Gold nanostructures: a class of multifunctional materials for biomedical applications
Claire M.
Cobley
,
Jingyi
Chen
,
Eun Chul
Cho
,
Lihong V.
Wang
and
Younan
Xia
*
Department of Biomedical Engineering, Washington University, St. Louis, MO 63130, USA. E-mail: xia@biomed.wustl.edu
First published on 6th September 2010
Abstract
Gold nanostructures have proven to be a versatile platform for a broad range of biomedical applications, with potential use in numerous areas including: diagnostics and sensing, in vitro and in vivo imaging, and therapeutic techniques. These applications are possible because of the highly favorable properties of gold nanostructures, many of which can be tailored for specific applications. In the first part of this tutorial review, we will discuss the most critical properties of gold nanostructures for biomedical applications: surface chemistry, localized surface plasmon resonance (LSPR), and morphology. In the second part of the review, we will discuss how these properties can be harnessed for a selection of biomedical applications, aiming to give the reader an overview of general strategies as well as highlight some recent advances in this field.
 Claire M. Cobley | Claire M. Cobley graduated with a BS in Chemistry from Brown University (2006) and a MS in Chemistry from the University of Washington (2007). She is currently pursuing her PhD in Biomedical Engineering with Professor Younan Xia at Washington University in St. Louis. Her research interests include the synthesis, analysis, and biomedical applications of metal nanostructures. |
 Jingyi Chen | Jingyi Chen received a BS in Chemistry from Sun Yat-Sen University in China (1997), a MA in Chemistry from SUNY College at Buffalo (2002), and a PhD in Analytical Chemistry with Professor Younan Xia from the University of Washington (2006). She then worked as a postdoctoral fellow at Brookhaven National Laboratory. She worked as a Research Assistant Professor of Biomedical Engineering at Washington University in St. Louis from May 2008 to July 2010. She is currently an Assistant Professor of Chemistry and Biochemistry at the University of Arkansas, Fayetteville. Her research interests include the synthesis and characterization of nanostructured materials for clean energy and biomedical applications. |
 Eun Chul Cho | Eun Chul Cho was born in Seoul, Korea. He received a BS in Polymer Science and Engineering from Sung Kyun Kwan University in 1998. He received both his MS and PhD in Polymer and Interface Science from Pohang University of Science and Technology (POSTECH) in 2000 and 2004, respectively, with Professor Kilwon Cho. From January 2004 to December 2007, he worked for the Amorepacific Corporation, a cosmetic company in Korea. Since January, 2008, he has been working with Professor Younan Xia as a postdoctoral fellow at Washington University in St. Louis. His research interests include the synthesis of noble metal nanostructures and polymer colloids, and their biomedical applications. |
 Lihong V. Wang | Lihong V. Wang received his PhD degree from Rice University, Houston, Texas under the tutelage of Drs Robert Curl, Richard Smalley, and Frank Tittel. He currently holds the Gene K. Beare Distinguished Professorship of Biomedical Engineering at Washington University in St. Louis and directs the Optical Imaging Laboratory. His book entitled “Biomedical Optics: Principles and Imaging,” one of the first textbooks in the field, won the 2010 Joseph W. Goodman Book Writing Award. He also coauthored a book on polarization, edited the first comprehensive book on photoacoustic tomography, and currently serves as the Editor-in-Chief of the Journal of Biomedical Optics. His laboratory invented or discovered many optical imaging techniques. His Monte Carlo model of photon transport in scattering media has been used worldwide. |
 Younan Xia | Younan Xia received a BS in Chemical Physics from the University of Science and Technology of China in 1987. He came to the United States in 1991, received a MS in Chemistry from UPenn (Alan MacDiarmid) in 1993, and a PhD in Physical Chemistry from Harvard (George Whitesides) in 1996. After staying as a postdoctoral fellow, he started as an Assistant Professor of Chemistry at the University of Washington in Seattle, and was promoted to Associated Professor and Professor in 2002 and 2004, respectively. He moved to Washington University in St. Louis in 2007 and is the James M. McKelvey Professor for Advanced Materials. His research interests include nanomaterials, nanomedicine, tissue engineering, colloidal science, and electrospinning. |
Introduction
While gold has many favorable properties as a bulk metal (such as high electrical conductivity, reflectivity, malleability, and resistance to corrosion and oxidation), new properties and potential applications emerge when it is finely divided into the nanoscale. One of the most dramatic and useful changes is in gold's interaction with light. When a gold nanoparticle is irradiated with light, strong absorption and/or scattering will occur at specific resonant wavelengths, which depend greatly on the morphology and dielectric environment of the gold nanoparticle.1 This phenomenon is known as localized surface plasmon resonance (LSPR), and is the origin of many of the new applications of nanoscale gold particles. This phenomenon has been used for hundreds of years to give a rich red color to stained glass, but systematic study only began in 1857 when Michael Faraday demonstrated the synthesis of gold colloids in an aqueous medium.2 The next major advances came when Gustav Mie solved Maxwell's equations for spherical particles, making it possible to predict and explain the optical properties of gold nanospheres, and in 1951 when John Turkevich pioneered a robust and simple synthesis method.3,4 Both Mie theory and the Turkevich synthesis are still widely used today. Building on these foundational studies, a resurgence of interest in gold nanostructures has occurred in recent years. With more powerful characterization tools and more refined synthetic methodologies, scientists have been able to achieve unprecedented control over the dimensions, morphology, and properties of gold nanostructures.5–7Along with this greater degree of control, a wide variety of new applications for gold nanostructures have been realized, particularly in the area of biomedicine.8–12 By tuning the LSPR from the visible into the near infrared (NIR) region, it is possible to harness the strong optical properties of gold nanostructures in biological environments, as absorption from water and hemoglobin is low at these wavelengths.13,14 Combined with the other favorable properties of gold nanostructures, including relatively bio-inert surfaces, easily modified surface chemistry, and a high degree of size and shape control, this has made gold nanostructures a strong platform for a broad spectrum of biomedical applications.5–12
In this tutorial review, we will first give an overview of the properties of gold nanostructures most critical to biomedical applications: controllable surface chemistry, LSPR, and morphology. In the following sections, we will explain how these properties can be exploited to create and improve biomedical diagnostic techniques and treatments, including discussions of cellular uptake, optical imaging techniques, drug delivery, and photothermal treatment.
Properties of gold nanostructures
Controllable surface chemistry
A number of the critical attributes that make gold nanostructures such a promising platform for biomedical applications are related to the surface chemistry of gold. First of all, the non-reactive and relatively bio-inert nature of gold makes this metal a good candidate for both in vitro and in vivo applications.15 The low cytotoxicity of gold nanostructures has been demonstrated in a number of studies, and initial in vivo and clinical biocompatibility studies have also shown favorable results.15–17Gold colloids were studied in humans in the 1950's as radiotracers for sentinel lymph node biopsy, and a phase I clinical trial has recently been completed for gold nanosphere-based drug carriers.16–18 Despite the involvement of high doses of a toxic agent (tumor necrosis factor-alpha, TNF), minimal side effects were observed from treatment with gold nanospheres.17 Nevertheless, it is important that we continue to study the biocompatibility of these materials, as their suitability for different applications will depend heavily on a number of factors, including the size and shape of the particles, the surface coatings, and the method of administration.11,15Though non-reactive in most situations, the surface of gold is well-known for forming strong, stable gold–thiolate bonds (Au–S, ∼50 kcal/mol) to molecules with thiol (–SH) or disulfide groups (S–S).19 This binding has been extensively studied with self-assembled monolayers (SAMs), where molecules (typically long-chain alkanethiols) generate highly ordered monolayers when they adsorb onto a gold surface.19 This well-characterized binding has been used extensively to add functionality to gold surfaces and gold nanostructures (Fig. 1A).20–22 By carefully choosing the functional group at the distal end of the molecule, it is possible to design and generate a well-defined interface to interact (or not interact) with cells and biomolecules in specific ways. Gold nanostructures can also adsorb biological molecules in a non-specific manner. This binding can be used to add specific functionality, but is also an important factor to consider when examining the cell/gold interaction in a biological environment, as a variety of proteins will adsorb to a non-protected gold surface when it is transferred into a biological medium. The adsorbed proteins can affect the surface properties of gold nanostructures and thus their cellular uptake properties.22
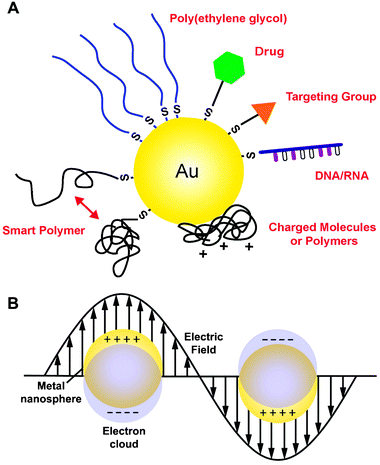 | ||
| Fig. 1 (A) Gold nanostructures can be conjugated with a wide variety of functional moieties, both through the gold–thiolate bond and by passive adsorption. (B) Localized surface plasmon resonance (LSPR) is another critical property of gold nanostructures that results from the collective oscillation of delocalized electrons in response to an external electric field. | ||
For in vivo applications, most gold nanostructures are conjugated with poly(ethylene glycol) (PEG). PEG is a biocompatible polymer that helps prevent particle aggregation, non-specific protein adsorption, and the uptake of circulating gold nanostructures by the reticuloendothelial system (RES), allowing for longer circulation in the bloodstream and consequently greater accumulation of gold nanostructures in tumors through passive targeting.23 Passive targeting is a simple and effective means to concentrate nanostructures in tumors for cancer therapy or imaging. Due to the rapid growth of blood vessels in tumors, the blood vessel walls are typically more porous and leaky than those found in healthy tissue. For this reason, it is easier for appropriately sized nanostructures to move from the bloodstream into tumors.23 Additionally, the lymphatic drainage from tumors is reduced compared to healthy tissue, making it harder for nanostructures to leave the tumor once they are there. Combined together, these effects are referred to as passive targeting or the enhanced permeability and retention (EPR) effect.
When the surfactants, capping agents, or stabilizers used for nanoparticle synthesis bind less strongly compared with gold–thiolate interactions, their replacement with a thiol-terminated PEG is relatively straightforward, requiring simple incubation of gold nanostructures with an excess amount of PEG for a few hours.24 Alternatively, a layer of PEG (or another polymer) can be conjugated in an additional layer on top of the surface of the as-synthesized particles.25
It is also possible to create gold nanostructures with active targeting capabilities via careful surface modification.23,24 Active targeting takes advantage of the fact that rapidly growing cancer cells over-express certain receptors on their surface. If nanostructures are conjugated with a complimentary moiety (e.g., antibodies, peptides, and folate, among others), then they will bind to cancer cells as they circulate, increasing their concentration in the desired region. The binding of the targeting moiety can either occur directly to the surface of the nanostructure, to the terminal end of an attached PEG chain with an appropriate tail group (e.g., a carboxylic acid-based group to bind to amine groups found in antibodies), or through click chemistry.25
Thiolate binding and polymer adsorption can also be employed to attach a number of other functional groups to the surface of gold nanostructures.17,20,21 For example, it is possible to control the charge of a gold nanostructure by adsorbing specific polymers to the surface, which is important as surface charges can have a significant effect on both the cellular uptake and biodistribution of nanostructures. Positively charged nanostructures have been shown to have enhanced affinity to negatively charged cell surfaces and are the most likely to cross cellular membranes and enter the cytoplasm of cells, an important aspect to consider in delivery applications.22,26 It is also possible to use the thiol group to attach oligonucleotides for sensing applications, smart polymers or drugs for stealth delivery, as well as a wide variety of other types of molecules.17,20,21 A few examples of different functional groups that can be attached will be discussed in the application section below.
Localized surface plasmon resonance
One of the most interesting and powerful properties of gold nanostructures is localized surface plasmon resonance (LSPR). When a gold nanostructure encounters electromagnetic radiation of an appropriate wavelength, the delocalized conduction electrons of the metal will begin to oscillate collectively relative to the lattice of positive nuclei with the frequency of the incoming light. Fig. 1B illustrates this phenomenon for a gold nanosphere. This process can be divided into two types of interactions: scattering, in which the incoming light is re-radiated at the same wavelength in all directions, and absorption, in which the energy is transferred into vibrations of the lattice (i.e., phonons), typically observed as heat. Together, these processes are referred to as extinction (extinction = absorption + scattering). In addition, LSPR also generates strong electric near fields close to the surface of the particle. All three of these interactions can be harnessed for biomedical applications, which will be discussed in greater detail in the second half of this review.The specifics of the LSPR response of gold nanostructures depend on a number of variables, particularly the size, shape, and morphology of the nanostructure, as well as the dielectric environment.1,27,28 Consequently, controlling the morphology of gold nanostructures is a powerful route to control the LSPR response. Even small changes in aspect ratio or corner sharpness can have a large impact.27 For biomedical applications, scientists are typically most interested in gold nanostructures that absorb and scatter strongly in the near-infrared region (NIR, 650–900 nm), as light can penetrate deeply into tissue in this region due to low absorption from blood and water, and scattering from tissue.13 A typical gold nanosphere has a LSPR wavelength around 520 nm. By changing the size it is possible to shift this peak slightly, but to achieve strong extinction in the NIR region it is necessary to synthesize new morphologies with more tunable plasmonic properties.14 Two general strategies are used to shift the LSPR ratio into the NIR region: careful control of the aspect ratio for solid structures or fine-tuning of the wall thickness of hollow or core–shell structures.14,29–31
Morphological effects
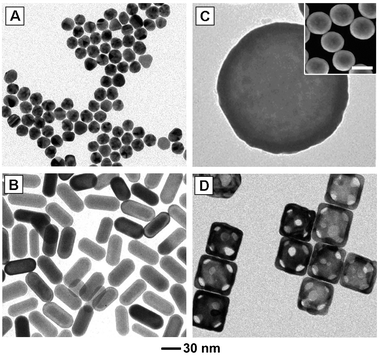 | ||
| Fig. 2 Commonly studied gold nanostructures: (A) multiply twinned gold nanoparticles (often referred to as nanospheres), (B) gold nanoshells (silica beads coated with a polycrystalline gold layer), (C) gold nanorods, and (D) gold nanocages. The scale bar in the inset is 200 nm. Modified with permission from ref. 53 (A, B) and ref. 63 (C), copyright 2010 Wiley and 2007 American Chemical Society, respectively. | ||
Gold nanospheres are the simplest structure to synthesize, and there are now well-developed techniques to produce particles of high uniformity with a variety of sizes.20 In a typical reaction, a dissolved gold salt (such as HAuCl4) is reduced to gold atoms in the presence of a capping agent or surfactant to prevent aggregation. In the most commonly used method, the reducing agent and capping agent are the same: citrate ions. Depending on the size of the nanosphere, strong LSPR extinction will occur between 500–600 nm. Detailed tuning of spectra and theoretical analysis of the size dependence of LSPR peaks are available for gold nanospheres and all the nanostructures discussed herein in a review by our group in a previous issue of this journal.14
Gold nanorods typically have two LSPR peaks, one for the transverse mode around 515 nm and the other for the longitudinal mode whose position depends strongly on the aspect ratio of the rod. These structures are typically synthesized by a seed-mediated, solution-phase method, where small gold seeds are added to a series of growth solutions.30 By adjusting the synthesis parameters (e.g., the concentrations of key reagents and the size of the initial gold seeds), it is possible to synthesize nanorods with strong LSPR-based absorption across the visible spectrum and into the NIR.
The LSPR of gold nanoshells and gold nanocages can also be tuned into the NIR by adjusting the thickness of gold walls surrounding a dielectric or hollow core. However, different synthetic methods are used to achieve this tuning in each type of structure. Nanoshells are created by depositing small gold nanoparticles onto the surface of a silica sphere, followed by deposition of more gold through chemical reduction.31 The final LSPR depends on the ratio between the diameter of the particle (typically ∼120 nm), and the thickness (∼10 nm) of the deposited gold layer.
Gold nanocages are created by hollowing out the interior of a sacrificial template of silver nanocubes (as well as silver nanocrystals with other shapes). When silver nanocubes are titrated with a gold salt (typically HAuCl4), a galvanic replacement reaction occurs, causing gold atoms to be deposited on the surface of the nanocube and silver atoms to be dissolved from a small pit in the surface.32 This pit gradually expands, hollowing out the interior until a uniform shell of gold–silver alloy is formed. If more HAuCl4 is titrated at this point, silver will be selectively removed from the alloy, and the resulting vacancies in the lattice will coalesce into pores on the surface of the nanocages. These pores will form preferentially on the corners if nanocubes with truncated corners are used, allowing for precise control over their sizes, positions, and uniformity.33 The hollow porous structure of nanocages enables additional applications, due to their ability to deliver drugs and other effectors stored in the interior of the cages.21Gold nanocages can be created with a wide variety of sizes (i.e. 30 to 150 nm or more) by changing the size of the initial silver nanocubes.
Beyond LSPR tuning, the specific morphology of gold nanostructures can also have a significant impact on the cellular uptake, in vivo distribution, and pharmacokinetics, key attributes to control for biological applications.24,34–36 For example, the size of nanostructures will affect both the amount of time they circulate in the blood, where they accumulate, and whether they can be easily cleared from the body. Initial experimental and theoretical studies with gold nanospheres have suggested that ∼50 nm is the ideal size for cellular uptake in an in vitro environment, though the optimum particle size will also depend on the type of surface coating employed (as this has a strong effect on the effective size in an aqueous medium), and the optimum size for in vivo experiments may also be different from those determined from in vitro studies.24,34–36
Typically, the relative contributions of these cross sections are calculated theoretically using either Mie theory or the discrete-dipole approximation (DDA).3,40 Mie theory provides an exact solution by directly solving Maxwell's equations, but is limited to spheres, spheroids, shells, and infinite cylinders. The DDA method can be used for any structure, as it approximates a nanostructure as an array of polarizable points, which interact with both incoming light and each other. While both are useful techniques to model the expected properties, it is more desirable to have an experimental method to directly measure these contributions. For this reason, a technique has been developed that combines UV-Vis spectroscopy and photoacoustic imaging to experimentally determine σa.41 This technique has been used to compare the contributions of σa and σs to the optical properties of most of the gold nanostructures shown in Fig. 2. The general technique is as follows: σe is obtained using a UV-vis-NIR spectrometer, together with the well-known Beer–Lambert law while photoacoustic imaging is used to measure σa, and then the ratio between the two is calculated.41
Photoacoustic imaging is based on the photoacoustic effect, where absorption of pulsed laser radiation results in transient heat. The resulting thermo-elastic expansion generates an acoustic signal, which can be directly measured. As the photoacoustic signal is generated due to the optical absorption of a material, the signal is expected to be directly proportional to the absorption coefficient (μa).42,43 The relationship between the photoacoustic signal and μa can be calibrated using an organic dye, making it possible to determine σa for different nanostructures by dividing the photoacoustically measured μa by the concentration of particles.
Fig. 3A shows a setup of the photoacoustic imaging system used for these optical measurements.41 The light travels through an optical condenser and is focused on tubes embedded in an optically scattering medium and filled with aqueous suspensions of gold nanostructures. The photoacoustic signals are detected by an ultrasound scanning transducer placed inside the optical condenser. Fig. 3B shows a typical photoacoustic image of gold nanocages at different concentrations, showing the proportional relationship between the concentration of particles and the photoacoustic signal.
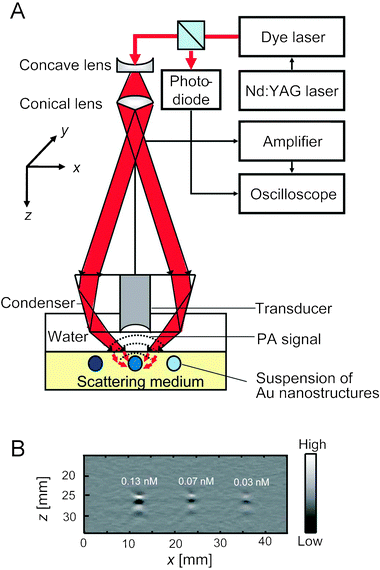 | ||
| Fig. 3 (A) An experimental setup of a photoacoustic (PA) imaging system. (B) A typical depth-resolved B-scan PA image (x–z scan) of a gold nanocage suspension at three different concentrations. Reproduced with permission from ref. 41, copyright 2009 American Chemical Society. | ||
Table 1 shows the optical cross sections of different types of gold nanostructures obtained with both photoacoustic and theoretical methods. For gold nanospheres, calculations were performed with Mie theory, while calculations for gold nanocages and nanorods were carried out using the DDA method.3,40 Each type of nanostructure has its own characteristic σa/σe ratio, which is essentially the same as that predicted by calculation. Some themes emerge from this data (and other studies) that are useful to keep in mind when designing gold nanostructures for biomedical applications. In general, optical cross sections increase as the dimensions of a nanostructure increase. On the other hand, as the size increases, the ratio of σa to σe decreases.9,41 Consequently, to create gold nanostructures with the highest absorption cross section it is necessary to find a balance between these two effects.
| Cross-section (10−15 m2) | σ a /σe | |||
|---|---|---|---|---|
| Extinction (σe) | Absorption (σa) | |||
| a All the values were obtained at 638 nm. b Not detected with photoacoustic imaging. | ||||
| Nanocages (45.0 nm) | Experimental | 7.26 ± 0.06 | 5.96 ± 0.25 | 0.82 ± 0.04 |
| Calculated | 19.88 | 16.26 | 0.82 | |
| Nanocages (32.0 nm) | Experimental | 2.99 ± 0.04 | 3.05 ± 0.12 | 1 ± 0.04 |
| Calculated | 6.39 | 6.02 | 0.94 | |
| Nanorods (44.0 × 19.8 nm) | Experimental | 2.16 ± 0.02 | 1.87 ± 0.23 | 0.87 ± 0.11 |
| Calculated | 2.15 | 1.83 | 0.85 | |
| Nanospheres (150 nm) | Experimental | 145 ± 14.76 | —b | — |
| Calculated | 99.96 | 5.73 | 0.056 | |
Biomedical applications of gold nanostructures
The interesting properties described above have led to strong interest in the use of gold nanostructures for a variety of biomedical applications. Gold nanostructures have been used for fundamental studies of how nanomaterials behave in both in vitro and in vivo environments, have enabled or enhanced a number of diagnostic techniques, and show great potential in targeted therapeutics.8–12,44Table 2 summarizes the connections between the properties listed in the first section of this review and some of the most extensively explored applications.| Diagnostic sensing | In vitro imaging | In vivo imaging | Drug delivery | Controlled release | Photothermal treatment | |
|---|---|---|---|---|---|---|
| Biocompatibility | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Surface modification | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Scattering | ✓ | ✓ | ✓ | |||
| Absorption | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Near-fields | ✓ | |||||
| Photoluminescence | ✓ | ✓ |
Straightforward surface modification plays an important role in many applications. The ability to optimize the uptake of nanostructures and direct them to particular locations in the body through passive and active targeting is particularly advantageous, as many current treatments for diseases such as cancer affect the body broadly, leading to harsh side effects that could hopefully be eliminated with more focused treatments. In addition to having a strong effect on the biodistribution and targeting of gold nanostructures, the gold–thiolate bond makes it possible to add new functionality to gold nanostructures. For example, thermosensitive polymers bound to the surface can allow gold nanostructures to serve as drug delivery vehicles that release their payload in response to temperature.21,45Conjugation with pH sensitive molecules can also transform gold nanostructures into all-optical pH sensors, and may be a route to selectively deliver drugs to the interior of cells.46 It is also possible to conjugate certain therapeutics directly to the surface of the nanostructures, using the stealth-like properties of PEG coatings to transport highly toxic drugs to the tumor site while minimizing side effects.16,17
The strong optical response of gold nanostructures due to LSPR is also critical to many of their biomedical applications.14 The different optical responses that result from this phenomenon and the corresponding applications are shown in Scheme 1. Light scattering is used primarily in imaging techniques such as optical coherence tomography (OCT) where the presence of particles with large scattering cross sections enhances image contrast, and dark-field microscopy, where transmitted light is blocked and scattered light is collected, showing scattering particles with a bright color against a dark background.8–11 Absorption is used in a variety of techniques that take advantage of the fact that strongly absorbing gold nanostructures can become highly localized heat sources when irradiated with a laser through the photothermal effect. The generated heat can be used to initiate release in drug delivery systems, enhance contrast in optical imaging techniques like photoacoustic imaging, and provide photothermal treatment.8–11,21,39,45,47,48 A selection of these techniques will be described in greater detail below.
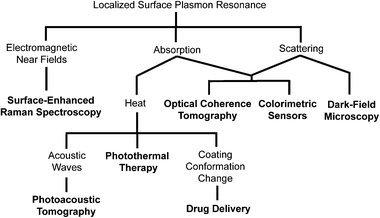 | ||
| Scheme 1 Localized surface plasmon resonance (LSPR) is the origin of a number of interesting phenomena observed with gold nanostructures. These properties have enabled a number of applications, which are indicated in bold. | ||
The LSPR of gold nanostructures has also enabled a number of different sensing techniques, which we will only discuss briefly as they have been covered extensively by previous reviews in this journal.49 The strong local fields from LSPR can enhance the Raman signals of molecules near the surface of a metallic nanostructure by many orders of magnitude. This technique is known as surface-enhanced Raman spectroscopy (SERS), and can enable ultrasensitive detection of biomarkers. Interested readers are directed to ref. 50 and 51 for more information about this technique. The sensitivity of LSPR to changes in dielectric environment and aggregation has also been harnessed to fabricate colorimetric sensors.20,52 A classic example is detection of a target DNA strand by conjugating gold nanospheres with the complimentary oligonucleotide. When the target strand is present, the gold nanospheres will aggregate, resulting in a dramatic color change from red to blue due to coupling between the plasmons of the individual particles.52
In this review, we will focus on three general areas. First, we will discuss the cellular uptake of gold nanostructures, including some recent studies of mixtures of particles. Secondly, we will discuss how gold nanostructures can be used for imaging and diagnostics in both in vitro and in vivo settings. Finally, we will discuss some potential therapies based on gold nanostructures: stealth delivery of a toxic drug through gold nanostructure conjugation, controlled release of effectors from gold nanocages, and photothermal therapy, where heat from LSPR absorption is used to directly destroy cancer cells.
Cellular uptake studies
Gold nanostructures are an ideal system to study cellular uptake of nanomaterials for a number of reasons: it is possible to synthesize a wide variety of particle morphologies with similar sizes, the effect of different surface modifications/targeting mechanisms can be studied due to strong gold–thiolate bonds, and the total amount of gold taken up by cells can be determined with high accuracy and sensitivity by inductively-coupled plasma mass spectrometry (ICP-MS).14,24 Initial studies with gold nanospheres have shown that 50 nm is an ideal size for cellular uptake, fitting with theoretical predictions.35,36 However, the presence of PEG layers and other changes in surface chemistry will also have a considerable effect on uptake results.22,24 In particular, positive charges and targeting ligands can significantly increase cellular uptake.Although ICP-MS provides accurate quantitative data for the cellular uptake of a single type of particle, it cannot differentiate different types of gold nanostructures in a mixture, making it impossible to study interference or competitive uptake using this method. Recently, a technique has been developed that instead relies on the different LSPR properties of gold nanostructures to determine cellular uptake.53
Gold nanospheres and nanorods with similar diameters but different aspect ratios were incubated independently and then co-incubated with human breast cancer SK-BR-3 cells (Fig. 4A). Three surface coatings were studied for each nanostructure: unmodified (with citrate or CTAB left from synthesis), PEGylated, or antibody-conjugated (anti-HER2-PEG). Fig. 4B illustrates how this spectroscopic method can determine the concentration of gold nanospheres and nanorods from a mixture. Gold nanorods have two LSPR peaks, a transverse peak at ∼515 nm, and a longitudinal peak that depends strongly on the length of the rod (652 nm for this example). While the peak at 515 nm overlaps with the peak of the nanospheres at 520 nm, the concentration of gold nanorods can be determined through a calibration curve at 652 nm. The relative contribution of nanorods to the peak at 515–520 nm can then be subtracted before calculating the concentration of nanospheres.
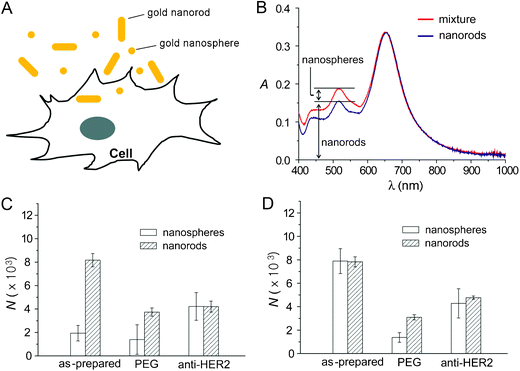 | ||
| Fig. 4 Cellular uptake of gold nanostructures. (A) Schematic of cellular uptake of gold nanostructures. (B) Detection method to distinguish between uptake of nanospheres and nanorods. (C) Cellular uptake of nanospheres and nanorods by SK-BR-3 cells when the gold nanospheres and nanorods were mixed (60 pM each) and then incubated with the cells at 37 °C for 24 h. (D) Cellular uptake of nanospheres and nanorods by SK-BR-3 cells when the gold nanostructures (60 pM) were incubated independently at 37 °C for 24 h. N is the number of gold nanostructures taken up per cell. Modified with permission from ref. 53, copyright 2010 Wiley. | ||
Fig. 4C shows the cellular uptake of nanospheres and nanorods when they were mixed and then incubated with the cells. For comparison, the uptake of gold nanospheres and nanorods when they were independently incubated with the cells is shown in Fig. 4D. These figures indicate, in some cases, that the uptake of gold nanospheres was significantly reduced when co-incubated with gold nanorods, as compared with the independent incubation. No clear trends were found relating the shape of gold nanostructures to their uptake, implying that surface chemistry is likely a more important factor affecting the uptake of gold nanostructures.22
Two-photon microscopy
Optical imaging methods are currently some of the most powerful tools available for in vitro studies, and a number of optical techniques are also being investigated as low cost, portable, non-ionizing alternatives to current in vivo techniques like computed tomography (CT) and magnetic resonance imaging (MRI).42,43 One powerful optical technique is two-photon microscopy that has gained popularity due to its ability to penetrate deeper into tissue than comparable high resolution imaging techniques like confocal microscopy. Typical depth penetration is on the order of 100's of microns. While originally used to image NIR fluorescence dyes, it has been recently demonstrated as a method to image gold nanostructures in cultured cells, tissue phantoms, and in vivo circulation for blood vessels near the skin surface, such as those in a mouse ear.54,55 The resulting signal has been reported to be 60–140 times greater than seen with conventional organic dyes.55 When gold nanostructures are excited with a femto-second pulsed laser, the absorption of two photons excites electrons from the d-band to the sp-band, creating electron-hole pairs. When these electron-hole pairs recombine, luminescence is emitted in the visible region. This process is significantly enhanced when the LSPR of the nanostructure is in the NIR region, where it matches well with the wavelength of the photons used to excite the luminescence. When compared with fluorescent dyes, this technique is not as susceptible to photobleaching, a significant advantage.Two-photon microscopy was used to demonstrate the in vitro targeting abilities of antibody-conjugated gold nanocages.24Fig. 5 shows the results after anti-EGFR conjugated nanocages (A–C) and PEGylated nanocages (D–F) were incubated with U87MGwtEGFR cells for 3 h. The first column shows two-photon luminescence from gold nanocages, the second column shows standard fluorescence imaging of cell membranes and endosomes with FM4-64 dye, and the third column shows an overlay of the two. It is clear from these images that the gold nanocages with anti-EGFR significantly increased the cellular binding and uptake.
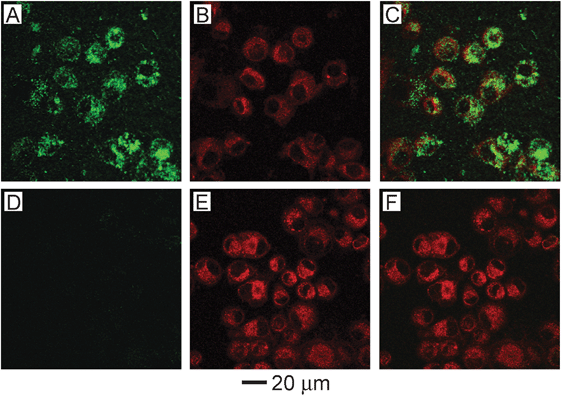 | ||
| Fig. 5 (A–C) U87MGwtEGFR cells after incubation with gold nanocages functionalized with EGFR antibodies. (D–F) Cells after incubation with non-targeted gold nanocages (functionalized with mPEG). (A, D) Two-photon images of gold nanocages showing little uptake without targeting. (B, E) Fluorescence images of SK-BR-3 cells with FM4-64 dye used to stain the membranes and endosomes. (C, F) Overlay of images of nanocages and images of cells, showing clear overlap between the two for the targeted nanocages. Modified with permission from ref. 24, copyright 2010 American Chemical Society. | ||
Photoacoustic imaging
Photoacoustic imaging can offer non-invasive optical imaging at even greater depths than two-photon microscopy.39,42,43 Both the spatial resolution and the imaging depth depend on the ultrasonic detection frequency, which can be adjusted depending on the desired application. The spatial resolution can be varied from 5 to 150 μm and the imaging depth can be varied from 0.7 to 30 mm.42 When tissue is irradiated with a pulsed laser source, the resulting heat creates an acoustic signal that can be measured and converted into an image by a scanning transducer. This signal can be enhanced by the use of contrast agents that strongly absorb light, such as gold nanostructures.42Fig. 6 shows the depth capability of photoacoustic imaging when gold nanocages were used as a contrast agent for in vivo sentinel lymph node detection (one of the first steps in breast cancer staging).39 In each successive panel, an additional layer of chicken breast tissue was placed on top of the region being imaged to demonstrate the feasibility of using this method to detect sentinel lymph nodes for breast cancer staging in humans. A distinct image of the sentinel lymph node could be obtained as deep as 33 mm, though this limitation was actually imposed by the memory of the acquisition system. This depth is significantly greater than the ∼12 mm depth of sentinel lymph nodes in humans, making this a promising way to detect sentinel lymph nodes without the invasive procedures that are currently necessary.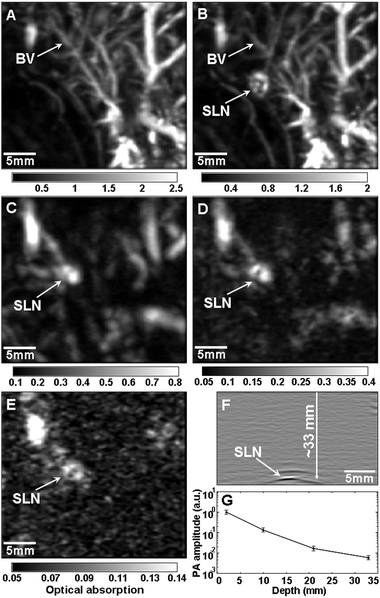 | ||
| Fig. 6 Depth capability of sentinel lymph node (SLN) mapping with gold nanocages for non-invasive in vivo breast cancer staging. Photoacoustic images taken (A) before and (B–E) after the injection: (A) control image; (B) 28 min; (C) 126 min with a layer of chicken breast tissue placed on auxillary region, total imaging depth was 10 mm; (D) 165 min with the second layer of chicken breast tissue, total imaging depth was 21 mm; (E) 226 min with the third layer of chicken breast tissue, total imaging depth was 33 mm. (F) Photoacoustic B-scan with 20 times signal averaging, showing the SLN located 33 mm deep. Memory of the acquisition system limited the record length in depth. (G) The amplitude variations of photoacoustic signals over imaging depths. Data were scaled down to 0 dB and normalized by the maximum. Error bar represents standard deviation. (F) Color bars represent the optical absorption. BV, blood vessel. Reproduced with permission from ref. 39, copyright 2009 American Chemical Society. | ||
Drug delivery
Drug delivery is a prime example of how gold nanostructures can be used for therapy in addition to diagnostics.17,20,21,56 Most work in this area has centered on cancer treatment, as targeted therapies are greatly needed in this field due to the harsh side effects of current broad-based treatments. Through both passive and active targeting, the concentration of drug can be increased at the tumor site while limiting the exposure of (and consequent toxicity to) healthy tissue.23 Stage I clinical trials have been completed for the delivery of tumor necrosis factor (TNF) bound to the surface of gold nanospheres.17TNF is a potent cytokine that has shown antitumor properties in isolation and even greater effects as part of a combined therapy.57,58 Though the mechanism for action is complex, results suggest that TNF is able to significantly increase the permeability of the tumor vasculature, eventually causing destruction of the vascular lining, and also allowing for greater delivery of other chemotherapeutic agents.57,58 Unfortunately, the practical dosage is limited by the severe toxicity of TNF. However, when TNF is conjugated to gold nanospheres, the toxicity can be greatly reduced, allowing for significantly higher dosages of TNF to be delivered to the tumor site.16Fig. 7 shows some of the results from in vivo mouse studies of TNF delivery and the resulting toxicity.16 In these studies, ∼30 nm gold nanospheres were conjugated with a combination of TNF and PEG. TNF is thought to function as an active targeting ligand in addition to being the therapeutic payload. Fig. 7A shows the concentration of TNF in the tumor after treatment with either native TNF (i.e. free in solution) or gold nanospheres conjugated with both PEG and TNF (Au-TNF). The presence of PEG was found to be critical to long circulation times and increased accumulation of TNF in tumors, as unprotected particles were rapidly cleared by RES. While the concentration of native TNF inside the tumor was relatively constant, the concentration of Au-TNF continued to increase and was over 6-fold higher than the native TNF after 3 h. Mice treated with Au-TNF also had smaller tumor volumes (when comparing the same TNF dosage) while suffering much lower rates of toxicity after 16 days (Fig. 7B). While 30–50% of the mice injected with 15 μg of native TNF suffered severe toxicity and died within 24 h of treatment, 100% of the mice treated with equal amounts of Au-TNF survived. Interestingly, though the unprotected particles (no PEG) could still be seen in the liver after a month due to a size larger than the renal clearance limit, there were no signs of distress. The initial results from stage I clinical trials using these drug-conjugated particles also showed minimal side effects in humans, and stage II trials are being planned.17
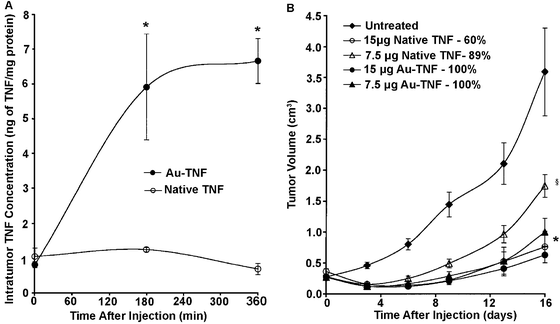 | ||
| Fig. 7 (A) Intratumor TNF accumulation following an intravenous injection of TNF either free in solution (native) or conjugated to PEGylated gold nanoparticles (Au-TNF) (* indicates p < 0.05 versus native). (B) Antitumor efficacy of native TNF and Au-TNF (Au-TNF). Percentages are mouse survival rates (* or § indicates p < 0.05 versus untreated controls). Modified with permission from ref. 16, copyright 2004 Taylor and Francis Inc. | ||
Controlled release from gold nanocages
By using smart polymers, it is possible to create drug delivery systems that not only deliver their payload to a specific place, but deliver it in response to outside stimulus.21,45Fig. 8A shows a schematic of a drug delivery system that combines the photothermal properties of gold nanocages with thermosensitive polymers.21 The strong binding between gold and thiol groups makes it straightforward to attach poly(N-isopropylacrylamide) (pNIPAAm) to the surface of the gold nanocages by using a disulfide initiator (Fig. 8B). When the gold nanocages are irradiated with a laser, the strongly absorbed light is converted into heat through the photothermal effect. When the temperature rises above a certain threshold (the low critical solution temperature, LCST), the pNIPAAm coating undergoes a conformational change. When the polymer is collapsed, the pores of the nanocage are exposed, allowing for effectors pre-loaded in the interior to be released (though some release will also occur from effectors trapped in the polymer layer). The loading process is simply the reverse—pNIPAAM-coated gold nanocages are mixed with a solution of effector at a temperature above the LCST, then quickly cooled below the LCST to trap the contents inside. Conveniently, the LCST of pNIPAAM can be tuned from 32–50 °C through the incorporation of different amounts of acrylic acid (AAm) during the polymerization. The studies presented here used a temperature of 39 °C as this temperature is above body temperature (37 °C), but below the threshold where thermal damage can occur in biological systems (42 °C).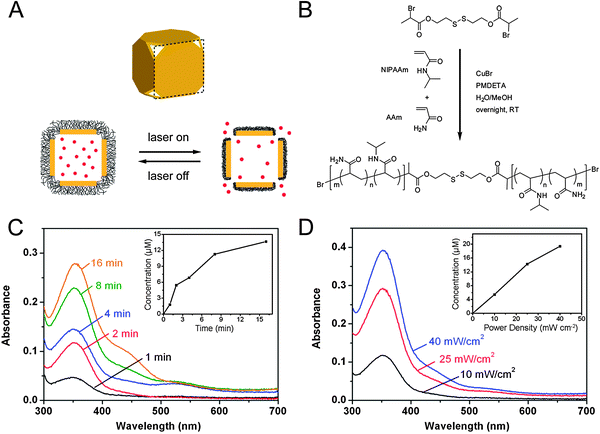 | ||
| Fig. 8 (A) Schematic illustrating the release mechanism for gold nanocages coated with smart polymer chains. (B) Atom transfer radical polymerization of NIPAAm and AAm monomers as initiated by a disulfide initiator and in the presence of a Cu(I) catalyst. (C–D) Controlled release from the gold nanocages covered by a smart polymer with an LCST at 39 °C (pNIPAAm-co-pAAm). (C, D) Absorption spectra of alizarin-PEG released from the copolymer-covered Au nanocages (C) by exposure to a pulsed NIR laser at a power density of 10 mW cm−2 for 1, 2, 4, 8 and 16 min and (D) by exposure to the NIR laser for 2 min at 10, 25 and 40 mW cm−2. The insets show the concentrations of alizarin-PEG released from the nanocages under different conditions. Modified with permission from ref. 21, copyright 2009 Nature Publishing Group. | ||
Fig. 8C and D, shows the release profiles of a PEG-conjugated alizarin dye as the laser irradiation time and laser power were increased, respectively.21 By adjusting these parameters, it is possible to controllably release the loaded effectors both in solution and in vitro, as the amount of dye release increases with both time and laser power. This system is versatile, and has also been demonstrated to release both chemotherapeutic drugs and enzymes, which retained ∼80% of their bioactivity after the release process.
Photothermal therapy
The heat generated from photothermal effect can also be used directly for therapy due to a process known as hyperthermia. When cells are exposed to a temperature above 42 °C (typically for several minutes), they can be irreversibly damaged due to protein denaturing and membrane disruption.59 This technique was originally developed using NIR dyes, but gold nanostructures can have absorption cross sections 4–5 orders of magnitude stronger and can exhibit greater selectivity through both passive and active targeting, making them ideal candidates for this type of therapy.48,60–61 Most studies have focused on the use of gold nanorods, nanoshells, or nanocages for photothermal treatment, as the NIR light used to excite their LSPR can penetrate deeper into tissue than the visible light that excites gold nanospheres.13Fig. 9A to J, shows a comparison of the temperature increase with time when the tumor of a mouse injected with either a suspension of nanocages (B–E) or a sham treatment of saline solution (F–I) was irradiated with a NIR continuous wave (CW) laser.48 The nanocages were injected via the tail vein and allowed to accumulate in the tumor through passive targeting for 72 h before irradiation. It is clear that in the presence of highly absorbing nanocages the laser irradiation resulted in a significant increase in temperature, while almost no change was observed for the sham treatment. To confirm that this temperature change led to tumor destruction, the metabolism of the tumor was measured before and after treatment with [18F] fluorodeoxyglucose positron emission tomography (18F-FDG PET).62 The metabolic activity was normalized by comparing it with a non-treated tumor on the same mouse. As can be seen in Fig. 9K, only the nanocage-treated mouse experienced a large decline in tumor metabolic activity.
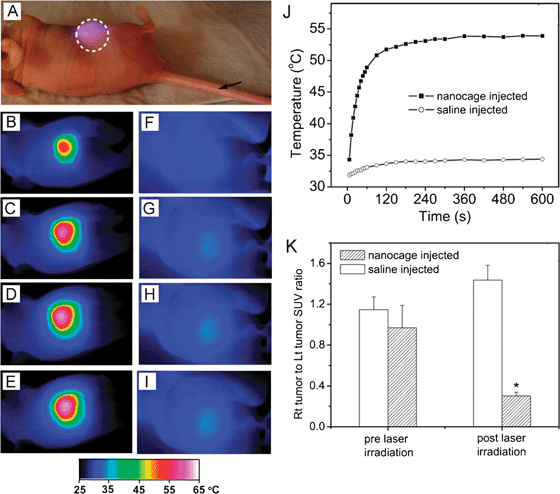 | ||
| Fig. 9 In vivo photothermal treatment with gold nanocages. (A) Photograph of a tumor-bearing mouse undergoing photothermal treatment. PEGylated gold nanocages or saline were administrated intravenously through the tail vein as indicated by the arrow. After the nanocages had been cleared from circulation (72 h after injection), the tumor on the right flank was irradiated by a diode laser at 0.7 W/cm2 with a beam size indicated by the dashed circle. (B–I) Thermographic images of (B–E) nanocage-injected and (F–I) saline-injected tumor-bearing mice at different time points: (B, E) 1 min, (C, F) 3 min, (D, G) 5 min, and (E, I) 10 min. (J) Plot of the average temperature within the tumors (dashed circle) as a function of irradiation time. (K) Plot showing the ratios of the metabolic activity between a laser-treated tumor and a non-treated tumor on the same mouse, measured with 18F-FDG PET/CT (SUV, standardized uptake values, * indicates P < 0.001). Modified with permission from ref. 48, copyright Wiley 2010. | ||
Concluding remarks
The tunable surface chemistry, morphology, and optical properties of gold nanostructures make them ideal for a variety of biomedical applications. Great progress has been made in this field in recent years, and we now have a better understanding on how we can optimize the properties of gold nanostructures for a specific application through size/shape-controlled synthesis, precise surface modification, and molecular targeting. In this review, we have discussed a range of the most interesting applications, including the use of gold nanostructures as optical imaging probes both in vitro and in vivo, their use as drug delivery and photothermal therapeutic agents, and how they can be used to probe the many variables that affect the cellular uptake of nanomaterials. Though strides have been made in this direction, the greatest challenge now is to continue to enhance our understanding of the behavior and fate of gold nanostructures in complex in vivo environments, and truly make these techniques clinically relevant.References
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer Series in Material Science 25, Springer, Berlin, 1995 Search PubMed.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- G. Mie, Anal. Phys., 1908, 25, 376–455 Search PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- M. Grzelczak, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- S. E. Skrabalak, J. Chen, L. Au, X. Lu, X. Li and Y. Xia, Adv. Mater., 2007, 19, 3177–3184 CrossRef CAS.
- S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842–51 CrossRef CAS.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–86 CrossRef CAS.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721–30 CrossRef CAS.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–62 CrossRef CAS.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–7 CrossRef CAS.
- M. Hu, J. Chen, Z.-Y. Li, L. Au, G. V. Hartland, X. Li, M. Marquez and Y. Xia, Chem. Soc. Rev., 2006, 35, 1084–94 RSC.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS.
- G. F. Paciotti, L. Myer, D. Weinreich, D. Goia, N. Pavel, R. E. McLaughlin and L. Tamarkin, Drug Delivery, 2004, 11, 169–183 CrossRef CAS.
- Cytimmune Science Inc. http://www.cytimmune.com/go.cfm?do=Page.View&pid=26 (accessed 23 April 2010).
- A. I. Sherman and M. Ter-pogossian, Cancer, 1953, 6, 1238–40 CrossRef CAS.
- J. Love, L. Estroff, J. Kriebel, R. Nuzzo and G. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–60 CrossRef CAS.
- L. Au, Q. Zhang, C. M. Cobley, M. Gidding, A. G. Schwartz, J. Chen and Y. Xia, ACS Nano, 2010, 4, 35–42 CrossRef CAS.
- A. Gole and C. J. Murphy, Langmuir, 2008, 24, 266–272 CrossRef CAS.
- E. C. Cho, L. Au, Q. Zhang and Y. Xia, Small, 2010, 6, 517–22 CrossRef CAS.
- K. Kelly, E. Coronado, L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- B. J. Wiley, S. H. Im, Z.-Y. Li, J. McLellan, A. R. Siekkinen and Y. Xia, J. Phys. Chem. B, 2006, 110, 15666–75 CrossRef CAS.
- S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587–95 CrossRef CAS.
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff, J. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857–70 CrossRef CAS.
- N. J. Halas, MRS Bull., 2005, 30, 362–367 CAS.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–901 CrossRef CAS.
- J. Chen, J. McLellan, A. Siekkinen, Y. Xiong, Z.-Y. Li and Y. Xia, J. Am. Chem. Soc., 2006, 128, 14776–7 CrossRef CAS.
- S.-D. Li and L. Huang, Mol. Pharmaceutics, 2008, 5, 496–504 CrossRef CAS.
- H. Gao, W. Shi and L. B. Freund, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9469–74 CrossRef CAS.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–48 CrossRef CAS.
- M. A. El-Sayed, Acc. Chem. Res., 2001, 34, 257–64 CrossRef CAS.
- K. H. Song, C. Kim, C. M. Cobley, Y. Xia and L. V. Wang, Nano Lett., 2009, 9, 183–188 CrossRef CAS.
- B. Draine and P. Flatau, J. Opt. Soc. Am. A, 1994, 11, 1491–1499 CrossRef.
- E. Cho, C. Kim, F. Zhou, C. Cobley, K. Song, J. Chen, Z. Li, L. Wang and Y. Xia, J. Phys. Chem. C, 2009, 113, 9023–9028 CrossRef CAS.
- C. Kim, C. Favazza and L. V. Wang, Chem. Rev., 2010, 110, 2756–82 CrossRef CAS.
- L. V. Wang and H. Wu, Biomedical Optics: Principles and Imaging, John Wiley and Sons, 1st edn, 2007 Search PubMed.
- R. A. Sperling, P. R. Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–908 RSC.
- S. R. Sershen, S. L. Westcott, N. J. Halas and J. L. West, J. Biomed. Mater. Res., 2000, 51, 293–8 CrossRef CAS.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–54 CrossRef CAS.
- J. Chen, C. Glaus, R. Laforest, Q. Zhang, M. Yang, M. Gidding, M. J. Welch and Y. Xia, Small, 2010, 6, 811–817 CrossRef CAS.
- R. Wilson, Chem. Soc. Rev., 2008, 37, 2028–45 RSC.
- C. L. Haynes, A. D. McFarland and R. P. V. Duyne, Anal. Chem., 2005, 77, 338A–346A CAS.
- M. Rycenga, P. H. C. Camargo, W. Li, C. H. Moran and Y. Xia, J. Phys. Chem. Lett., 2010, 1, 696–703 Search PubMed.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–81 CrossRef CAS.
- E. C. Cho, Y. Liu and Y. Xia, Angew. Chem. Int. Edit., 2010, 49, 1976–1980 CAS.
- L. Tong, Q. Wei, A. Wei and J.-X. Cheng, Photochem. Photobiol., 2009, 85, 21–32 CrossRef CAS.
- J. Park, A. Estrada, K. Sharp, K. Sang, J. A. Schwartz, D. K. Smith, C. Coleman, J. D. Payne, B. A. Korgel, A. K. Dunn and J. W. Tunnell, Opt. Express, 2008, 16, 1590–9 CrossRef CAS.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–15 CrossRef.
- R. van Horssen, T. L. M. ten Hagen and A. M. M. Eggermont, Oncologist, 2006, 11, 397–408 CrossRef CAS.
- J. M. Farma, M. Puhlmann, P. A. Soriano, D. Cox, G. F. Paciotti, L. Tamarkin and H. R. Alexander, Int. J. Cancer, 2007, 120, 2474–80 CrossRef CAS.
- L. O. Svaasand and J. S. Nelson, J. Biomed. Opt., 2004, 9, 353–61 CrossRef.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Lasers Med. Sci., 2008, 23, 217–28 CrossRef.
- R. R. Anderson and J. A. Parrish, Science, 1983, 220, 524–7.
- C. Pöttgen, S. Levegrün, D. Theegarten, S. Marnitz, S. Grehl, R. Pink, W. Eberhardt, G. Stamatis, T. Gauler, G. Antoch, A. Bockisch and M. Stuschke, Clin. Cancer Res., 2006, 12, 97–106 CrossRef.
- H. Wang, D. W. Brandl, P. Norlander and N. J. Halas, Acc. Chem. Res., 2007, 40, 53–62 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |