Molecular clips and tweezers hosting neutral guests†
Marie
Hardouin–Lerouge
,
Piétrick
Hudhomme
* and
Marc
Sallé
*
Laboratoire MOLTECH-Anjou, Université d'Angers, CNRS UMR 6200, 2 Boulevard Lavoisier, 49045 Angers, France. E-mail: pietrick.hudhomme@univ-angers.fr; marc.salle@univ-angers.fr
First published on 1st November 2010
Abstract
Intense current interest in supramolecular chemistry is devoted to the construction of molecular assemblies displaying controlled molecular motion associated to recognition. On this ground, molecular clips and tweezers have focused an increasing attention. This tutorial review points out the recent advances in the construction of always more sophisticated molecular clips and tweezers, illustrating their remarkably broad structural variety and focusing on their binding ability towards neutral guests. A particular attention is brought to recent findings in dynamic molecular tweezers whose recognition ability can be regulated by external stimuli. Porphyrin-based systems will not be covered here as this very active field has been recently reviewed.
 Piétrick Hudhomme, Marie Hardouin-Lerouge and Marc Sallé | Marie Hardouin-Lerouge received her Master Degree from the University of Angers in Molecular Engineering and Nanosciences in 2008. She is presently a PhD student in organic chemistry under the supervision of Prof. Piétrick Hudhomme. Her research interest is the synthesis and properties of electroactive molecular clips for supramolecular recognition. |
Piétrick Hudhomme received his PhD from the University of Nantes in 1990 under the supervision of Prof. Guy Duguay for research on multi-step asymmetric synthesis of cephalosporin antibiotics. After post-doctoral research with Prof. Douglas W. Young (University of Sussex), he became Maître de Conférences at the University of Nantes (1991). His research activities moved to the synthesis of organic materials and he was appointed Professor at the University of Angers (1998) in the laboratory of Prof. Alain Gorgues. His research interests encompass the organic synthesis of functional electroactive molecules for applications in molecular electronics, solar energy conversion and supramolecular chemistry. |
Prof. Marc Sallé studied chemistry at the Ecole Nationale Supérieure de Chimie de Rennes (ENSCR) and at the University of Paris VI. He received his PhD in Organic Chemistry (University of Angers, 1991) with Prof. Alain Gorgues and received the Dina Surdin prize in 1992. After a postdoc with Prof. M. R. Bryce (University of Durham, UK), he became Maître de Conférences at the University of Angers, and became Professor in 1998. He was elected as a junior member of the Institut Universitaire de France (IUF) in 2003. His research interests concern organic chemistry of molecular materials and supramolecular chemistry. |
1. Introduction
Supramolecular chemistry continues its expansion to include understanding and mimicking biological processes, molecular recognition, molecular self-assembly, catalysis, materials and medicinal chemistries, but also dynamic covalent chemistry.1 In particular, efforts were recently devoted to the synthesis of molecular machines that function through host–guest recognition. Such systems are designed to achieve a specific function and considerable efforts are currently focused on the construction of molecular switches and devices in which external stimuli are used to induce molecular motion.2,3 Therefore, the design of molecular systems capable of controlled molecular-level motion has become an area of growing interest, and a key issue in this field concerns the so-called molecular tweezers, clips and clefts.Such systems are characterized by open cavities capable of binding guests using various supramolecular interactions including hydrogen bonding, metal coordination, hydrophobic forces, van der Waals forces, electrostatic effects and/or π–π interactions. Consequently, such molecular receptors are designed with two ‘arms’ or ‘tips’ chosen for their propensity to sandwich a given guest, and which are connected through a central platform.
The term ‘molecular tweezer’ was introduced by Whitlock,4 defining a molecular receptor characterized by two flat, generally aromatic, identical pincers separated by some more or less rigid tether. Subsequently, some criteria were established to define a molecular tweezer.5,6 These are: (i) the presence of a spacer that prevents self-association, (ii) a spacer that establishes a distance of ca. 7 Å between the pincers (plane to plane or centroid to centroid), suitable for the inclusion of a single aromatic guest molecule, and (iii) a spacer that holds the pincers rigidly in a syn conformation. In their original publication, Chen and Whitlock described the flexible molecular tweezer 1 involving two caffeine chromophores separated by a diyne spacer (Fig. 1).
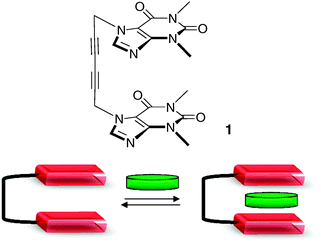 | ||
| Fig. 1 First molecular tweezer described by Chen and Whitlock and its principle of supramolecular recognition. | ||
Compound 1 can bind aromatic guests by sandwiching them between two more or less parallel aromatic side-walls. Since then, a large number of synthetic host systems based on this principle have been designed. Their structure potentially varies according to two parameters: (i) the chemical nature of the pincer, which is the active unit in terms of molecular recognition, and (ii) the nature of the connecting platform between both pincers, whose attribute is to fix the relative location of the latter.
F. G. Klärner et al. developed di- to tetramethylene-bridged compounds 2–4 and defined these systems as molecular clips and tweezers, respectively (Fig. 2).7 These architectures are characterized by “bond angle distortions which require little energy and, therefore, should induce a certain flexibility, allowing the receptor ‘arms’to be expanded and compressed during the substrate complexation”. Nevertheless, it is worth noting that although the concepts of molecular tweezers and molecular clips are sometimes considered separately, there seems to be relatively little to distinguish the two groups. Therefore, we have considered in this review, the so-called molecular clefts, clips, and tweezers at the same level.
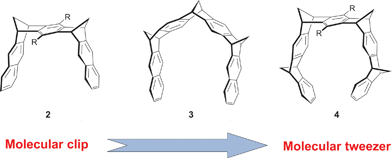 | ||
| Fig. 2 Molecular clip to molecular tweezer as defined by Klärner with di-, tri- and tetramethylene-bridged polyaromatic systems. | ||
Several reviews have already been published in the past on this subject, but they all focus on a specific aspect: Zimmerman in 19918 and 19935 on rigid molecular tweezers, Nolte in 19959 and 199910 on glycoluril-based molecular clips, Klärner in 2003 on polyarene molecular clips and tweezers,7 Harmata in 2004 on chiral molecular tweezers,6 and very recently, in 2010, Martín on molecular tweezers specifically designed for fullerene binding.11 Therefore, no recent general review is available. At this stage, one should mention that given a remarkable combination of photo- and electrochemical properties, the aromatic porphyrin unit has focused a tremendous interest for the construction of tweezer-like architectures, through the preparation of covalently linked cofacial bisporphyrins. This very active field has been recently reviewed12 and despite their interest, porphyrin-based systems will not be covered in the present tutorial review.
In this tutorial review, recent work on molecular tweezers, clips or clefts, organized around a rigidified platform and capable to form host–guest complexes with neutral molecules such as aromatic or fullerene guests by using π–π stacked aromatic interactions will be presented. Are also considered in this review, dynamic molecular tweezers whose recognition ability can be regulated through large-scale open–close movement induced by different kind of stimuli. This class of synthetic receptors is emerging in the literature and recent examples involved in the binding of neutral guests will be presented.
2. Rigid molecular clips
2.1 The glycoluril scaffold
The popularity of glycoluril as a building block in supramolecular chemistry can be traced to the pioneering work on molecular clips of Nolte et al. who discovered that the reaction of diphenylglycoluril 5 with formaldehyde gave a clip-shaped molecule 6 with a rigid U-shaped cavity (Fig. 3).9,10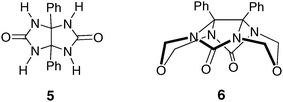 | ||
| Fig. 3 Structure of the glycoluril scaffold 6. | ||
The introduction of two phenyl rings afforded the clip-shaped molecule 7 (Fig. 4); a great variety of phenylglycoluril-based host derivatives possessing various functionalities has been synthesized from this platform. Consequently, diphenylglycoluril molecular clips were found to be attractive because of: (i) a straightforward synthetic access to a wide range of derivatives; (ii) a suitable preorganization of the aromatic binding sites for accommodating an aromatic moiety in the cleft.
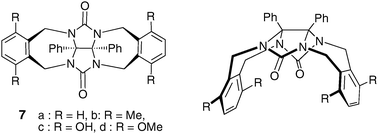 | ||
| Fig. 4 General structure of glycoluril-based molecular clips. | ||
The X-ray structure of the tetramethoxy derivative 7d shows that the o-xylylene walls define a cavity with an angle of 39.5° between both aromatic walls and a distance of 6.67 Å between the centers of the benzene rings. The two fused five-membered rings of glycoluril form an electron rich base, with two hydrogen bond acceptor sites.13 Complexation experiments showed that molecular clips 7 are good receptors for phenols, dihydroxybenzene or dihydroxynaphthalene derivatives. The binding process involves cooperative hydrogen bonding and π–π stacking interactions and high binding constants are found (Ka = 7100 M−1 in CDCl3 for clip 7d and 2,7-dihydroxynaphtalene). The binding ability appears very low with molecular clip 8 presenting 1,4-dimethoxynaphthalene tips. The corresponding X-ray crystal structure shows that all methoxy groups point into the cavity, preventing thus any complexation process (Fig. 5). On the other hand, when naphthalene walls are connected in the 1,8 positions, glycoluril 9 exists in solution as a mixture of three conformers which slowly interconvert on the NMR time scale. Only one conformer contains a cavity which can bind an aromatic guest (Ka = 185 M−1 in CDCl3 for 1,4-dicyanobenzene).14
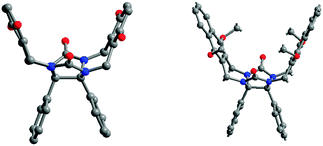 | ||
| Fig. 5 X-Ray structures of diphenylglycoluril molecular clips 7d (left) and 8 (right). | ||
Different types of sidewalls have been introduced in order to establish some structure—properties relationships (Fig. 6). For example, the monophenanthroline clip 10 and dipyridine clip 11 are able to complex olivetol (5-pentylresorcinol) with moderate binding constants (Ka = 210 M−1 and 125 M−1, respectively, CDCl3) whereas the diphenanthroline clip 12 does not present this host property (Ka < 1 M−1).15 For this series, steric effects are considered to be unfavourable for high binding constants. This is also illustrated by the fact that simple molecular clip 7d shows a good affinity for olivetol (Ka = 1500 M−1). On the other hand, the recently described glycoluril clip 13 containing arene-terminated sidewalls was recently shown to provide a deep cavity suitable for efficiently binding resorcinol.16
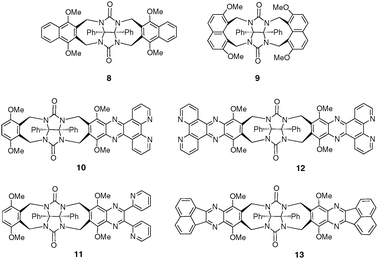 | ||
| Fig. 6 Molecular clips based on the glycoluril scaffold. | ||
Recently, molecular clip 14 based on glycoluril scaffold in which both phenyl groups have been replaced by two ester functionalities was synthesized (Fig. 7).17 Two extended ethynylated o-xylylene sidewalls provide the formation of a deep cavity. The high sensitivity and low limit detection of fluorescence spectroscopy was used to demonstrate the highly selective recognition properties of this fluorescent molecular clip towards 4-nitrophenol (Ka = 1.09 × 104 M−1 in THF–MeOH 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Beside a cavity effect, this binding is driven by a combination of hydrogen bonding and π–π stacking interactions between the electron-poor aromatic guest and the electron-rich sidewalls of the host, which induces a fluorescence quenching.
:1). Beside a cavity effect, this binding is driven by a combination of hydrogen bonding and π–π stacking interactions between the electron-poor aromatic guest and the electron-rich sidewalls of the host, which induces a fluorescence quenching.
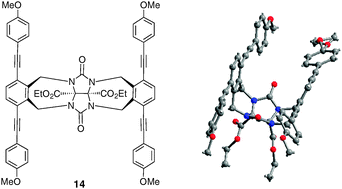 | ||
| Fig. 7 Fluorescent glycoluril-based molecular clip 14 and X-ray crystal structure. | ||
The ability of methylene-bridged glycoluril dimers 15 to undergo strong dimerization (Ka > 106 M−1 in CDCl3) was recently investigated to prepare robust, functionalizable, self-sorting supramolecular systems (Fig. 8).18 Such systems tend to self-organize in dimers resulting from the combination of hydrogen-bonds and π–π interactions.
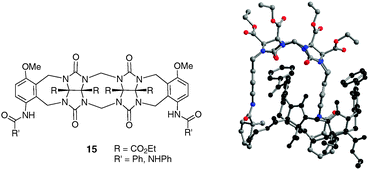 | ||
| Fig. 8 Dimeric glycoluril molecular clip 15 and its self-association observed by X-ray study. | ||
One original feature of glycoluril-based molecular clips was recently described by Chiu and Coll. with the synthesis of compound 16, in which strong electron donor tetrathiafulvalene (TTF) units are introduced as sidewalls of the clip (Fig. 9).19,20TTF units are located face-to-face at a suitable interplanar distance to allow the inclusion of guest species such as pyridinium motifs. Although this paraquat analogue is not a neutral molecule, this recent finding opens new development for glycolurile based molecular clips. Because of charge–transfer interactions and C–H⋯O bonding, clip 16 was found to form complexes with paraquat (Ka = 5600 M−1 in CD3CN) or bipyridinium-based macrocycle (Ka = 1600 M−1). The threaded and unthreaded states can be distinguished by their absorption spectra that correspond to a color change which can be readily seen by the naked eye. Interestingly, the exchange between threaded and unthreaded states can be controlled either by recognition of K+ or NH4+, by heating or by oxidation of the TTF moieties, which can lead to the dissociation of the macrocycle/molecular-clip complex. The latter complex operates as a “not-or” (NOR)-functioning molecular logic gate.
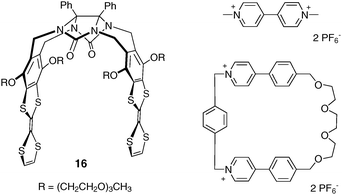 | ||
| Fig. 9 Glycoluril-based molecular clip 16 with TTF sidewalls, and complementary bipyridinium guests. | ||
Molecular clip 16 can also operate in the presence of a two-station [2]rotaxane, as a pH-controllable molecular switch which can be monitored by naked eye.21 When solutions of colourless [2]rotaxane and yellow molecular clip 16 are mixed, the solution turns green because of a charge transfer band (λ = 709 nm) resulting from the formation of [2]rotaxane/clip complex (Ka = 4100 M−1 in CD3CN/CDCl3 5:1) (Fig. 10). This complex is dissociated by addition of Et3N and can be reformed upon introduction of CF3CO2H.
![Formation of the [2]rotaxane/clip 16 complex and corresponding pH controllable switching (reproduced with permission from ref. 21. Copyright Royal Society of Chemistry 2010).](/image/article/2011/CS/b915145c/b915145c-f10.gif) | ||
| Fig. 10 Formation of the [2]rotaxane/clip 16 complex and corresponding pH controllable switching (reproduced with permission from ref. 21. Copyright Royal Society of Chemistry 2010). | ||
2.2 Rigidified polyarene platforms
Zimmerman and co-workers designed molecular tweezers presenting a high degree of pre-organization with a spacer that maintains a syn conformation of the binding arms.5 Representative examples are shown with compounds 17–19. Such systems exhibit a certain degree of conformational mobility since both pincers can potentially rotate around the bond connecting them to the central platform (Fig. 11). Such rotation changes the cavity size, allowing some optimization to take place upon inclusion of guest species.![Molecular tweezers containing a dibenz[c,h]acridine U-shaped spacer; X-ray crystal structure of compound 17 (R,R′ = OMe, R′′ = 4-tBu)](/image/article/2011/CS/b915145c/b915145c-f11.gif) | ||
| Fig. 11 Molecular tweezers containing a dibenz[c,h]acridine U-shaped spacer; X-ray crystal structure of compound 17 (R,R′ = OMe, R′′ = 4-tBu) | ||
In the dibenz[c,h]acridine U-shaped spacer, chromophores are attached at the C-2 and C-12 positions, which are separated by 7.24 Å. That is slightly larger than the 6.8 Å interchromophore separation, which is optimum for sandwiching an aromatic guest. The tweezer exhibits conformational mobility and can readily adjust the size of this aromatic cleft for binding an aromatic guest.22 The two acridine units of such molecular tweezers show interesting cooperativity in sandwiching 2,4,7-trinitrofluorenone (TNF) guest for 17 (R,R′′ = tBu; R′ = H) (Ka = 172 M−1 in CDCl3)23 and 2,4,5,7-tetranitrofluorenone (TENF) for 17 (R,R′ = OMe; R′′ = 3,5-di-tBu) (Ka = 3400 M−1).24 The minor role played by the spacer was evidenced in that series and the affinity for TENF guest can be increased by substituting acridines with methoxy groups or replacing the acridine of tweezer 17 by one to two anthracene ring in tweezers 18 and 19 respectively (Ka = 900 M−1 in CDCl3 for 17, Ka = 2800 M−1 for 18 and Ka = 20000 M−1 for 19).5 An extension of this tweezers family lies on the introduction of an active carboxylic acid site oriented toward the binding cleft, resulting in an increasing the C-2 to C-12 distance to 8.2 Å (Fig. 12). A very high affinity was found for tweezer 20a (Ka = 25000 M−1 in CDCl3) for 9-adenine.8 The critical role of hydrogen bonding in the binding process was demonstrated by the absence of any complexation with ester 20b (Ka < 5 M−1).
![Dibenz[c,h]acridine-based molecular tweezers containing an active carboxylic acid site.](/image/article/2011/CS/b915145c/b915145c-f12.gif) | ||
| Fig. 12 Dibenz[c,h]acridine-based molecular tweezers containing an active carboxylic acid site. | ||
As shown above, these tweezers can modulate the cavity shape through rotation of the tips. Another family of molecular tweezers has been designed by Klärner and co-workers, who developed highly rigidified polyarene architectures (Fig. 13).
 | ||
| Fig. 13 Two families of molecular tweezers with different degrees of rigidification. | ||
Hydrocarbon-based architectures di-, tri- and tetramethylene-bridged compounds 2, 3 and 4, respectively, were synthesized through a convergent synthesis based on a molecular “Lego” set, consisting of bisdienophiles and dienes.7 These are arranged in a belt-like concave–convex topography, so that an aromatic guest might be sandwiched via multiple π–π and CH–π interactions. To fit with the substrate topography, the size and shape of the receptor cavity can be tuned by varying the number and size of the spacer units.
Dimethylene-bridged architectures 2 have been referred as molecular clips, because they form complexes by “clipping” an aromatic substrate inside the receptor cavity. The distance between both sidewalls can be tuned using benzene- or naphthalene-spaced receptors (Fig. 14).
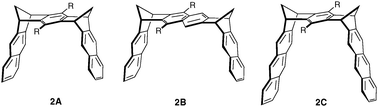 | ||
| Fig. 14 Dimethylene-bridged molecular clips. | ||
The distance between the tweezer pincers in system 2A (R = OMe) is ca. 10 Å in the solid state before guest complexation, and reaction with 1,4-dinitrobenzene (p-DNB) produces a host–guest complex in which this distance was reduced to 7.6 Å (Fig. 15).25
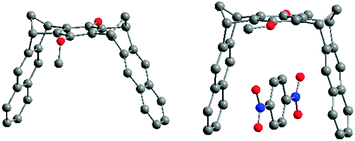 | ||
| Fig. 15 Single-crystal structures of molecular clip 2A (left) and complex p-DNB@2A (right). | ||
Thanks to larger van der Waals contact surfaces, molecular clips 2C with anthracene sidewalls form more stable host–guest complexes with electron-deficient aromatic and quinoid molecules, such as the inclusion complex TCNB@2C (R = OH, Ka = 12800 M−1 in CDCl3).26,27 Moreover, complexes TCNB@2C and TNF@2C (TNF = 2,4,7-trinitro-9-fluorenone) were described as the first examples of charge-transfer luminescence from host–guest complexes. These act as potential chemical sensors with characteristic color changes observed upon complex formation, from colourless to red or purple with TCNB, and from yellow to green with TNF.
An interesting extension of model derivative 2A corresponds to clip 2A′ bearing two chiral (−)menthyl groups at the extremity of the tips. The corresponding inherently molecular clips, pseudoenantiomeric anti and anti′, as well as mesoid syn, were separated by chiral HPLC (Fig. 16). Remarkably, compound 2A′ in its anti′ form is able to discriminate between L- and D-tryptophane methyl esters with a relative binding constant ratio KD/KL = 3.5 with KD = 13600 M−1 in THF–MeOH–H2O (4:1:5).28
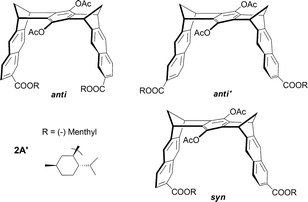 | ||
| Fig. 16 Molecular clips 2A′ designed for supramolecular chiral discrimination. | ||
Trimethylene-bridged molecular clips 3A and 3B present a narrower shape and a remarkably high Ka value (5.4 × 106 M−1 in CHCl3) was observed for host 3B, with TCNB guest (Fig. 17).27
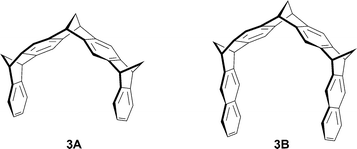 | ||
| Fig. 17 Some representative structures of trimethylene-bridged molecular tweezers. | ||
The concept defined by Whitlock is slightly modified in compound 4 which is not far from being a macrocycle, and the pincers being not parallel, the guest can be considered as encapsulated by the host (Fig. 18). Consequently, contrary to the above-mentioned tweezers for which an aromatic guest is sandwiched in-between both tips, this one is arranged nearly parallel to the central naphthalene spacer unit in the case of receptors 4. Therefore, though guest 4 cannot be strictly considered as sandwiched, this case appears relevant with the topic of this review. Tweezer 4B (R = H) is a better receptor for substrate such as TCNQ (Ka > 105 M−1 in CDCl3) than 4A (R = H) (Ka = 1100 M−1 in CDCl3) since the complexation by the latter requires a substantial distortion of the receptor geometry (Fig. 18).7
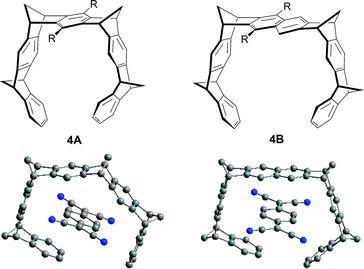 | ||
| Fig. 18 Structures of tetramethylene-bridged molecular tweezers 4A and 4B. X-Ray structures of complexes TCNQ@4A (left) and TCNQ@4B (right). | ||
The same tendency was observed with the 1,2,4,5-tetracyanobenzene (TCNB) guest as shown by respective X-ray crystal structures of TCNB@2C and TCNB@4B (Fig. 19).29
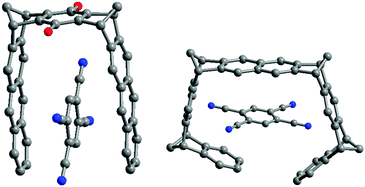 | ||
| Fig. 19 X-Ray crystal structures of TCNB@2C and TCNB@4B complexes. | ||
These clips and tweezers containing naphthalene or anthracene sidewalls form luminescent host–guest complexes with TCNB in CHCl3 solution resulting from predominant charge transfer interactions. Association/dissociation barriers formation (ΔG = −7.9 kcal mol−1 for TCNB@4B and −9.0 kcal mol−1 for TCNB@3B) were determined as well as dissociation values of host–guest complexes (ΔG# = 15.7 kcal mol−1 for TCNB@4B and 12.4 kcal mol−1 for TCNB@3B). These observations are consistent with the close topology of tweezers 4B which makes a kinetically rather difficult process the inclusion of a guest molecule. Contrariwise, clip 3B presenting a more opened shape allows an easier inclusion of the guest molecules, which is accompanied by a lower activation barrier.27
The versatility of the synthesis which is supported by cycloaddition processes, allowed an access to water-soluble systems. Corresponding receptors exhibit a good selectivity in substrate binding in aqueous solution. For instance, tweezers 4B and clip 2C (R = OP(CH3)O2−Li+) form highly stable host–guest complexes with the enzyme cofactor model N-methylnicotinamide iodide (NMNA) in methanol (Ka = 11.9 × 104 M−1 in CD3OD for 4B and 1.04 × 104 M−1 for 2C) and water as well.30
Triptycene was also found to be a useful three-dimensional Y-shaped building block for the construction of a new class of rigidified polyarene receptors. This is for example illustrated with the triptycene-based bis(crown ether) host 21, which incorporates two dibenzo-24-crown-8 ether moieties, and which was shown to form stable clip-shaped complex with paraquat (Fig. 20).31 A charge transfer from the electron-rich host to the electron poor paraquat (Ka = 16300 M−1 in CD3CN) is observed. X-Ray crystal structure shows that the complexation process involves multiple CH⋯O hydrogen bonds between the paraquat unit and ether oxygen atoms, but also π-stacking interactions between paraquat ring and aromatic rings of the receptor.
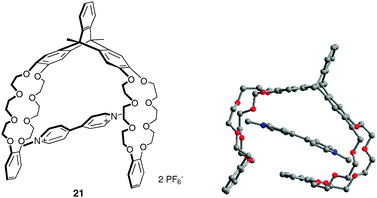 | ||
| Fig. 20 Complexation of paraquat with triptycene-based molecular clip 21 and corresponding X-ray crystal structure (anions omitted). | ||
A recent extension to the H-shaped scaffold pentiptycene-based bis(crown ether) host 22 incorporating two dibenzo-24-crown-8 ether moieties has been proposed.32 It exhibits an efficient complexation ability toward cyclobis(paraquat-p-phenylene) in solution (Ka = 2650 M−1 in CD3CN) (Fig. 21).
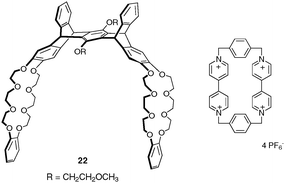 | ||
| Fig. 21 Structures of pentiptycene-based molecular clip 22 and cyclobis(paraquat-p-phenylene). | ||
2.3 Rigid Kagan's ethers and chiral Tröger's bases
The perpendicular arrangement (93° angle) of two aromatic rings found in Kagan's ether 23 constitutes a powerful scaffold for constructing receptors capable of aromatic guests binding with both face-to-face and face-to-edge interactions. On this basis, a class of molecular tweezers was initially developed with analogs of Kagan's ether.33 The complex between tweezers 24 and 1,3,5-trinitrobenzene (TNB) was crystallized showing the TNB guest is parallel to the dibenzofurane units with a distance of 3.32 Å in agreement with a stacking interaction between the two systems34 (Ka = 2000 M−1 in CHCl3).6 Fukazawa and Coll developed the dioxa[2,2]orthocyclophane 25 which presents a tweezers-type arrangement with two terminal aromatic rings separated by 6.5 Å in the syn conformation.35 This was demonstrated by the binding affinity of host 26 for TCNQ guest. This one is suitably positioned for combining stacking interactions with the terminal naphthalene and phenanthrene chromophores, and an edge-to-face interaction with the central durene bridge (Fig. 22).36 The highest association constants were obtained with pyromellitic dianhydride (PDA) or TCNB (Ka = 1000 M−1 for PDA@26 and TCNB@26).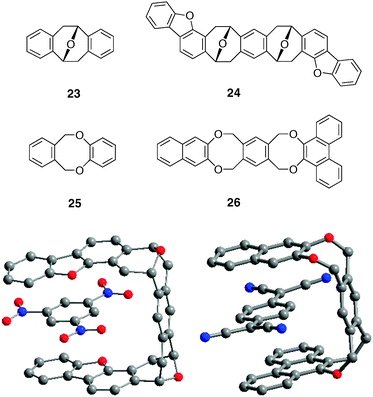 | ||
| Fig. 22 X-Ray crystal structures of TNB@24 (left) and TCNQ@26 (right) complexes. | ||
The development of chiral molecular tweezers is of particular interest because they will contribute to new research avenues in chiral supramolecular chemistry and related applications.6 Tröger's base provides a relatively rigid chiral skeleton with the requirements for the construction of molecular tweezers. Dolenský and Coll. recently described large-pincered molecular tweezers 27 based on a bis-Tröger's base for which the calculated lateral pincers lie almost parallel to each other (32°) with a distance of 6.7–8 Å between them (Fig. 23).37 The syn isomer, unlike the anti isomer, was shown to form a charge-transfer complex with tetracyanoethylene (TCNE) characterized by a color change from colourless (anti) to darker (syn) purple solution. It should be noted that the inherent chirality in the cleft has not been yet exploited in molecular recognition studies, although enantioselective recognition is possible.38
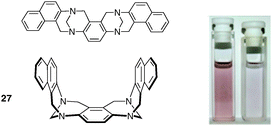 | ||
| Fig. 23 Structure of Tröger's base molecular tweezer and color change observed with the complexation of TCNE by 27syn (reproduced with permission from ref. 37. Copyright American Chemical Society 2010). | ||
2.4 Concave polycyclic aromatic hydrocarbons for fullerene recognition
Beside usual host systems which are designed for recognition of planar guests between pincers of a molecular tweezer, a novel type of molecular receptors has been recently introduced with curved conjugated carbon networks as bowl-shaped polycyclic aromatic hydrocarbons. Their accessible concave surfaces appear to be ideal recognizing curved-surface fullerenes through concave–convex π–π interactions.39 The first experimental evidence was provided by the formation of a stable inclusion complex of C60 with the “buckycatcher” 28, a molecular clip with two corannulene pincers and a tetrabenzocyclooctatetraene tether (Fig. 24). The inclusion complex C60@28 was crystallized showing a distance between corannulene units and C60 surface close to 3.1 Å and complexation was also evidenced in solution (Ka = 8600 M−1 in toluene-d8).40 Molecular clip 29, a more flexible analog of the buckycatcher 28, was shown to encapsulate only solvating molecules of nitrobenzene in the solid state.41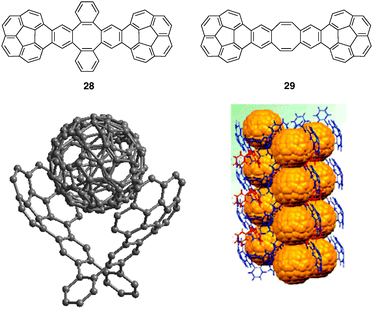 | ||
| Fig. 24 X-Ray crystal structure of C60@28 and corresponding crystal packing pattern (reproduced with permission from ref. 40. Copyright American Chemical Society 2010). | ||
3. Tweezers involving flexible platforms
The platform which bridges both pincers of the tweezers can be flexible by its own, or through an intermediate linker, if any. For such systems, both binding sites do not necessarily adopt a cofacial arrangement, and an external stimulus, such as a binding event, can induce a conformational rearrangement. This process often requires large reorganization energies which generally result in lower association constants in host–guest complexes compared to what occurs with rigid tweezers. Conversely, such flexible systems are often more straightforward to synthesize and they can adjust to the size of the substrate. Significant new inputs have been described in this family of tweezers, involving in particular electron-rich tips.3.1 Electron-rich flexible tweezers
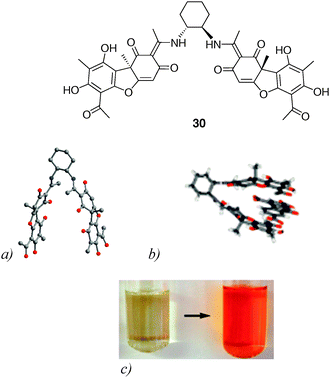 | ||
| Fig. 25 X-Ray crystal structures of (a) molecular tweezer 30; (b) the corresponding TNF complex and (c) color change upon TNF binding by tweezer 30 (reproduced with permission from ref. 42. Copyright American Chemical Society 2010). | ||
300 M−1 in CHCl3/hexafluoro-propan-2-ol (6:1)), which is manifested by a deep red colour change. It is worth noting that the high binding ability results from an original combination of mutually complementary π–π stacking interactions between the tweezers and the macrocyclic guest, in addition to NH⋯O and CH⋯O hydrogen bonds (Fig. 26). Interestingly, such set of interactions between tweezer 31 and complementary accepting moieties, could be extended to the recognition of specific monomers sequences in copolyimides macromolecules.44
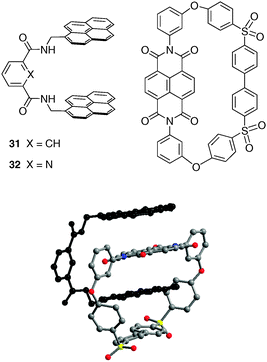 | ||
| Fig. 26 Structures of tweezers 31 and 32; X-ray crystal structure of a 1:1 complex between tweezer 31 and a macrocyclic ether-imide-sulfone. | ||
In order to increase the number of binding interactions, an approach developed by the same group was to design new electrodeficient molecular tweezers 33 bearing two naphtalenediimide (NDI) units, complementary to the above-mentioned electron-rich systems 31. A 1:1 tweezer-tweezer complex is formed by mixing 31 and 33, each of the π–electron poor diimide units being able to maintain face-to-face strong binding interactions with the π-electron rich pyrene guest (Fig. 27).45 A characteristic deep red solution corresponding to the charge transfer was formed with a binding constant of Ka = 3500 M−1 (CHCl3).
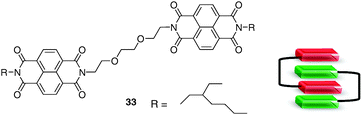 | ||
| Fig. 27 Bis-NDI tweezer 33 and association model for donor–acceptor tweezers 31–33. | ||
Chiral molecular tweezers using a bile acid scaffold and pyrene-sidewalls have also been designed to bind electron-deficient aromatic species (Fig. 28).46 The two pyrene units in compound 34 are separated by ca. 6–6.5 Å on the C-3 and C-12 positions of the rigid bile acid backbone. The resulting clip allows moderate association constants with polynitroaromatic compounds (e.g.Ka = 220 M−1 for TNF in CDCl3).
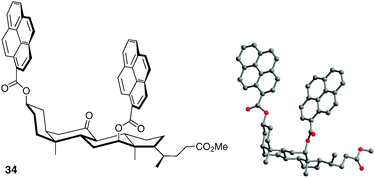 | ||
| Fig. 28 Structure of chiral bile acid molecular tweezer 34. | ||
In the active field of molecular receptors designed for recognition of fullerenes in solution, the concept of concave receptors appears particularly promising besides the well-described poly-porphyrinic systems.11 As studied by Martín and Coll., a prototypal unit is proposed with the concave aromatic π-extended TTF which presents a good shape complementarity with the convex buckyball.48 A straightforward preparation of tweezers molecules using the flexible isophthalic platform has been developed recently.49 These systems incorporate extended-tetrathiafulvalene (exTTF) units as binding sites (Fig. 29). Tweezer molecule 35 exhibits a good binding ability for C60 (Ka ≈ 3000 M−1 in chlorobenzene).50 Depending on the solvent, the resulting 1:1 complex appears either as one fullerene sandwiched between two exTTF units (chlorobenzene) or to exist as a supramolecular tetramer (CHCl3–CS2).
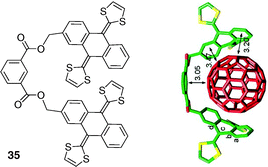 | ||
| Fig. 29 Molecular tweezer 35 and the calculated 35·C60 complex at the BH&H/6-31G** level. (Reproduced with permission from ref. 50. Copyright Royal Society of Chemistry 2010.) | ||
As an extension to this system, fullerene-exTTF tweezer monomer 36 was designed to self-assemble in a head-to-tail fashion through recognition of the C60 moiety by the exTTF pincer-like receptor (Fig. 30).51 The recognizing units are complementary both at the structural and electronic levels and self-associate to produce a linear head-to-tail supramolecular polymer (Fig. 30). This arrangement prevents C60 aggregation and provides a promising new entry into D–A devices.
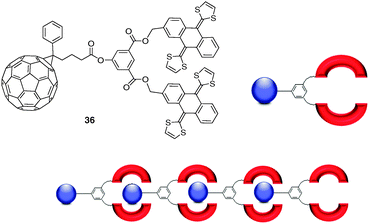 | ||
| Fig. 30 Chemical structure of molecular tweezer 36 and illustration of the resulting supramolecular donor–acceptor polymer. | ||
Concave–convex interactions in host–guest complexes from 35 (or 36) and C60 unit seem to play a role in the binding process.39 This contribution in stabilizing the supramolecular system has been illustrated by a comparative study of the C60 binding ability of tweezers 35, 37–39. These are all built from an isophthalic diester spacer grafted with two recognizing units,50i.e. concave donating units (35), concave accepting unit (37), planar accepting units (38) and planar donating units (39) (Fig. 31). Such a collection of compounds allowed an investigation of the relative contributions of π–π, van der Waals, electrostatic, and concave–convex interactions upon recognizing C60. On the basis of 1H NMR titration experiments and density functional theory (DFT) calculations, it appears that concave–convex complementarity contributes to the binding process, even if quantitatively small. This is illustrated in particular with compound 35 which presents the highest affinity for C60 (Ka = 3000 M−1, CDCl3–CS2 1:1). The case of receptors 37 and 38 is also informative. In spite of the more electron-poor character of 37 when compared to 38, its binding constant towards C60 is larger (37: Ka = 1 540 M−1, 38: Ka = 790 M−1). Finally, no C60 binding is observed with 39, presenting planar donating units. Note that extension of this strategy can be applied to non-symmetrical tweezers able to bind carbon nanotubes.52,53
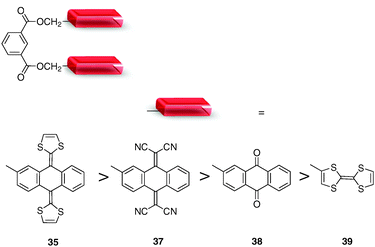 | ||
| Fig. 31 Chemical structures of molecular tweezers 35, 37–39, associating an isophthalic diester spacer with various recognizing units. | ||
At this stage, it is worth noting that a Pt(II)-assembled monopyrroloTTF (MPTTF) dimer 40 has been very recently described,54 and shows a good affinity for C60 (Ka = 1400 M−1, CH2Cl2). Such affinity is in this case assigned to a greater π-extension of the MPTTF donating framework related to the TTF backbone in 39, and to the presence of the metallic cation (Fig. 32).
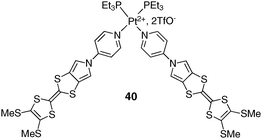 | ||
| Fig. 32 Chemical structure of monopyrroloTTF dimer 40. | ||
J. O. Jeppesen et al. described recently molecular tweezer 41 which assembles two MPTTF units and two thiophene moieties within a calix ring (Fig. 33).55 Whereas this system adopts an extended conformation in the solid state, a tweezer-like 1,3-alternate conformation with one acceptor TCNQ sandwiched between the two electron-rich MPTTF units was observed. This complexation results from donor–acceptor interactions, and is characterized in the solid-state by an interplanar distance between the two MPTTF units of ca. 6.7 Å, and 3.3 Å between MPTTF and TCNQ units. Solution studies (UV-vis-NIR and ESR, CH2Cl2) also showed that a charge transfer takes place between the MPTTF units and TCNQ.
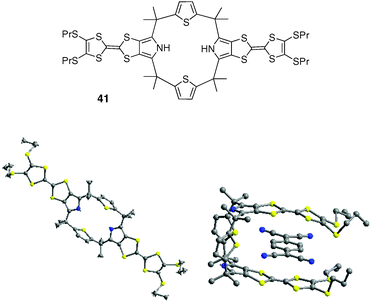 | ||
| Fig. 33 X-Ray structures of compound 41 (left) and corresponding sandwich TCNQ complex (right) (solvent molecule and a second TCNQ molecule located between two adjacent 41 molecules are omitted). | ||
According to a similar strategy as for compound 26 involving the dioxa[2.2]orthocyclophane skeleton,36 a flexible system incorporating redox-active TTF units has been recently proposed by Azov and Coll with compound 42 (Fig. 34).56 Two TTF moieties are in this case connected to the opposite sides of a 1,2,4,5-substituted benzene scaffold via 8-membered dithia rings. Such architecture is relatively rigid and conformational analysis of cycloocta-1,5-diene-like ring systems indicates the existence of three low energy boat, chair, and skew conformations. Only the cis-boat-boat conformation shows a tweezer-like arrangement, with the TTF planes positioned almost parallel at ca. 6 Å away from each other (Fig. 35). All possible combinations of boat, chair and skew conformations have been shown to roughly lie within a 2–4 kcal mol−1 energy window.
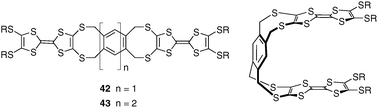 | ||
| Fig. 34 Bis-TTF based molecular tweezers developed by Azov and Coll. | ||
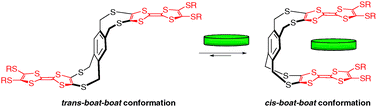 | ||
| Fig. 35 Equilibrium between the trans boat-boat conformation (among other conformations) of 42, and the closed cis boat-boat conformation suitable for sandwiching electron-poor aromatic guests. | ||
An extension of this system has been proposed by the same authors by incorporating a naphthalene backbone instead of a benzene ring (compound 43), thus controlling the intra TTF–TTF distance within the clip.57 Molecular recognition experiments carried out with 42 and 43 show relatively low binding properties with 2,4,7-trinitro-9-fluorenylidenemalonitrile (Ka = 16 M−1 and 22 M−1 for 42 and 43, respectively, CDCl3), whereas only tweezer 43 exhibits a very weak binding with TCNB (Ka = 6 M−1). Therefore, these clip-like architectures present a non pre-defined topology and sufficient conformational freedom to adopt the cis-conformation required for host recognition thanks to a large-scale open-close molecular movement. In a closed conformation the two tips of the tweezers overlay each other, whereas in an opened conformation the tips are located opposite to each other.
Another interesting feature of such kind of clips relies on their electrochemically-driven dynamic properties.56,57Oxidation of 42 at the first TTF redox level is characterized on the cyclic voltammogram, by two TTF units in close interaction. Conversely, oxidation at the TTF dication state shows that the two redox-active units are no longer in interaction and at a larger distance. This observation results from coulombic repulsive interactions between the two doubly positively charged TTF units. Consequently, whereas a closed (cis) conformation is likely preferred at least when the system is oxidized at the mono-oxidized state, a conformational transition to an opened conformation is obtained at the fully oxidized state. This observation is supported by variable temperature NMR studies led on the –SCH2groups. Such architectures offer promising perspective for the construction of electrochemically responsive molecular machines endowed with a controlled movement.
3.2 Tunable flexible tweezers
Molecules that can change their conformations (rotation, shuttling, flipping, bending) in response to various stimuli offer a potential to be used in constructing molecular machines or devices, which is the case of molecular tweezers.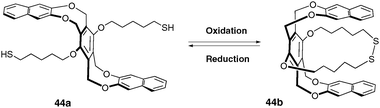 | ||
| Fig. 36 Opened and closed conformations of compound 44. | ||
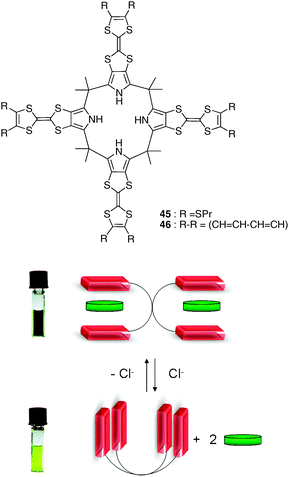 | ||
| Fig. 37 Dynamic behaviour of receptor 45 with an electron-poor guest and chloride anion (reproduced with permission from ref. 59. American Chemical Society 2010). | ||
These results were recently extended with a second generation of TTF-calix[4]pyrroles compounds such as 46 which, thanks to an additional fused aromatic ring on each TTF tip, presents a higher π-surface compared with 45. This compound was designed to better match flat electron-deficient guests in terms of size and shape. The authors could demonstrate that such systems exhibit a positive homotropic allosteric effect upon guest binding, and a significantly enhanced colorimetric response to nitroaromatic compounds (TNB, picric acid, TNP, and 2,4,6-trinitrotoluene TNT), positioning this system as highly efficient chemosensors for nitroaromatic explosives (Fig. 38).61
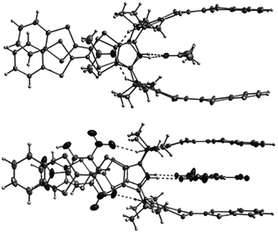 | ||
| Fig. 38 X-Ray crystal structures of 46·2Me2CO (top) and 46·2TNP (bottom) (reproduced with permission from ref. 61. Copyright Wiley-VCH 2010). | ||
3.2.3 Flexible tweezers triggered by metal cation binding
Lehn and Coll have designed a series of dynamic devices undergoing ion-induced conformational changes that modulate affinity for the substrate. These bistable receptors are built either around a terpyridine scaffold or a pyridine–pyrimidine–pyridine sequence, which self-converts from a W to a U shape (U to W, respectively) upon cation coordination. These systems present a tweezer-like behaviour under the U form which is well-suited for guest intercalation. Therefore, the binding (or release) of a given substrate is controlled through a shape switching of the receptor which is induced by metal–ion coordination. For instance, compound 47 adopts a U-shaped tweezer-like conformation with two tips (anthracene units) well positioned to sandwich a substrate (Fig. 39).62 A colour change due to charge transfer complexation is instantaneously observed upon introduction of electron-acceptors (TNF, TCNQ), and intercalation is evidenced in solution (1H NMR study) as well as in the solid state. The crystal structure shows that the two anthracene donors are separated by 7 Å and are almost parallel (Fig. 40). The introduction of Cu(CH3CN)4, BF4 to this solution leads to a switching from the tweezer-form (U) to the W conformation, with a releasing of the substrate which can be monitored by 1H NMR and a loss of the charge transfer colour.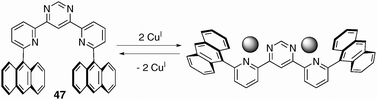 | ||
| Fig. 39 Cation-driven two-stage U (left)/W (right) molecular shape switching. | ||
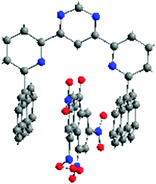 | ||
| Fig. 40 X-Ray crystal structure of the complex 47. TNF. | ||
Conversely, replacing the central pyrimidine ring with a pyridine unit leads to a complementary behaviour, with the U-shape occurring upon cation complexation. An interesting example is provided with compound 48 (Fig. 41). Mixing this compound with one equivalent of a coordinating ligand (1,10-phenanthroline) in presence of a zinc triflate salt, produces a sandwich complex in which the metal cation induces the U-form and contributes also to bind the phenanthroline guest by coordination (Fig. 42). Noteworthy, no binding was observed for 48 with non-coordinating electron-poor guests such as TNF and TCNQ. This is presumably due to a pinching of the terpyridine backbone upon metal complexation and by obstruction of the cavity space by the solvation sphere of the octahedral cation, which prevent intercalation of any guest between the tips. This is not any more the case with analog 49, possessing an amide spacer, and ensuring a higher spatial separation between the coordination site and the intercalation pocket. When the Zn(II) complex is titrated with TCNQ or TNF, intercalation occurs and the expected charge transfer is now observed.
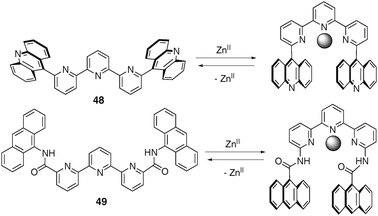 | ||
| Fig. 41 Cation-driven two-stage W (left)/U (right) molecular shape switching. | ||
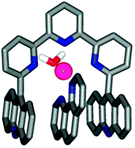 | ||
| Fig. 42 X-Ray crystal structure of the sandwich complex 48·Zn(II)·Phen (reproduced with permission from ref. 62. American Chemical Society 2010). | ||
The tritopic ligand 50, which combines one terpyridine and two pyridine subunits was used by Barboiu and Coll to reach self-complementary molecular clefts which are stabilized by strong π–π stacking interactions. Such supramolecular assemblies result from the binding of octahedral metal ions Co2+ or Pb2+ ions (Fig. 43)63 through orthogonal-terpyridine and linear-pyridine metal-coordination. Different cleft entities are observed in both solution and solid state, and may be interconverted as a function of metal/ligand stoichiometry. In particular, robust self-complementary duplex cleft structures are generated with the pyridine side wall of one clip molecule filling the cavity of the other and vice versa, generating different zipper architectures.
![Preparation and X-ray crystal structures of duplex cleft complexes [Co2(50)2(CH3CN)4]4+(left) and [Pb2(50)2(CF3SO3)2]2+ (right).](/image/article/2011/CS/b915145c/b915145c-f43.gif) | ||
| Fig. 43 Preparation and X-ray crystal structures of duplex cleft complexes [Co2(50)2(CH3CN)4]4+(left) and [Pb2(50)2(CF3SO3)2]2+ (right). | ||
Another example of a dynamic tweezer-type receptor triggered by metal coordination is proposed with compound 51 (Fig. 44).64 The metallo-tweezer is constructed in this case from a pyridine bisimidazole platform, bearing two appended anisole groups. In this tweezer-like complex, the copper atom organizes a binding pocket ideal for guest recognition with concomitant participation of the metal (coordination) and of the electron rich side walls. Intercalation of various aromatic heterocycles, including adenine (Ka = 8.8 × 104 M−1, acetone) or fluorescent dimethylaminostyrylpyridine (Ka = 1.1 × 105 M−1) could be demonstrated.
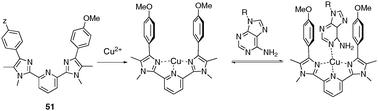 | ||
| Fig. 44 Tweezer 51 and metal-assisted recognition of adenine. | ||
Very recently, a series of new dynamic molecular tweezers whose recognition ability can be regulated by a metal-induced W to U switching has been described (e.g. compound 52) (Fig. 45).65,66 They differ from the previous electron-rich systems 47–49 by incorporating electron-poor NDI (1,4,5,8-naphthalenetetracarboxylic diimide) binding sites, prone to interact through π-stacking with electron-rich substrates. Such tweezer molecules have been, in particular, incorporated in dynamic libraries leading to functional constitutional dynamic networks.67 The metal coordinating subunit in this case corresponds to a pyridine–hydrazone–pyridine framework. Whereas free ligand 52 displays a W-shaped conformation, the metal complex 52·M (M = Zn, Hg, Pb) presents a U-conformation, suitable for aromatic guests intercalation. The cavity size and shape are defined by the size of the metal cation, since a smaller cation should decrease the distance between both NDI walls, but also should lead to a severe pinching of the clip, preventing the insertion of a substrate. For instance, in the case of the Pb2+ complex, the addition of the electron-rich pyrene was accompanied with a colour change of the solution which turned from yellow to red with a Ka value of 320 M−1 (CDCl3/CD3CN 6:4), whereas no binding was observed in the case of the smaller Zn2+ complex.
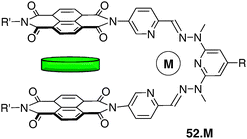 | ||
| Fig. 45 Electron-poor tweezer 52·M molecule intercalating an electron-rich polyaromatic substrate. | ||
4. Conclusion and outlook
This tutorial review focuses on current trends in the design of molecular tweezers/clips through representative examples, and on their neutral guests binding properties. Such systems have known a growing interest in the last period, a consequence of new synthetic methodologies as well as a response to an increasing demand in the front of molecular machinery. Design optimization has been led on both the platform subunit, whose role is to control the spatial pre-organization and the rigid/flexible character of the system, as well as by varying the pair of binding units. Molecular tweezers presenting a distance of ca. 6.5–7 Å between both binding arms obviously correspond to the most usual case appropriate for sandwiching aromatic planar guests. They generally support π–π stacking interactions, which are eventually coupled to hydrogen bond or attractive CH–π interactions. Moreover, molecular electrostatic potentials have been shown recently to be powerful tools for the interpretation and the prediction of supramolecular interactions. On these grounds, the preferential binding and significant through-space interactions with the substituents can be rationalized based on computed electrostatic potentials of the clips and the guest molecules.68 Whereas most tweezers are electron-rich, new complementary examples appeared recently, involving electron-poor pincers, and exhibiting a good affinity for electron-rich guests. Recent efforts have also been produced in generating pincers programmed for receiving spherical guests like fullerenes, which is favored through concave–convex host–guest complementarity. New promising potentialities have appeared with dynamic molecular clips and tweezers that can change their conformations in response to external stimuli. Such controllable dynamic features can represent a new step towards “smart” adaptive nanodevices. Emergence of such systems is of strong potential interest in host–guest chemistry, chiral recognition,28 but also for supramolecular catalysis and for the development of complex architectures that exhibit behaviours typically reserved for natural systems, paving the way to nanomechanical chemical systems and opening new research avenues in supramolecular chemistry. Finally, the development of molecular clips and tweezers soluble in water where the hydrophobic interactions govern the supramolecular phenomenon, are of particular interest to reach bioactive agents able to pinch biomolecules such as nucleobases,69 potential drug candidates,70 or enzyme inhibitors.71,72Acknowledgements
The authors gratefully acknowledge ANR-PNANO (TTF-Based Nanomat) for financial support.Notes and references
- J. W. Steed and J. L. Atwood, in Supramolecular Chemistry, John Wiley & Sons Ltd, 2nd edn, 2009 Search PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS.
- Special Issue: Molecular Machines and Switches Adv. Funct. Mater. 2007, 17, 671-840.
- C.-W. Chen and H. W. Whitlock Jr., J. Am. Chem. Soc., 1978, 100, 4921–4922 CrossRef CAS.
- S. C. Zimmerman, Top. Curr. Chem., 1993, 165, 71–102 CAS.
- M. Harmata, Acc. Chem. Res., 2004, 37, 862–873 CrossRef CAS.
- F.-G. Klärner and B. Kahlert, Acc. Chem. Res., 2003, 36, 919–932 CrossRef.
- S. C. Zimmerman, Bioorganic Chem. Frontiers, 1991, 2, 33–71 Search PubMed.
- R. P. Sijbesma and R. J. M. Nolte, Top. Curr. Chem., 1995, 175, 25–56 CAS.
- A. E. Rowan, J. A. A. W. Elemans and R. J. M. Nolte, Acc. Chem. Res., 1999, 32, 995–1006 CrossRef CAS.
- E. M. Pérez and N. Martín, Pure Appl. Chem., 2010, 82, 523–533 CrossRef CAS.
- P. D. Harvey, C. Stern, C. P. Gros and R. Guilard, Coord. Chem. Rev., 2007, 251, 401–428 CrossRef CAS.
- R. P. Sijbesma, A. P. M. Kentgens, E. T. G. Lutz, J. H. van der Maas and R. J. M. Nolte, J. Am. Chem. Soc., 1993, 115, 8999–9005 CrossRef CAS.
- R. P. Sijbesma, S. S. Wijmenga and R. J. M. Nolte, J. Am. Chem. Soc., 1992, 114, 9807–9813 CrossRef CAS.
- J. N. H. Reek, J. A. A. W. Elemans and R. J. M. Nolte, J. Org. Chem., 1997, 62, 2234–2243 CrossRef.
- P. Polavarapu, H. Melander, V. Langer, A. Gogoll and H. Grennberg, New J. Chem., 2008, 32, 643–651 RSC.
- N. She, M. Gao, L. Cao, A. Wu and L. Isaacs, Org. Lett., 2009, 11, 2603–2606 CrossRef CAS.
- S. Ghosh, A. Wu, J. C. Fettinger, P. Y. Zavalij and L. Isaacs, J. Org. Chem., 2008, 73, 5915–5925 CrossRef CAS.
- P.-N. Chen, P.-T. Chiang and S.-H. Chiu, Chem. Commun., 2005, 1285–1287 RSC.
- P.-T. Chiang, P.-N. Chen, C.-F. Lin, Y.-H. Liu, C.-C. Lai, S.-M. Peng and S.-H. Chiu, Chem.–Eur. J., 2006, 12, 865–876 CrossRef CAS.
- K.-W. Cheng, C.-C. Lai, P.-T. Chiang and S.-H. Chiu, Chem. Commun., 2006, 2854–2856 RSC.
- S. C. Zimmerman, C. M. VanZyl and G. S. Hamilton, J. Am. Chem. Soc., 1989, 111, 1373–1381 CrossRef CAS.
- S. C. Zimmerman and C. M. VanZyl, J. Am. Chem. Soc., 1987, 109, 7894–7896 CrossRef CAS.
- S. C. Zimmerman, M. Mrksich and M. Baloga, J. Am. Chem. Soc., 1989, 111, 8528–8530 CrossRef CAS.
- F.-G. Klärner, J. Panitzky, D. Bläser and R. Boese, Tetrahedron, 2001, 57, 3673–3687 CrossRef CAS.
- F.-G. Klärner, B. Kahlert, R. Boese, D. Bläser, A. Juris and F. Marchioni, Chem.–Eur. J., 2005, 11, 3363–3374 CrossRef.
- F. Marchioni, A. Juris, M. Lobert, U. P. Seelbach, B. Kahlert and F.-G. Klärner, New J. Chem., 2005, 29, 780–784 RSC.
- G. Fukuhara, S. Madenci, J. Polkowska, F. Bastkowski, F.-G. Klärner, Y. Origane, M. Kaneda, T. Mori, T. Wada and Y. Inoue, Chem.–Eur. J., 2007, 13, 2473–2479 CrossRef CAS.
- F.-G. Klärner, U. Burkert, M. Kamieth, R. Boese and J. Benet-Buchholz, Chem.–Eur. J., 1999, 5, 1700–1707 CrossRef CAS.
- F.-G. Klärner, B. Kahlert, A. Nellesen, J. Zienau, C. Ochsenfeld and T. Schrader, J. Am. Chem. Soc., 2006, 128, 4831–4841 CrossRef.
- T. Han and C.-F. Chen, Org. Lett., 2006, 8, 1069–1072 CrossRef CAS.
- J. Cao, Y. Jiang, J.-M. Zhao and C.-F. Chen, Chem. Commun., 2009, 1987–1989 RSC.
- J. Kagan, S.-A. Chen and D. A. Agdeppa Jr., Tetrahedron Lett., 1977, 18, 4469–4470 CrossRef.
- M. Harmata and C. L. Barnes, J. Am. Chem. Soc., 1990, 112, 5655–5657 CrossRef CAS.
- H. Kurebayashi, T. Mine, K. Harada, S. Usui, T. Okajima and Y. Fukazawa, Tetrahedron, 1998, 54, 13495–13504 CrossRef CAS.
- H. Kurebayashi, M. Sakaguchi, T. Okajima, T. Haino, S. Usui and Y. Fukazawa, Tetrahedron Lett., 1999, 40, 5545–5548 CrossRef CAS.
- M. Havlík, V. Král, R. Kaplánek and B. Dolenský, Org. Lett., 2008, 10, 4767–4769 CrossRef CAS.
- J. J. Turner and M. M. Harding, Supramol. Chem., 2005, 17, 369–375 CrossRef CAS.
- T. Kawase and H. Kurata, Chem. Rev., 2006, 106, 5250–5273 CrossRef CAS.
- A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842–3843 CrossRef CAS.
- L. Kobryn, W. P. Henry, F. R. Fronczek, R. Sygula and A. Sygula, Tetrahedron Lett., 2009, 50, 7124–7127 CrossRef CAS.
- B. Legouin, P. Uriac, S. Tomasi, L. Toupet, A. Bondon and P. van de Weghe, Org. Lett., 2009, 11, 745–748 CrossRef CAS.
- H. M. Colquhoun, Z. Zhu and D. J. Williams, Org. Lett., 2003, 5, 4353–4356 CrossRef CAS.
- H. M. Colquhoun and Z. Zhu, Angew. Chem., Int. Ed., 2004, 43, 5040–5045 CrossRef CAS.
- B. G. Greenland, S. Burattini, W. Hayes and H. M. Colquhoun, Tetrahedron, 2008, 64, 8346–8354 CrossRef CAS.
- L. J. D'Souza and U. Maitra, J. Org. Chem., 1996, 61, 9494–9502 CrossRef CAS.
- D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC.
- E. M. Pérez and N. Martín, Chem. Soc. Rev., 2008, 37, 1512–1519 RSC.
- E. M. Pérez, L. Sánchez, G. Fernández and N. Martín, J. Am. Chem. Soc., 2006, 128, 7172–7173 CrossRef CAS.
- E. M. Pérez, A. L. Capodilupo, G. Fernández, L. Sánchez, P. M. Viruela, R. Viruela, E. Ortí, M. Bietti and N. Martín, Chem. Commun., 2008, 4567–4569 RSC.
- G. Fernández, E. M. Pérez, L. Sánchez and N. Martín, Angew. Chem., Int. Ed., 2008, 47, 1094–1097 CrossRef CAS.
- M. A. Herranz, C. Ehli, S. Campidelli, M. Gutiérrez, G. L. Hug, K. Ohkubo, S. Fukuzumi, M. Prato, N. Martín and D. M. Guldi, J. Am. Chem. Soc., 2008, 130, 66–73 CrossRef.
- C. Ehli, D. M. Guldi, M. A. Herranz, N. Martín, S. Campidellici and Maurizio Prato, J. Mater. Chem., 2008, 18, 1498–1503 RSC.
- J.-Y. Balandier, M. Chas, P. I. Dron, S. Goeb, D. Canevet, A. Belyasmine, M. Allain and M. Sallé, J. Org. Chem., 2010, 75, 1589–1599 CrossRef CAS.
- T. Poulsen, K. A. Nielsen, A. D. Bond and J. O. Jeppesen, Org. Lett., 2007, 9, 5485–5488 CrossRef CAS.
- V. A. Azov, R. Gómez and J. Stelten, Tetrahedron, 2008, 64, 1909–1917 CrossRef CAS.
- M. Skibiński, R. Gómez, E. Lork and V. A. Azov, Tetrahedron, 2009, 65, 10348–10354 CrossRef CAS.
- H. Iwamoto, Y. Hidaka and Y. Fukazawa, Tetrahedron Lett., 2008, 49, 277–280 CrossRef CAS.
- K. A. Nielsen, W.-S. Cho, J. O. Jeppesen, V. M. Lynch, J. Becher and J. L. Sessler, J. Am. Chem. Soc., 2004, 126, 16296–16297 CrossRef CAS.
- K. A. Nielsen, W.-S. Cho, G. H. Sarova, B. M. Petersen, A. D. Bond, J. Becher, F. Jensen, D. M. Guldi, J. L. Sessler and J. O. Jeppesen, Angew. Chem., Int. Ed., 2006, 45, 6848–6853 CrossRef CAS.
- J. S. Park, F. Le Derf, C. M. Bejger, V. M. Lynch, J. L. Sessler, K. A. Nielsen, C. Johnsen and J. O. Jeppesen, Chem.–Eur. J., 2010, 16, 848–854 CAS.
- A. Petitjean, R. G. Khoury, N. Kyritsakas and J.-M. Lehn, J. Am. Chem. Soc., 2004, 126, 6637–6647 CrossRef CAS.
- M. Barboiu, E. Petit and G. Vaugnan, Chem.–Eur. J., 2004, 10, 2263–2270 CrossRef CAS.
- J. P. Plante and T. E. Glass, Org. Lett., 2006, 8, 2163–2166 CrossRef CAS.
- S. Ulrich and J.-M. Lehn, Chem.–Eur. J., 2009, 15, 5640–5645 CrossRef CAS.
- S. Ulrich, A. Petitjean and J.-M. Lehn, Eur. J. Inorg. Chem., 2010, 13, 1913–1928.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- S. E. Wheeler and K. N. Houk, J. Chem. Theory Comput., 2009, 5, 2301–2312 CrossRef CAS.
- T. Schrader, Chem.–Eur. J., 1997, 3, 1537–1541 CAS.
- S. T. Weiss, N. R. McIntyre, M. L. McLaughlin and D. J. Merkler, Drug Discovery Today, 2006, 11, 819–824 CrossRef CAS.
- P. Talbiersky, F. Bastkowski, F. G. Klarner and T. Schrader, J. Am. Chem. Soc., 2008, 130, 9824–9828 CrossRef CAS.
- M. Kirsch, P. Talbiersky, J. Polkowska, F. Bastkowski, T. Schaller, H. de Groot, F. G. Klarner and T. Schrader, Angew. Chem., Int. Ed., 2009, 48, 2886–2890 CrossRef CAS.
Footnote |
| † In memory of Professor Michel Jubault. |
| This journal is © The Royal Society of Chemistry 2011 |