Main-chain supramolecular block copolymers
Si Kyung
Yang
ab,
Ashootosh V.
Ambade
c and
Marcus
Weck
*b
aSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA
bDepartment of Chemistry and Molecular Design Institute, New York University, New York, NY 10003, USA. E-mail: marcus.weck@nyu.edu; Fax: +1 212 995 4895; Tel: +1 212 992 7968
cNational Chemical Laboratory, Dr Homi Bhabha Road, Pune-411008, India
First published on 15th November 2010
Abstract
Block copolymers are key building blocks for a variety of applications ranging from electronic devices to drug delivery. The material properties of block copolymers can be tuned and potentially improved by introducing noncovalent interactions in place of covalent linkages between polymeric blocks resulting in the formation of supramolecular block copolymers. Such materials combine the microphase separation behavior inherent to block copolymers with the responsiveness of supramolecular materials thereby affording dynamic and reversible materials. This tutorial review covers recent advances in main-chain supramolecular block copolymers and describes the design principles, synthetic approaches, advantages, and potential applications.
 Si Kyung Yang | Si Kyung Yang received his BS degree in 2001 from Hankuk University of Foreign Studies and his MS degree in 2003 from Korea University. He moved to the United States in 2005 to attend Georgia Tech, obtaining his PhD degree in Chemistry in the summer of 2009. While there he worked under the direction of Professor Marcus Weck studying supramolecular and polymer chemistry. He then joined the Zimmerman group at the University of Illinois at Urbana-Champaign for his postdoctoral studies where he is currently working on the synthesis of water-soluble dendrimers and their application to fluorescence imaging. |
 Ashootosh V. Ambade | Ashootosh Ambade received his PhD from IIT Bombay, India and worked with Professor Thayumanavan at UMass Amherst during his first post-doc. He then joined Professor Marcus Weck's group as post-doctoral associate and worked on main-chain supramolecular polymers. He is currently a Scientist Fellow at National Chemical Laboratory, India. |
 Marcus Weck | Marcus Weck obtained his Diploma working with Helmut Ringsdorf and in 1998 his PhD degree in Organic Chemistry from the California Institute of Technology under the direction of Robert H. Grubbs. After a two-year postdoctoral stay at Harvard University with George M. Whitesides, he joined the faculty at Georgia Tech in 2000. In 2007, he moved to NYU where he is currently a Professor in the Department of Chemistry and the Associate Director of the Molecular Design Institute. His research interests are in organic and polymer chemistry as well as materials science. The main focus of his group is on supported catalysis and the introduction of complexity through the use of orthogonal functionalization methods to synthesize polymers, organized assemblies, and nanostructures. |
1. Introduction
Since the seminal discoveries by Staudinger and Carothers, synthetic polymers, commonly known as ‘plastics’ have found a place in almost all areas of human life; from waste bags to advanced electronic and medical devices. Today, it is possible to prepare tailor-made polymers with desirable properties for a wide range of applications. Often, these conventional polymers have high melt viscosities and are difficult to process which potentially limits the use of such materials in important applications, for example, as thermally sensitive substrates. The combination of supramolecular chemistry and polymer science has emerged in the form of supramolecular polymer chemistry that has the ability to improve material characteristics of conventional polymers thereby overcoming the shortcomings described above.Supramolecular polymers can be classified into two types-main-chain and side-chain functionalized polymers. When recognition units in the side-chains of a polymer are functionalized in a noncovalent fashion, side-chain supramolecular polymers are obtained. This class of polymers has been studied extensively with random as well as block copolymer architectures and several reviews have summarized these findings.1–3
There are two ways in which main-chain supramolecular polymers can be synthesized. The first approach is analogous to a typical polycondensation (Fig. 1a) where repeat units are connected by covalent bonds, however, instead of covalent bonds, noncovalent linkages are holding monomers together (Fig. 1b). In this approach, termed ‘supramolecular polymerization’, small molecules are functionalized with two (self-) complementary recognition units affording the monomers. Self-assembly of these monomers then results in a polymer chain.4–7 If non-self-complementary units are being employed, the stoichiometry of the two monomers plays an important role in polymer formation.
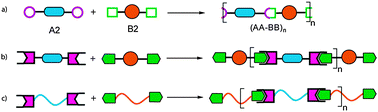 | ||
| Fig. 1 Schematic representation of polymerizations involving formation of covalent bonds and that of noncovalent bonds. (a) traditional condensation polymerization of small molecules involving the formation of covalent bonds, (b) supramolecular polymerization of small molecules involving the formation of noncovalent bonds, (c) formation of noncovalent bonds between telechelic polymers towards main-chain supramolecular multi-block copolymers. | ||
The second approach utilizes telechelic polymers as the building blocks wherein conventional polymers—homopolymers as well as random/block copolymers—are modified with recognition units at their chain-ends.8Self-assembly of the resulting monotelechelic polymers affords supramolecular block copolymers whereas ditelechelic homopolymers or block copolymers connected with noncovalent linkers present more complex supramolecular structures.
In this article, we focus solely on main-chain supramolecular polymers obtained by the second approach, that is, via the self-assembly of telechelic polymers. This system has certain advantages over small molecule monomer-based supramolecular polymers in that it combines the characteristic features of block copolymers such as microphase separation with the reversibility and tunability of supramolecular materials. Another benefit is that copolymer syntheses can be carried out in a modular fashion, i.e. a large variety of copolymers with tunable properties can be assembled easily without the need for tedious synthesis. However, introduction of the recognition moieties in small molecules is easier when compared to the preparation of noncovalently functionalized telechelic polymers, which requires careful design of synthetic strategies involving both organic and polymer chemistry.
Mainly two types of noncovalent interactions have been used to assemble main-chain supramolecular polymers: hydrogen bonding and metal coordination. Hydrogen bonding appears to be the most suitable interaction as it provides tunable reversibility and, when used as the noncovalent linkage, the resulting polymers may behave as thermoplastic elastomers. The association constants of metal coordination interactions are higher resulting in often lower flexibility, however, it can also be tuned by using different metal ions. The metal-linked polymers are more stable and can be utilized potentially as catalysts, luminescent materials and sensors.
In this tutorial review, using selected examples from the literature, we present recent advances in the field of main-chain supramolecular polymers that will be categorized into multiblock, diblock, and triblock copolymers assembled from ditelechelic, monotelechelic, and both di- and monotelechelic polymers, respectively.
2. Supramolecular multiblock copolymers
Supramolecular multiblock copolymers are of particular interest since unprecedented polymeric blends with tunable physical properties can be obtained by simply mixing polymeric building blocks. Significant variations in the copolymer composition can be obtained easily without the need for elaborate synthesis. The supramolecular polymerization of macromolecular repeating units affords a novel class of materials that not only maintains attractive features of both conventional and supramolecular polymers but also circumvents the limited formation of high molecular weight species in small molecule-based supramolecular polymers. Supramolecular multiblock copolymers can be prepared by three distinct self-assembly mechanisms: (i) assembly of single homotelechelic polymersvia either self-complementary hydrogen bonding or addition of metal ions, (ii) assembly between two different homotelechelic polymers, and (iii) assembly of single heterotelechelic polymersvia two orthogonal binding interactions.2.1 Single homotelechelic polymer-based systems
In supramolecular polymer chemistry, hydrogen bonding has been used widely for creating well-defined supramolecular block copolymersvia main-chain self-assembly of telechelic polymers containing terminal recognition units. For the self-assembly between polymeric constituents, hydrogen bonding interactions should have optimized association constants and directionality to hold individual chains together. If the association constant is too high (Ka ∼ 1012 M−1) the reversibility will be poor whereas if it is too low (Ka ∼ 102 M−1) polymer chains fall apart preventing the formation of high molecular weight material. Therefore, single or double hydrogen bonds are unsuitable for the preparation of polymeric assemblies due to insufficient stability of the resulting block copolymer. The strength and directionality of hydrogen bonding can be enhanced by combining multiple hydrogen bonds in a unit and by employing a specific arrangement of the hydrogen bond donors and acceptors. Therefore, numerous efforts have focused on the use of multiple hydrogen bonding motifs in supramolecular polymer chemistry. The triple hydrogen bond arrays derived from the nucleobase pairs of DNA, have been employed extensively as functional handles in polymeric materials. While highly attractive for the preparation of side-chain functionalized polymers,1–3 the triple hydrogen bonding pairs are often not strong enough for main-chain self-assembly of telechelic polymers where multivalency effects are absent unlike side-chain functionalization. Over the past decade, the research focus on hydrogen-bonded main-chain supramolecular polymers has therefore moved to quadruple and larger hydrogen bonding pairs that yield very stable complexes between polymeric blocks.One of the best-known examples is the self-complementary quadruple hydrogen bonding unit based on ureidopyrimidone (UPy) that dimerizes with a strong self-association constant (Kdim > 107 M−1 in CHCl3) using a self-complementary DDAA (donor–donor–acceptor–acceptor) array.4 Such binding motifs were first introduced by Meijer and coworkers, who demonstrated their use for the assembly of supramolecular multiblock copolymers. For example, two UPy functionalities were incorporated in both chain-ends of a wide range of symmetrical telechelic polymers, allowing for the formation of supramolecular multi- and homoblock copolymers with improved materials properties (Fig. 2A).4,9–12 These polymers exhibit not only the mechanical properties of conventional polymers, but also the low melt viscosity of small organic molecules, making them easily processable and highly useful for applications including adhesives, inks, coatings, and cosmetics. Recently, Aliev and Hailes reported a UPy-like, ureido-substituted cytosine-based quadruple hydrogen bonding motif that avoids the formation of tautomeric isomers by the replacement of heterocyclic NH moieties with CH or NR, but strongly dimerizes with a comparable self-association constant.13 The cytosine derivative-functionalized homotelechelic poly(ethylene glycol) (PEG) has been synthesized and used to form supramolecular multi- and homoblock copolymers with a high degree of polymerization (DP) at higher concentrations (Fig. 2B).
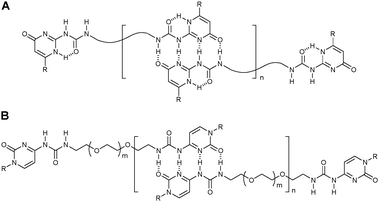 | ||
| Fig. 2 Supramolecular polymersvia self-complementary quadruple hydrogen bonding. | ||
In addition, metal coordination has been used for the self-assembly polymerization of single homotelechelic polymers by many research groups.14–17 The Sijbesma group synthesized a polymeric diphosphane ligand and subsequently obtained high molecular weight coordination polymers by the addition of palladium(II) ions (Fig. 3A).15 The reversibility of the coordinative interactions was demonstrated by the sonochemical degradation studies showing the continuous reformation of the molecular weight distributions of the original polymer chains. The Lehn group reported a series of neutral dynamic metallo-supramolecular polymers prepared by a self-assembly polymerization that involves three steps: (i) tridentate ligand formation by subunit condensation, (ii) chain extension of polymeric ligands by multiple metal coordination, and (iii) neutral coordination center formation by deprotonation (Fig. 3B).16 They also demonstrated that these dynamic coordination polymers further undergo ligand exchange and recombination both in solution and neat, yielding a variety of polymeric blends with new mechanical and optical properties. Recently, the effect of metal–ligand complexes present in metallo-supramolecular polymers containing conjugated polymeric repeating units on their optical properties has been investigated by the Rowan and Weder groups.17 The 2,6-bis(1′-methylbenzimidazolyl)pyridine (Mebip) ligands were attached to both chain-ends of poly(2,5-dialkoxy-p-phenylene ethynylene) (PPE) macromonomer by nonconjugated hexamethylene spacers so as to minimize electronic interactions between the PPE and metal-Mebip ligand complexes (Fig. 3C). Their study revealed that the optoelectronic properties of the self-assembled polymers, in which the metal–ligand complexes are electronically decoupled from the conjugated PPE, are similar to those of the parent PPE, demonstrating the potential application of such self-assembly polymerization strategies in the fabrication of electronic devices.
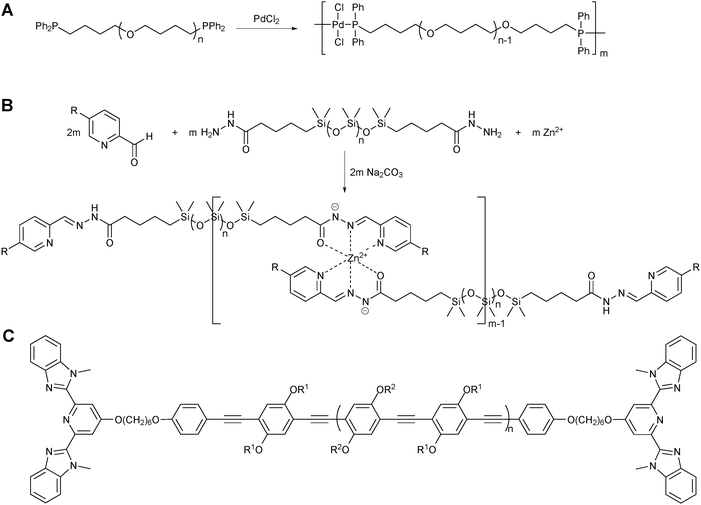 | ||
| Fig. 3 Examples of supramolecular multiblock copolymers assembled from single homotelechelic polymers. | ||
2.2 Complementary homotelechelic polymer-based systems
Another strategy towards supramolecular multiblock copolymers is the self-assembly of two homotelechelic polymers bearing complementary recognition units, in which two chemically different, sometimes incompatible, polymeric blocks can be incorporated, allowing for the formation of alternating block copolymers.Binder and coworkers reported the synthesis of telechelic poly(isobutylene)s (PIBs) functionalized with thymine or diaminopyridine-substituted isophthalamide (the Hamilton receptor) and poly(etherketone)s (PEKs) functionalized with triazine or barbituric acid at both polymer chain-ends (Fig. 4A).18–22 By combining the two strongly phase-separating polymers (PIBs and PEKs) via complementary hydrogen bonding, they obtained multiblock copolymers and studied their thermal behavior. The weaker thymine-triazine hydrogen bonding could stabilize the microphase-separated material up to temperatures corresponding to the Tg of the PEK component, while the stronger interactions between the Hamilton receptor and barbituric acid led to the stabilization up to 80 °C above the Tg of the PEK. This demonstrates that connecting incompatible polymers by noncovalent linkers can be useful for creating materials with a variety of tunable properties. Zimmerman and co-workers have recently developed a complementary hydrogen bonding motif, ureidoguanosine (UG), which forms a highly stable complex with 2,7-diamino-1,8-naphthyridine (DAN/Napy) (Ka ∼ 107 M−1) via quadruple hydrogen bonding arrays.23 They prepared hydrogen-bonded alternating multiblock copolymers (poly(styrene) (PS) and PEG) in solution based on the UG-DAN interactions in which the DP could be tuned by the concentration and ratio of the blocks in the mixture (Fig. 4B). Furthermore, the thermo-reversible property of these multiblock copolymers was demonstrated.
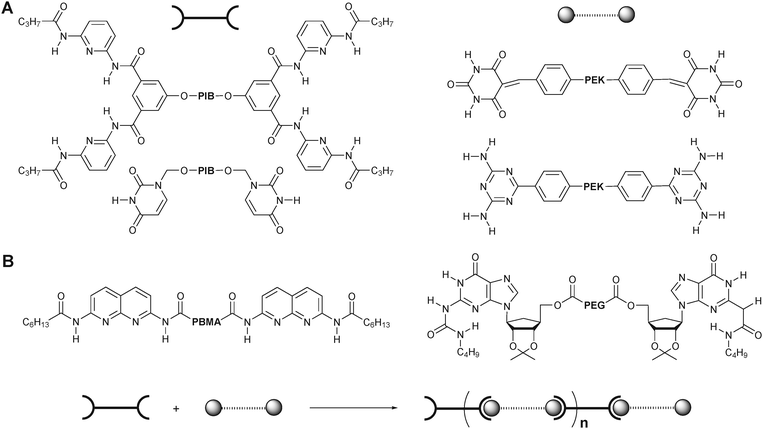 | ||
| Fig. 4 Self-assembly of complementary homotelechelic polymers into supramolecular multiblock copolymers. PIB = poly(isobutylene); PEK = poly(etherketone); PBMA = poly(butyl methacrylate); PEG = poly(ethylene glycol). | ||
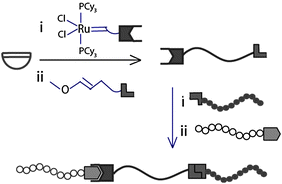
Most synthetic strategies towards multiblock copolymers, including the above examples, rely on post-polymerization functionalization steps to introduce the molecular recognition unit, resulting in often nonquantitative conversions and requiring reaction conditions that may be incompatible with other functional groups along the polymer. The successful synthesis of telechelic polymers with complementary binding motifs that circumvents the need for post-polymerization functionalization is a desirable prerequisite for the easy and high-yielding preparation of functionalized and well-defined supramolecular block copolymers. The ring-opening metathesis polymerization (ROMP) of cyclic olefins in the presence of bifunctional chain-transfer agents (CTAs) is an efficient strategy for the incorporation of supramolecular functionalities onto both chain-ends of a polymer. The Weck group employed this methodology to introduce hydrogen bonding and metal coordination moieties at the chain-ends of cyclooctene-based homopolymers allowing for the complete incorporation of the recognition units in situ (Fig. 5).24 The resulting telechelic polymers were self-assembled into supramolecular multiblock copolymersviahydrogen bonding or metal coordination between corresponding terminal recognition units. Meijer and co-workers also synthesized bifunctional telechelic polymers through ROMP in the presence of CTAs functionalized with complementary quadruple hydrogen bonding motifs, UPy and Napy.25 The formation of multiblock copolymers was demonstrated by self-assembling the resulting telechelic polymers in both solution and the bulk, leading to stable, microphase-separated copolymer morphologies.
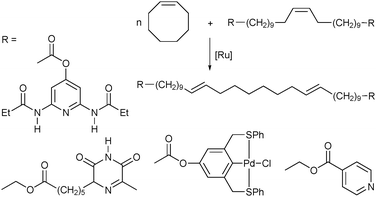 | ||
| Fig. 5 Synthesis of telechelic polymers by ring-opening metathesis polymerization in the presence of functionalized chain-transfer agents. | ||
Although highly advantageous over traditional post-polymerization functionalization due to complete incorporation of almost any terminal recognition motif in situ, the ROMP-CTA strategy limits control over the obtained telechelic polymer properties because it relies on the nonliving polymerization of functionalized cyclooctenes. The Weck group recently reported a methodology for the synthesis of symmetrically end-functionalized polymers with full control over the properties in a single step by means of ROMP using a bimetallic ruthenium initiator and functionalized chain-terminators (CTs) (Fig. 6).26 The combination of a living/controlled polymerization of norbornene monomers functionalized with hydrophobic or hydrophilic groups and a Pd(II) SCS pincer ligand-based metal coordination allowed for the formation of well-defined supramolecular alternating multiblock copolymers. Furthermore, the bulk properties of the multiblock copolymers could be tuned by the controlled addition of the activating agent AgBF4.
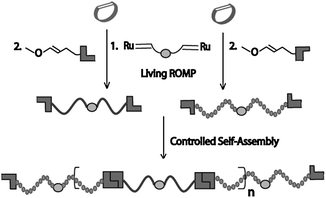 | ||
| Fig. 6 Schematic representation of the synthetic strategy towards well-defined supramolecular alternating block copolymers. | ||
2.3 Heterotelechelic polymer-based systems
The material properties of multiblock copolymers can be improved by introducing multiple orthogonal supramolecular interactions in the polymer main-chains. In particular, the incorporation of different types of interactions, such as hydrogen bonding and metal coordination, allows for the polymers to be selectively responsive to solvent, concentration, or temperature and competitive ligands or redox chemistry, respectively, opening a pathway towards highly switchable materials. For instance, the Schubert group synthesized a heterotelechelic polymer by installing a UPy unit and a terpyridine ligand at the two chain-ends of poly(ε-caprolactone). The UPy and terpyridine unit are capable of undergoing self-complementary hydrogen bonding and metal coordination, respectively (Fig. 7).27 Subsequently, the dimerization of UPy afforded diblock copolymers whereas addition of a metal ion resulted in high molecular weight supramolecular polymers. These materials based on two orthogonal interactions exhibit tunable properties depending on concentration, temperature, and the metal and counterions used. The reversibility of the complex formation was also demonstrated by employing competitive ligands. In order to address control over the polymeric structure, Schubert and co-workers reported a methodology for the one-step preparation of well-defined heterotelechelic supramolecular polymersvianitroxide-mediated radical polymerization (NMP) initiated by a bifunctional NMP initiator.28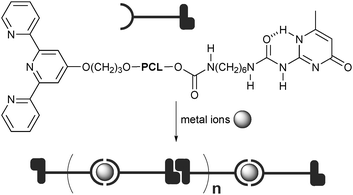 | ||
| Fig. 7 Supramolecular multiblock copolymers containing alternating terpyridine metal complexes and self-complementary hydrogen bonding. PCL = poly(ε-caprolactone). | ||
3. Supramolecular diblock copolymers
Another class of supramolecular assemblies is diblock copolymers assembled from monotelechelic polymers. For example, Long and co-workers reported the synthesis of UPy-functionalized monotelechelic polymers which can dimerize to form di- and homoblock copolymers, and the relationship between the end-group structure and physical properties.29 In this study, the well-defined monotelechelic PS, poly(isoprene) (PI), and PS-b-PI block copolymers have been prepared via living anionic polymerization followed by an end-capping reaction. Quantitative incorporation of the terminal functional groups cannot be guaranteed via the post-polymerization modification methods. This limitation can be overcome by controlled polymerization initiated by recognition unit-functionalized initiators. In this manner, Scherman and co-workers employed UPy-containing initiators for the ring-opening polymerization (ROP) to afford UPy functionalized monotelechelic poly(ε-caprolactone)s (PCLs) capable of forming dimerized structures in chloroform (Fig. 8).30 This method provides for the complete incorporation of a UPy functionality at one chain-end of the PCLs as well as avoids any undesired interactions of the UPy units with Sn(oct)2 catalysts by UPy dimerization during the polymerization, allowing for full control over polymer properties.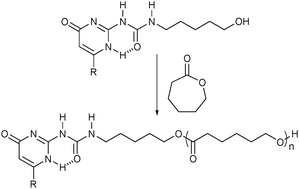 | ||
| Fig. 8 Ring-opening polymerization of ε-caprolactone with a UPy-functionalized initiator. | ||
The noncovalent connection of two chemically different monotelechelic polymers leads to the formation of supramolecular AB diblock copolymers, which have received much attention due to their various ordered micro- and nanophase morphologies. The self-assembly of two incompatible homopolymers towards AB diblock copolymers requires very strong, complementary hydrogen bonding recognition pairs. Li and co-workers reported that a tautomer of the UPy unit forms a strong and selective heterocomplex with Napy by means of ADDA-DAAD quadruple hydrogen bond arrays.31 This complementary UPy-Napy hydrogen bonding has been shown to be effective in the preparation of main-chain supramolecular polymers through the self-assembly of telechelic polymers. The Hawker and Meijer groups have synthesized UPy- or Napy-functionalized initiators for atom transfer radical polymerization (ATRP) to obtain the corresponding monotelechelic polymers.32Self-assembly of the resulting polymers affords supramolecular diblock copolymers that display a direct correlation between the terminal recognition units and physical properties (Fig. 9A). Bulk blending studies of these materials revealed that the phase behavior can be controlled by the nature of the hydrogen bonding units present in the blend. For example, complementary UPy-Napy complexes result in a significant reduction in phase separation, while self-complementary UPy-UPy pairs lead to a slight miscibility of the homopolymers. The use of UPy-Napy pairs for blending poly(benzyl methacrylate) (PbnMA) with poly(n-butyl acrylate) (PnBA) allows for the blend interfaces to contain some diblock copolymers which serve as compatibilizers and provide stable nanostructured materials.
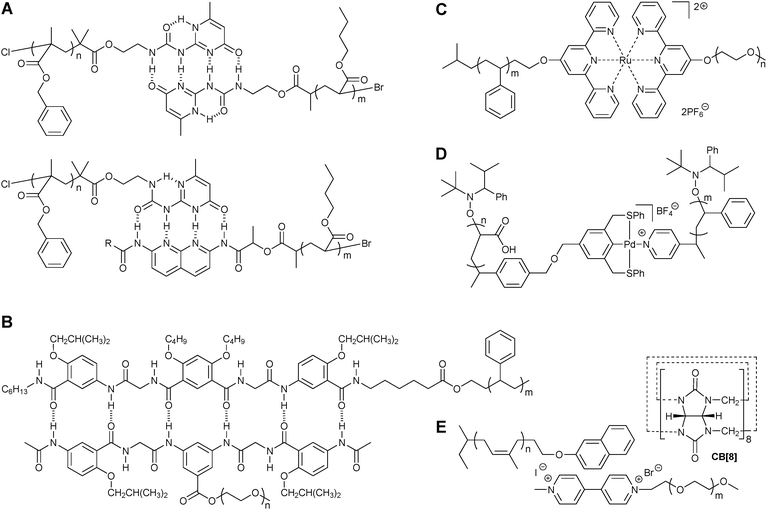 | ||
| Fig. 9 Examples of supramolecular AB diblock copolymers. | ||
A very strong oligoamide-based sextuple hydrogen-bonded heteroduplex with an association constant of Ka > 109 M−1 in chloroform has been developed by Gong and co-workers.33 This complementary binding motif was introduced in the chain-ends of PS and PEG (Fig. 9B), and the self-assembly between the resulting monotelechelic polymers led to supramolecular AB diblock copolymers, exhibiting typical PS-PEG diblock copolymer-like microphase separation that depends on the molecular weights of the blocks.
Another major class of noncovalent interactions employed in supramolecular AB diblock copolymers is metal coordination. In particular, terpyridine and pincer ligands have been used extensively because of their significantly strong, directional, and reversible interactions.34,35 The Schubert group has synthesized terpyridine-terminated polymers based on PS, PEG, or poly(ethylene-co-butylene) (PEB) that can form stable complexes by means of a variety of transition metal ions.36 In particular, the addition of ruthenium to the two different monotelechelic polymers affords asymmetric complexes, allowing for the preparation of a library of metallo-supramolecular AB diblock copolymers (Fig. 9C). This synthetic strategy has been extended to the formation of amphiphilic supramolecular block copolymer-based micelles where the ruthenium-terpyridine complexes could be disassembled upon the addition of a strong competitive ligand.37 In addition to ruthenium, cobalt and nickel have been used recently to synthesize terpyridine-based AB diblock copolymers.38 Moughton and O'Reilly also afforded asymmetric amphiphilic metallo-supramolecular diblock copolymers by self-assembling a Pd(II) SCS pincer ligand-terminated poly(acrylic acid) block with a pyridine end-functionalized PS block (Fig. 9D).39 The resulting block copolymers were further self-assembled to form well-defined, monodisperse micelles which were then stabilized by selectively cross-linking the hydrophilic shell. Finally, a hollow polymeric nanocage with interior metal functionality was prepared by removing the hydrophobic core domain. It was suggested that this ‘nanocage’ may be potentially useful in pseudo-homogeneous supported Heck coupling reactions.
Host–guest complexes have been shown to be efficient linking units for polymeric chains.40,41 For example, cucurbit[8]uril (CB[8]) as a host molecule is capable of selectively binding two guest molecules (e.g. viologen derivatives and hydroxynaphthalenes) with a high association constant of Ka > 1011 M−1 in an aqueous environment.42 By combining this dynamic linker CB[8] with two guest polymers based on PEG and PI, Scherman and co-workers have synthesized an amphiphilic diblock copolymer that further self-assembles into a vesicle or a micelle (Fig. 9E).40 The hydrophilic CB[8]-PEG part can be used to enhance the solubility of other hydrophobic polymeric chains in water making this system potentially useful for biological applications.
4. Supramolecular triblock copolymers
Supramolecular AB diblock copolymers can be modified by the replacement of one of the monotelechelic blocks with a homotelechelic block to afford ABA triblock copolymers allowing for a high degree of complexity. Weck and co-workers reported side-chain functionalized ABA triblock copolymers, in which the polymeric main-chains are held by hydrogen bonding between cyanuric acid (CA) and the Hamilton receptor while the side-chains are functionalized using pincer-pyridine metal coordination.43 It was shown that the main-chain self-assembly is independent of side-chain functionalization and the final self-assembled structure can be attained irrespective of the sequence followed. The Jo group also reported polymer blends containing ABA triblock copolymers prepared by self-assembling a homotelechelic PS block with two monotelechelic PI blocks using ionic interactions between sulfonic acid and amine.44 The Schubert group has extended their terpyridine-based strategies from AB to ABA block copolymers by the employment of a small, bifunctional terpyridine molecule-based supramolecular polymer as a central B block.45ABC triblock copolymers are a more challenging type of supramolecular block copolymers. Here two strong, different, and orthogonal complementary noncovalent interactions are required to linearly connect three different homopolymer blocks. The incorporation of another functional block to the AB diblock copolymers described in section 3 not only allows for the generation of novel materials with a greater variety of tunable morphologies but also augments the usefulness in the fabrication of highly functional nanodevices.
The key to the construction of supramolecular ABC triblock copolymers is the central heterotelechelic polymer containing two orthogonal recognition units. Weck and co-workers reported a novel methodology for the synthesis of the unsymmetrically end-functionalized polymerviaROMP using a functionalized ruthenium initiator and a functionalized CT in a single step without any post-polymerization modifications. The stepwise self-assembly of this heterotelechelic polymer with complementary homopolymers led to the formation of a supramolecular ABC triblock copolymer (Fig. 10).46 The supramolecular interactions employed were the Hamilton receptor-CA hydrogen bonding at one end and palladated pincer-pyridine metal coordination at the other end. However, due to incomplete orthogonality of the two interactions used, the triblock copolymer could be self-assembled only via a particular sequence. This limitation has been successfully overcome by the Weck group by employing two distinct and orthogonal hydrogen bonding receptor pairs (Fig. 11).47 In this study, the metal coordination present in the previous system was replaced with a UG-DAN hydrogen bonding interaction that was shown to be fully orthogonal to the Hamilton receptor-CA interaction. They demonstrated the first one-pot synthesis of a supramolecular ABC triblock copolymer by main-chain self-assembly of a heterotelechelic polymer containing the Hamilton receptor and DAN at the chain-ends with the other two CA- and UG- terminated monotelechelic homopolymers. The triblock copolymer could also be assembled in two steps starting from either end of the B block demonstrating the versatility of this approach.
 | ||
| Fig. 10 Schematic representation of the synthesis of a heterotelechelic polymer from functionalized initiators and chain-terminators and its self-assembly into supramolecular ABC triblock copolymers. | ||
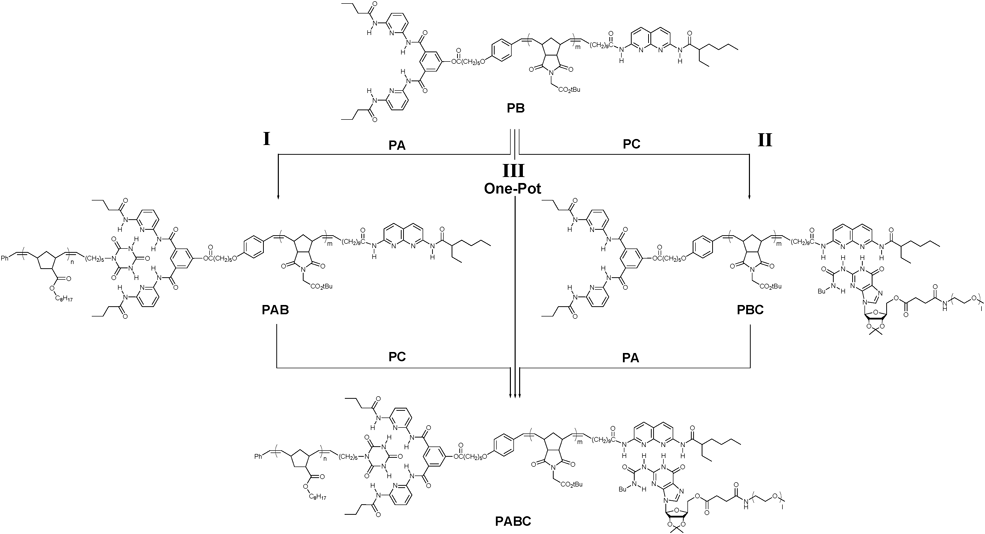 | ||
| Fig. 11 ABC triblock copolymer formation through stepwise and one-pot self-assembly. | ||
5. Conclusions
In the last decade, the concept of noncovalently connecting repeat units of a polymer chain has been extended successfully from small molecule-based building blocks to main-chain supramolecular block copolymers constructed from telechelic polymers. With the emergence of functionalized initiators and chain terminators for different types of controlled polymerizations, the need for post-polymerization functionalization can be circumvented and synthesis of well-defined telechelic polymers has become possible.Several commercially important backbones such as PS, PEO, PEK, PMMA, PCL, and poly(norbornene)s have been used in the preparation of supramolecular block copolymers and network like structures. A modular approach for supramolecular synthesis has afforded a library of copolymer architectures giving rise to materials with tunable mechanical and thermal properties while involving little synthetic effort. Supramolecular block copolymers with immiscible blocks have exhibited different morphologies both in solution and the solid state. The next goal could be to create higher order nanostructures by hierarchical organization of these supramolecular block copolymers.
Although the initial purpose of introducing multiple hydrogen bonding groups in polymers was to obtain easily processable materials several promising applications such as self-healing materials and biomaterials have emerged that exploit temperature-dependent responsiveness. This property has also been exploited in the preparation of ‘gels’ and can be used in designing materials for controlled release applications. The hydrogen bonded system has also been proposed as a candidate for improving recyclability of commercial plastics. When metal coordination has been employed as the noncovalent interaction, inherent properties of the metal center such as catalysis, redox chemistry, sensing, and luminescence can be utilized for exploring responsive-functional organic/inorganic hybrid materials. A combination of hydrogen bonding and metal-coordination for preparation of supramolecular polymers has been studied in a few examples but holds promise for materials with a wide range of tunability.
Now that the synthetic strategies for main-chain supramolecular block copolymers are in place the focus started to shift to establish the structure-property relationships of this new class of materials and to incorporate extra functionalities aimed at designing practical and useful advanced materials. This is an exciting time for research in this field of polymer chemistry and the future of the supramolecular polymeric materials looks promising.
Acknowledgements
We gratefully acknowledge the support and enthusiasm of former and current group members and colleagues. Financial support has been provided by the National Science Foundation (ChE-0239385 and ChE-0911460). This work was supported partially by the MRSEC Program of the National Science Foundation under Award Number DMR-0820341.References
- J. M. Pollino and M. Weck, Chem. Soc. Rev., 2005, 34, 193 RSC.
- C. R. South, C. Burd and M. Weck, Acc. Chem. Res., 2007, 40, 63 CrossRef CAS.
- M. Weck, Polym. Int., 2007, 56, 453 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS.
- T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171 CrossRef CAS.
- L. S. Shimizu, Polym. Int., 2007, 56, 444 CrossRef CAS.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS.
- C.-A. Fustin, P. Guillet, U. S. Schubert and J.-F. Gohy, Adv. Mater., 2007, 19, 1665 CrossRef CAS.
- J. H. K. K. Hirschberg, F. H. Beijer, H. A. van Aert, P. C. M. M. Magusin, R. P. Sijbesma and E. W. Meijer, Macromolecules, 1999, 32, 2696 CrossRef.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874 CrossRef CAS.
- H. M. Keizer, R. van Kessel, R. P. Sijbesma and E. W. Meijer, Polymer, 2003, 44, 5505 CrossRef CAS.
- D. J. M. van Beek, M. A. J. Gillissen, B. A. C. van As, A. R. A. Palmans and R. P. Sijbesma, Macromolecules, 2007, 40, 6340 CrossRef CAS.
- V. G. H. Lafitte, A. E. Aliev, P. N. Horton, M. B. Hursthouse, K. Bala, P. Golding and H. C. Hailes, J. Am. Chem. Soc., 2006, 128, 6544 CrossRef CAS.
- S. Schmatloch, A. M. J. van den Berg, A. S. Alexeev, H. Hofmeier and U. S. Schubert, Macromolecules, 2003, 36, 9943 CrossRef CAS.
- J. M. J. Paulusse and R. P. Sijbesma, Angew. Chem., Int. Ed., 2004, 43, 4460 CrossRef CAS.
- C.-F. Chow, S. Fujii and J.-M. Lehn, Angew. Chem., Int. Ed., 2007, 46, 5007 CrossRef CAS.
- M. Burnworth, J. D. Mendez, M. Schroeter, S. J. Rowan and C. Weder, Macromolecules, 2008, 41, 2157 CrossRef CAS.
- W. H. Binder, S. Bernstorff, C. Kluger, L. Petraru and M. J. Kunz, Adv. Mater., 2005, 17, 2824 CrossRef CAS.
- W. H. Binder, M. J. Kunz and E. Ingolic, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 162 CrossRef CAS.
- M. J. Kunz, G. Hayn, R. Saf and W. H. Binder, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 661 CrossRef CAS.
- W. H. Binder, M. J. Kunz, C. Kluger, G. Hayn and R. Saf, Macromolecules, 2004, 37, 1749 CrossRef CAS.
- W. H. Binder, L. Petraru, T. Roth, P. W. Groh, V. Pálfi, S. Keki and B. Ivan, Adv. Funct. Mater., 2007, 17, 1317 CrossRef CAS.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 13986 CrossRef CAS.
- M. N. Higley, J. M. Pollino, E. Hollembeak and M. Weck, Chem.–Eur. J., 2005, 11, 2946 CrossRef CAS.
- O. A. Scherman, G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11850 CrossRef CAS.
- S. K. Yang, A. V. Ambade and M. Weck, Chem.–Eur. J., 2009, 15, 6605 CrossRef CAS.
- H. Hofmeier, R. Hoogenboom, M. E. L. Wouters and U. S. Schubert, J. Am. Chem. Soc., 2005, 127, 2913 CrossRef CAS.
- U. Mansfeld, M. D. Hager, R. Hoogenboom, C. Ott, A. Winter and U. S. Schubert, Chem. Commun., 2009, 3386 RSC.
- K. Yamauchi, J. R. Lizotte, D. M. Hercules, M. J. Vergne and T. E. Long, J. Am. Chem. Soc., 2002, 124, 8599 CrossRef CAS.
- A. D. Celiz and O. A. Scherman, Macromolecules, 2008, 41, 4115 CrossRef CAS.
- X.-Z. Wang, X.-Q. Li, X.-B. Shao, X. Zhao, P. Deng, X.-K. Jiang, Z.-T. Li and Y.-Q. Chen, Chem.–Eur. J., 2003, 9, 2904 CrossRef CAS.
- K. E. Feldman, M. J. Kade, T. F. A. de Greef, E. W. Meijer, E. J. Kramer and C. J. Hawker, Macromolecules, 2008, 41, 4694 CrossRef CAS.
- X. Yang, F. Hua, K. Yamato, E. Ruckenstein, B. Gong, W. Kim and C. Y. Ryu, Angew. Chem., Int. Ed., 2004, 43, 6471 CrossRef CAS.
- J.-F. Gohy, Coord. Chem. Rev., 2009, 253, 2214 CrossRef CAS.
- A. O. Moughton and R. K. O'Reilly, Macromol. Rapid Commun., 2010, 31, 37 CrossRef CAS.
- B. G. G. Lohmeijer and U. S. Schubert, Angew. Chem., Int. Ed., 2002, 41, 3825 CrossRef CAS.
- J.-F. Gohy, B. G. G. Lohmeijer, S. K. Varshney, B. Décamps, E. Leroy, S. Boileau and U. S. Schubert, Macromolecules, 2002, 35, 9748 CrossRef CAS.
- C. Mugemana, P. Guillet, S. Hoeppener, U. S. Schubert, C.-A. Fustin and J.-F. Gohy, Chem. Commun., 2010, 46, 1296 RSC.
- A. O. Moughton and R. K. O'Reilly, J. Am. Chem. Soc., 2008, 130, 8714 CrossRef CAS.
- U. Rauwald and O. A. Scherman, Angew. Chem., Int. Ed., 2008, 47, 3950 CrossRef CAS.
- Q. Yan, J. Yuan, Z. Cai, Y. Xin, Y. Kang and Y. Yin, J. Am. Chem. Soc., 2010, 132, 9268 CrossRef CAS.
- H.-J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526 CrossRef CAS.
- A. V. Ambade, C. Burd, M. N. Higley, K. P. Nair and M. Weck, Chem.–Eur. J., 2009, 15, 11904 CrossRef CAS.
- J. Huh, H. J. Park, K. H. Kim, K. H. Kim, C. Park and W. H. Jo, Adv. Mater., 2006, 18, 624 CrossRef CAS.
- M. A. R. Meier, D. Wouters, C. Ott, P. Guillet, C.-A. Fustin, J.-F. Gohy and U. S. Schubert, Macromolecules, 2006, 39, 1569 CrossRef CAS.
- A. V. Ambade, S. K. Yang and M. Weck, Angew. Chem., Int. Ed., 2009, 48, 2894 CrossRef CAS.
- S. K. Yang, A. V. Ambade and M. Weck, J. Am. Chem. Soc., 2010, 132, 1637 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |