function analysis, ed. G. G. Harrigan and R. Goodacre, Kluwer Academic Publishers, Boston, 1st edn., 2003, 239–256 Search PubMed.
- A. A. Freitas, Data mining and knowledge discovery with evolutionary algorithms, Springer-Verlag, 2002 Search PubMed.
- J. Handl and J. Knowles, International Joint Conference on Neural Networks, 2006, 2, pp. 217–238 Search PubMed.
- J. Handl, D. B. Kell and J. Knowles, IEEE/ACM Trans. Comput. Biol. Bioinf., 2007, 4, 279 Search PubMed.
- D. S. Broomhead and D. Lowe, Complex Syst., 1988, 2, 312 Search PubMed.
- R. Goodacre, J. Exp. Bot., 2005, 56, 245 CAS.
- D. B. Kell, Expert Rev. Mol. Diagn., 2007, 7, 329 CrossRef CAS.
- T. M. D. Ebbels and R. Cavill, Prog. Nucl. Magn. Reson. Spectrosc., 2009, 55, 361 CrossRef CAS.
- S. Bijlsma, I. Bobeldijk, E. R. Verheij, R. Ramaker, S. Kochhar, I. A. Macdonald, B. van Ommen and A. K. Smilde, Anal. Chem., 2006, 78, 567 CrossRef CAS.
- K. Wongravee, N. Heinrich, M. Holmboe, M. L. Schaefer, R. R. Reed, J. Trevejo and R. G. Brereton, Anal. Chem., 2009, 81, 5204 CrossRef CAS.
- R. Cavill, H. C. Keun, E. Holmes, J. C. Lindon, J. K. Nicholson and T. M. D. Ebbels, Bioinformatics, 2009, 25, 112 CAS.
- D. Broadhurst, R. Goodacre, A. Jones, J. J. Rowland and D. B. Kell, Anal. Chim. Acta, 1997, 348, 71 CrossRef.
- R. M. Jarvis and R. Goodacre, Bioinformatics, 2005, 21, 860 CAS.
- P. Smialowski, D. Frishman and S. Kramer, Bioinformatics, 2010, 26, 440 CrossRef CAS.
- J. A. Westerhuis, H. C. J. Hoefsloot, S. Smit, D. J. Vis, A. K. Smilde, E. J. J. van Velzen, J. P. M. van Duijnhoven and F. A. van Dorsten, Metabolomics, 2008, 4, 81 CrossRef CAS.
- A. Linden, Journal of Evaluation in Clinical Practice, 2006, 12, 132 Search PubMed.
- C. E. Metz, Semin. Nucl. Med., 1978, 8, 283 CrossRef CAS.
- W. B. Dunn, D. I. Broadhurst, S. M. Deepak, M. H. Buch, G. McDowell, I. Spasic, D. I. Ellis, N. Brooks, D. B. Kell and L. Neyses, Metabolomics, 2007, 3, 413 CrossRef CAS.
- K. A. Janes and M. B. Yaffe, Nat. Rev. Mol. Cell Biol., 2006, 7, 820 CrossRef CAS.
- D. B. Kell, FEBS J., 2006, 273, 873 CrossRef CAS.
- D. B. Kell, Curr. Opin. Microbiol., 2004, 7, 296 CrossRef CAS.
- S. Lee, Spiderman, Amazing Fantasy #15, Marvel Comics, 1962 Search PubMed.
- B. Efron and R. J. Tibshirani, Introduction to the bootstrap, Chapman and Hall, 1993 Search PubMed.
- J. P. Ioannidis, JAMA, J. Am. Med. Assoc., 2005, 294, 218 CrossRef CAS.
- J. P. Ioannidis and T. A. Trikalinos, J. Clin. Epidemiol., 2005, 58, 543 CrossRef.
- J. P. Ioannidis, T. A. Trikalinos, E. E. Ntzani and D. G. Contopoulos-Ioannidis, Lancet, 2003, 361, 567 CrossRef.
- F. K. Kavvoura, M. B. McQueen, M. J. Khoury, R. E. Tanzi, L. Bertram and J. P. A. Ioannidis, Am. J. Epidemiol., 2008, 168, 855 CrossRef.
- J. T. Leek and J. D. Storey, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18718 CrossRef CAS.
- D. Donoho and J. S. Jin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14790 CrossRef CAS.
- D. F. Ransohoff, Nat. Rev. Cancer, 2004, 4, 309 CrossRef CAS.
- A. E. P. Heazell, M. Brown, W. B. Dunn, S. A. Worton, I. P. Crocker, P. N. Baker and D. B. Kell, Placenta, 2008, 29, 691 CrossRef CAS.
- C. Denkert, J. Budczies, W. Weichert, G. Wohlgemuth, M. Scholz, T. Kind, S. Niesporek, A. Noske, A. Buckendahl, M. Dietel and O. Fiehn, Mol. Cancer, 2008, 7, 72 CrossRef.
- W. R. Wikoff, E. Kalisak, S. Trauger, M. Manchester and G. Siuzdak, J. Proteome Res., 2009, 8, 3578 CrossRef CAS.
- H. G. Gika, G. A. Theodoridis, J. E. Wingate and I. D. Wilson, J. Proteome Res., 2007, 6, 3291 CrossRef CAS.
- R. S. Plumb, P. D. Rainville, W. B. Potts, K. A. Johnson, E. Gika and I. D. Wilson, J. Proteome Res., 2009, 8, 2495 CrossRef CAS.
- D. Monleon, J. M. Morales, A. Barrasa, J. A. Lopez, C. Vazquez and B. Celda, NMR Biomed., 2009, 22, 342 CrossRef CAS.
- E. Holmes, T. M. Tsang, J. T. J. Huang, F. M. Leweke, D. Koethe, C. W. Gerth, B. M. Nolden, S. Gross, D. Schreiber, J. K. Nicholson and S. Bahn, PLoS Med., 2006, 3, 1420 CAS.
- I. Takeda, C. Stretch, P. Barnaby, K. Bhatnager, K. Rankin, H. Fu, A. Weljie, N. Jha and C. Slupsky, NMR Biomed., 2009, 22, 577 CrossRef CAS.
- L. Botros, D. Sakkas and E. Seli, Mol. Hum. Reprod., 2008, 14, 679 CrossRef CAS.
- C. J. Nelson, J. P. Otis, S. L. Martin and H. V. Carey, Physiol. Genomics, 2009, 37, 43 Search PubMed.
- J. J. Xu, J. Zhang, J. Y. Dong, S. H. Cai, J. Y. Yang and Z. Chen, Anal. Bioanal. Chem., 2009, 393, 1657 CrossRef CAS.
- A. Backshall, D. Allferez, F. Telchert, I. D. Wilson, R. W. Wilkinson, R. A. Goodlad and H. C. Keun, J. Proteome Res., 2009, 8, 1423 CrossRef CAS.
- F. P. J. Martin, Y. L. Wang, N. Sprenger, E. Holmes, J. C. Lindon, S. Kochhar and J. K. Nicholson, J. Proteome Res., 2007, 6, 1471 CrossRef CAS.
- J. C. Lin, M. M. Su, X. Y. Wang, Y. P. Qiu, H. K. Li, J. Hao, H. Z. Yang, M. M. Zhou, C. Yan and W. Jia, J. Sep. Sci., 2008, 31, 2831 CrossRef CAS.
- C. A. Sellick, R. Hansen, A. R. Maqsood, W. B. Dunn, G. M. Stephens, R. Goodacre and A. J. Dickson, Anal. Chem., 2009, 81, 174 CrossRef CAS.
- S. V. Vulimiri, M. Misra, J. T. Hamm, M. Mitchell and A. Berger, Chem. Res. Toxicol., 2009, 22, 492 CrossRef CAS.
- R. Pandher, C. Ducruix, S. A. Eccles and F. I. Raynaud, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1352 CrossRef CAS.
- H. Mizuno, N. Tsuyama, S. Date, T. Harada and T. Masujima, Anal. Sci., 2008, 24, 1525 CrossRef CAS.
- A. N. Lane, T. W. M. Fan, R. M. Higashi, J. L. Tan, M. Bousamra and D. M. Miller, Exp. Mol. Pathol., 2009, 86, 165 CrossRef CAS.
- G. G. Cezar, J. A. Quam, A. M. Smith, G. J. M. Rosa, M. S. Piekarczyk, J. F. Brown, F. H. Gage and A. R. Muotri, Stem Cells Dev., 2007, 16, 869 CrossRef CAS.
- W. B. Dunn, M. Brown, S. A. Worton, I. P. Crocker, D. Broadhurst, R. Horgan, L. Kenny, P. N. Baker, D. B. Kell and A. E. P. Heazell, Placenta, 2009, 30, 974 CrossRef CAS.
- K. A. Lawton, A. Berger, M. Mitchell, K. E. Milgram, A. M. Evans, L. N. Guo, R. W. Hanson, S. C. Kalhan, J. A. Ryals and M. V. Milburn, Pharmacogenomics, 2008, 9, 383 CrossRef CAS.
- E. M. Lenz, J. Bright, I. D. Wilson, A. Hughes, J. Morrisson, H. Lindberg and A. Lockton, J. Pharm. Biomed. Anal., 2004, 36, 841 CrossRef CAS.
- G. L. Jones, E. Sang, C. Goddard, R. J. Mortishire-Smith, B. C. Sweatman, J. N. Haselden, K. Davies, A. A. Grace, K. Clarke and J. L. Griffin, J. Biol. Chem., 2005, 280, 7530 CAS.
- R. M. Salek, M. L. Maguire, E. Bentley, D. V. Rubtsov, T. Hough, M. Cheeseman, D. Nunez, B. C. Sweatman, J. N. Haselden, R. D. Cox, S. C. Connor and J. L. Griffin, Physiol. Genomics, 2007, 29, 99 Search PubMed.
- C. B. Clish, E. Davidov, M. Oresic, T. N. Plasterer, G. Lavine, T. Londo, M. Meys, P. Snell, W. Stochaj, A. Adourian, X. Zhang, N. Morel, E. Neumann, E. Verheij, J. T. Vogels, L. M. Havekes, N. Afeyan, F. Regnier, J. van der Greef and S. Naylor, OMICS, 2004, 8, 3 CrossRef CAS.
- J. Y. Wu, H. J. Kao, S. C. Li, R. Stevens, S. Hillman, D. Millington and Y. T. Chen, J. Clin. Invest., 2004, 113, 434 CAS.
- H. J. Kao, C. F. Cheng, Y. H. Chen, S. L. Hung, C. C. Huang, D. Millington, T. Kikuchi, J. Y. Wu and Y. T. Chen, Hum. Mol. Genet., 2006, 15, 3569 CrossRef CAS.
- M. Mayr, Y. L. Chung, U. Mayr, X. K. Yin, L. Ly, H. Troy, S. Fredericks, Y. H. Hu, J. R. Griffiths and Q. B. Xu, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 2135 CrossRef CAS.
- J. L. Griffin, E. Sang, T. Evens, K. Davies and K. Clarke, FEBS Lett., 2002, 530, 109 CrossRef CAS.
- A. S. Plump, J. D. Smith, T. Hayek, K. Aalto-Setala, A. Walsh, J. G. Verstuyft, E. M. Rubin and J. L. Breslow, Cell (Cambridge, Mass.), 1992, 71, 343 CrossRef CAS.
- D. L. Coleman and K. P. Hummel, Am. J. Physiol., 1969, 217, 1298 Search PubMed.
- K. P. Hummel, M. M. Dickie and D. L. Coleman, Science, 1966, 153, 1127 CrossRef CAS.
- K. Sharma, P. McCue and S. R. Dunn, Am. J. Physiol. Renal Physiol., 2003, 284, F1138 CAS.
- M. E. Dumas, S. P. Wilder, M. T. Bihoreau, R. H. Barton, J. F. Fearnside, K. Argoud, L. D'Amato, R. H. Wallis, C. Blancher, H. C. Keun, D. Baunsgaard, J. Scott, U. G. Sidelmann, J. K. Nicholson and D. Gauguier, Nat. Genet., 2007, 39, 666 CrossRef CAS.
- J. Xu, G. Xiao, C. Trujillo, V. Chang, L. Blanco, S. B. Joseph, S. Bassilian, M. F. Saad, P. Tontonoz, W. N. Lee and I. J. Kurland, J. Biol. Chem., 2002, 277, 50237 CrossRef CAS.
- H. J. Atherton, N. J. Bailey, W. Zhang, J. Taylor, H. Major, J. Shockcor, K. Clarke and J. L. Griffin, Physiol. Genomics, 2006, 27, 178 Search PubMed.
- G. Medina-Gomez, S. L. Gray, L. Yetukuri, K. Shimomura, S. Virtue, M. Campbell, R. K. Curtis, M. Jimenez-Linan, M. Blount, G. S. Yeo, M. Lopez, T. Seppanen-Laakso, F. M. Ashcroft, M. Oresic and A. Vidal-Puig, PLoS Genet., 2007, 3, e64 Search PubMed.
- G. Medina-Gomez, L. Yetukuri, V. Velagapudi, M. Campbell, M. Blount, M. Jimenez-Linan, M. Ros, M. Oresic and A. Vidal-Puig, Dis. Models Mech., 2009, 2, 582 Search PubMed.
- M. Kolak, J. Westerbacka, V. R. Velagapudi, D. Wagsater, L. Yetukuri, J. Makkonen, A. Rissanen, A. M. Hakkinen, M. Lindell, R. Bergholm, A. Hamsten, P. Eriksson, R. M. Fisher, M. Oresic and H. Yki-Jarvinen, Diabetes, 2007, 56, 1960 CrossRef CAS.
- K. H. Pietilainen, J. Naukkarinen, A. Rissanen, J. Saharinen, P. Ellonen, H. Keranen, A. Suomalainen, A. Gotz, T. Suortti, H. Yki-Jarvinen, M. Oresic, J. Kaprio and L. Peltonen, PLoS Med., 2008, 5, e51 CrossRef.
- C. B. Newgard, J. An, J. R. Bain, M. J. Muehlbauer, R. D. Stevens, L. F. Lien, A. M. Haqq, S. H. Shah, M. Arlotto, C. A. Slentz, J. Rochon, D. Gallup, O. Ilkayeva, B. R. Wenner, W. S. Yancy, Jr., H. Eisenson, G. Musante, R. S. Surwit, D. S. Millington, M. D. Butler and L. P. Svetkey, Cell Metab., 2009, 9, 311 CrossRef CAS.
- M. Ala-Korpela, Clin. Chem. Lab. Med., 2008, 46, 27 CrossRef CAS.
- L. C. Kenny, D. Broadhurst, M. Brown, W. B. Dunn, C. W. G. Redman, D. B. Kill and P. N. Baker, Reproductive Sciences, 2008, 15, 591 Search PubMed.
- L. C. Kenny, W. B. Dunn, D. I. Ellis, J. Myers, P. N. Baker and D. B. Kell, Metabolomics, 2005, 1, 227 CrossRef.
- A. T. Turer, R. D. Stevens, J. R. Bain, M. J. Muehlbauer, J. van der Westhuizen, J. P. Mathew, D. A. Schwinn, D. D. Glower, C. B. Newgard and M. V. Podgoreanu, Circulation, 2009, 119, 1736 CrossRef CAS.
- J. L. Griffin, C. K. Cemal and M. A. Pook, Physiol. Genomics, 2004, 16, 334 Search PubMed.
- J. L. Griffin and J. P. Shockcor, Nat. Rev. Cancer, 2004, 4, 551 CrossRef CAS.
- T. M. Tsang, J. L. Griffin, J. Haselden, C. Fish and E. Holmes, Magn. Reson. Med., 2005, 53, 1018 CrossRef CAS.
- J. L. Griffin, K. K. Lehtimaki, P. K. Valonen, O. H. Grohn, M. I. Kettunen, S. Yla-Herttuala, A. Pitkanen, J. K. Nicholson and R. A. Kauppinen, Cancer Res., 2003, 63, 3195 CAS.
- K. K. Lehtimaki, P. K. Valonen, J. L. Griffin, T. H. Vaisanen, O. H. Grohn, M. I. Kettunen, J. Vepsalainen, S. Yla-Herttuala, J. Nicholson and R. A. Kauppinen, J. Biol. Chem., 2003, 278, 45915 CrossRef.
- A. R. Tate, C. Majos, A. Moreno, F. A. Howe, J. R. Griffiths and C. Arus, Magn. Reson. Med., 2003, 49, 29 CrossRef CAS.
- J. L. Griffin, H. J. Williams, E. Sang, K. Clarke, C. Rae and J. K. Nicholson, Anal. Biochem., 2001, 293, 16 CrossRef CAS.
- C. Ohdoi, W. L. Nyhan and T. Kuhara, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2003, 792, 123 CrossRef CAS.
- S. Prabakaran, J. E. Swatton, M. M. Ryan, S. J. Huffaker, J. T. Huang, J. L. Griffin, M. Wayland, T. Freeman, F. Dudbridge, K. S. Lilley, N. A. Karp, S. Hester, D. Tkachev, M. L. Mimmack, R. H. Yolken, M. J. Webster, E. F. Torrey and S. Bahn, Mol. Psychiatry, 2004, 9, 684 CAS.
- S. Rozen, M. E. Cudkowicz, M. Bogdanov, W. R. Matson, B. S. Kristal, C. Beecher, S. Harrison, P. Vouros, J. Flarakos, K. Vigneau-Callahan, T. D. Matson, K. M. Newhall, M. F. Beal, R. H. Brown and R. Kaddurah-Daouk, Metabolomics, 2005, 1, 101 CrossRef CAS.
- T. M. Tsang, B. Woodman, G. A. McLoughlin, J. L. Griffin, S. J. Tabrizi, G. P. Bates and E. Holmes, J. Proteome Res., 2006, 5, 483 CrossRef CAS.
- E. Holmes, T. M. Tsang, J. T. Huang, F. M. Leweke, D. Koethe, C. W. Gerth, B. M. Nolden, S. Gross, D. Schreiber, J. K. Nicholson and S. Bahn, PLoS Med., 2006, 3, e327 CrossRef.
- A. Subramanian, A. Gupta, S. Saxena, A. Gupta, R. Kumar, A. Nigam, R. Kumar, S. K. Mandal and R. Roy, NMR Biomed., 2005, 18, 213 CrossRef CAS.
- A. J. Sinclair, M. R. Viant, A. K. Ball, M. A. Burdon, E. A. Walker, P. M. Stewart, S. Rauz and S. P. Young, NMR Biomed., 2010, 23, 123 CAS.
- R. Kaddurah-Daouk, PLoS Med., 2006, 3, e363 CrossRef.
- S. Prabakaran, J. E. Swatton, M. M. Ryan, S. J. Huffaker, J. T. J. Huang, J. L. Griffin, M. Wayland, T. Freeman, F. Dudbridge, K. S. Lilley, N. A. Karp, S. Hester, D. Tkachev, M. L. Mimmack, R. H. Yolken, M. J. Webster, E. F. Torrey and S. Bahn, Mol. Psychiatry, 2004, 9, 684 CAS.
- C. L. Florian, N. E. Preece, K. K. Bhakoo, S. R. Williams and M. Noble, NMR Biomed., 1995, 8, 253 CrossRef CAS.
- L. L. Cheng, I. W. Chang, D. N. Louis and R. G. Gonzalez, Cancer Res., 1998, 58, 1825 CAS.
- F. A. Howe, S. J. Barton, S. A. Cudlip, M. Stubbs, D. E. Saunders, M. Murphy, P. Wilkins, K. S. Opstad, V. L. Doyle, M. A. McLean, B. A. Bell and J. R. Griffiths, Magn. Reson. Med., 2003, 49, 223 CrossRef CAS.
- C. Denkert, J. Budczies, T. Kind, W. Weichert, P. Tablack, J. Sehouli, S. Niesporek, D. Konsgen, M. Dietel and O. Fiehn, Cancer Res., 2006, 66, 10795 CrossRef CAS.
- L. L. Cheng, C. Wu, M. R. Smith and R. G. Gonzalez, FEBS Lett., 2001, 494, 112 CrossRef CAS.
- D. G. Robertson, Toxicol. Sci., 2005, 85, 809 CrossRef CAS.
- H. C. Keun, Pharmacol. Ther., 2006, 109, 92 CrossRef CAS.
- M. Coen, E. Holmes, J. C. Lindon and J. K. Nicholson, Chem. Res. Toxicol., 2008, 21, 9 CrossRef CAS.
- M. E. Bollard, E. G. Stanley, J. C. Lindon, J. K. Nicholson and E. Holmes, NMR Biomed., 2005, 18, 143 CrossRef CAS.
- J. C. Lindon, J. K. Nicholson, E. Holmes, H. Antti, M. E. Bollard, H. Keun, O. Beckonert, T. M. Ebbels, M. D. Reilly, D. Robertson, G. J. Stevens, P. Luke, A. P. Breau, G. H. Cantor, R. H. Bible, U. Niederhauser, H. Senn, G. Schlotterbeck, U. G. Sidelmann, S. M. Laursen, A. Tymiak, B. D. Car, L. Lehman-McKeeman, J. M. Colet, A. Loukaci and C. Thomas, Toxicol. Appl. Pharmacol., 2003, 187, 137 CrossRef CAS.
- J. C. Lindon, H. C. Keun, T. M. D. Ebbels, J. M. T. Pearce, E. Holmes and J. K. Nicholson, Pharmacogenomics, 2005, 6, 691 CrossRef CAS.
- T. M. D. Ebbels, H. C. Keun, O. P. Beckonert, M. E. Bollard, J. C. Lindon, E. Holmes and J. K. Nicholson, J. Proteome Res., 2007, 6, 4407 CrossRef CAS.
- S. C. Connor, M. P. Hodson, S. Ringeissen, B. C. Sweatman, P. J. McGill, C. J. Waterfield and J. N. Haselden, Biomarkers, 2004, 9, 364 CrossRef CAS.
- J. Delaney, M. P. Hodson, H. Thakkar, S. C. Connor, B. C. Sweatman, S. P. Kenny, P. J. McGill, J. C. Holder, K. A. Hutton, J. N. Haselden and C. J. Waterfield, Arch. Toxicol., 2005, 79, 208 CrossRef CAS.
- S. Ringeissen, S. C. Connor, H. R. Brown, B. C. Sweatman, M. P. Hodson, S. P. Kenny, R. I. Haworth, P. McGill, M. A. Price, M. C. Aylott, D. J. Nunez, J. N. Haselden and C. J. Waterfield, Biomarkers, 2003, 8, 240 CrossRef CAS.
- T. A. Clayton, J. C. Lindon, J. R. Everett, C. Charuel, G. Hanton, J. L. Le Net, J. P. Provost and J. K. Nicholson, Arch. Toxicol., 2003, 77, 208 CAS.
- R. J. Mortishire-Smith, G. L. Skiles, J. W. Lawrence, S. Spence, A. W. Nicholls, B. A. Johnson and J. K. Nicholson, Chem. Res. Toxicol., 2004, 17, 165 CrossRef CAS.
- http://www.lipidmaps.org/ .
- F. Spener, M. Lagarde, A. Geloen and M. Record, Eur. J. Lipid Sci. Technol., 2003, 105, 481 CrossRef.
- C. X. Hu, R. van der Heijden, M. Wang, J. van der Greef, T. Hankemeier and G. W. Xua, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 2836 CrossRef CAS.
- M. M. Wiest and S. M. Watkins, Curr. Opin. Lipidol., 2007, 18, 181 CrossRef CAS.
- A. Z. Fernandis and M. R. Wenk, Curr. Opin. Lipidol., 2007, 18, 121 CrossRef CAS.
- L. D. Roberts, G. McCombie, C. M. Titman and J. L. Griffin, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 871, 174 CrossRef CAS.
- M. R. Wenk, Nat. Rev. Drug Discovery, 2005, 4, 594 CrossRef CAS.
- T. W. Mitchell, H. Pham, M. C. Thomas and S. J. Blanksby, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 2722 CrossRef CAS.
- A. Carrasco-Pancorbo, N. Navas-Iglesias and L. Cuadros-Rodriguez, TrAC, Trends Anal. Chem., 2009, 28, 263 CrossRef CAS.
- K. Schmelzer, E. Fahy, S. Subramaniam and E. A. Dennis, in Methods in Enzymology, Vol. 432, ed. H. A. Brown, Academic Press, San Diego, 1st edn., 2007, pp. 171–183 Search PubMed.
- M. Oresic, Eur. J. Lipid Sci. Technol., 2009, 111, 99 CrossRef CAS.
- X. Su, X. L. Han, D. J. Mancuso, D. R. Abendschein and R. W. Gross, Biochemistry, 2005, 44, 5234 CrossRef CAS.
- A. Giovane, A. Balestrieri and C. Napoli, J. Cell. Biochem., 2008, 105, 648 CrossRef CAS.
- E. J. Lesnefsky, P. Minkler and C. L. Hoppel, J. Mol. Cell. Cardiol., 2009, 46, 1008 CrossRef CAS.
- R. H. Houtkooper and F. M. Vaz, Cell. Mol. Life Sci., 2008, 65, 2493 CrossRef CAS.
- P. M. Kochanek, R. P. Berger, H. Bayir, A. K. Wagner, L. W. Jenkins and R. S. B. Clark, Curr. Opin. Crit. Care, 2008, 14, 135 CrossRef.
- R. M. Adibhatla and J. F. Hatcher, Future Lipidol., 2007, 2, 403 Search PubMed.
- C. N. Serhan, Y. Lu, S. Hong and R. Yang, in Methods in Enzymology, Vol. 432, H. A. Brown, Academic Press, San Diego, 1st edn., 2007, pp. 275–317 Search PubMed.
- T. P. Malan and F. Porreca, Prostaglandins Other Lipid Mediators, 2005, 77, 123 CrossRef CAS.
- I. M. Cristea and M. Degli Esposti, Chem. Phys. Lipids, 2004, 129, 133 CrossRef CAS.
- J. T. Smilowitz, M. M. Wiest, S. M. Watkins, D. Teegarden, M. B. Zemel, J. B. German and M. D. Van Loan, J. Nutr., 2009, 139, 222 CAS.
- K. R. Ong, A. H. Sims, M. Harvie, M. Chapman, W. B. Dunn, D. Broadhurst, R. Goodacre, M. Wilson, N. Thomas, R. B. Clarke and A. Howell, Cancer Prev. Res., 2009, 2, 720 Search PubMed.
- J. B. German, M. A. Roberts, L. Fay and S. M. Watkins, J. Nutr., 2002, 132, 2486 CAS.
- G. Fave, M. E. Beckmann, J. H. Draper and J. C. Mathers, Genes Nutr., 2009, 4, 135 Search PubMed.
- M. Jenab, N. Slimani, M. Bictash, P. Ferrari and S. A. Bingham, Hum. Genet., 2009, 125, 507 CrossRef.
- A. N. Lane, T. W. M. Fan and R. M. Higashi, in Methods in Cell Biology, Vol. 84, ed. J. Correia, Academic Press, London, 1st edn., 2008, vol. 84, pp. 541–588 Search PubMed.
- N. Zamboni, S. M. Fendt, M. Ruhl and U. Sauer, Nat. Protoc., 2009, 4, 878 Search PubMed.
- T. W. M. Fan, A. N. Lane, R. M. Higashi, M. A. Farag, H. Gao, M. Bousamra and D. M. Miller, Mol. Cancer, 2009, 8, 41 CrossRef.
- N. Zamboni and U. Sauer, Curr. Opin. Microbiol., 2009, 12, 553 CrossRef CAS.
- N. Zamboni, in Topics in Current Genetics, ed. J. Nielsen and M. Jewett, Springer, Berlin, 2007, pp. 129–157 Search PubMed.
- K. Noh, K. Gronke, B. Luo, R. Takors, M. Oldiges and W. Wiechert, J. Biotechnol., 2007, 129, 249 CrossRef.
- J. G. Jones, R. Naidoo, A. D. Sherry, F. M. H. Jeffrey, G. L. Cottam and C. R. Malloy, FEBS Lett., 1997, 412, 131 CrossRef CAS.
- E. D. Lewandowski and D. L. Johnston, Am. J. Physiol., 1990, 258, H1357 CAS.
- P. Morris and H. Bachelard, NMR Biomed., 2003, 16, 303 CrossRef CAS.
- N. R. Sibson, A. Dhankhar, G. F. Mason, K. L. Behar, D. L. Rothman and R. G. Shulman, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2699 CrossRef CAS.
- J. Munger, B. D. Bennett, A. Parikh, X. J. Feng, J. McArdle, H. A. Rabitz, T. Shenk and J. D. Rabinowitz, Nat. Biotechnol., 2008, 26, 1179 CrossRef CAS.
- R. Shroff, L. Rulisek, J. Doubsky and A. Svatos, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10092 CrossRef.
- J. S. Fletcher, Analyst, 2009, 134, 2204 RSC.
- S. Mas, R. Perez, R. Martinez-Pinna, J. Egido and F. Vivanco, Proteomics, 2008, 8, 3735 CrossRef CAS.
- Z. Takats, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471 CrossRef CAS.
- T. R. Northen, O. Yanes, M. T. Northen, D. Marrinucci, W. Uritboonthai, J. Apon, S. L. Golledge, A. Nordstrom and G. Siuzdak, Nature, 2007, 449, 1033 CrossRef CAS.
- L. M. De Leon-Rodriguez, A. J. M. Lubag, C. R. Malloy, G. V. Martinez, R. J. Gillies and A. D. Sherry, Acc. Chem. Res., 2009, 42, 948 CrossRef CAS.
- R. Powers, Comb. Chem. High Throughput Screening, 2007, 10, 676 CrossRef CAS.
- D. R. Elias, D. L. J. Thorek, A. K. Chen, J. Czupryna and A. Tsourkas, Cancer Biomarkers, 2008, 4, 287 Search PubMed.
- C. Gieger, L. Geistlinger, E. Altmaier, M. H. de Angelis, F. Kronenberg, T. Meitinger, H. W. Mewes, H. E. Wichmann, K. M. Weinberger, J. Adamski, T. Illig and K. Suhre, PLoS Genet., 2008, 4, e1000282 Search PubMed.
- T. Shlomi, M. N. Cabili and E. Ruppin, Mol. Syst. Biol., 2009, 5, 263.
- D. Ziogas, T. Liakakos, E. Lykoudis, E. Fatourou and D. H. Roukos, Radiother. Oncol., 2009, 90, 161 CrossRef CAS.
- J. L. Markley, E. L. Ulrich, H. M. Berman, K. Henrick, H. Nakamura and H. Akutsu, J. Biomol. NMR, 2008, 40, 153 CrossRef CAS.
- Q. Cui, I. A. Lewis, A. D. Hegeman, M. E. Anderson, J. Li, C. F. Schulte, W. M. Westler, H. R. Eghbalnia, M. R. Sussman and J. L. Markley, Nat. Biotechnol., 2008, 26, 162 CrossRef CAS.
- F. Zhang, L. Bruschweiler-Li, S. L. Robinette and R. Brushweiler, Anal. Chem., 2008, 80, 7549 CrossRef CAS.
- S. G. Villas-Boas, D. G. Delicado, M. Akesson and J. Nielsen, Anal. Biochem., 2003, 322, 134 CrossRef.
- K. Bryan, L. Brennan and P. Cunningham, BMC Bioinformatics, 2008, 9, 470 CrossRef.
- J. G. Xia, T. C. Bjorndahl, P. Tang and D. S. Wishart, BMC Bioinformatics, 2008, 9, 507 CrossRef.
- S. Bocker and F. Rasche, Bioinformatics, 2008, 24, i49 CrossRef.
- D. P. Overy, D. P. Enot, K. Tailliart, H. Jenkins, D. Parker, M. Beckmann and J. Draper, Nat. Protoc., 2008, 3, 471 Search PubMed.
- S. Rogers, R. A. Scheltema, M. Girolami and R. Breitling, Bioinformatics, 2009, 25, 512 CrossRef CAS.
- T. Kind and O. Fiehn, BMC Bioinformatics, 2007, 8, 105 CrossRef.
- L. W. Sumner, A. Amberg, D. Barrett, M. H. Beale, R. Beger, C. A. Daykin, T. W. M. Fan, O. Fiehn, R. Goodacre, J. L. Griffin, T. Hankemeier, N. Hardy, J. Harnly, R. Higashi, J. Kopka, A. N. Lane, J. C. Lindon, P. Marriott, A. W. Nicholls, M. D. Reily, J. J. Thaden and M. R. Viant, Metabolomics, 2007, 3, 211 CrossRef CAS.
- http://pubchem.ncbi.nlm.nih.gov/ .
- http://www.chemspider.com/ .
- A. Marston and K. Hostettmann, Planta Med., 2009, 75, 672 CrossRef CAS.
- J. Kopka, N. Schauer, S. Krueger, C. Birkemeyer, B. Usadel, E. Bergmuller, P. Dormann, W. Weckwerth, Y. Gibon, M. Stitt, L. Willmitzer, A. R. Fernie and D. Steinhauser, Bioinformatics, 2005, 21, 1635 CAS.
- N. Schauer, D. Steinhauser, S. Strelkov, D. Schomburg, G. Allison, T. Moritz, K. Lundgren, U. Roessner-Tunali, M. G. Forbes, L. Willmitzer, A. R. Fernie and J. Kopka, FEBS Lett., 2005, 579, 1332 CrossRef CAS.
- T. Kind, G. Wohlgemuth, D. Lee, Y. Lu, M. Palazoglu, S. Shahbaz and O. Fiehn, Anal. Chem., 2009, 81, 10038 CrossRef CAS.
- A. W. T. Bristow, W. F. Nichols, K. S. Webb and B. Conway, Rapid Commun. Mass Spectrom., 2002, 16, 2374 CrossRef CAS.
- A. W. T. Bristow, K. S. Webb, A. T. Lubben and J. Halket, Rapid Commun. Mass Spectrom., 2004, 18, 1447 CrossRef CAS.
- H. Jenkins, N. Hardy, M. Beckmann, J. Draper, A. R. Smith, J. Taylor, O. Fiehn, R. Goodacre, R. J. Bino, R. Hall, J. Kopka, G. A. Lane, B. M. Lange, J. R. Liu, P. Mendes, B. J. Nikolau, S. G. Oliver, N. W. Paton, S. Rhee, U. Roessner-Tunali, K. Saito, J. Smedsgaard, L. W. Sumner, T. Wang, S. Walsh, E. S. Wurtele and D. B. Kell, Nat. Biotechnol., 2004, 22, 1601 CrossRef CAS.
- O. Fiehn, D. Robertson, J. Griffin, M. van der Werf, B. Nikolau, N. Morrison, L. W. Sumner, R. Goodacre, N. W. Hardy, C. Taylor, J. Fostel, B. Kristal, R. Kaddurah-Daouk, P. Mendes, B. van Ommen, J. C. Lindon and S. A. Sansone, Metabolomics, 2007, 3, 175 CrossRef CAS.
- J. L. Griffin, A. W. Nicholls, C. A. Daykin, S. Heald, H. C. Keun, I. Schuppe-Koistinen, J. R. Griffiths, L. L. Cheng, P. Rocca-Serra, D. V. Rubtsov and D. Robertson, Metabolomics, 2007, 3, 179 CrossRef CAS.
- M. J. van der Werf, R. Takors, J. Smedsgaard, J. Nielsen, T. Ferenci, J. C. Portais, C. Wittmann, M. Hooks, A. Tomassini, M. Oldiges, J. Fostel and U. Sauer, Metabolomics, 2007, 3, 189 CrossRef CAS.
- O. Fiehn, L. W. Sumner, S. Y. Rhee, J. Ward, J. Dickerson, B. M. Lange, G. Lane, U. Roessner, R. Last and B. Nikolau, Metabolomics, 2007, 3, 195 CrossRef CAS.
- N. Morrison, D. Bearden, J. G. Bundy, T. Collette, F. Currie, M. P. Davey, N. S. Haigh, D. Hancock, O. A. H. Jones, S. Rochfort, S. A. Sansone, D. Stys, Q. Teng, D. Field and M. R. Viant, Metabolomics, 2007, 3, 203 CrossRef CAS.
- D. V. Rubtsov, H. Jenkins, C. Ludwig, J. Easton, M. R. Viant, U. Guenther, J. L. Griffin and N. Hardy, Metabolomics, 2007, 3, 223 CrossRef CAS.
- R. Goodacre, D. Broadhurst, A. K. Smilde, B. S. Kristal, J. D. Baker, R. Beger, C. Bessant, S. Connor, G. Calmani, A. Craig, T. Ebbels, D. B. Kell, C. Manetti, J. Newton, G. Paternostro, R. Somorjai, M. Sjostrom, J. Trygg and F. Wulfert, Metabolomics, 2007, 3, 231 CrossRef CAS.
- N. W. Hardy and C. F. Taylor, Metabolomics, 2007, 3, 243 CrossRef CAS.
- S. A. Sansone, D. Schober, H. J. Atherton, O. Fiehn, H. Jenkins, P. Rocca-Serra, D. V. Rubtsov, I. Spasic, L. Soldatova, C. Taylor, A. Tseng and M. R. Viant, Metabolomics, 2007, 3, 249 CrossRef CAS.
- I. Spasic, D. Schober, S. A. Sansone, D. Rebholz-Schuhmann, D. B. Kell and N. W. Paton, BMC Bioinf., 2008, 9(S5) Search PubMed.
- I. Spasic, W. B. Dunn, G. Velarde, A. Tseng, H. Jenkins, N. Hardy, S. G. Oliver and D. B. Kell, BMC Bioinformatics, 2006, 7, 281 CrossRef.
- E. Urbanczyk-Wochniak, A. Luedemann, J. Kopka, J. Selbig, U. Roessner-Tunali, L. Willmitzer and A. R. Fernie, EMBO Rep., 2003, 4, 989 CrossRef CAS.
- P. H. Bradley, M. J. Brauer, J. D. Rabinowitz and O. G. Troyanskaya, PLoS Comput. Biol., 2009, 5, e1000270 CrossRef.
| This journal is © The Royal Society of Chemistry 2011 |