Osmium-free direct syn-dihydroxylation of alkenes
Carole J. R.
Bataille
and
Timothy J.
Donohoe
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: carole.bataille@chem.ox.ac.uk; Fax: +44 (0)1865 285 002; Tel: +44 (0)1865 285 117, +44 (0)1865 275 695
First published on 3rd November 2010
Abstract
Numerous synthetic protocols for producing syn-diols from the corresponding alkenes have been developed and published over recent years. It is the intent of the following tutorial review to present a concise summary of the main methods used to prepare syn-diol fragments directly from alkene precursors, and that do not make use of osmium oxo complexes as catalysts.
 Carole J. R. Bataille | Carole J. R. Bataille was an undergraduate in Organic Chemistry at the University of Rouen and obtained her PhD with R. C. D. Brown at the University of Southampton. She then undertook two consecutive post-doctoral positions with T. J. Donohoe and J. M. Brown in the Chemistry Research Laboratory, Oxford. She is currently working as a post-doctoral researcher for A. J. Russell and S. G. Davies, at Oxford. Her research interests are mainly focussed on asymmetric oxidation and reduction reactions and Medicinal Chemistry. |
 Timothy J. Donohoe | Timothy J. Donohoe was an undergraduate in Chemistry at the University of Bath and then obtained his DPhil with S. G. Davies at Oxford (1992). Following a postdoctoral stay with P. D. Magnus in the USA, he joined the University of Manchester in 1994 as a lecturer and was promoted to Reader in 2000. In 2001, he joined the Dyson Perrins Laboratory, Oxford, as Lecturer in Chemistry and Fellow of Magdalen College, and in 2004, he was appointed Professor of Chemistry at Oxford University. His research interests encompass asymmetric synthesis, the synthesis of aromatic compounds, total synthesis, catalysis and redox reactions. |
1. Introduction
1,2-syn-Diols are a common motif in many biologically active natural products and important intermediates in many syntheses. Therefore, numerous synthetic approaches to 1,2-syn-diols have been developed and published over the years based mainly on the direct osmium-catalysed syn-dihydroxylation of alkenes, which is an extremely reliable and powerful reaction.1 In fact, it is fair to say that this syn-dihydroxylation is one of the cornerstone reactions of organic synthesis, especially when one considers the asymmetric variants that exist.However, the use of osmium tetroxide is not without its drawbacks, most notably the expense, toxicity and, related to this, the volatility of the catalyst. In response several research groups have investigated alternative reagents for the syn-dihydroxylation of alkenes, aimed at mimicking the outcome and benefits of osmium catalysed reactions but without the drawbacks. We feel that the time is now right to review and summarises the main osmium-free synthetic routes that have been used for the preparation of 1,2-syn-diols directly from alkene precursors, and outline their advantages and disadvantages.
2. Ruthenium-mediated syn-dihydroxylation
After OsO4, one of the most commonly used complexes in direct syn-dihydroxylation is RuO4 and due to the plethora of literature available, this review will only depict the major conditions used in the ruthenium-promoted oxidation of alkenes.2The very high oxidative potential of ruthenium(VIII) is one of the major reasons why its use is not more widespread within the synthetic community. It is often difficult to selectively terminate the oxidation at the diol stage and RuO4 is therefore more commonly employed in ketohydroxylation and oxidative cleavage reactions.3
2.1 Ruthenium tetroxide based methods
The principle that ruthenium tetroxide would add stereospecifically (cis) to alkenes was first reported by Sharpless and Akashi, however, the yields were low (ca. 20%) and the reaction was therefore deemed impractical.4 This report was later followed by an interesting and early example of syn-dihydroxylation of an alkene using RuO4 reported by Sica et al. in 1993.5 The authors observed the formation of syn-diols in the oxidation of monoene steroids using stoichiometric RuO4 as the oxidant; the diols were obtained in moderate yields alongside the corresponding hydroxyketones resulting from over-oxidation (Scheme 1).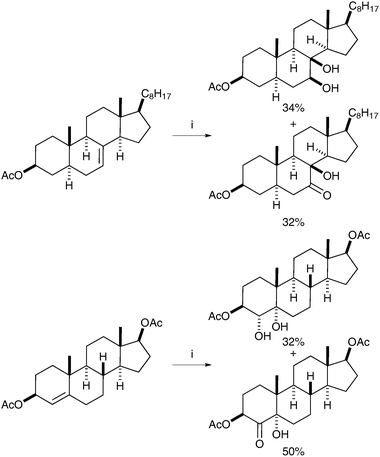
Following this study, Shing et al. published a catalytic version of this reaction, which was termed “flash dihydroxylation”, due to its very short reaction time.6 They described the successful syn-dihydroxylation of a large number of alkenes, using RuCl3·3H2O (7 mol%) and NaIO4 as reoxidant. The products were obtained in moderate to good yields in a maximum reaction time of 3 min (Scheme 2). These conditions were particularly successful for substrates that were difficult to oxidise with OsO4.7
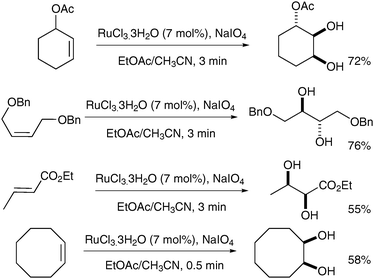 | ||
| Scheme 2 | ||
In recent years, Plietker has investigated the oxidation with RuO4 and attempted to circumnavigate the different issues linked to the use of RuO4 (over-oxidation/oxidative cleavage).8 Mechanistic studies of the dihydroxylation led to the conclusion that the turnover-limiting step is the hydrolysis of the intermediate ruthenate ester (Fig. 1).9
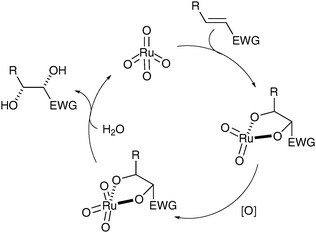 | ||
| Fig. 1 | ||
This hypothesis was supported by an acceleration of the reaction, which was observed when adding a catalytic amount of a Brönsted acid, H2SO4; this also allowed Plietker et al. to decrease the catalyst loading to 0.5 mol% (Scheme 3).3a
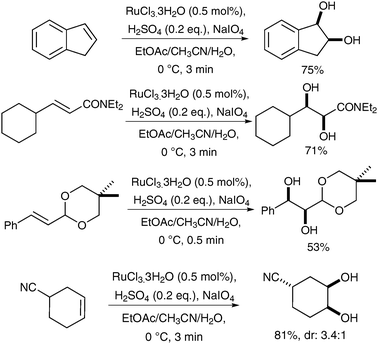 | ||
| Scheme 3 | ||
Subsequently, further investigation was undertaken, which resulted in an improved protocol: H2SO4 was replaced by CeCl3 as a redox-active Lewis acid, which leads to the in situ formation of Ce(IV)–periodato complexes upon addition of NaIO4.8,10 These complexes do not catalyse glycol cleavage as readily as the previous methods, and therefore allow for a longer reaction time. A larger array of alkenes could therefore be converted to the corresponding syn-diols using 0.25 mol% of RuO4 in good to excellent yields (Scheme 4).
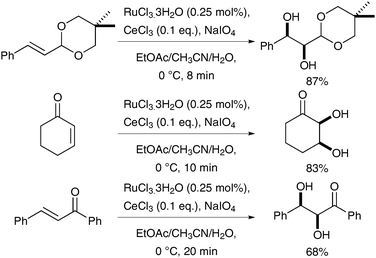 | ||
| Scheme 4 | ||
Notably, Che et al. have reported the preparation of a supported ruthenium catalyst, which was used successfully in the syn-dihydroxylation of alkenes.11 Following Fiévet's protocol, ruthenium nanoparticles were prepared by the reduction of RuCl3·3H2O with sodium acetate and immobilized by treatment of the resulting colloidal solution with calcium hydroxyapatite. The ruthenium nanoparticles obtained were grafted onto hydroxyapatite (nano-RuHAP) and then used in the dihydroxylation of a range of alkenes, following the conditions optimised by Plietker (Scheme 5). The catalyst could be used up to four times without a significant decrease in the yield.
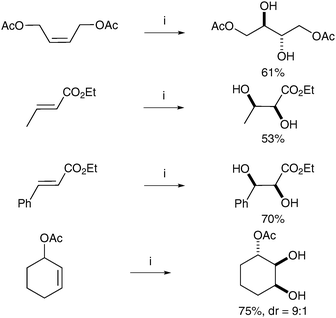 | ||
| Scheme 5 Reagents and conditions: (i) nano-RuHAP (1 mol%), H2SO4 (0.2 eq.), NaIO4, EtOAc/CH3CN/H2O, 0 °C, 30 min. | ||
2.2 Tandem metathesis oxidation procedures
The literature contains a few reports of so-called tandem metathesis/dihydroxylation, where a substituted alkene is formed in situ by ring-closing or cross metathesis and then cis-dihydroxylated utilising the same source of ruthenium (after appropriate oxidation of the metal). This sequential reaction was first described in 2006 by Blechert et al.;12NaIO4/YbCl3 were used to prepare RuO4 from the Ru–carbene catalystin situ. After an initial study of commercially available metathesis catalysts, it was found that Grubbs I gave the best results and this catalyst was then used against a large array of dienes in an RCM/dihydroxylation tandem reaction. The dienes were treated with Grubbs I catalyst in the first instance before addition of YbCl3·6H2O and NaIO4; the resulting diols were obtained in moderate to good yields (Scheme 6).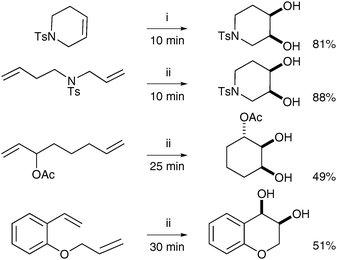 | ||
| Scheme 6 Reagents and conditions: (i) Grubbs I (2 mol%), NaIO4, YbCl3·6H2O, EtOAc/CH3CN/H2O, 0 °C; (ii) Grubbs I (2 mol%), CH2Cl2, reflux, 60 min; then NaIO4, YbCl3·6H2O, EtOAc/CH3CN/H2O, 0 °C. | ||
It is noteworthy that these conditions can also be applied to a cross metathesis/dihydroxylation sequence, although here Hoveyda-Grubbs II was used as a catalyst. The overall yields of dihydroxylated products were lower, probably due to the lower efficiency of the catalyst in promoting the dihydroxylation reaction upon oxidation (Scheme 7).
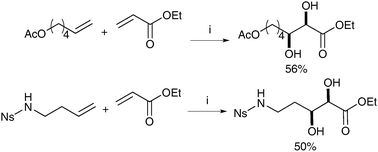 | ||
| Scheme 7 Reagents and conditions: (i) Hoveyda-Grubbs II (2.5 mol%), CH2Cl2, reflux, 120 min; then NaIO4, YbCl3·6H2O, EtOAc/CH3CN/H2O, 0 °C, 10 min. | ||
Snapper et al. later improved this protocol, making it more practical by eliminating the solvent switch, which also resulted in increased yields for several examples.13 Treatment of a range of dienes with Grubbs II catalyst and subsequent addition of CeCl3·7H2O and NaIO4 (following Plietker's improved conditions) led to the corresponding cyclic diols in good yields (Scheme 8). Cross metathesis was also investigated and the corresponding acyclic diols were obtained in moderate yields under the same conditions.
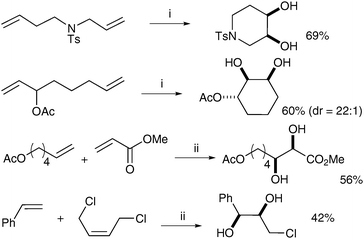 | ||
| Scheme 8 Reagents and conditions: (i) Grubbs II (5 mol%), EtOAc, rt; then NaIO4, CeCl3·7H2O, CH3CN/H2O; (ii) Grubbs II (5 mol%), CH2Cl2, reflux, 120 min; then NaIO4, CeCl3·7H2O, EtOAc/CH3CN/H2O. | ||
2.3 Use of ruthenium complexes for oxidation
More recently, Che et al. have investigated novel, alternative ruthenium complexes that can also be used in cis-dihydroxylation (Fig. 2).14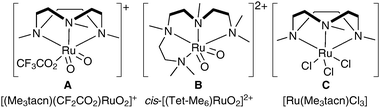 | ||
| Fig. 2 Me3tacn: 1,4,7-trimethyl-1,4,7-triazacyclononane. | ||
The addition of bulky ligands to the ruthenium allows the oxidative power of RuO4 to be modulated; these complexes are therefore interesting alternatives for substrates where over-oxidation or cleavage reactions are prevalent.
The ruthenium complex A was prepared in moderate overall yield by sequential treatment of [Me3tacnRuIIICl3] with CF3SO3Ag in aqueous TfOH followed by (NH4)2CeIV(NO3)6 and NaClO4. Complex A was used stoichiometrically in a tert-butanol/water mixture to oxidise a diverse set of alkenes, affording the corresponding syn-diols in good yields (Scheme 9).
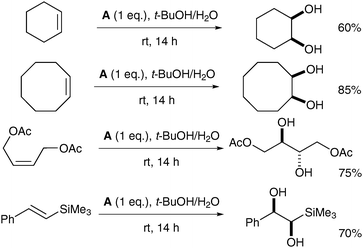 | ||
| Scheme 9 | ||
The reaction medium plays a crucial role in the selectivity between cis-dihydroxylation and oxidative cleavage: in anhydrous MeCN, the complex A led almost exclusively to cleavage of the double bond (Scheme 10).
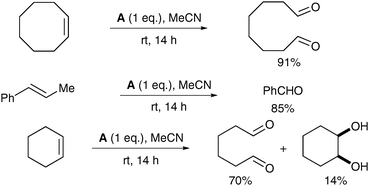 | ||
| Scheme 10 | ||
Further mechanistic studies were undertaken in order to confirm that the reaction does indeed proceed via the formation of a [3+2] adduct and to explain the dramatic role of the medium in this reaction. These showed that, in the absence of water, intermediate E was not formed and the pathway led to the oxidative cleavage of intermediate D instead (Scheme 11).
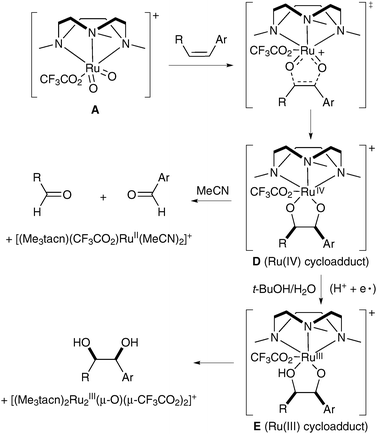 | ||
| Scheme 11 | ||
Che et al. have recently reported a catalytic variant of these conditions.15 Complex C (Fig. 2) was prepared in a similar manner to complex A; 1 mol% of C in the presence of H2O2 as a co-oxidant and Al2O3 and NaCl as additives was used to oxidise a large range of alkenes to the corresponding syn-diols in good to excellent yields (Scheme 12). Additionally, a small amount of the trans-dihydroxylated product was obtained in some cases.
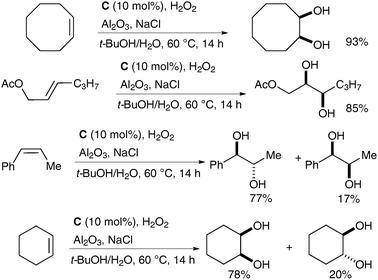 | ||
| Scheme 12 | ||
2.4 Asymmetric dihydroxylation
There are only a few RuO4 mediated asymmetric dihydroxylation reactions reported in the literature, and all are based on a chiral auxiliary approach. Lee et al. reported the first ruthenium catalysed asymmetric dihydroxylation reaction in 1999, using Oppolzer's (S)-sultam as a chiral auxiliary (Fig. 3).16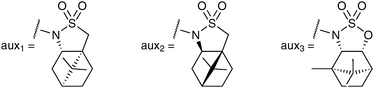 | ||
| Fig. 3 | ||
The alkenes bearing the chiral auxiliary, prepared from the NH-sultam and the corresponding acid chlorides, were treated under Shing's protocol, RuCl3·3H2O (7 mol%) and NaIO4 as a reoxidant to afford the syn-diols, which were converted in situ to the corresponding acetals in good overall yields and good to excellent diastereoselectivities (Scheme 13).
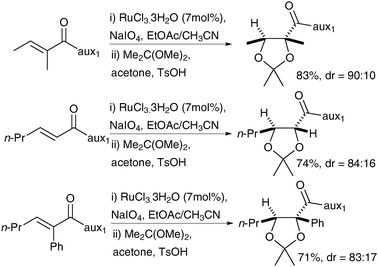 | ||
| Scheme 13 | ||
Neisius and Plietker have also reported the asymmetric synthesis of syn-diols using a tandem cross metathesis/dihydroxylation sequence, with one of the alkene substrates bearing a chiral auxiliary.17 The metathesis catalyst, here Grubbs-Hoyveda, is converted in situ into RuO4 in order to perform the dihydroxylation step. After investigating a range of auxiliaries, they decided to use (R)-sultam (aux 2) to access (R,S)-diols and sulfamidate (aux 3) for the (S,R)-diols (Fig. 3). A large range of olefins underwent the cross metathesis/dihydroxylation tandem reaction and the resulting diols were converted in situ to the corresponding methyl esters, which could be obtained in moderate to good yields and in good to excellent enantioselectivities (Scheme 14).
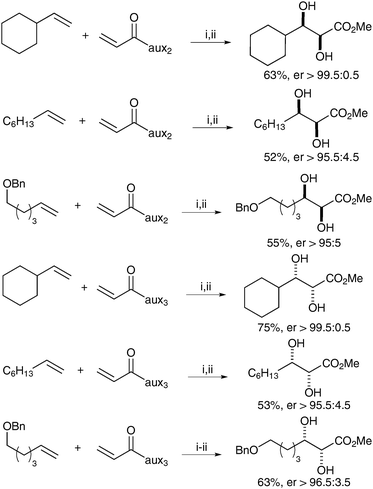 | ||
| Scheme 14 Reagents and conditions: (i) Grubbs Hoveyda (2.5 mol%), CuCl, CH2Cl2, 12 h, reflux; then Bu4NIO4, CeCl3·7H2O, NaIO4, CH3CN/acetone/H2O, 0 °C 30 min; (ii) MeMgBr, MeOH, 0 °C, 10 min. | ||
Plietker et al. then applied this methodology to the synthesis of anthopleurine and ent-anthopleurine; the two enantiomers were obtained in moderate yields but excellent stereoselectivities (Scheme 15).
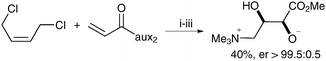 | ||
| Scheme 15 Two step preparation of anthopleurine. Reagents and conditions: (i) Grubbs Hoveyda (2.5 mol%), CuCl, CH2Cl2, 12 h, reflux; then Bu4NIO4, CeCl3·7H2O, NaIO4, CH3CN/acetone/H2O, 0 °C, 30 min; (ii) MeMgBr, MeOH, 0 °C, 10 min; (iii) Me3N, H2O, 12 h, then Dowex 550A. | ||
ent-Anthopleurine was obtained in a similar manner starting from the sulfamidate auxiliary in 36% overall yield and er > 99.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶0.5.
∶0.5.
3. Manganese-mediated syn-dihydroxylation
The permanganate ion (MnO4−) is a common oxidant in organic chemistry and has been used routinely in dihydroxylation reactions for some time.18 The wealth of literature reviewing and describing permanganatesyn-dihydroxylation resembles the high research interest it has received and, therefore, only the fundamental principles of this approach and the latest examples of its use will be highlighted in this review. Like ruthenium tetroxide, permanganate has a high oxidative potential and can lead to over-oxidation and oxidative cleavage.19Permanganate has also to be used stoichiometrically, which leads to problems, especially due to the formation of stoichiometric amounts of MnO2 by-product in the reaction, which can require removal by elaborate work-up procedures.3.1 Permanganate syn-dihydroxylation
Permanganate dihydroxylation in alkaline medium has been known for over a century and was first reported by Kekulé and Anschütz in 1881 with the oxidation of maleic and fumaric acids (Scheme 16) to the corresponding meso-tartaric and DL-tartaric acids in moderate yields.20 In 1973, Simàndi et al. published a mechanistic investigation explaining the syn-character of this addition using the same pair of acids.21 A comprehensive review, which examines the oxidation of alkenes with permanganate from a mechanistic point of view was published in 2009.22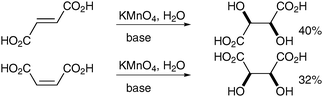 | ||
| Scheme 16 The products were isolated as their potassium salts. | ||
Since these initial studies, manganese-containing reagents have been studied and used extensively. The examples reported in the literature can be divided in three major categories:
• homogeneous oxidation using potassium permanganate or tetralkylammonium permanganate
• heterogeneous conditions involving a phase-transfer catalyst
• catalytic homogeneous conditions with manganese complexes in the presence of a reoxidant.
 | ||
| Scheme 17 PMP: p-methoxyphenyl. | ||
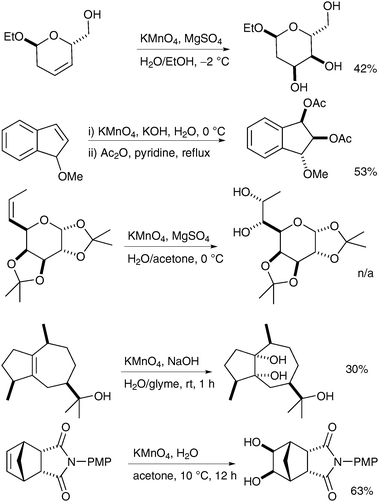
The main scenarios in which permanganate dihydroxylation reactions can be useful are cases where OsO4 dihydroxylation was either unsuccessful or merely led to mediocre yields. For example, Salamci et al. applied aqueous permanganate oxidation to the synthesis of (±)-proto-quercitol, and obtained the desired diol from the substituted cyclohexene in moderate yield and excellent selectivity (Scheme 18) (osmium-based conditions did not work well, although the yield was not reported).28
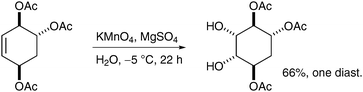 | ||
| Scheme 18 | ||
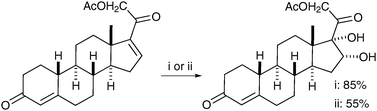
Katzenellenbogen et al. prepared a key precursor to fluoro-furanyl norprogesterone using aqueous KMnO4 oxidation in good yield; again, stoichiometric OsO4 in pyridine was reported to furnish the desired product in only moderate yield (Scheme 19).29
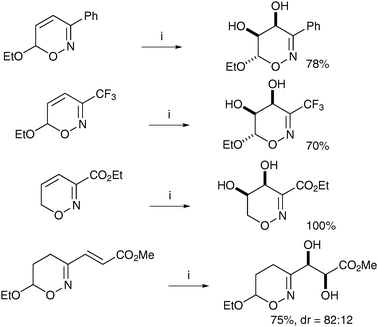
Reissig et al. have applied these conditions to the dihydroxylation of oxazines and to the practical formation of pyrrolizidinones.30 Readily available oxazines were treated with KMnO4 in the presence of MgSO4 in EtOH and the corresponding diols were obtained in good yields and excellent diastereoselectivities (Scheme 20).
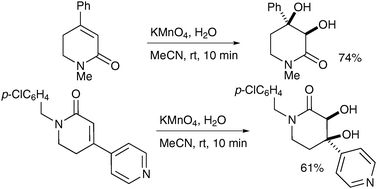
Azines can also be oxidised successfully to the corresponding diols as shown by Soldatenkov et al. in 2000 (Scheme 21).31 Contrary to typical behaviour, the pyridine ring is not oxidised to the corresponding N-oxide under these conditions.
 | ||
| Scheme 21 | ||
Notably, different experimental techniques have been implemented to increase the yield of these reactions. Hydorn et al. have reported that the use of a continuous-flow permanganate oxidation process, which allows the synthesis of dihydroxylated steroids in very good yields and on a large scale (up to 10 kg) (Scheme 22).32
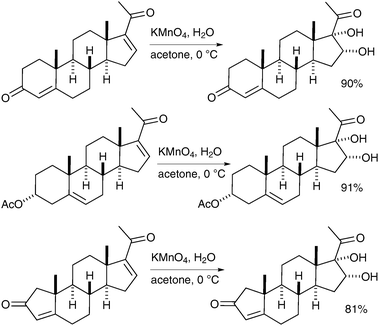 | ||
| Scheme 22 | ||
Another means to increase the yield of the desired diols was described by Varma and Naicker in 1998, who used sonication to accelerate the permanganate oxidation.33 A range of monosubstituted alkenes was treated with aqueous potassium permanganate with a sonic probe inserted in the reaction mixture and the corresponding diols were obtained in good yields; some interesting examples are depicted in Scheme 23.
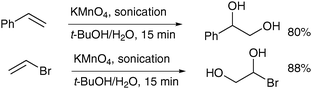 | ||
| Scheme 23 | ||
Interestingly Ayari et al. have shown that the stirring efficiency of the reaction has a dramatic effect on the yields and generation of by-products (Scheme 25, vide infra).34 Taylor et al. have investigated this observation further and combined effective turbulent stirring with slow permanganate addition and continuous extraction to increase the yield of cis-dihydroxylation.35 A range of alkenes was subjected to these improved conditions and the corresponding diols were obtained in moderate to good yields, even on large scale (from 0.2 to 1 mol of substrates) (Scheme 24).
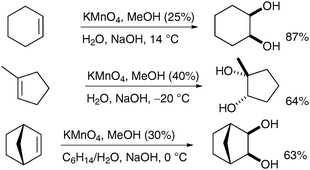 | ||
| Scheme 24 | ||
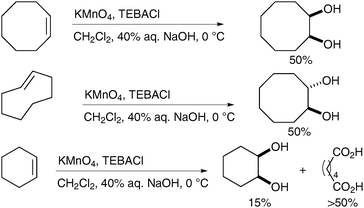 | ||
| Scheme 25 | ||
Foglia et al. then applied these heterogeneous conditions to the oxidation of olefins bearing hydrophobic chains, which are not soluble in aqueous media.38 Either quaternary ammonium halides or crown ethers (such as DC6: dicyclohexyl-18-crown-6) were used as phase transfer catalysts. The authors observed that the reaction rates and yields increased under these heterogeneous conditions (Scheme 26).
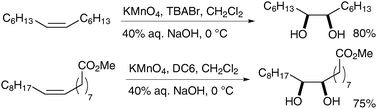 | ||
| Scheme 26 | ||
Heterogeneous reaction conditions were applied to the synthesis of bicyclic lactams by Moloney et al. in 2004;3918-crown-6 was used as a phase transfer catalyst and the desired diol obtained in moderate yield and as a single diastereomer (Scheme 27).
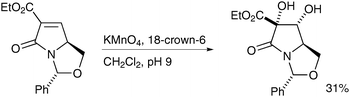 | ||
| Scheme 27 | ||
Ogino and Mochizuki described, in 1979, that the oxidation does not require a biphasic medium;40 treatment of diverse alkenes with potassium permanganate in the presence of TEBACl in CH2Cl2 and subsequent basic hydrolysis led to the formation of the corresponding diols in good yields (Scheme 28). The stable organomanganese intermediates formed under these anhydrous conditions were quenched upon addition of a basic or neutral aqueous solution. It is noteworthy that if the organomanganese complex derived from cyclooctene is treated with an aq. NaOH solution, it furnishes a 1∶1 mixture of the desired diol and the bis-aldehyde, resulting from oxidative cleavage.
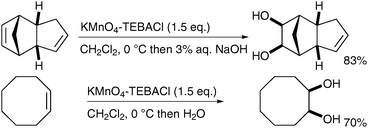 | ||
| Scheme 28 | ||
The same set of conditions was also used in the synthesis of a brassinosteroid by Rivera et al. in 2004;41 the alkenes were oxidised to the corresponding syn-diols in good yields (Scheme 29).
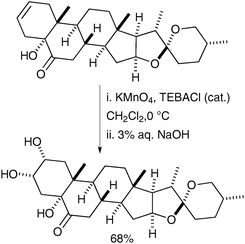 | ||
| Scheme 29 | ||
Chandrasekaran et al. described the synthesis of a tetralkylammonium permanganate species, which can then be used in homogeneous anhydrous syn-dihydroxylation reactions. Addition of cetyltrimethylammonium bromide (CTABr) to potassium permanganate led to the formation of cetyltrimethylammonium permanganate (CTAP).42Alkenes were then treated with CTAP in CH2Cl2 or t-BuOH (Scheme 30).
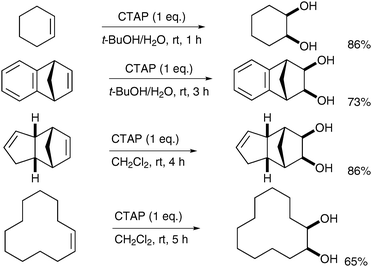 | ||
| Scheme 30 | ||
Ayari et al. have performed comparative studies of the homogeneous potassium and tetralkylammonium permanganate conditions, and heterogeneous conditions with a phase-transfer catalyst on perfluorinated substrates.34 They found that for these substrates, the tetralkylammonium permanganate conditions led to the best results in all cases (Scheme 31).
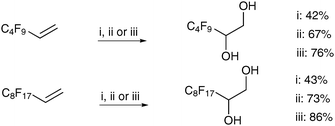 | ||
| Scheme 31 Reagents and conditions: (i) KMnO4, Aliquat 336 (5 mol%), MeOH (30%), H2O, NaOH, 24 h; (ii) KMnO4, acetone, 0 °C, 5 h; (iii) CTAP, CH2Cl2, 0 °C to rt, 20 h. | ||
Hazra et al. combined the phase transfer catalyst BTMAOH (benzyltrimethylammonium hydroxide) and TDTAP (tetradecyltrimethylammonium permanganate) in their synthesis of homobrassinolide.43 The syn-dihydroxylation was totally chemoselective for oxidation of the cyclohexene ring and afforded the desired diol in good yield (Scheme 32).
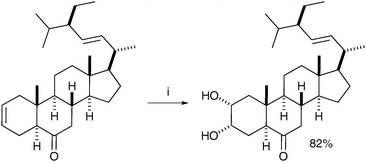 | ||
| Scheme 32 Reagents and conditions: (i) TDTAP, BTMAOH, t-BuOH, CH2Cl2, 3 °C TO 28 °C, 2 h. | ||
Caycho et al. have reported the dihydroxylation of a rigid tricyclic diene using TEBAP (triethylbenzylammonium permanganate) in CH2Cl2 to afford the desired diol in good yield;44 the rigidity of the structure led to unexpected epoxidation of the second double bond to afford a diol epoxide as a by-product (Scheme 33). The formation of the epoxide can be rationalised by formation of an oxomanganate intermediate, which can attack the second double bond, in an intramolecular manner.
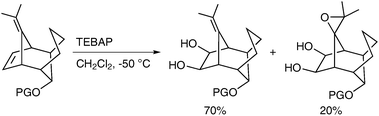 | ||
| Scheme 33 | ||
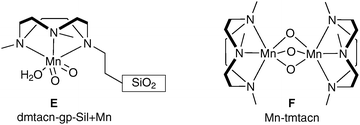 | ||
| Fig. 4 | ||
This is the first example of a manganese-catalysed dihydroxylation (Scheme 34).
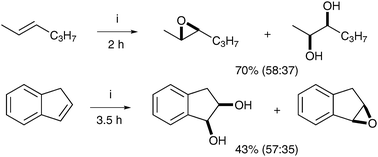 | ||
| Scheme 34 Reagents and conditions: (i) dmtacn-gp-SiI + Mn, H2O2, CH3CN, 0 °C. | ||
After this initial report, a number of co-catalysts and manganese complexes have been investigated in order to afford a diol-selective reaction.46 In 2002, de Vos et al. showed that using complex F and catalytic quantities of GMHA (glycidyl methacrylate hyaluronic acid) added to the reaction led to an increase in the amount of cis-diols.47 It is noteworthy that the diol/epoxide ratio is substrate dependent, with the ring size having an especially profound influence (Scheme 35).
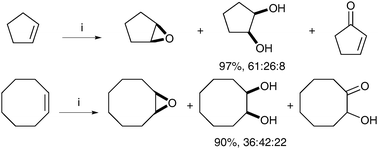 | ||
| Scheme 35 Reagents and conditions: (i) Mn-tmtacn (0.1 mol%), GMHA (25 mol%), H2O2, MeCN, 0 °C, 5 h. | ||
de Boer et al. have demonstrated that carboxylic acids could also favourably promote cis-dihydroxylation over epoxidation.48 Treatment of a range of alkenes with complex F in the presence of substoichiometric quantities of substituted benzoic acids showed that 2,6-dichlorobenzoic acid was the best additive for cis-dihydroxylation (cyclooctene with 2-methoxybenzoic acid led to a 1.5∶1 ratio and 47% yield, whereas 3-methoxybenzoic acid led to 1.7∶1 ratio and 26% yield). The corresponding diols were obtained as the major product in moderate to good yield (Scheme 36).
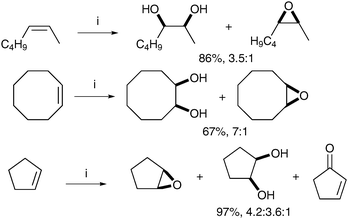 | ||
| Scheme 36 Reagents and conditions: (i) Mn-tmtacn (0.1 mol%), 2,6-dichlorobenzoic acid (3 mol%), H2O2 (1.3 eq.), MeCN, 0 °C, 5 h. | ||
3.2 Asymmetric dihydroxylation
In a similar manner to the ruthenium example described previously, the literature offers only a sparse number of asymmetric examples of manganese dihydroxylation; only two methods have been described up to date. In 2002, Brown et al. reported interesting work, which showed that good levels of enantioselectivity could be achieved via asymmetric phase-transfer dihydroxylation.49 A range of alkenes was treated with stoichiometric powdered potassium permanganate in the presence of chiral phase-transfer reagent G in CH2Cl2; the corresponding cis-diols were obtained in moderate yields and moderate to good enantioselectivities (Scheme 37).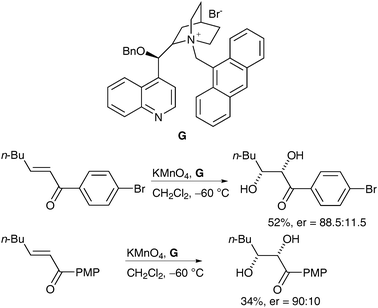 | ||
| Scheme 37 | ||
Feringa et al. have also applied their novel complexes to an asymmetric variant of the dihydroxylation reaction.50 The authors decided to use carboxylic acids from the chiral pool to induce enantioenrichment. 2,2-Dimethylchromene was dihydroxylated using catalytic amounts of complex F (0.4 mol%) and a chiral carboxylic acid (4 mol%) in the presence of H2O2 as a reoxidant, to produce the corresponding diol in moderate to excellent yields and moderate enantioselectivities (Scheme 38).
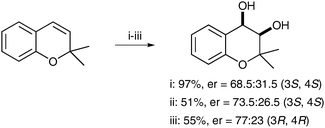 | ||
| Scheme 38 Reagents and conditions: (i) F (2.5 mol%), Boc-Phg-OH (25 mol%), H2O2 (30 mol%), CH3CN, 0 °C, 5 h; (ii) F (2.5 mol%), Boc-Phg-OH (25 mol%), H2O2 (30 mol%), CH3CN, −20 °C, 5 h; (iii) F (0.4 mol%), Ac-D-Phg-OH (4 mol%), H2O2 (30 mol%), CH3CN, −20 °C, 5 h. | ||
4. Microbiological-enzymatic syn-dihydroxylation
The enzymatic dihydroxylation of alkenes was first reported in 1908 by Stomer, with his description of the consumption of toluene and xylene by a microorganism, Bacillus hexacarovorum. Since that time, the enzymatic enantioselective syn-dihydroxylation of a double bond in mono- and polycyclic arenes, using molecular oxygen as a reoxidant has been described in the literature on many occasions (Scheme 39).51 | ||
| Scheme 39 | ||
The author is directed towards some comprehensive reviews that have been published very recently, one dealing especially with biocatalysis and enzymatic dihydroxylation and another one on mononuclear non-heme iron enzymes and their applications.52
5. Iron-mediated syn-dihydroxylation
5.1 Non-enantioselective dihydroxylation
In Nature, an important class of nonheme iron enzyme, Rieske dioxygenases, allow the asymmetric cis-dihydroxylation of double bonds present in an arene. Unfortunately, as often encountered with enzymes, they are only reactive towards a small range of substrates which presents a major obstacle to their applicability. Biomimetic models of these enzymes have been developed and studied in the last few years and they have shown great potential in both racemic and asymmetric cis-dihydroxylation. It should be noted that studies have so far demonstrated that bio-inspired non-heme iron catalysis could mimic natural enzymes, but have not yet proven their synthetic utility.The first example was reported by Que et al. in 2002, in their comparison of the reactivity of iron complexes towards the dihydroxylation and epoxidation of alkenes.53 The complexes were modulated in order to achieve a higher ratio of diol/epoxide, although yields and turn-over were normally low (Fig. 5).
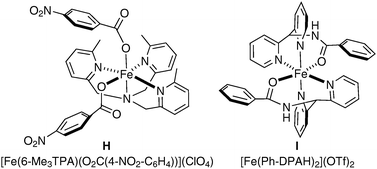 | ||
| Fig. 5 | ||
Alkenes could be oxidised with a catalytic amount of complex H in the presence of H2O2 as a reoxidant, in acetonitrile; the desired diols were obtained as the major product albeit in low yields (Scheme 40).
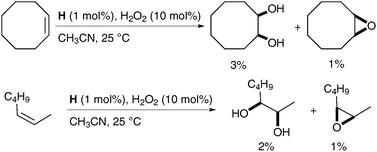 | ||
| Scheme 40 | ||
Further studies on the catalyst led to the discovery of complex I, which gave slightly increased yields (Scheme 41).54
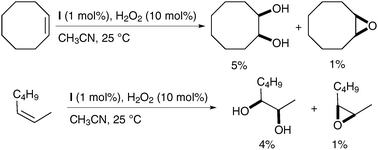 | ||
| Scheme 41 | ||
Pleasingly, Que et al. have very recently reported the first example of dihydroxylation of an aromatic double bond by means of a biomimetic iron complex (Fig. 6).55
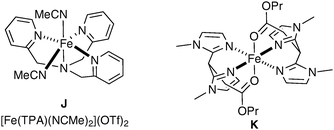 | ||
| Fig. 6 | ||
Naphthalene was oxidised using a substoichiometric amount of complex J in the presence of H2O2 as a reoxidant in CH3CN and the corresponding naphthalene diol was obtained as the major product, albeit in low yield (Scheme 42).
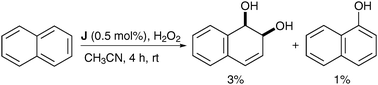 | ||
| Scheme 42 | ||
The mechanistic details of this reaction and elucidation of the reactive intermediates leading to cis-diol formation was reported in 2007 by Comba et al.56 Gebbink et al. have investigated alternative complexes (K, Fig. 6), which favour epoxide formation over dihydroxylation; however, the yields are again very low (Scheme 43).57
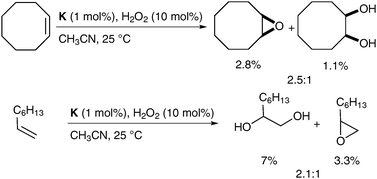 | ||
| Scheme 43 | ||
Rutledge et al. have also looked at alternative iron complexes based on the imidazole motif and successfully obtained a slight increase in yield.58 Complex L was prepared in situ using Fe(OAc)2 as the source of iron and H2O2 as a reoxidant in methanol (Fig. 7).
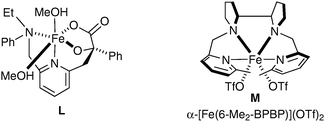 | ||
| Fig. 7 | ||
The corresponding diols were obtained as the major product over the epoxide although in poor yields (Scheme 44). It is noteworthy that the absence of epoxide in the cyclopentene example shown is probably due to the high volatility of cyclopentene oxide.
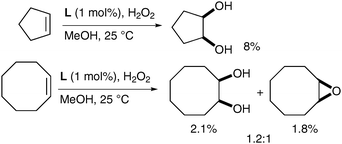 | ||
| Scheme 44 | ||
5.2 Asymmetric dihydroxylation
To the best of our knowledge, there is currently only one example in the literature of asymmetric cis-dihydroxylation using iron complexes, which was reported by Que et al. in 2008.59 The iron complex bears chiral ligands, allowing obtention of a high level of enantioenrichment (up to er = 98.5∶1.5) (Fig. 7). Olefins were treated with a catalytic amount of complex M using H2O2 as a reoxidant in CH3CN; the corresponding diols were obtained in moderate to excellent enantioselectivities and poor yields (Scheme 45).
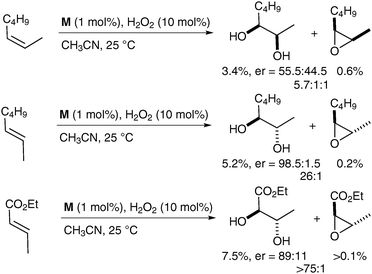 | ||
| Scheme 45 | ||
6. Other osmium-free methods of syn-dihydroxylation
Other direct syn-dihydroxylation procedures have been reported in the literature, although, to a lesser extent than the previously described methods. These can be divided into two categories, metal-mediated dihydroxylations and metal free-mediated ones.6.1 Metal-mediated dihydroxylation
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh), readily available from CpMo(CO)3Cl, was used in a catalytic amount (2 mol%) in the presence of a reoxidant (typically H2O2 or TBHP in tert-butanol) to prepare cis-cyclohexane-1,2-diol from cyclohexene in good yield (Scheme 46).
CPh), readily available from CpMo(CO)3Cl, was used in a catalytic amount (2 mol%) in the presence of a reoxidant (typically H2O2 or TBHP in tert-butanol) to prepare cis-cyclohexane-1,2-diol from cyclohexene in good yield (Scheme 46).
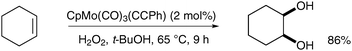 | ||
| Scheme 46 | ||
The epoxide by-product was not observed and the catalyst could be re-used up to five times without loss of activity. Biradar et al. rationalised the formation of the cis-diol by a [4+2] cycloaddition similar to the likely OsO4 mechanism. However, it is unclear whether this combination of reagents will prove to be a reliable method to achieve stereoselective syn-dihydroxylation in the future.
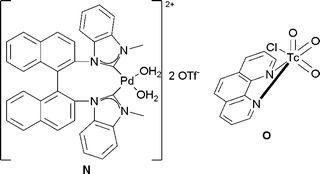 | ||
| Fig. 8 | ||
Treatment of a range of alkenes with a catalytic amount of bis-(NHC)–Pd(II) complex N in the presence of PhI(OAc)2, as a reoxidant in acetic acid/water led to the corresponding protected diols in good yields and good diastereoselectivities (Scheme 47).
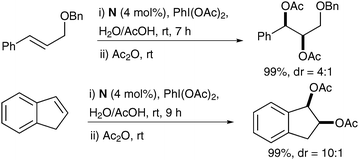 | ||
| Scheme 47 | ||
Notably, a mixture of mono and diacetylated diols was obtained from the reaction; therefore, the authors decided to push the acetylation further by addition of acetic anhydride in their study.
 | ||
| Scheme 48
Reagents and conditions: (i) I2 (0.5 eq.), CAN (0.5 eq.), AcOH/H2O (9∶1), Δ, 10 h. | ||
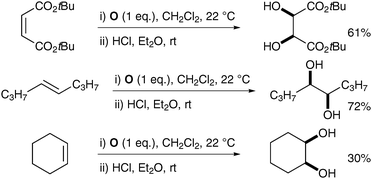 | ||
| Scheme 49 | ||
6.2 Metal free-mediated dihydroxylation
 | ||
| Scheme 50 Reagents and conditions: (i) NaIO4 (30 mol%), LiBr (20 mol%), AcOH, 95 °C, 18 h; (ii) K2CO3, MeOH, rt, 24 h. | ||
Typical trans-dihydroxylation was observed when PhI(OAc)2 was employed in place of NaIO4.
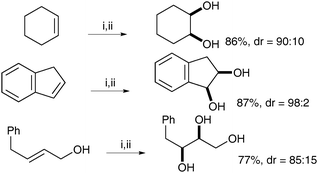
 | ||
| Scheme 51 | ||
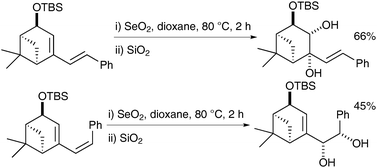
Interestingly this reaction is chemoselective in as much as when the exocyclic double bond is cis, it undergoes direct syn-dihydroxylation, whereas when the exocyclic double bond is trans, the endocyclic double bond undergoes selective oxidation (Scheme 52).
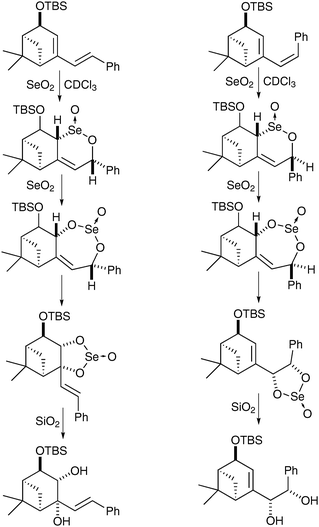 | ||
| Scheme 52 | ||
The authors rationalised those findings by studies of the mechanism and isolation of the selenino lactone and cyclic selenite intermediates in both cases
Tiecco et al. have reported the one-pot syn-dihydroxylation of olefins using PhSeOSO3H, prepared in situ by oxidation of Ph2Se2 with (NH4)2S2O8 or H2O2.66 A range of alkenes was treated with Ph2Se2 and the oxidising reagent in MeCN/water and the cis-diols were obtained in good yields and moderate to excellent diastereoselectivites, which depend upon the structure of the substrates (Scheme 53).
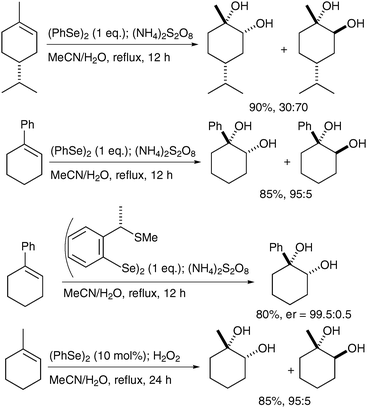 | ||
| Scheme 53 | ||
After an investigation of the mechanism, they confirmed that the reaction proceeds via the corresponding trans-oxo-selenite species.
 | ||
| Scheme 54 | ||
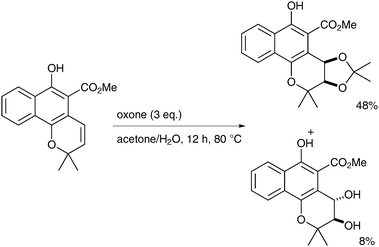
The reaction was investigated en-route to the synthesis of cis-3,4-dihydroxy-3,4-dihydromollugin. The reaction was not fully stereoselective but variation of the reaction conditions can favour cis over trans-diols. The presence of the alcohol group on the substrate is important for the cis-selectivity, as protected alcohols led to the trans-diol under the same conditions, which render this methodology extremely substrate-dependent.
7. Conclusions
The direct syn-dihydroxylation of alkenes to the corresponding 1,2-cis-diols has received a significant amount of attention from the chemical community. Although a large part of the literature is dedicated to osmium-mediated syn-dihydroxylation, it was the aim of this tutorial review to highlight that a plethora of alternative osmium-free methods are also available with notable renewed interest being shown in the last decade. Clearly, there are many obstacles still to be overcome before a reagent emerges that will have the high yield, stereospecificity, high functional group tolerance and prospects for enantioselectivity that one currently expects from osmium. However, the pool of reagents from which to refine and develop syn-dihydroxylation is impressively large and we expect an ever increasing improvement towards these challenging targets in the future.Notes and references
- (a) A. B. Zaitsev and H. Adolfsson, Synthesis, 2006, 1725–1756 CAS; (b) M. Beller, C. Doebler and G. Mehltretter, WO Pat. W2000064848, 2000.
- T. Naota, H. Takaya and S. I. Murahashi, Chem. Rev., 1998, 98, 2599–2660 CrossRef CAS.
- (a) B. Plietker, M. Niggemann and A. Pollrich, Org. Biomol. Chem., 2004, 2, 1116–1124 RSC; (b) M. Pagliaro, S. Campestrini and R. Ciriminna, Chem. Soc. Rev., 2005, 34, 837–845 RSC; (c) C. M. Che, W. P. Yip and W. Y. Yu, Chem.–Asian J., 2006, 1, 453–458 CrossRef CAS.
- K. B. Sharpless and K. Akashi, J. Am. Chem. Soc., 1976, 98, 1986–1987 CrossRef CAS.
- V. Piccialli, D. M. A. Smaldone and D. Sica, Tetrahedron, 1993, 49, 4211–4228 CrossRef CAS.
- (a) T. K. M. Shing, V. M. F. Tai and E. K. W. Tam, Angew. Chem., Int. Ed. Engl., 1994, 33, 2312–2313 CrossRef; (b) T. K. M. Shing and E. K. W. Tam, Tetrahedron Lett., 1999, 40, 2179–2180 CrossRef CAS; (c) T. K. M. Shing, E. K. W. Tam, V. W. F. Tai, I. H. F. Chung and Q. Jiang, Chem.–Eur. J., 1996, 2, 50–57 CrossRef CAS.
- M. Lang and T. Ziegler, Eur. J. Org. Chem., 2007, 768–776 CrossRef CAS.
- B. Plietker, Synthesis, 2005, 2453–2472 CrossRef CAS.
- B. Plietker and M. Niggemann, Org. Lett., 2003, 5, 3353–3356 CrossRef CAS.
- (a) P. Tiwari and A. K. Misra, J. Org. Chem., 2006, 71, 2911–2913 CrossRef CAS; (b) B. Plietker and M. Niggemann, J. Org. Chem., 2005, 70, 2402–2405 CrossRef CAS.
- C. M. Ho, W. Y. Yu and C. M. Che, Angew. Chem., Int. Ed., 2004, 43, 3303–3307 CrossRef CAS.
- S. Beligny, S. Eibauer, S. Maechling and S. Blechert, Angew. Chem., Int. Ed., 2006, 45, 1900–1903 CrossRef CAS.
- A. A. Scholte, M. H. An and M. L. Snapper, Org. Lett., 2006, 8, 4759–4762 CrossRef CAS.
- W. P. Yip, W. Y. Yu, N. Y. Zhu and C. M. Che, J. Am. Chem. Soc., 2005, 127, 14239–14249 CrossRef CAS.
- W. P. Yip, C. M. Ho, N. Zhu, T. C. Lau and C. M. Che, Chem.–Asian J., 2008, 3, 70–77 CrossRef CAS.
- A. W. M. Lee, W. H. Chan, W. H. Yuen, P. F. Xia and W. Y. Wong, Tetrahedron: Asymmetry, 1999, 10, 1421–1424 CrossRef CAS.
- N. M. Neisius and B. Plietker, J. Org. Chem., 2008, 73, 3218–3227 CrossRef CAS.
- (a) N. Singh and D. G. Lee, Org. Process Res. Dev., 2001, 5, 599–603 Search PubMed; (b) D. G. Lee and T. Chen, J. Am. Chem. Soc., 1989, 111, 7534–7538 CrossRef CAS.
- (a) H. B. Henbest, W. R. Jackson and B. C. G. Robb, J. Chem. Soc. B, 1966, 803–807 RSC; (b) K. B. Wiberg and K. A. Saegebarth, J. Am. Chem. Soc., 1957, 79, 2822–2824 CrossRef CAS.
- (a) A. Kekulé and R. Anschütz, Ber. Dtsch. Chem. Ges., 1881, 14, 713–717 CrossRef; (b) A. Kekulé and R. Anschütz, Ber. Dtsch. Chem. Ges., 1880, 13, 2150–2152 CrossRef; (c) S. Tanatar, Ber. Dtsch. Chem. Ges., 1879, 12, 2293–2298 CrossRef.
- (a) M. Jaky, L. I. Simàndi, L. Maros and I. Molnarpe, J. Chem. Soc., Perkin Trans. 2, 1973, 1565–1569 RSC; (b) L. I. Simàndi and M. Jaky, J. Chem. Soc., Perkin Trans. 2, 1973, 1856–1860 RSC; (c) L. I. Simàndi and M. Jaky, J. Chem. Soc., Perkin Trans. 2, 1973, 1861–1866 RSC.
- S. Dash, S. Patel and B. K. Mishra, Tetrahedron, 2009, 65, 707–739 CrossRef CAS.
- A. J. Fatiadi, Synthesis, 1987, 85–127 CrossRef CAS.
- J. K. Cha and N. S. Kim, Chem. Rev., 1995, 95, 1761–1795 CrossRef CAS.
- M. Hudlicky, Oxidations in Organic Chemistry, in ACS Monograph, No. 186, ed. M. Hudlicky, American Chemical Society, Washington, DC, USA, Illus, 1990 Search PubMed.
- Modern Oxidation Methods, ed. J.-E. Bäckvall, Wiley-VCH Verlag GmbH & Co. KGaA, 2004 Search PubMed.
- (a) G. B. Howarth, W. A. Szarek and J. K. N. Jones, J. Chem. Soc. C, 1970, 2218–2225 RSC; (b) R. Annunziata, M. Cinquini, F. Cozzi and L. Raimondi, Tetrahedron, 1988, 44, 6897–6902 CrossRef CAS; (c) B. T. Bagmanov, Russ. J. Org. Chem. (Transl. of Zh. Org. Khim.), 2007, 43, 1635–1641 CrossRef CAS; (d) R. E. K. Winter, R. J. Zehr, M. Honey and W. Vanarsdale, J. Org. Chem., 1981, 46, 4309–4312 CrossRef CAS; (e) J. Cheng, Z. J. Fang, S. Li, B. H. Zheng and Y. H. Jiang, Carbohydr. Res., 2009, 344, 2093–2095 CrossRef CAS.
- (a) E. Salamci, H. Secen, Y. Sutbeyaz and M. Balci, Synth. Commun., 1997, 27, 2223–2234 CrossRef CAS; (b) E. Salamci, H. Secen, Y. Sutbeyaz and M. Balci, J. Org. Chem., 1997, 62, 2453–2457 CrossRef CAS.
- D. Vijaykumar, W. Mao, K. S. Kirschbaum and J. A. Katzenellenbogen, J. Org. Chem., 2002, 67, 4904–4910 CrossRef CAS.
- (a) R. Zimmer, M. Collas, R. Czerwonka, U. Hain and H. U. Reissig, Synthesis, 2008, 237–244 CrossRef CAS; (b) R. Zimmer, K. Homann, J. Angermann and H. U. Reissig, Synthesis, 1999, 1223–1235 CrossRef CAS.
- A. T. Soldatenkov, A. V. Temesgen, I. A. Bekro, S. A. Soldatova, N. I. Golovtsov and N. D. Sergeeva, Khim. Geterotsikl. Soedin., 2000, 402, 1661–1666.
- A. E. Hydorn, J. N. Korzun and J. R. Moetz, Steroids, 1964, 3, 493–504 CrossRef CAS.
- R. S. Varma and K. P. Naicker, Tetrahedron Lett., 1998, 39, 7463–7466 CrossRef CAS.
- A. Ayari, S. Szonyi, E. Rouvier and A. Cambon, J. Fluorine Chem., 1990, 50, 67–75 CrossRef CAS.
- J. E. Taylor, T. E. Janini and O. C. Elmer, Org. Process Res. Dev., 1998, 2, 147–150 Search PubMed.
- C. M. Starks, J. Am. Chem. Soc., 1971, 93, 195–199 CrossRef CAS.
- (a) W. P. Weber and J. P. Shepherd, Tetrahedron Lett., 1972, 13, 4907–4908 CrossRef; (b) D. G. Lee and V. S. Chang, J. Org. Chem., 1978, 43, 1532–1536 CrossRef CAS.
- T. A. Foglia, P. A. Barr and A. J. Malloy, J. Am. Oil Chem. Soc., 1977, 54, A858–A861 CrossRef.
- I. F. Cottrell, P. J. Davis and M. G. Moloney, Tetrahedron: Asymmetry, 2004, 15, 1239–1242 CrossRef CAS.
- T. Ogino and K. Mochizuki, Chem. Lett., 1979, 443–446 CAS.
- D. G. Rivera, O. Pando, V. Leliebre-Lara, D. Coll, F. Leon and F. Coll, J. Chem. Res., 2004, 53–54 Search PubMed.
- V. Bhushan, R. Rathore and S. Chandrasekaran, Synthesis, 1984, 431–433 CrossRef CAS.
- B. G. Hazra, T. P. Kumar and P. L. Joshi, Liebigs Ann./Recl., 1997, 1997, 1029–1034 Search PubMed.
- J. R. Caycho, F. Garcia-Tellado, P. de Armas and J. J. Marrero-Tellado, Chem. Lett., 1998, 25–26 CrossRef CAS.
- D. E. De Vos, S. de Wildeman, B. F. Sels, P. J. Grobet and P. A. Jacobs, Angew. Chem., Int. Ed., 1999, 38, 980–983 CrossRef CAS.
- (a) B. F. Sels, A. L. Villa, D. Hoegaerts, D. E. De Vos and P. A. Jacobs, Top. Catal., 2000, 13, 223–229 CrossRef CAS; (b) N. J. Schoenfeldt, A. W. Korinda and J. M. Notestein, Chem. Commun., 2010, 46, 1640–1642 RSC.
- J. Brinksma, L. Schmieder, G. van Vliet, R. Boaron, R. Hage, D. E. De Vos, P. L. Alsters and B. L. Feringa, Tetrahedron Lett., 2002, 43, 2619–2622 CrossRef CAS.
- J. W. de Boer, J. Brinksma, W. R. Browne, A. Meetsma, P. L. Alsters, R. Hage and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 7990–7991 CrossRef CAS.
- R. A. Bhunnoo, Y. L. Hu, D. I. Laine and R. C. D. Brown, Angew. Chem., Int. Ed., 2002, 41, 3479–3480 CrossRef CAS.
- J. W. de Boer, W. R. Browne, S. R. Harutyunyan, L. Bini, T. D. Tiemersma-Wegman, P. L. Alsters, R. Hage and B. L. Feringa, Chem. Commun., 2008, 3747–3749 RSC.
- (a) R. Kourist, P. D. de Maria and U. T. Bornscheuer, ChemBioChem, 2008, 9, 491–498 CrossRef CAS; (b) F. Fabris, J. Collins, B. Sullivan, H. Leisch and T. Hudlicky, Org. Biomol. Chem., 2009, 7, 2619–2627 RSC; (c) D. J. Aberhart, J. Org. Chem., 1980, 45, 5218–5220 CrossRef CAS; (d) N. I. Bowers, D. R. Boyd, N. D. Sharma, M. A. Kennedy, G. N. Sheldrake and H. Dalton, Tetrahedron: Asymmetry, 1998, 9, 1831–1834 CrossRef CAS; (e) D. R. Boyd, N. D. Sharma, T. Belhocine, J. F. Malone, S. McGregor and C. C. R. Allen, Chem. Commun., 2006, 4934–4936 RSC; (f) D. R. Boyd, N. D. Sharma, N. I. Bowers, I. N. Brannigan, M. R. Groocock, J. E. Malone, G. McConville and C. C. R. Allen, Adv. Synth. Catal., 2005, 347, 1081–1089 CrossRef CAS; (g) D. R. Boyd, N. D. Sharma, N. I. Bowers, H. Dalton, M. D. Garrett, J. S. Harrison and G. N. Sheldrake, Org. Biomol. Chem., 2006, 4, 3343–3349 RSC; (h) D. R. Boyd, N. D. Sharma, S. A. Haughey, M. A. Kennedy, J. F. Malone, S. D. Shepherd, C. C. R. Allen and H. Dalton, Tetrahedron, 2004, 60, 549–559 CrossRef CAS; (i) D. R. Boyd, N. D. Sharma, J. F. Malone and C. C. R. Allen, Chem. Commun., 2009, 3633–3635 RSC; (j) D. R. Boyd, N. D. Sharma, P. J. Stevenson, J. Chima, D. J. Gray and H. Dalton, Tetrahedron Lett., 1991, 32, 3887–3890 CrossRef CAS.
- (a) T. Hudlicky and J. W. Reed, Synlett, 2009, 685–703 CrossRef CAS; (b) T. Hudlicky and J. W. Reed, Chem. Soc. Rev., 2009, 38, 3117–3132 RSC; (c) P. C. A. Bruijnincx, G. van Koten and R. Gebbink, Chem. Soc. Rev., 2008, 37, 2716–2744 RSC.
- (a) K. Chen, M. Costas, J. H. Kim, A. K. Tipton and L. Que, J. Am. Chem. Soc., 2002, 124, 3026–3035 CrossRef CAS; (b) J. Y. Ryu, J. Kim, M. Costas, K. Chen, W. Nam and L. Que, Chem. Commun., 2002, 1288–1289 RSC; (c) M. Costas and L. Que, Angew. Chem., Int. Ed., 2002, 41, 2179–2181 CrossRef CAS.
- P. D. Oldenburg, A. A. Shteinman and L. Que, J. Am. Chem. Soc., 2005, 127, 15672–15673 CrossRef CAS.
- Y. Feng, C. Y. Ke, G. Q. Xue and L. Que, Chem. Commun., 2009, 50–52 RSC.
- J. Bautz, P. Comba, C. L. de Laorden, M. Menzel and G. Rajaraman, Angew. Chem., Int. Ed., 2007, 46, 8067–8070 CrossRef CAS.
- P. C. A. Bruijnincx, I. L. C. Buurmans, S. Gosiewska, M. A. H. Moelands, M. Lutz, A. L. Spek, G. van Koten and R. Gebbink, Chem.–Eur. J., 2008, 14, 1228–1237 CrossRef.
- (a) S. M. Barry and P. J. Rutledge, Synlett, 2008, 2172–2174 CAS; (b) V. J. Dungan, Y. Ortin, H. Mueller-Bunz and P. J. Rutledge, Org. Biomol. Chem., 2010, 8, 1666–1673 RSC.
- K. Suzuki, P. D. Oldenburg and L. Que, Angew. Chem., Int. Ed., 2008, 47, 1887–1889 CrossRef CAS.
- A. V. Biradar, B. R. Sathe, S. B. Umbarkar and M. K. Dongare, J. Mol. Catal. A: Chem., 2008, 285, 111–119 CrossRef CAS.
- W. F. Wang, F. J. Wang and M. Shi, Organometallics, 2010, 29, 928–933 CrossRef CAS.
- C. A. Horiuchi, G. Dan, M. Sakamoto, K. Suda, S. Usui, O. Sakamoto, S. Kitoh, S. Watanabe, T. Utsukihara and S. Nozaki, Synthesis, 2005, 2861–2864 CrossRef CAS.
- R. M. Pearlstein and A. Davison, Polyhedron, 1988, 7, 1981–1989 CrossRef CAS.
- L. Emmanuvel, T. M. A. Shaikh and A. Sudalai, Org. Lett., 2005, 7, 5071–5074 CrossRef.
- T. M. Nguyen and D. Lee, Org. Lett., 2001, 3, 3161–3163 CrossRef CAS.
- (a) C. Santi, M. Tiecco, L. Testaferri, C. Tomassini, S. Santoro and G. Bizzoca, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 956–960 Search PubMed; (b) S. Santoro, C. Santi, M. Sabatini, L. Testaferri and M. Tiecco, Adv. Synth. Catal., 2008, 350, 2881–2884 CrossRef CAS.
- S. Rani and Y. D. Vankar, Tetrahedron Lett., 2003, 44, 907–909 CrossRef CAS.
- N. V. S. Mudiganti, S. Claessens, P. Habonimana and N. de Kimpe, J. Org. Chem., 2008, 73, 3867–3874 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |