Visible light photoredox catalysis: applications in organic synthesis
Jagan M. R.
Narayanam
and
Corey R. J.
Stephenson
*
Department of Chemistry, Boston University, Boston, Massachusetts 02215, USA. E-mail: crjsteph@bu.edu
First published on 8th June 2010
Abstract
The use of visible light sensitization as a means to initiate organic reactions is attractive due to the lack of visible light absorbance by organic compounds, reducing side reactions often associated with photochemical reactions conducted with high energy UV light. This tutorial review provides a historical overview of visible light photoredox catalysis in organic synthesis along with recent examples which underscore its vast potential to initiate organic transformations.
 Jagan M. R. Narayanam | Jagan M. R. Narayanam was born in Peteru (AP), India, in 1979. He received an MSc from the University of Hyderabad in 2001 and a PhD in 2008 from ETH Zürich working under the direction of Prof. A. Vasella. Since May 2008, he is working as a Swiss National Science Foundation postdoctoral fellow in the group of Prof. Corey Stephenson. His research in Stephenson's group focused on the development of visible light photoredox catalysis in organic synthesis and its application in the synthesis of complex natural products. |
 Corey R. J. Stephenson | Corey R. J. Stephenson was born in Collingwood, Ontario, Canada, in 1974. He received a BSc degree in 1998 from the University of Waterloo working with J. Michael Chong, and a PhD in 2004 from the University of Pittsburgh working under the direction of Peter Wipf. After carrying out postdoctoral research in the Laboratory of Organic Chemistry at the ETH Zürich, Switzerland, with Erick M. Carreira, he joined the faculty at Boston University as an assistant professor of chemistry in 2007. |
Introduction
Synthetic organic chemists' pursuit towards new reactions and chemoselective transformations under mild and green conditions is a continuous process. Nearly a century ago, Ciamician realized that “light” is an abundant and renewable energy source for performing green chemical reactions.1 Since then, photochemistry and photocatalysis (via photoinduced-electron-transfer, PET) have found broad utility in organic synthesis.2,3 However, the lack of visible light absorption by many organic molecules has limited the application of photochemical synthesis. Hence, employing visible light absorbing photocatalysts and utilizing their electron/energy transfer processes to sensitize organic molecules to carry out required photochemical reactions would serve as a valuable tool for overcoming this barrier.Nature's ability to use various visible light absorbing chromophores/photocatalysts for converting solar energy to chemical energy has inspired various research groups to develop a plethora of hosts involving photoredox systems in an effort to mimic natural photosynthesis.4–7 These systems have provided a platform to understand and elucidate the electron or energy transfer pathways involved in natural photosynthesis. Among these, photoredox catalysts are of notable importance due to their applications in water splitting, solar energy storage, proton coupled electron transfer, and photovoltaics.8–11 In particular, Ru(II)polypyridine complexes are quite interesting due to their ease of synthesis, stability at room temperature, and excellent photoredox properties. Among these complexes, Ru(bpy)3Cl2, a commercially available complex, is one of the most widely employed photocatalyst. In this tutorial review, we intend to highlight recent advances and potential applications of photoredox catalysts in organic synthesis.
Photophysical properties of Ru(bpy)32+
Irradiation of Ru(bpy)3Cl2 with visible light (λmax = 452 nm) populates the excited state Ru(bpy)32+* via metal to ligand charge transfer (MLCT). Relative to the ground state species, this excited species can be easily oxidized or reduced (Fig. 1). Several oxidative and reductive quenchers of the excited state are known and some of them are shown in Fig. 1. Oxidative quenching of Ru(bpy)32+* provides Ru(bpy)33+, a strong oxidant (1.29 V vs.SCE = Standard Calomel Electrode, in CH3CN), while reductive quenching provides Ru(bpy)3+, a strong reducing agent (−1.33 V vs.SCE in CH3CN). Hence, depending upon the conditions employed and the proper selection of the quencher, Ru(bpy)32+* can be utilized as a single electron oxidant or reductant.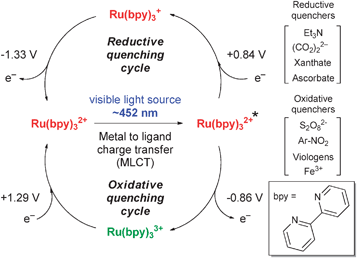 | ||
| Fig. 1 General photoredox paradigm of Ru(bpy)3Cl2. | ||
Photoredox catalysis in organic synthesis
Despite the excellent photoredox properties and their ease of preparation from commercially available precursors, tris(bipyridine) ruthenium complexes have attracted very little attention of synthetic organic chemists. In 1984, Cano-Yelo and Deronzier reported one of the first examples, demonstrating a photocatalytic Pschorr reaction for the synthesis of phenanthrene and substituted phenanthrenes (Scheme 1).12 The Pschorr reaction involves an intramolecular arylation upon reduction of a diazonium salt by a reducing agent, electrochemical reduction, or simple heating.13 The authors have chosen stilbenediazonium ion 1 to promote the photocatalytic Pschorr reaction. Visible light irradiation of 1 in the presence of Ru(bpy)32+ in acetonitrile produced phenanthrene carboxylic acid 2 in quantitative yield.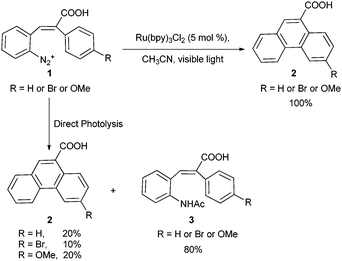 | ||
| Scheme 1 Photocatalytic Pschorr reaction. | ||
The proposed mechanism of this reaction is outlined in Scheme 2. Excitation of Ru(bpy)32+ by visible light generates Ru(bpy)32+* (Ru(bpy)33+/Ru(bpy)32+* −0.86 V vs.SCE) which transfers an electron to 1 (E1/2 = −0.1 V vs.SCE in CH3CN) to produce aryl radical 4. Intramolecular radical arylation furnishes radical 5 which undergoes oxidation by Ru(bpy)33+ and subsequent deprotonation to give 2 while regenerating the catalyst Ru(bpy)32+.
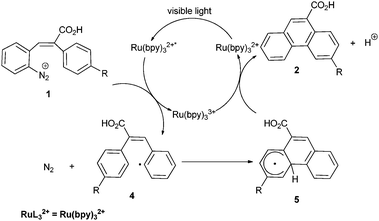 | ||
| Scheme 2 Proposed mechanism for the photocatalytic Pschorr reaction. | ||
More interestingly, the direct photolysis of 1 yielded only 10–20% of 2. The remainder of the reactant was converted to the acetamide 3, obtained by trapping of the aryl cation with acetonitrile followed by hydrolysis. These observations also suggest that the intramolecular arylation does not occur via a cationic pathway. In subsequent studies, the authors found that para substituted aryl diazonium salts can also oxidatively quench the excited state species Ru(bpy)32+* to provide Ru(bpy)33+ and corresponding aryl radicals, which are reduced to arenes.14 Inspired by their initial success, Cano-Yelo and Deronzier extended the application of photoredox catalysis to the oxidation of benzylic alcohols to the corresponding aldehydes (Scheme 3).15
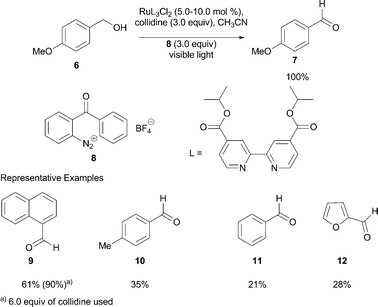 | ||
| Scheme 3 Oxidation of benzylic alcohols using photocatalysis. | ||
Visible light irradiation of benzylic alcohol 6 in the presence of RuL32+, 8, and collidine in acetonitrile provided the aldehyde 7 in quantitative yield. The yield of the aldehyde decreased as the half-wave oxidation potential of alcohol increased. On the other hand, addition of excess base improved the yield of aldehyde (yield of 9 improved from 61% to 90% by addition of 3 additional equiv. of collidine). In the absence of collidine, the yield of aldehyde was markedly decreased (30% of 7 and <10% of 9 formed from their corresponding alcohols). In all of the reported reactions, a 3 ∶ 1 ratio of benzophenone and fluorenone were formed as byproducts.
In accordance with their observations, the authors proposed a mechanism initiated by quenching of Ru(bpy)32+* by the aryldiazonium ion 8 to generate Ru(bpy)33+ and aryl radical 13. Although the authors did not provide a clear mechanism for the oxidation of alcohol to aldehyde, presumably the first step might be the donation of a single electron to Ru(bpy)33+ from benzylic alcohol 16 to form the radical cation 18 while regenerating Ru(bpy)32+. Hydrogen atom abstraction by 13 from the radical cation 18 followed by deprotonation gives the aldehyde 17 (Scheme 4). Fluorenone 15 was observed as a byproduct of the previously described Pschorr reaction (see Scheme 4).
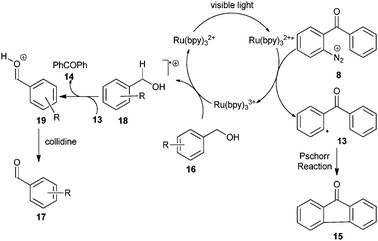 | ||
| Scheme 4 Proposed mechanism for the oxidation of benzylic alcohols. | ||
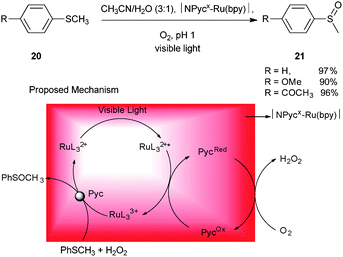
Nearly 20 years after Deronzier's first application of photoredox catalysis for the synthesis of organic compounds, Zen and co-workers reported the selective and efficient photocatalytic oxidation of sulfides to sulfoxides.16 For this oxidation, a Nafion membrane doped with a lead ruthenate pyrochlore (Pyc) catalyst and Ru(bpy)32+ (designated as |NPycx–Ru(bpy)|) was used. Irradiation of the sulfide 20 with visible light in the presence of |NPycx–Ru(bpy)| in CH3CN/H2O and continuous purging with O2 for 3 h afforded the sulfoxide 21 in 97% yield (Scheme 5). It is important to note that no over oxidation of the sulfoxide to the corresponding sulfone, a common problem in classical approaches, was observed. This photocatalytic oxidation reaction is highlighted by its high sulfoxide selectivity, environmentally benign reagents, ease of purification, and excellent yields.
 | ||
| Scheme 5 Photocatalytic oxidation of sulfides to sulfoxides. | ||
A proposed reaction mechanism is shown in Scheme 5, where Pyc plays a dual catalytic role as the reducing agent in the oxygen reduction reaction (ORR) to produce H2O2 and also as an electron acceptor from the excited Ru(bpy)32+* to form Ru(bpy)33+. Hydrogen peroxide generated in situ oxidizes the sulfide to sulfoxide while regenerating Ru(bpy)32+.
All of the examples highlighted thus far have dealt with the oxidative quenching of Ru(bpy)32+* to Ru(bpy)33+ and utilization of its high oxidation potential for chemical transformations. Fukuzumi and co-workers demonstrated the reduction of phenacylbromides using dihydroacridines and Ru(bpy)32+, either in the presence or absence of acid.17 During these studies, Fukuzumi and co-workers demonstrated the modulation of the excited state quenching mechanism by varying the concentration of Brønsted acid. Although the same product is formed in both cases, in the absence of acid, the reaction proceeds via reductive quenching of Ru(bpy)32+* to Ru(bpy)3+ (Scheme 6, Path A), while at high acid concentration, the reaction proceeds via oxidative quenching of Ru(bpy)32+* to Ru(bpy)33+ since Acr-H2 is proposed to be protonated (Scheme 6, Path B). These results further display the ability of modulating the reactivity of photoredox complexes by varying the reaction conditions.
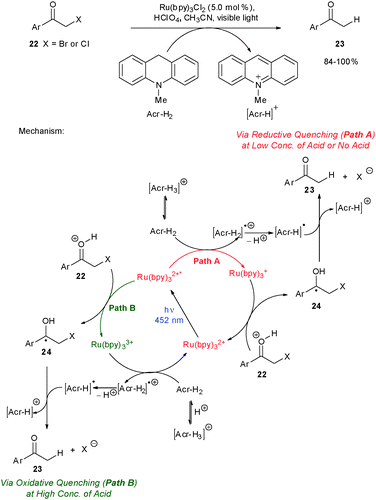 | ||
| Scheme 6 Reductive dehalogenation of phenacyl halides. | ||
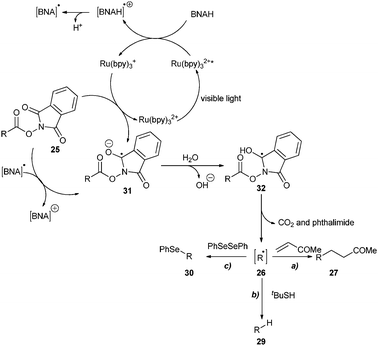
Subsequently, the reductive quenching pathway was utilized by Okada, Oda and co-workers for the decarboxylation of acyloxyphthalimides to generate alkyl radicals (Scheme 7). Visible light irradiation of acyloxyphthalimide 25 in the presence of Ru(bpy)32+ and BNAH (1-benzyl-1,4-dihydronicotinamide) in aqueous THF furnished the alkyl radical 26 which was trapped by a α,β-unsaturated ketone or ester to produce 27 and 28, respectively, through C–C bond formation.18 Delighted with this success, the authors have further expanded the scope of the reaction to form C–H19 and C–Se20,21 bonds. Use of tBuSH in the reaction gave the alkane 29, whereas the reaction in the presence of PhSeSePh furnished the phenylselenyl ether 30.
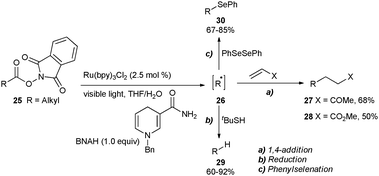 | ||
| Scheme 7 Photoredox-mediated decarboxylation of acyloxyphthalimides. | ||
The proposed mechanism depicted in Scheme 8 begins with single electron transfer to the visible light excited Ru(bpy)32+* from BNAH to provide the strong reductant Ru(bpy)3+ and BNAH radical cation, which further decomposes to BNA radical and a proton. The single electron reduction of acyloxyphthalimide either by Ru(bpy)3+ or BNA radical provides the radical anion 31. Protonation generates the radical 32 and removal of CO2 and phthalimide provides the alkyl radical 26, which could be trapped by radical acceptors to form C–C, C–H and C–Se bonds.
 | ||
| Scheme 8 Proposed mechanism for the decarboxylation of acyloxyphthalimides using photoredox catalysis. | ||
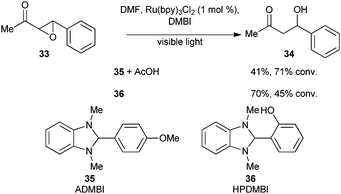
In 2006, Hasegawa and co-workers reported the photocatalytic reductive opening of Cα–O bond of ketoepoxides to afford β-hydroxy ketones.22 In this report, the authors used N,N-dimethylbenzimidazolines (Scheme 9, 35 or 36) as sacrificial electron and hydrogen atom donors. The low oxidation potentials of ADMBI 35 (+0.28 V vs.SCE) and HPDMBI 36 (+0.30 V vs.SCE) suggest that the reaction is occurring via reductive quenching of Ru(bpy)32+* to Ru(bpy)3+. Protonation of the ketone enables single electron transfer from Ru(bpy)3+ to the π* of carbonyl of 33. Subsequent opening of epoxide and hydrogen atom abstraction afforded the observed β-hydroxy ketone 34. Although the yields and conversion are moderate, this reaction served to demonstrate the potential of Ru(bpy)3+ as a useful reducing agent.
 | ||
| Scheme 9 Photocatalytic reductive opening of epoxides. | ||
In 2008, Yoon and co-workers reported the photoredox-mediated highly diastereoselective [2+2] cycloaddition of enones.23 Pioneering studies by Bauld and Krische demonstrated that bis-enones could undergo copper- and cobalt-mediated single electron transfer initiated formal [2+2] cycloaddition reactions.24–26
Formation of [2+2] cycloadducts under electrochemical conditions also supported the intermediacy of radical anion intermediates.27,28 This inspired Yoon and co-workers to speculate that the formation of a similar radical anion could be feasible by using Ru(bpy)3+ intermediates generated using visible light irradiation and reductive quenching of Ru(bpy)32+* by an electron donor. Irradiation of an aryl dienone 37 with visible light in the presence of Ru(bpy)32+, iPr2NEt, and LiBF4 in acetonitrile gave the desired intramolecular formal [2+2] cycloaddition product 38 in 89% yield with a >10 ∶ 1 diastereomeric ratio (Scheme 10). A current limitation to this methodology is that one of the cycloaddition partners for this reaction must be an aryl enone to initiate the single electron reduction to form the required radical anion. Aryl enones bearing both electron-withdrawing or -donating groups and a variety of α,β-unsaturated carbonyl compounds were shown to be suitable partners for this reaction. In all the intramolecular cycloadditions reported, the cis-cyclobutanes were obtained preferentially. This reaction also facilitates the formation of quaternary centers in the case of α-substituted enones. In contrast, intermolecular cycloaddition reaction of aryl enones provided the homo-dimerization products (45 and 46) in very good yields and high diastereomeric ratio (dr). Unlike intramolecular reactions, this reaction provided all trans-cyclobutanes (meso-isomers) in preference to all cis. However, as mentioned above, this reaction is limited to only aryl enones and no reaction was observed in the case of alkyl enones. Omission of any of the reagents, including light, resulted in no observed [2+2] cycloaddition product 38. In addition, the absence of product formation without the addition of iPr2NEt suggests that the cycloaddition is not initiated by electron transfer from the photoexcited Ru(bpy)32+* to the enone, rather it is initiated by Ru(bpy)3+ that is generated by the reductive quenching of Ru(bpy)32+* by iPr2NEt. Also, no reaction occurred in the absence of LiBF4, a result which has been attributed to its Lewis acidic nature and ability to improve the solubility of Ru(bpy)3Cl2 in acetonitrile. On the basis of these observations, the authors proposed the mechanism shown in Scheme 11.
![Visible light photocatalyzed [2+2] cycloadditions.](/image/article/2011/CS/b913880n/b913880n-s10.gif) | ||
| Scheme 10 Visible light photocatalyzed [2+2] cycloadditions. | ||
![Proposed mechanism for Yoon's formal [2+2] cycloaddition of enones.](/image/article/2011/CS/b913880n/b913880n-s11.gif) | ||
| Scheme 11 Proposed mechanism for Yoon's formal [2+2] cycloaddition of enones. | ||
The authors proposed that excitation of Ru(bpy)32+ by visible light generates the photoexcited state Ru(bpy)32+*, which is followed by single electron transfer from iPr2NEt forming Ru(bpy)3+, a strong reductant (−1.33 V vs.SCE). A single electron reduction of lithium coordinated enone 49 produces the radical 51, eventually leading to the cyclobutane product 48. Presumably, a 1,4-addition of radical 50 to another molecule of 49 (for intramolecular, another enone or α,β-unsaturated carbonyl), followed by radical cyclization, furnishes the cyclobutane radical 52. Oxidation of 52 can be achieved either by excited Ru(bpy)32+* or by the iPr2NEt radical cation and produces the cyclobutane 48 along with Ru(bpy)3+ or iPr2NEt.29
Expanding upon the intermolecular homo-dimerization of aryl enones (see 45 and 46 in Scheme 10), Du and Yoon reported crossed intermolecular formal [2+2] cycloadditions.30 Similar to the intramolecular cycloadditions, one of the reaction partners must be an aryl enone. To overcome the major undesired pathway of homodimerization of the aryl enone, the authors strategically selected more reactive Michael acceptors as the second reaction partner (Scheme 12).
![Yoon's intermolecular [2+2] cycloaddition of enones.](/image/article/2011/CS/b913880n/b913880n-s12.gif) | ||
| Scheme 12 Yoon's intermolecular [2+2] cycloaddition of enones. | ||
A mixture of methyl vinyl ketone 54 and aryl enone 53 (2.5 ∶ 1), Ru(bpy)32+ (5 mol%), iPr2NEt, and LiBF4 was subjected to visible light irradiation for 4 h and provided the crossed [2+2] cycloadduct 55 (84%, >10 ∶ 1 dr) in a highly chemoselective fashion. Only trace amounts of homo-coupled product derived from 53 were observed. Both electron-rich and -poor substrates are compatible under these conditions. Variation of the substituents at the β-position was possible; however the reaction is sensitive to steric bulk. Substitution at the β-position with primary and secondary alkyl groups proceeds smoothly, whereas tert-butyl substitution provided a very low yield of 60. α,β-Unsaturated thioesters proved to be very good Michael acceptor partners to provide good yields of hetero-coupled product. Formation of quaternary centers also proceeded smoothly with reasonably good yield and diastereoselectivity (63, 57%, dr 5 ∶ 1). On the basis of the previous results, as expected, the reaction between two alkyl enones did not provide any of the cycloaddition products (64, 0%).
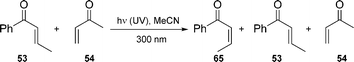
In addition, the authors have shown that both the inter- and intramolecular reactions proceed smoothly even in ambient sunlight and in multigram scales. Interestingly, irradiation of 53 and 54 with UV light failed to provide any observable cycloaddition products; the only products observed in the reaction mixture arise from isomerization of enone 53 (eqn (1)).
 | (1) |
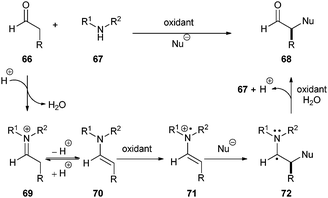
Organocatalysis has rapidly evolved over the last decade by providing more than 130 chemical reactions that facilitate enantioselective C–C, C–O, C–N, and C–X (X = halogen) bond formation.31–35 The central dogma of all these reactions involves a two electron pathway; a filled HOMO of electron-rich substrate reacts with an empty LUMO of an electron-poor or -deficient species. MacMillan and co-workers only recently introduced the concept of organo-SOMO-catalysis, a one electron mode of activation that has enabled the development of several useful transformations.36–39 A general scheme for describing organo-SOMO-catalysis is shown in Scheme 13. A single electron oxidation of an enamine 70, derived from an aldehyde 66 and a secondary amine 67 by an oxidant (typically CAN), results in the formation of the corresponding enamine radical cation 71. Addition of nucleophiles to the α-carbon of the enamine followed by oxidation of 72 and hydrolysis provides 68.
 | ||
| Scheme 13 General mechanism for SOMO catalysis reaction. | ||
With the success of organo-SOMO catalysis, Nicewicz and MacMillan reported the merging of photoredox catalysis with organocatalysis in the pursuit of enantioselective alkylation of aldehydes (Scheme 14).40 Nicewicz and MacMillan hypothesized that Fukuzumi's catalytic formation of electron-deficient radicals by the reductive cleavage of C–X bonds using Ru(bpy)3+ (see Scheme 6)17 (E1/2 for phenacylbromide = −0.49 V vs.SCE in CH3CN)41 could be coupled with the organocatalytic formation of enamines. Electron-deficient radicals are known to react with electron-rich olefins to form C–C bonds.42,43 In this case, the reaction of the radical with the chiral, electron-rich enamine would provide the new C–C bond with enantiofacial control. Indeed, the authors observed that irradiation of an aldehyde 66, a secondary amine 75 (20 mol%), Ru(bpy)3Cl2 (0.5 mol%), and 2,6-lutidine in DMF with visible light for 6 h provided the α-alkylated aldehyde 74 in excellent yields and enantioselectivities. The reaction also proceeds well with sterically demanding substrates (77 and 78), and forms quaternary centers in high ee (80). The reaction is highlighted by its mild conditions, high functional group tolerance, and lack of observed epimerization.
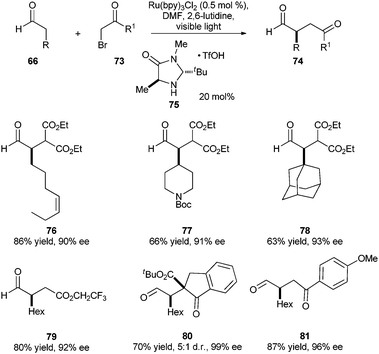 | ||
| Scheme 14 Photoredox organocatalysis: asymmetric α-alkylation of aldehydes. | ||
The proposed mechanism of the photoredox organocatalysis alkylation of aldehyde 66 is shown in Scheme 15. The reaction consists of two cooperative catalytic cycles: first is the excitation of Ru(bpy)32+ with visible light irradiation to its excited state Ru(bpy)32+*, which will be reductively quenched by enamine 83 (proposed as a sacrificial quantity, 1 mol% derived from aldehyde 66 and amine 75) to form Ru(bpy)3+. Single electron transfer from Ru(bpy)3+ to the α-bromocarbonyl compound 73 furnishes the electron-deficient radical 82 and regenerates Ru(bpy)32+ to complete the first catalytic cycle.
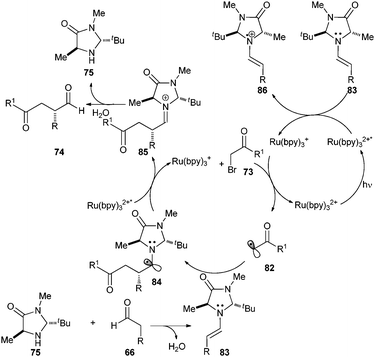 | ||
| Scheme 15 Proposed mechanism for asymmetric α-alkylation of aldehydes. | ||
The second catalytic cycle begins with the coupling of electron deficient radical 82 with the SOMOphilic (electron rich) enamine 83 and forms the key C–C bond and generates an α-amino radical 84. The low oxidation potential of this single electron species 84 (−0.92 to −1.12 V vs.SCE in CH3CN)44 allows it to be oxidized by Ru(bpy)32+* to provide the iminium cation 85 and the reductant Ru(bpy)3+, which will re-engage in the first catalytic cycle. Hydrolysis of 85 regenerates the amine catalyst 75 while delivering the requisite enantioenriched α-alkylated aldehyde 74 (Scheme 15).
Following their success in the asymmetric alkylation of aldehydes using photoredox organocatalysis, MacMillan and co-workers very recently reported the extension of asymmetric alkylation to the trifluoromethylation of aldehydes.45Organofluorine compounds, in particular trifluoromethylated compounds, are important in medicinal chemistry due to their high metabolic stability, binding selectivity, and lipophilicity.46–48 Similar to the catalytic asymmetric alkylation of aldehydes, asymmetric perfluoro alkylation has also been an elusive reaction.49–52
To solve this problem, MacMillan and co-workers utilized photoredox organocatalysis. In their initial experiments with Ru(bpy)32+ and CF3I (−1.22 V vs.SCE),53,54amine 89, and octanal under visible light irradiation gave only 52% of 88 with no enantioselectivity. Changing the photocatalyst to Ir(ppy)2(dtbbpy)PF655 provided better yields (85%), but no ee was observed. However, performing the reaction at lower temperature (−20 °C) provided enantioinduction up to 52% ee. Furthermore, optimization revealed that amine catalyst 75 was superior to 89 and gave almost perfect enantiocontrol in the trifluoromethylation [79%, 99% ee, with Ru(bpy)32+ as photocatalyst; 87%, 67% ee]. The first catalytic, enantioselective trifluoromethylation of various aldehydes was successfully achieved with good yields and high ee's (Scheme 16). Notably, trifluoromethylation of bulky aldehydes (91, 92), benzylic trifluoromethylation (93), and perfluoroalkylation of aldehydes (96, 97) proceeded smoothly without any loss of ee. Surprisingly, use of CF3Br instead of CF3I did not provide the trifluoromethylation, instead bromodifluoromethylation occurred (98). Similar to the asymmetric alkylation, trifluoromethylation reaction also proceeds with the same mechanism as shown in Scheme 15. The only difference is the change of photocatalyst from Ru(bpy)32+ to Ir(ppy)2(dtbbpy)PF6. Excitation of Ir(ppy)2(dtbbpy)+ with visible light to its excited state Ir(ppy)2(dtbbpy)+*, followed by single electron reduction, provides Ir(ppy)2(dtbbpy) (−1.51 V vs.SCE in CH3CN), which is a stronger reducing agent than Ru(bpy)3+ (−1.33 V vs.SCE in CH3CN).
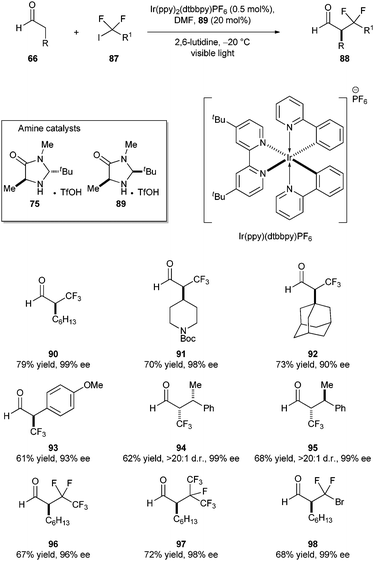 | ||
| Scheme 16 Photoredox organocatalysis: asymmetric perfluoroalkylation of aldehydes. | ||
Subsequently, Koike and Akita disclosed the oxyamination of enamines and aldehydes.56 Similar to MacMillan's approach, a mixture of aldehyde and a secondary amine (morpholine) with TEMPO and Ru(bpy)3Cl2 under visible light irradiation furnished the oxyamination product (Scheme 17).
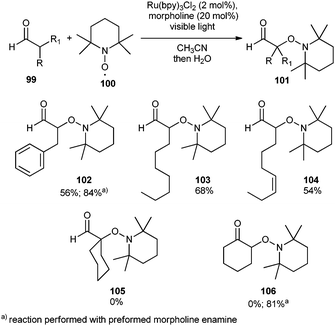 | ||
| Scheme 17 Photocatalytic oxyamination of aldehydes. | ||
Moderate yields were obtained for the mono-substituted aldehydes (102, 103, and 104), whereas no reaction was observed for the α,α-disubstituted aldehydes (105) and ketones (106). These results suggested that the efficient formation of the enamine is necessary for an efficient oxyamination reaction using photoredox catalysis. Similar reactivity was observed in MacMillan's asymmetric alkylation reactions where no racemization of the products (alkylated aldehydes) occurs due to the inertness of secondary amines towards α-hindered aldehydes. However, the oxyamination catalyzed by photocatalysis proceeded smoothly with the preformed enamines, derived from both aldehydes (84% of 102) and ketones (81% of 106).
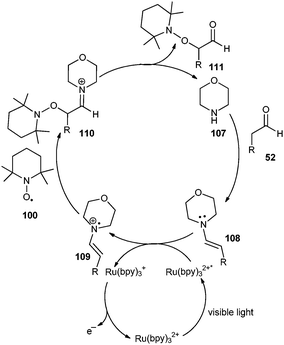
The mechanism proposed by the authors is shown in Scheme 18. Reductive quenching of Ru(bpy)32+* by enamine 108 generates Ru(bpy)3+ and an enamine radical cation. The authors propose direct radical coupling of TEMPO and enamine radical cation to form the C–O bond and the corresponding iminium 110. Hydrolysis of 110 produces 111 and regenerates the secondary amine. The electron acceptor responsible for catalyst turnover from Ru(bpy)3+ to Ru(bpy)32+ remains unclear at this time. A similar asymmetric process was described by Sibi and Hasegawa using SOMO catalysis.57
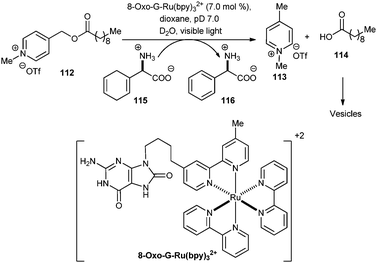
Another application of photoredox catalysis is photo removal of protecting groups (PRPG's).58,59 Recently two publications appeared with the similar concept of reductive cleavage of picolinium esters. Firstly, DeClue and co-workers demonstrated the photocatalytic continuous and controlled reductive cleavage of decanoic acid picolinium esters 112 (reduction potential −1.06 V vs.SCE) to produce bilayer-forming molecules (vesicles, Scheme 19).60 The authors hypothesized that a strong reductant like Ru(bpy)3+ could cleave C–O bond to produce a decanoate anion and a picolinium radical, which is subsequently trapped by a hydrogen atom donor. To achieve this, the authors have used a photocatalyst consisting of a tethered 8-oxoguanidine linked to Ru(bpy)32+ (8-oxo-G-Ru(bpy)32+). It differs from the previously discussed photocatalysts in that it contains a built-in electron donor, i.e., both Ru(bpy)32+ and the electron donor are regenerated during the course of the reaction.
 | ||
| Scheme 19 Nucleobase mediated photocatalytic vesicle formation from an ester precursor. | ||
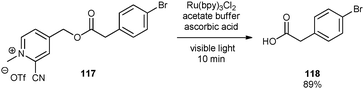
Borak and Falvey demonstrated that 2-cyanopicolinium esters could be reductively cleaved using Ru(bpy)32+ and ascorbic acid under visible light irradiation.61 In this case, ascorbic acid (0.29 V vs.SCE) acts as both electron and hydrogen donor. Substitution of electron-withdrawing groups on the picolinium group lowers the reduction potential (−0.63 V vs.SCE), thereby facilitating the reductive cleavage of C–O bond (eqn (2)).
 | (2) |
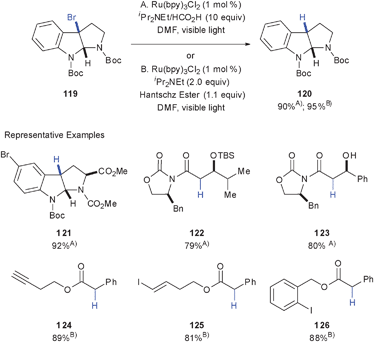
Photoredox-mediated reductive dehalogenation, first observed by Fukuzumi for α-haloacetophenones,17 has been further developed by Stephenson and co-workers into a synthetically useful transformation.62Halogens α- to a carbonyl, electron withdrawing or aryl group could be selectively reduced under these conditions. Conceptually, this reaction also proceeds via reductive quenching of photoexcited Ru(bpy)32+* to Ru(bpy)3+ and subsequent electron transfer to C–X bond α- to an electron-withdrawing group, followed by hydrogen abstraction. For this transformation, the authors have described two sets of conditions in DMF (Scheme 20): A. Ru(bpy)3Cl2 (1.0 mol%), iPr2NEt (10 equiv.) and HCO2H (10 equiv.); or B. Ru(bpy)3Cl2 (1.0 mol%), iPr2NEt (2.0 equiv.) and Hantschz Ester (1.1 equiv.). Both conditions provided reductive dehalogenation in excellent yields and could be chosen based on the specific substrate. In particular, conditions B were utilized for less activated halogen substrates for which the SN2 substitution of the formate ion was competitive with the reductive dehalogenation when using conditions A.
 | ||
| Scheme 20 Reductive dehalogenation using photoredox catalysis. | ||
These conditions proved to be very mild and efficient for reductive dehalogention of C–Br and C–Cl bonds in the presence of free hydroxyls (123), olefins (125), and alkynes (124). Furthermore, excellent chemoselectivity was observed by selective reduction of alkyl bromides and chlorides α- to electron-withdrawing groups over aryl bromides (121), iodides (126) and vinyl iodides (125). Excellent protecting group tolerance was also observed, including silyl ethers (122) and carbamates (119, 121). This alternative to currently available methods for radical reduction is marked by mild reaction conditions, excellent chemoselectivity and functional group tolerance, and avoids the use of toxic or hazardous reagents (cf. Bu3SnH/AIBN). Furthermore, the radical nature of the reaction was supported by the cyclopropyl ring fragmentation of α-bromo-α-cyclopropyl imide 127 to α,β-unsaturated imide 128 (52%, eqn (3)).
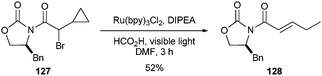 | (3) |
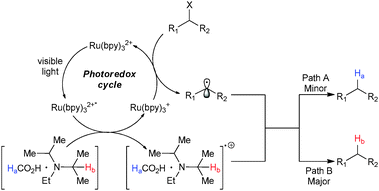 | ||
| Scheme 21 Plausible mechanism for the reductive dehalogenation. | ||
Reductive quenching of visible light excited Ru(bpy)32+* to Ru(bpy)3+ by iPr2NEt·HCO2H and subsequent electron transfer to cleave the C–X bond α- to an electron withdrawing group generates the alkyl radical. Reduction of this radical could be achieved via two pathways: (a) hydrogen abstraction from formic acid (Ha, minor); or (b) hydrogen abstraction from iPr2NEt (Hb, major).
Although the reductive dehalogenation using photoredox catalysis at this stage is only limited to activated halogen compounds, it can be extended to the reduction of unactivated halogen substrates by simply modifying redox potentials of the photocatalysts.64 Furthermore, photocatalytic generation of a radical by the reduction of a C–X bond has great potential as an alternative for C–C bond forming reactions via the chemistry of alkyl free-radicals.
In view of utilizing photoredox catalysis for C–C bond forming reactions, Stephenson and co-workers demonstrated the functionalization of indoles and pyrrolesvia intramolecular radical cyclizations.65 The functionalization of indoles and pyrroles has attracted much attention due to their high abundance in biologically and medicinally active compounds. In this regard, the authors hypothesized that the radical generated by reductive cleavage of C–X bonds α- to electron-withdrawing groups could be trapped by electron-rich arenes such as indoles and pyrroles. The authors have used indole or pyrrole tethered bromomalonates as substrates, under conditions similar to the reductive dehalogenation reaction, with the notable omission of formic acid (see Scheme 22). The use of Et3N instead of iPr2NEt as sacrificial electron donor provided the best yields. A broad range of functionalized indoles and pyrroles efficiently undergo intramolecular radical additions using photoredox catalysis.
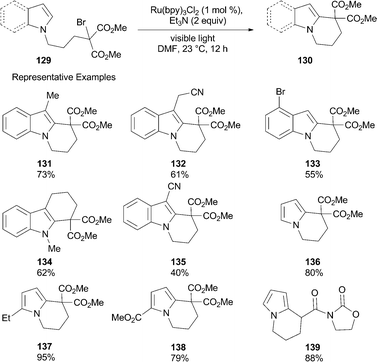
The efficiency for this transformation seems to be dependent upon the electronic properties of the nucleophile, i.e., indole or pyrrole. Substitution of indoles with electron-withdrawing groups lowers the yields of the cyclized product (135). In particular, the more electron-rich pyrrole subunits typically outperform indoles. Similar to reductive dehalogenation, the radical cyclization reaction also proceeds with high chemoselectivity (133).
In addition, the authors have demonstrated the utility of photoredox catalysis by extending this reaction to cascade cyclizations. As an example, bromomalonate 140 irradiated with visible light under conditions identical to the radical cyclization underwent sequential radical cyclizations to provide the tetracyclic compound 141 in good yield as a single diastereoisomer (eqn (4)).
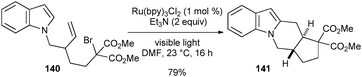 | (4) |
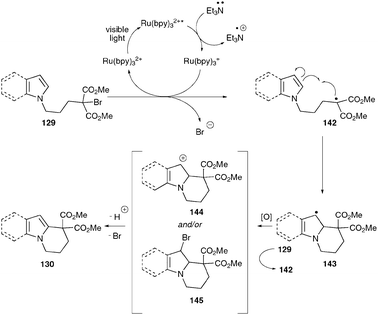
The mechanism starts with single electron transfer to Ru(bpy)32+* from Et3N, forming Ru(bpy)3+ and the radical cation of triethylamine. Reduction of the C–Br bond by Ru(bpy)3+ forms radical 142 while regenerating Ru(bpy)32+. Intramolecular cyclization of the electron-rich aromatic ring (indole or pyrrole) with the electrophilic radical 142 produces the benzylic radical 143. This radical could be oxidized by either Ru(bpy)32+* or the bromomalonate 129 to provide intermediates 144 or 145 respectively. Deprotonation of 144 or elimination of HBr from 145 affords the tricyclic compound 130. Alternative methods for the oxidation of this benzylic radical such as the action of adventitious oxygen or the ammonium radical cation cannot be excluded at this time. The mild conditions used in photoredox catalysis provide an advantage over the current methods available in the literature for similar transformations, particularly for use with highly functionalized molecules.
On the basis of their mechanistic hypotheses, Stephenson and co-workers anticipated that the iminium ion generated under photoredox conditions could be intercepted with an appropriate nucleophile.66 Preliminary studies indicated that simple nucleophiles such as CH3OH could trap the iminium ion generated from N-aryltetrahydroisoquinolines 146 in the presence of Ir(ppy)2(dtbbpy)PF6 (1 mol%) under visible light irradiation. Interestingly, similar to the studies of Koike and Akita,56catalyst turnover was accomplished without an apparent external oxidant. Simply switching the solvent from CH3OH to CH3NO2 provided the aza-Henry products in excellent yields for a variety of substrates (Scheme 24). Although the reaction is much slower, N-phenylpyrrolidine was also a competent substrate in this reaction (27% yield, 40% conversion after 72 h).
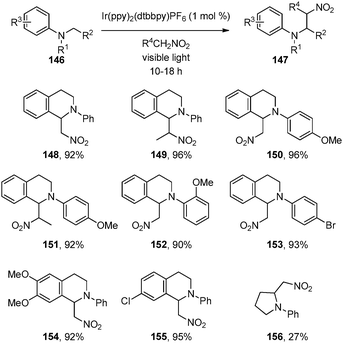 | ||
| Scheme 24 Oxidative aza-Henry reactions using photoredox catalysis. | ||
Both light and a photocatalyst were found to be necessary for the efficient conversion to the aza-Henry products, while the exclusion of oxygen from the reaction resulted in a diminished rate. On the basis of luminescence quenching experiments, the following mechanism was proposed (Scheme 25): the radical cation of the amine 157 is formed upon visible light excitation of the Ir3+ catalyst and reductive quenching. While the mechanism for catalyst turnover is not clear at this time, the authors propose that adventitious oxygen likely plays a critical role in the reaction. The corresponding radical anion may abstract a hydrogen atom from the trialkylammonium radical cation to form the desired iminium ion. Nucleophilic attack of 160 onto the iminium provides the observed product 161.
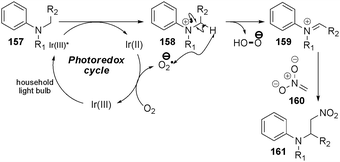 | ||
| Scheme 25 Proposed mechanism of the aza-Henry reaction. | ||
Conclusions and outlook
The dual reactivity of the photoexcited state of catalysts such as Ru(bpy)32+ and their applications in organic synthesis discussed here provide an introduction to photoredox catalysis as it has been utilized in organic synthesis. The early work of Deronzier showcased the utility of Ru(bpy)33+, derived from the oxidative quenching of photoexcited Ru(bpy)32+* by aryldiazonium salts, and its application to the Pschorr reaction and oxidation of benzylic alcohols. Recent efforts by Yoon, MacMillan and Stephenson demonstrate the utility of reductive quenching of Ru(bpy)32+* and its application to various synthetic transformations. Yoon's formal [2+2] cycloadditions of enones represent ‘visible light’ as a reagent for chemical reactions. MacMillan's photoredox organocatalysis demonstrates the control of facial selectivity in radical mediated asymmetric alkylations. Finally, Stephenson's work on photoredox catalysis provides a tin-free, environmentally benign method to access free radical intermediates for reductive dehalogenation and intramolecular radical addition to indoles and pyrroles and the mild generation of reactive iminium intermediates via C–H activation. Above all, use of visible light in organic synthesis makes photoredox catalysis an appealing and ‘green’ alternative for chemical synthesis. These recent developments in photoredox catalysis provide an early glimpse into the broad potential of these methods to mediate a variety of useful chemical transformations.References
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS.
- M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS.
- D. Gust and T. A. Moore, Science, 1989, 244, 35–41 CrossRef CAS.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 1993, 26, 198–205 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- V. Balzani, G. Bergamini, F. Marchioni and P. Ceroni, Coord. Chem. Rev., 2006, 250, 1254–1266 CrossRef CAS.
- X. Sala, I. Romero, M. Rodriguez, L. Escriche and A. Llobet, Angew. Chem., Int. Ed., 2009, 48, 2842–2852 CrossRef CAS.
- H. Cano-Yelo and A. Deronzier, J. Chem. Soc., Perkin Trans. 2, 1984, 1093–1098 RSC.
- R. M. Elofson and F. F. Gadallah, J. Org. Chem., 1971, 36, 1769–1771 CrossRef CAS.
- H. Cano-Yelo and A. Deronzier, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 3011–3019 RSC.
- H. Cano-Yelo and A. Deronzier, Tetrahedron Lett., 1984, 25, 5517–5520 CrossRef CAS.
- J. M. Zen, S. L. Liou, A. S. Kumar and M. S. Hsia, Angew. Chem., Int. Ed., 2003, 42, 577–579 CrossRef CAS.
- S. Fukuzumi, S. Mochizuki and T. Tanaka, J. Phys. Chem., 1990, 94, 722–726 CrossRef CAS.
- K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS.
- K. Okada, K. Okubo, N. Morita and M. Oda, Tetrahedron Lett., 1992, 33, 7377–7380 CrossRef CAS.
- K. Okada, K. Okubo, N. Morita and M. Oda, Chem. Lett., 1993, 2021–2024 CrossRef CAS.
- D. H. R. Barton, M. A. Csiba and J. C. Jaszberenyi, Tetrahedron Lett., 1994, 35, 2869–2872 CrossRef CAS.
- E. Hasegawa, S. Takizawa, T. Seida, A. Yamaguchi, N. Yamaguchi, N. Chiba, T. Takahashi, H. Ikeda and K. Akiyama, Tetrahedron, 2006, 62, 6581–6588 CrossRef CAS.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS.
- T. G. Baik, A. L. Luis, L. C. Wang and M. J. Krische, J. Am. Chem. Soc., 2001, 123, 6716–6717 CrossRef CAS.
- L. C. Wang, H. Y. Jang, Y. Roh, V. Lynch, A. J. Schultz, X. P. Wang and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 9448–9453 CrossRef CAS.
- J. K. Yang, D. F. Cauble, A. J. Berro, N. L. Bauld and M. J. Krische, J. Org. Chem., 2004, 69, 7979–7984 CrossRef CAS.
- Y. Roh, H. Y. Jang, V. Lynch, N. L. Bauld and M. J. Krische, Org. Lett., 2002, 4, 611–613 CrossRef CAS.
- J. K. Yang, G. A. N. Felton, N. L. Bauld and M. J. Krische, J. Am. Chem. Soc., 2004, 126, 1634–1635 CrossRef CAS.
- W. Buckel, Angew. Chem., Int. Ed., 2009, 48, 6779–6787 CrossRef CAS.
- J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604–14605 CrossRef CAS.
- A. Berkessel and H. Groger, Asymmetric Organo Catalysis, Wiley-VCH, Weinheim, 2005 Search PubMed.
- P. I. Dalko, Enantioselective Organocatalysis: Reactions and Experimental Procedures, Wiley-VCH, Weinheim, 2007 Search PubMed.
- G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79–87 CAS.
- S. G. Ouellet, A. M. Walji and D. W. C. MacMillan, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef CAS.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS.
- T. D. Beeson, A. Mastracchio, J. B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS.
- H. Y. Jang, J. B. Hong and D. W. C. MacMillan, J. Am. Chem. Soc., 2007, 129, 7004–7005 CrossRef CAS.
- H. Kim and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 398–399 CrossRef CAS.
- J. E. Wilson, A. D. Casarez and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 11332–11334 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS.
- D. D. Tanner and H. K. Singh, J. Org. Chem., 1986, 51, 5182–5186 CrossRef CAS.
- P. Renaud and S. Schubert, Synlett, 1990, 624–626 CrossRef.
- G. A. Russell and K. Wang, J. Org. Chem., 1991, 56, 3475–3479 CrossRef CAS.
- D. D. M. Wayner, J. J. Dannenberg and D. Griller, Chem. Phys. Lett., 1986, 131, 189–191 CrossRef CAS.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS.
- J. A. Ma and D. Cahard, J. Fluorine Chem., 2007, 128, 975–996 CrossRef CAS.
- N. Hoffmann, J. Photochem. Photobiol., C, 2008, 9, 43–60 CrossRef CAS.
- N. Shibata, S. Mizuta and H. Kawai, Tetrahedron: Asymmetry, 2008, 19, 2633–2644 CrossRef CAS.
- T. Billard and B. R. Langlois, Eur. J. Org. Chem., 2007, 891–897 CrossRef CAS.
- C. P. Andrieux, L. Gelis, M. Medebielle, J. Pinson and J. M. Saveant, J. Am. Chem. Soc., 1990, 112, 3509–3520 CrossRef CAS.
- S. M. Bonesi and R. Erra-Balsells, J. Chem. Soc., Perkin Trans. 2, 2000, 1583–1595 RSC.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS.
- T. Koike and M. Akita, Chem. Lett., 2009, 38, 166–167 CrossRef CAS.
- M. P. Sibi and M. Hasegawa, J. Am. Chem. Soc., 2007, 129, 4124–4125 CrossRef CAS.
- C. G. Bochet, J. Chem. Soc., Perkin Trans. 1, 2002, 125–142 RSC.
- A. P. Pelliccioli and J. Wirz, Photochem. Photobiol. Sci., 2002, 1, 441–458 RSC.
- M. S. DeClue, P. A. Monnard, J. A. Bailey, S. E. Maurer, G. E. Collis, H. J. Ziock, S. Rasmussen and J. M. Boncella, J. Am. Chem. Soc., 2009, 131, 931–933 CrossRef CAS.
- J. B. Borak and D. E. Falvey, J. Org. Chem., 2009, 74, 3894–3899 CrossRef CAS.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS.
- P. J. DeLaive, B. P. Sullivan, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1979, 101, 4007–4008 CrossRef CAS.
- J. W. Tucker, J. D. Nguyen, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Chem. Commun., 2010, 46 10.1039/c0cc00981d.
- J. W. Tucker, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Org. Lett., 2010, 12, 368–371 CrossRef CAS.
- A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |