Engineered hydrogen-bonded polymer multilayers: from assembly to biomedical applications
Georgina K.
Such
,
Angus P. R.
Johnston
and
Frank
Caruso
*
Centre for Nanoscience and Nanotechnology, Department of Chemical and Biomolecular Engineering, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: fcaruso@unimelb.edu.au
First published on 30th September 2010
Abstract
Over the last two decades the layer-by-layer (LbL) assembly technique has become a highly versatile platform for the synthesis of nanoengineered thin films and particles. The widespread need for highly functional and responsive materials for applications in biomedicine—such as drug and gene delivery—has recently led to considerable efforts in the assembly of LbL materials, particularly films that can be subsequently stabilised and functionalised through a range of chemistries. In this tutorial review, recent developments in hydrogen-bonded LbL-assembled materials will be discussed, focusing on the design of materials with enhanced stimuli-responsive characteristics. Emphasis will be given to materials engineered for biomedical applications, specifically films/capsules that afford controlled loading and release of therapeutic cargo for application in vitro and in vivo.
 Georgina K. Such | Georgina K. Such completed her PhD at the University of New South Wales under the supervision of Professor Tom Davis and Dr Richard Evans (CSIRO Molecular and Health Technologies). Her research focused on the use of living radical polymerisation to control photochromic properties. Since the completion of her PhD she has worked as a postdoctoral researcher in the Department of Chemical and Biomolecular Engineering at the University of Melbourne. Her research interests include polymer design, nanostructured materials and click chemistry. |
 Angus P. R. Johnston | Angus P. R. Johnston received his PhD on the preparation of novel materials for rapid DNA sequencing in 2006 from the University of Queensland under the supervision of Professor Matt Trau. He is currently a postdoctoral research fellow in the Department of Chemical and Biomolecular Engineering at the University of Melbourne. His current research interests include targeted drug delivery, nanoengineered thin films and molecular sensing techniques. |
 Frank Caruso | Professor Frank Caruso is Director of the Centre for Nanoscience and Nanotechnology in the Department of Chemical and Biomolecular Engineering at the University of Melbourne, and is an Australian Research Council Federation Fellow. He received his PhD degree in 1994 from the University of Melbourne, and then moved to the CSIRO Division of Chemicals and Polymers in Melbourne. He was an Alexander von Humboldt Research Fellow and then group leader at the Max Planck Institute of Colloids and Interfaces (Berlin, Germany) from 1997–2002. His research focuses on polymers at interfaces, colloidal systems, biomaterials and nanocomposite thin films. |
1. Introduction
The interface between nanotechnology and biomedical science has converged significantly over the last decade, largely driven by the potential of nanoengineered materials to revolutionise future healthcare and medicine.1–4 Central to advances in this emerging and rapidly growing field is the ability to engineer responsive materials with tailored properties. For drug and gene delivery, this requires versatile techniques that yield high-drug-content materials that can retain and specifically release the therapeutic cargo in response to specific stimuli in vivo. The materials must also be engineered for stealth characteristics in vivo to optimise the delivery of the therapeutic payload. A prominent method used to assemble films over the last two decades is based on layer-by-layer (LbL) assembly,5,6 where films can be formed through the alternate adsorption of materials through complementary interactions. Although originally applied to charged materials,7 where assembly is driven by electrostatic interactions, more recent studies have exploited hydrogen bonding,8,9DNA hybridisation,9 sequential chemical reactions10,11 and metal–ligand complexation11 to prepare multilayer films. Such versatility allows a diverse range of materials to be assembled into thin films, including synthetic or biological polymers (charged and/or uncharged), inorganic or metal nanoparticles, proteins and small molecules. Furthermore, LbL assembly can be adapted to a range of substrates/templates with different compositions and morphology, including membranes, fibres and particles (Fig. 1).12 The LbL assembly of materials on particles and the subsequent formation of capsules13,14 are of particular interest, principally because of the promise of these carrier particles in therapeutic delivery applications.15 A main feature of LbL assembly is that the properties of the resulting materials—films or capsules—can be precisely tuned by controlling the assembly conditions, the materials adsorbed at each layer, and subsequent processing steps for stabilisation and functionalisation. The expectation that such materials will find increasing application in biomedicine is only now starting to emerge.15–18 In this tutorial review, we highlight recent advances in the assembly and engineering of hydrogen-bonded films/capsules. We focus on hydrogen-bonded systems9 because they can be designed to exhibit stimuli-responsive properties under physiologically-relevant conditions, and allow the incorporation of uncharged polymers, which is important for developing stealth materials. We highlight the loading and release of therapeutic cargo from these materials with special emphasis on the application of these systems for drug/gene delivery, both in vitro and in vivo.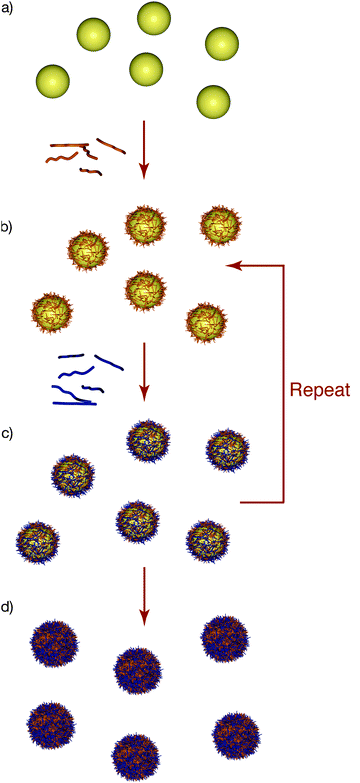 | ||
| Fig. 1 Layer-by-layer (LbL) assembly of polyelectrolytes on particles. (a) Particles with an inherent charge are incubated in a solution of oppositely charged polyelectrolyte. (b) Following several centrifugation/rinsing steps to remove excess polyelectrolyte, the coated particles exhibit charge reversal, which facilitates the adsorption of another layer of oppositely charged polyelectrolyte. (c) The process is repeated until the desired number of layers (and hence thickness) is achieved. (d) The particles can be subsequently removed to yield polyelectrolyte capsules. | ||
2. Layer-by-layer film assembly
2.1 Background
One of the key advantages of the LbL technique is the precision with which each layer of the film can be engineered. This can be achieved by varying either (a) the materials used, (b) the number of layers deposited, or (c) the conditions of deposition.7 For example, in charged polymer (polyelectrolyte) systems, increasing the salt concentration can significantly affect the thickness and density of the films, as the salt ions shield the polyelectrolyte charges, leading to a more coiled polyelectrolyte conformation and thicker layers.19 In the case of weak (i.e., pH-dependent) polyelectrolytes, film thickness and morphology can be tuned by both the salt concentration and pH.20 While there are many useful studies based on multilayers assembled from oppositely charged polyelectrolytes,18 it is challenging to design materials that are responsive within biologically-relevant conditions (e.g., pH 4–8 or at 37 °C) from these polymers. In addition, electrostatically assembled films are typically highly charged, which can lead to issues with fouling in biological systems. Therefore, recent interest has been given to the development of films based on other interactions, most notably hydrogen bonding.Hydrogen-bonded films are well known to be responsive to temperature and pH.21 However, early reported hydrogen-bonded multilayers are typically only stable at low pH,21 thereby limiting their in vitro and in vivo applicability. Recently, the use of a wider range of polymer building blocks and a variety of crosslinking techniques to stabilise such films has significantly increased the versatility of hydrogen-bonded materials. These significant advances in material design have led to developments in hydrogen-bonded films in areas such as drug delivery15 and microreactors.22
2.2 Fundamentals of hydrogen-bonding assembly
Hydrogen-bonding multilayer film assembly is based on the alternate deposition of polymers containing a hydrogen bond acceptor and a hydrogen bond donor, respectively. Commonly used acceptors include poly(ethylene glycol) (PEG), where the oxygen atoms on the polymer backbone act as hydrogen bond acceptors, and poly(N-vinylpyrrolidone) (PVPON) where the carbonyl group acts as the acceptor (Fig. 2). The most commonly used donors include polyacids, such as poly(methacrylic acid) (PMA) and poly(acrylic acid) (PAA), which contain carboxylic acid groups. When protonated, the carboxylic acid groups act as hydrogen bond donors. Early studies on hydrogen-bonded multilayers formed on planar surfaces were reported by the Rubner and Zhang groups.23,24 The study by Stockton and Rubner examined the use of polyaniline (PAni) alternately layered with uncharged polymers, such as PVPON, poly(vinyl alcohol) (PVA), poly(acrylamide) (PAAm), and PEG.23 Zhang et al. investigated multilayer systems of PAA and poly(vinylpyridine) (PVP) to demonstrate hydrogen bonding could be used to facilitate multilayer growth.24 In 2003, hydrogen-bonding assembly was extended to particle templates by using PVPON and m-methylphenol-formaldehyde as the hydrogen bond acceptor and donor, respectively.25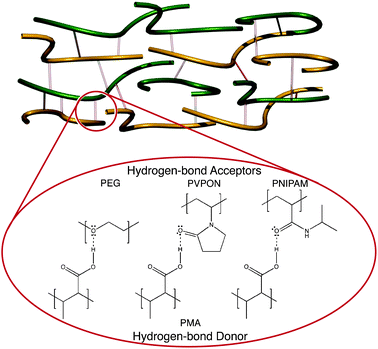 | ||
| Fig. 2 Examples of hydrogen bond interactions used in the formation of multilayer films: protonated PMA acts as a hydrogen bond donor to a range of hydrogen bond acceptors such as PEG, PVPON and PNIPAM. | ||
Compared to films based on electrostatic interactions, the pH range where hydrogen-bonding polymers can interact and form stable films is limited. This range is dependent on the pKa of the component polymers, as well as the strength of the interaction between the hydrogen-bonding pairs. For example, PMA/PEG and PMA/PVPON hydrogen-bonded multilayer films have vastly different disassembly properties: the former disassemble at pH ≈ 4, while the latter are significantly more stable and disassemble at pH ≈ 6.4.21 The strength of the hydrogen-bonding interactions also plays an important role in the thickness and morphology of the multilayer films assembled.21
Film assembly mediated by hydrogen bonding allows a range of uncharged materials to be incorporated into the multilayers. Polymers such as PEG, PVPON and PVA are important because they have well-documented low-fouling properties in vivo and thus are attractive polymers for biomedical applications. Another class of materials that can be incorporated in hydrogen-bonded multilayers are temperature-responsive macromolecules such as poly(N-isopropylacrylamide) (PNIPAM), poly(N-vinylcaprolactam) (PVCL) and poly(vinyl methyl ether) (PVME). These polymers have a low critical solution temperature (LCST), the temperature at which the polymers undergo a dramatic change in solubility.26 The most common example is PNIPAM, which has a LCST of 32 °C, which can be readily tuned by copolymerisation with a hydrophilic or hydrophobic comonomer. Incorporation of these polymers into LbL multilayer films offers the potential to engineer temperature-responsive films and capsules that undergo transitions close to body temperature (37 °C), making them of interest for biomedical applications. The thickness of multilayers based on thermally responsive polymers can be tuned by temperature during polymer deposition27 or after fabrication of the films.28
Although hydrogen-bonding film assembly has been well demonstrated for a variety of synthetic polymers such as PAA, PMA and PVPON, recent interest has also focused on the use of biopolymers. It is well known that DNA forms secondary and tertiary structures based on its capacity to hydrogen bond. These interactions can be exploited to assemble multilayer films of DNA29 and PNA (peptide nucleic acids).30 For these films, the oligonucleotides are engineered so that ‘sticky’ single-stranded ends are present on each oligomer, which facilitate multilayer film assembly based on the programmable interactions of specific DNA sequences. DNA films and capsules formed via this process have significantly different shrinking and swelling properties,31 depending on the type, length and order of the nucleic acid sequences used (e.g., homopolymers, diblock copolymers).
Hydrogen-bonded multilayers offer many different avenues for the development of responsive films and capsules. Firstly, the ability of these materials to disassemble within a mild pH range offers new possibilities for therapeutic delivery applications, as this disassembly pH can be readily tuned by varying the hydrogen-bonding pairs or the assembly conditions used. Secondly, stimuli-responsive swelling or cargo release can be achieved by incorporating polymers such as PNIPAM, which is temperature-responsive, and PMA, which is pH-sensitive. The versatility of hydrogen-bonded multilayers has allowed the development of materials with a range of properties tailored to biomedical applications. These include the potential for controllable disassembly, the capability to respond to stimuli such as pH and temperature, and the ability to load and release cargo specifically.
2.3 Approaches for stabilising hydrogen-bonded films
The inherent instability of hydrogen-bonded films can be utilised for some biomedical applications;32 however, many applications in this area require stabilisation of the films, particularly within the physiological pH range (pH 6–8). To achieve this, a number of covalent crosslinking approaches have been investigated, including chemical,33,34 thermal35 and photochemical36 techniques. The following section will highlight a number of crosslinking methods employed, focusing predominantly on chemical approaches.Hydrogen-bonded films are typically formed by the assembly of a polyacid with various hydrogen bond acceptors.8 In such systems, when the pH is raised above the pKa of the polyacid, the carboxylic acid group is deprotonated, thus disrupting the hydrogen bonding. When, for example, the polyacid is crosslinked, elevation of the pH above the pKa of the polyacid disrupts the hydrogen bonding and releases the second non-crosslinked component.8 This approach has been used to prepare a range of single-component, crosslinked films that (without additional film engineering) do not disassemble.8 However, subjecting them to stimuli such as pH or salt can lead to changes in film properties because of changes in the charge density of the polymers comprising the films (e.g., polyacids). For example, capsules can undergo pH-tuneable swelling and contraction due to the highly charged polymeric wall. This approach has been used extensively to form responsive polyacid films and capsules.37
One common crosslinking approach is based on reacting polyacids, such as PAA or PMA, with ethylenediamine using carbodiimide chemistry (Fig. 3). Kozlovskaya and Sukhishvili37 have extensively studied such polyacid systems assembled alternately with PVPON or PVCL. The synthesis of two-component capsules was also investigated by modifying either PVCL or PVPON with amines to crosslink these polymers within the multilayer. Both single and two-component capsules respond reversibly to changes in pH, causing a variation in size,37 permeability38 and mechanical stiffness.39 The use of thiol–ene chemistry to photochemically crosslink PMA multilayers was also demonstrated.40
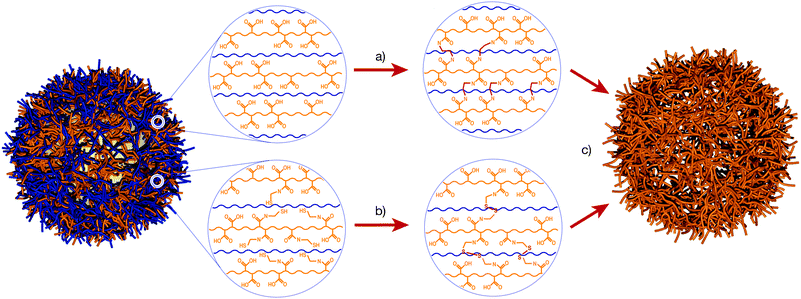 | ||
| Fig. 3 Examples of approaches used to stabilise hydrogen-bonded films. (a) Crosslinking of a polyacid (e.g., PMA) with ethylenediamine, which reacts with the carboxylic acid groups on the PMA. (b) Thiol-modified PMA (PMASH) can be oxidised to form redox-cleavable disulfide linkages. (c) When exposed to high pH, the PMA is deprotonated, disrupting the hydrogen bonding with the PVPON. The crosslinked PMA retains its structure and the PVPON is expelled from the film. PMA – orange, PVPON – blue. | ||
Carbodiimide chemistry can also be used to crosslink hydroxyl and carboxylic acid groups. In recent work, capsules were synthesised using hydroxypropylcellulose (HPC) layered alternately with PAA on 300 nm-diameter silica templates, followed by crosslinking using carbodiimide chemistry.41Capsules with both high and low degrees of crosslinking were formed. Both systems showed good stability up to pH 10, with increased swelling observed for the capsules with a low degree of crosslinking.
An alternative approach to capsule stabilisation was recently introduced.42,43 Here, thiol-modified PMA (PMASH) was assembled with PVPON to form PMASH/PVPON films. The thiol moieties were then crosslinked postassembly by oxidation of the thiols into disulfides using chloramine T (Fig. 3). At physiological pH, the hydrogen bonding between the deprotonated PMA and the PVPON is disrupted, and single-component PMASH capsules are formed. The disulfide linkages are expected to be stable in oxidising conditions, such as the bloodstream, and to disassemble in reducing conditions, such as the cytoplasm of the cell. This type of crosslinking offers the potential to design capsules that can degrade specifically in vivo (see later for details).9 Recently, a new approach to the synthesis of PMASH capsules was described, removing the need for harsh oxidising agents such as chloramine T, which can potentially damage sensitive encapsulated cargo. This approach was based on activation of thiol groups within the multilayer and then crosslinking using thiol-functionalised polymers, such as PEG and poly(hydroxypropyl methacylamide) (PHPMA).44
A general approach to stabilising hydrogen-bonded multilayer films has been demonstrated by combining ‘click’ chemistry with small molecule crosslinkers (Fig. 4).45,46 The hydrogen-bonding polymers are modified with a click functional moiety (such as an alkyne) and following film assembly are crosslinked with a diazide crosslinker. This technique has been demonstrated for a number of polymer systems (including poly(glutamic acid) (PGA) and PVPON) and has the significant advantage of enabling control over the degradation of the films by the use of different crosslinking molecules. In one study, alkyne-modified PVPON (PVPONAlk) was assembled alternately with PMA and then crosslinked with a diazide crosslinker that contained a disulfide linkage. Incubation of these capsules at pH 7 resulted in PMA removal, leaving single-component PVPON capsules. Such hollow capsules combine the advantages of low-fouling properties and degradation (within a cellular environment).45 A similar approach was also used to form PGA capsules by assembling PGAAlk with PVPON and then crosslinking using a non-degradable diazide linker.46 When incubated at pH 7, the PVPON was expelled, yielding single-component PGA capsules.
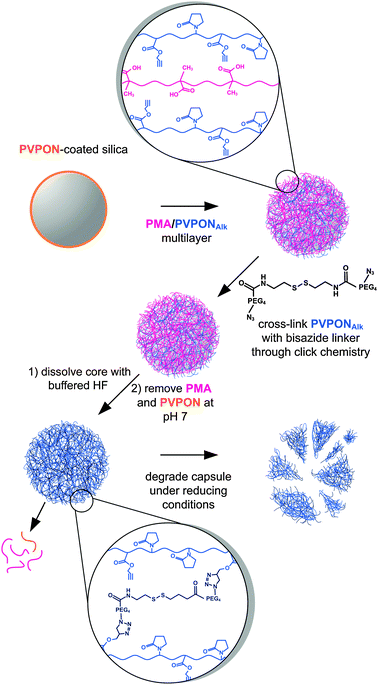 | ||
| Fig. 4 Hydrogen-bonded PMA/PVPONAlk multilayer assembly on colloidal particles. Free alkyne groups can then be used to crosslink the film using a diazide linker which, in this case, contains a disulfide bond. After removal of the template and release of PMA at pH 7, single-component, crosslinked PVPON capsules are formed.45 | ||
Films assembled from DNA typically show decreased stability at physiological conditions; however, techniques to stabilise these films have been developed.47DNA films can be engineered so that after assembly, single-stranded regions of DNA remain within the film structure. A crosslinking single-stranded DNA sequence can be added to the film that can hybridise to multiple single-stranded regions, thus crosslinking the film. Films assembled in this way are stable in physiological and low salt conditions.47
3. Towards applications of hydrogen-bonded multilayers
3.1 Encapsulation
Loading a therapeutic cargo into nanostructured LbL films is fundamental for developing these systems for drug and gene delivery applications.15 A wide range of molecules, including small anticancer drugs, oligonucleotides, plasmids, peptides and whole proteins, have been encapsulated within hydrogen-bonded LbL films. The vastly different chemical and physical properties of such therapeutics highlight the need for a number of different approaches for the successful encapsulation of each cargo. In particular, low molecular weight species (such as anticancer drugs) can freely diffuse through LbL multilayer films and therefore require advanced techniques for loading.48 The encapsulation techniques fall broadly into three categories: incorporating the cargo within the film during film assembly; postloading preformed films/capsules; or preloading the cargo onto the template (either planar or colloidal).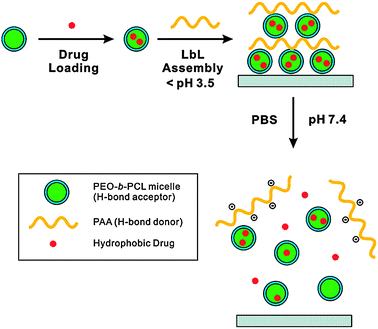 | ||
| Fig. 5 Polymer micelles with an outer layer of PEG act as hydrogen bond acceptors for PAA in multilayer film assembly. Hydrophobic drugs can be loaded into the micelle cores, and are only released when the pH is increased to 7.48 | ||
Similar loading techniques have been applied to the assembly of films on particles to form microcapsules containing drug-loaded liposomes. For example, liposomes have been incorporated into PMASH/PVPON multilayer films and capsules by using PMA modified with a cholesterol group (PMAC) as an anchor layer.52 The cholesterol moiety inserts into the hydrophobic shell of the liposomes, enabling the PMAC to assemble on top of the liposomes and providing a PMA surface for subsequent film growth. This approach has been used to demonstrate that both enzymes (β-lactamase)52 and anticancer drugs (thiocoraline)53 can be encapsulated within PMASH capsules and retain their activity.
An alternative technique for encapsulating low molecular weight cargo is to covalently attach the drug to the polymer through a degradable linkage. This has been demonstrated by covalently attaching cysteine-modified peptides to PMASH through a cleavable disulfide linkage and using this peptide-modified polymer as a prelayer in the assembly of PMASH capsules.54 In a similar approach, doxorubicin (DOX) has been covalently attached to PGAAlk using carbodiimide chemistry, and this polymer–drug conjugate then assembled alternately with PVPON.46 These capsules were then crosslinked using a bifunctional azide through click chemistry to form DOX-loaded PGA capsules. Using this technique, the position and degree of DOX loading within the capsules can be readily tailored.
A general limitation with the postloading process is the large excess of cargo required during incubation, which may limit its applicability for expensive cargo such as DNA, peptides or non-abundant proteins.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 30 bp single-stranded DNA, ∼1000 800 bp double-stranded DNA and ∼300 3 kbp plasmids) to be encapsulated by a single DNA adsorption step. Furthermore, higher cargo loadings can be achieved using porous particle templates.60 Even higher loadings can be achieved by directly layering on cargo that crystallises into particles (e.g., protein crystals);56,61 however, this technique is limited by the ability to generate a crystalline form of the cargo and the polydispersity of the crystals formed.
000 30 bp single-stranded DNA, ∼1000 800 bp double-stranded DNA and ∼300 3 kbp plasmids) to be encapsulated by a single DNA adsorption step. Furthermore, higher cargo loadings can be achieved using porous particle templates.60 Even higher loadings can be achieved by directly layering on cargo that crystallises into particles (e.g., protein crystals);56,61 however, this technique is limited by the ability to generate a crystalline form of the cargo and the polydispersity of the crystals formed.
3.2 Degradation and release
Another important aspect of developing films/capsules for therapeutic delivery applications is engineering release of the cargo from the multilayer systems. The ability to control cargo release is fundamental, especially for biomedical applications, where materials need to be engineered to release therapeutics within specific regions (e.g., in cells) and/or in response to specific triggers. There are three main approaches used to address this challenge in hydrogen-bonded systems: (a) disassembly of the multilayer film; (b) incorporating responsive components within the film; or (c) using degradable crosslinking agents that result in film disassembly.Sukhishvili and Granick also investigated the use of different materials for the tuneable disassembly of LbL capsules.64 Initial work used PMA assembled with either PEG or PVPON to form responsive polyacid capsules; however, these films were not stable at physiological pH. Subsequent work demonstrated that the pH range where the hydrogen-bonded multilayers are stable could be increased significantly by the use of polyacids, such as poly(ethylacrylic acid) (PEA) or tannic acid (TA), due to the higher pKa of both materials.65 Films were assembled from TA (pKa ≈ 8.5) alternately deposited with a series of hydrogen bond acceptors, and disassembly was found to occur above pH 8. Sukhishvili and coworkers also demonstrated tuning of the disassembly pH of a weakly interacting hydrogen-bonding system, PVCL/poly(L-aspartic acid) (PLAA), by combining this system with a strong hydrogen-bonding pair.66 Similar films have been used to release components from particles, as demonstrated by Mehrotra et al. using PAA/PEG multilayers to release lysozyme over periods greater than two weeks.50
In another approach, release of therapeutic cargo can be engineered through the responsive characteristics of the hydrogen-bonded film, such as temperature or pH-induced swelling, causing an increase in permeability. In one such study, Zhu and Sukhishvili designed a multilayer system containing PNIPAM-based polymer micelles that swelled reversibly with temperature.28 It was demonstrated that this structural change could retard cargo release from the micelles by a factor of two.
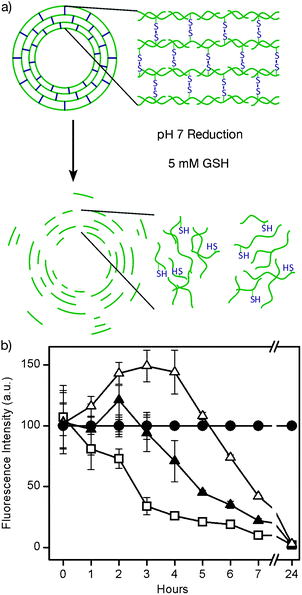 | ||
| Fig. 6 a) Schematic of PMASH capsules degraded by GSH. b) Degradation of PMASH capsules with varying degrees of thiolation upon exposure to 5 mM GSH. Open squares correspond to 10% thiolation, closed triangles to 15% thiolation, and open triangles to 20% thiolation. Closed circles correspond to capsules in PBS buffer.67 | ||
In related work, single-component PVPON capsules stabilised with cleavable disulfide bonds were formed by crosslinking PVPONAlk/PMA multilayers with a bifunctional azide linker containing a disulfide bond.45 The advantage of using a small linker is that the linker can be engineered for specific and controlled degradation. Examination of two linkers, one with a disulfide moiety and one without, showed that capsules stabilised with the disulfide linker underwent degradation in the presence of GSH, while the capsules with a PEG-based linker remained stable.
Enzymatically-induced degradation of films has also been investigated using DNA films (Fig. 7). Films assembled from DNA can have specific sequences, known as ‘cut sites’, engineered within the film. These regions are recognised by restriction enzymes,68 which hydrolyse the double-stranded DNA at specific points along the recognition sequence. Films and capsules with an EcoRI cut site have been shown to specifically degrade in the presence of the EcoRI enzyme, while films lacking the EcoRI sequence remained stable.68
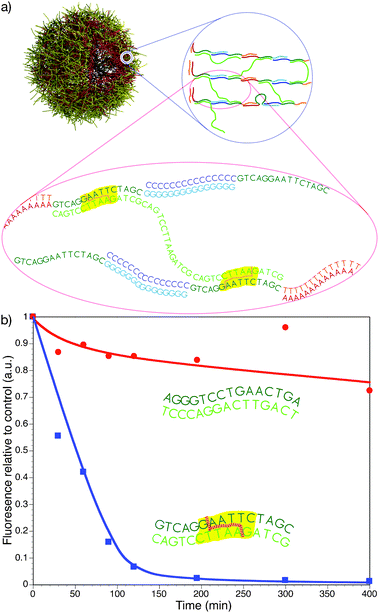 | ||
| Fig. 7 a) Schematic of an LbL-assembled triblock DNA capsule (A15X15G15/T15X15C15) (red–green–aqua and orange–green–blue, respectively) crosslinked with X15X15X15 (light green). The EcoRI cut sites are shown with yellow hatching. b) Degradation of 4-bilayer triblock DNA capsules containing EcoRI cut sites (squares) and capsules without EcoRI cut sites (circles).68 | ||
4. In vitro and in vivo studies
Over the last five years there has been a growing number of studies aimed at developing LbL-assembled delivery systems for applications in biomedicine. The following section highlights recent developments in this area.Rubner and coworkers have exploited the responsive ability of hydrogen-bonded multilayers to attach polymer patches to individual cells with the aim to use these systems for therapeutic or cell isolation applications.32 Multi-component, stratified films were assembled on a patterned planar surface, which was prepared using lithography so polymer assembly would only occur on 10–15 μm sized patches spaced approximately 50 μm apart. The films comprised of 3 stratified layers: a hydrogen-bonded ‘release’ region (assembled from PMA/PNIPAM); a ‘payload’ region (FITC-PAH/iron oxide nanoparticles); and a cell adhesion region (chitosan/hyaluronic acid). Both the payload and cell adhesion regions are held together by electrostatic interactions and are therefore stable across a wide pH and temperature range, although the hydrogen-bonded ‘release’ region of the film is only stable under more limited conditions. The PMA/PNIPAM layers interact and assemble at low pH (3); however, at higher pH (such as physiological pH) and below the LCST of PNIPAM (32 °C), the film disassembles. Above the LCST of PNIPAM, the interaction remains stable. Thus, if cells are incubated with the immobilised patches at 37 °C, the cells adhere to the adhesion layer of the patch. If the temperature is lowered to 4 °C, the interaction between the PMA and PNIPAM is disrupted and the ‘release’ region disassembles, releasing the patch and cells from the surface (Fig. 8). Using this technique, it has been demonstrated that the payload remains attached to the cells and does not significantly interfere with the cell viability or function. By incorporating magnetic nanoparticles into the patch, cells with a patch can be easily isolated from a mixed population of cells using a magnetic field.
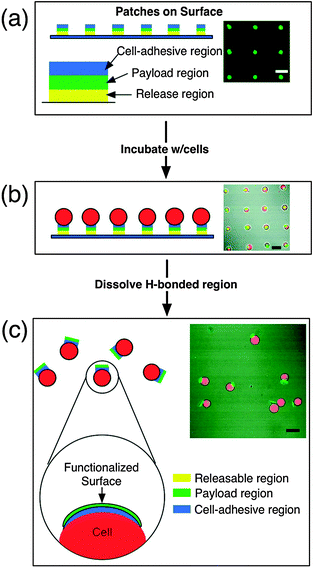 | ||
| Fig. 8 Schematic and confocal representations of: (a) an array of surface-bound patches spaced 50 μm apart (green fluorescence corresponds to the fluorescein in the payload region); (b) B-cell incubation and attachment of patches; c) after the temperature is reduced to 4 °C for 30 min, the patches are released from the surface while remaining attached to the cell membrane.32 | ||
Capsules assembled viahydrogen-bonding interactions have also been used to deliver therapeutic compounds.54,57 For effective drug delivery, it is critical that the encapsulated therapeutic can be released. As noted earlier, most hydrogen-bonded films are unstable in physiological conditions, so the films need to be stabilised to retain the cargo under physiological conditions, and release the cargo when internalised into cells. Relevant work in this area using PMASH capsules has demonstrated the delivery of immunologically active peptides both in vitro and in vivo and anticancer drugsin vitro.
Short polypeptides have significant potential as vaccines for a number of diseases, such as HIV and hepatitis C. However, these polypeptides are rapidly degraded by the body and as such have seen limited clinical success. By encapsulating the peptides within a polymer carrier, there is the potential to increase the lifetime of the peptide within the body to have a significant therapeutic effect. In recent work by De Rose et al., PMASH capsules have been shown to bind to, and be internalised by, dendritic cells (DCs) in whole human blood.54 DCs are professional antigen presenting cells that are key to generating effective immunity to disease. It was found that PMASH capsules effectively bind to DCs and other antigen presenting cellsin vitro and that they could successfully generate an immune response.
To gain a greater understanding of the immune response to the PMASH capsulesin vivo, whole ovalbumin (OVA) was encapsulated to study the presentation pathway (Fig. 9a and b).59 OVA contains peptide sequences that can be processed via both the MHC class I and class II pathways. Significantly higher class I and class II responses were observed for OVA-loaded capsules compared to free OVA; however, the class II response was significantly stronger than the class I response (Fig. 9c and d). MHC class II responses are typically associated with bacterial infections and the response seen with the capsules is consistent with the internalisation of large particulate material. For effective viral vaccines, an increased class I response would be desirable.
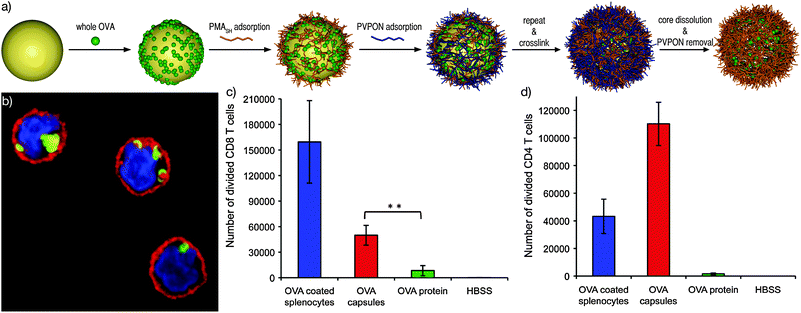 | ||
| Fig. 9 (a) Schematic of the loading and assembly of proteins into PMASH capsules. (b) CLSM image of dendritic cells with internalised capsules (nucleus—blue, cell membrane—red, capsules—green). Number of divided (c) CD8 T-cells (MHC class I response) and (d) CD4 T-cells (MHC class II response) treated with OVA-coated splenocytes (positive control), HBSS (saline—negative control), OVA-loaded capsules and free OVA protein.54,59 | ||
Stabilised capsules assembled viahydrogen bonding have also been used to deliver the anticancer drug DOX.46,57 As mentioned earlier, DOX is small enough to freely diffuse through polymer multilayers. To effect entrapment of DOX, two different techniques have been employed. Sivakumar et al. reported the encapsulation of DOX in oil-loaded PMASH capsules,57 while Ochs et al. covalently attached DOX to biodegradable PGA capsules stabilised using ‘click’ chemistry.46 Both techniques prevent leakage of DOX from the capsules and enable the encapsulation of ∼2 attograms and ∼1 picogram of DOX per capsule, respectively. When cancer cells were exposed to these capsulesin vitro, effective cell killing was observed. In the case of the oil-loaded PMASH capsules, the efficacy of the DOX-loaded capsules was more than 5 orders of magnitude greater than that of free DOX. Interestingly, the distribution of DOX within the cells when the drug was delivered using capsules was significantly different to that observed for free DOX. The free DOX accumulated almost exclusively in the nucleus of the cells, whereas DOX released from the oil-loaded PMASH capsules exhibited significantly higher DOX levels in the cytoplasm (Fig. 10).69
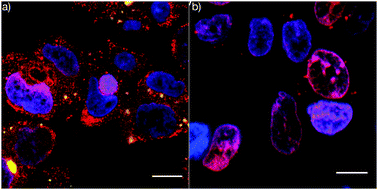 | ||
| Fig. 10 Confocal microscopy images of LIM1899 cancer cells treated with (a) DOX-loaded PMASH capsules and (b) free DOX. DOX (red), capsules (green) and nucleus (blue). Scale bar 10 μm.69 | ||
5. Conclusions and outlook
There has been significant development within the field of hydrogen-bonded multilayers in the last five to ten years, however, significant challenges still remain. The application of these materials in vivo has been only very recently explored and challenges associated with long-term biocompatibility, toxicity and the immunogenicity of these new materials are yet to be fully investigated. For particle-based delivery systems, maintaining a long circulation half-life within the bloodstream is a significant challenge. Organs like the liver, kidney and spleen are well adapted to filtering foreign material from the bloodstream and this clearance system represents a challenge that is yet to be addressed in detail. Nonetheless, the advances highlighted in this tutorial review have shown the potential of hydrogen-bonded films/capsules for effective therapeutic loading and release, and provides the impetus for further in vivo studies. The development of advanced methods for loading allows a range of therapeutics to be successfully encapsulated. Hydrogen-bonded films can now also be engineered to have specific degradation properties based on either control over polymer components or by using specifically engineered crosslinking approaches. In particular, the use of disulfide chemistry, in combination with other stabilisation approaches (e.g., click chemistry), is an important advance for the development of capsules to release therapeutics in cellular environments, as demonstrated by the examples highlighted. Building on such progress, the next five years are expected to deliver further significant advances in the design, assembly and application of hydrogen-bonded assemblies in biomedical applications.Notes and references
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS.
- J. R. Heath and M. E. Davis, Annu. Rev. Med., 2008, 59, 251–265 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- V. Wager, A. Dullaart, A. K. Bock and A. Zweck, Nat. Biotechnol., 2006, 24, 1211–1217 CrossRef CAS.
- G. Decher, J. D. Hong and J. Schmitt, Makromol. Chem., Macromol. Symp., 1991, 46, 321–327 Search PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- Multilayer Thin Films, ed. G. Decher and J. Schlenoff, Wiley-VCH, Weinheim, 2003 Search PubMed.
- E. Kharlampieva, V. Kozlovskaya and S. A. Sukhishvili, Adv. Mater. (Weinheim, Ger.), 2009, 21, 3053–3065 CrossRef CAS.
- J. F. Quinn, A. P. R. Johnston, G. K. Such, A. N. Zelikin and F. Caruso, Chem. Soc. Rev., 2007, 36, 707–718 RSC.
- D. E. Bergbrieter and K.-S. Liao, Soft Matter, 2009, 5, 23–28 RSC.
- W. Tong and C. Gao, J. Mater. Chem., 2008, 18, 3799–3812 RSC.
- Y. Wang, A. S. Angelatos and F. Caruso, Chem. Mater., 2008, 20, 848–858 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS.
- E. Donath, G. B. Sukhorukov, F. Caruso, S. A. Davis and H. Möhwald, Angew. Chem., Int. Ed., 1998, 37, 2202–2205 CrossRef.
- A. L. Becker, A. P. R. Johnston and F. Caruso, Small, 2010 DOI:10.1002/smll.201000379.
- T. Boudou, T. Crouzier, K. Ren, G. Blin and C. Picart, Adv. Mater. (Weinheim, Ger.), 2010, 22, 441–467 CrossRef CAS.
- L. L. del Merceto, P. Rivera-Gil, A. Z. Abbasi, M. Ochs, C. Ganas, I. Zins, C. Sönnichsen and W. J. Parak, Nanoscale, 2010, 2, 458–467 RSC.
- B. G. De Geest, N. N. Sanders, G. B. Sukhorukov, J. Demeester and S. C. De Smedt, Chem. Soc. Rev., 2007, 36, 636–639 RSC.
- G. Decher and J. Schmitt, Prog. Colloid Polym. Sci., 1992, 89, 160–164 CAS.
- S. S. Shiratori and M. F. Rubner, Macromolecules, 2000, 33, 4213–4219 CrossRef CAS.
- E. Kharlampieva and S. A. Sukhishvili, J. Macromol. Sci., Polym. Rev., 2006, 46, 377–395 CAS.
- B. Städler, A. D. Price, R. Chandrawati, L. Hosta-Rigau, A. N. Zelikin and F. Caruso, Nanoscale, 2009, 1, 68–73 RSC.
- W. B. Stockton and M. F. Rubner, Macromolecules, 1997, 30, 2717–2725 CrossRef CAS.
- L. Wang, Z. Q. Wang, X. Zhang, J. C. Shen, F. Chi and H. Fuchs, Macromol. Rapid Commun., 1997, 18, 509–514 CrossRef CAS.
- Y. Zhang, Y. Guan, S. Yang, J. Xu and C. C. Han, Adv. Mater. (Weinheim, Ger.), 2003, 15, 832–835 CrossRef CAS.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS.
- J. F. Quinn and F. Caruso, Langmuir, 2004, 20, 20–22 CrossRef CAS.
- Z. Zhu and S. A. Sukhishvili, ACS Nano, 2009, 3, 3595–3605 CrossRef CAS.
- A. P. R. Johnston, E. S. Read and F. Caruso, Nano Lett., 2005, 5, 953–956 CrossRef CAS.
- A. L. Becker, A. P. R. Johnston and F. Caruso, Macromol. Biosci., 2010, 10, 488–495 CAS.
- A. P. R. Johnston and F. Caruso, Angew. Chem., Int. Ed., 2007, 46, 2677–2680 CrossRef CAS.
- A. J. Swiston, C. Cheng, S. H. Um, D. J. Irvine, R. E. Cohen and M. F. Rubner, Nano Lett., 2008, 8, 4446–4453 CrossRef CAS.
- S. Y. Yang, D. Lee, R. E. Cohen and M. F. Rubner, Langmuir, 2004, 20, 5978–5981 CrossRef CAS.
- V. Kozlovskaya, S. Ok, A. Sousa, M. Libera and S. A. Sukhishvili, Macromolecules, 2003, 36, 8590–8592 CrossRef CAS.
- S. Y. Yang and M. F. Rubner, J. Am. Chem. Soc., 2002, 124, 2100–2101 CrossRef CAS.
- J. Chen and W. Cao, Chem. Commun., 1999, 1711–1712 RSC.
- V. Kozlovskaya and S. A. Sukhishvili, Macromolecules, 2006, 39, 5569–5572 CrossRef CAS.
- V. Kozlovskaya, A. Shamaev and S. A. Sukhishvili, Soft Matter, 2008, 4, 1499–1507 RSC.
- N. Elsner, V. Kozlovskaya, S. A. Sukhishvili and A. Fery, Soft Matter, 2006, 2, 966–972 RSC.
- L. A. Connal, C. R. Kinnane, A. N. Zelikin and F. Caruso, Chem. Mater., 2009, 21, 27–30 CrossRef.
- G. Guan, Y. Zhang, T. Zhou and S. Zhou, Soft Matter, 2009, 5, 842–849 RSC.
- A. N. Zelikin, J. F. Quinn and F. Caruso, Biomacromolecules, 2006, 7, 27–30 CrossRef CAS.
- A. N. Zelikin, Q. Li and F. Caruso, Angew. Chem., Int. Ed., 2006, 45, 7743–7745 CrossRef CAS.
- S.-F. Chong, R. Chandrawati, B. Städler, J. Park, J. H. Cho, Y. Wang, Z. F. Zhia, V. Bulmus, T. P. Davis, A. N. Zelikin and F. Caruso, Small, 2009, 5, 2601–2610 CrossRef CAS.
- C. R. Kinnane, G. K. Such, G. Antequera-Garcia, Y. Yan, S. J. Dodds, L. M. Liz-Marzan and F. Caruso, Biomacromolecules, 2009, 10, 2839–2846 CrossRef CAS.
- C. J. Ochs, G. K. Such, Y. Yan, M. P. van Koeverden and F. Caruso, ACS Nano, 2010, 4, 1653–1663 CrossRef CAS.
- A. P. R. Johnston and F. Caruso, Small, 2008, 4, 612–618 CrossRef CAS.
- B. Kim, S. W. Park and P. T. Hammond, ACS Nano, 2008, 2, 386–392 CrossRef CAS.
- J. Zhang and D. M. Lynn, Adv. Mater. (Weinheim, Ger.), 2007, 19, 4218–4223 CrossRef CAS.
- S. Mehrotra, D. Lynam, R. Maloney, K. M. Pawelec, M. H. Tuszynski, I. Lee, C. Chan and J. Sakamoto, Adv. Funct. Mater., 2010, 20, 247–258 CrossRef CAS.
- Y. Zhao, J. Bertrand, X. Tong and Y. Zhao, Langmuir, 2009, 25, 13151–13157 CrossRef CAS.
- B. Städler, R. Chandrawati, A. D. Price, S.-F. Chong, K. Breheney, A. Postma, L. A. Connal, A. N. Zelikin and F. Caruso, Angew. Chem., Int. Ed., 2009, 48, 4359–4362 CrossRef CAS.
- L. Hosta-Rigau, B. Städler, Y. Yan, E. C. Nice, J. K. Heath, F. Albericio and F. Caruso, Adv. Funct. Mater., 2010, 20, 59–66 CrossRef CAS.
- R. De Rose, A. N. Zelikin, A. P. R. Johnston, A. Sexton, S.-F. Chong, C. Cortez, W. Mulholland, F. Caruso and S. J. Kent, Adv. Mater. (Weinheim, Ger.), 2008, 20, 4698–4703 CrossRef CAS.
- Z. Ding, Y. Guan, Y. Zhang and X. X. Zhu, Polymer, 2009, 50, 4205–4211 CrossRef CAS.
- A. Wang, C. Tao, Y. Cui, L. Duan, Y. Yang and J. Li, J. Colloid Interface Sci., 2009, 332, 271–279 CrossRef CAS.
- S. Sivakumar, V. Bansal, C. Cortez, S.-F. Chong, A. N. Zelikin and F. Caruso, Adv. Mater. (Weinheim, Ger.), 2009, 21, 1820–1824 CrossRef CAS.
- A. N. Zelikin, A. L. Becker, A. P. R. Johnston, K. L. Wark, F. Turatti and F. Caruso, ACS Nano, 2007, 1, 63–69 CrossRef CAS.
- A. Sexton, P. G. Whitney, S.-F. Chong, A. N. Zelikin, A. P. R. Johnston, R. De Rose, A. G. Brooks, F. Caruso and S. J. Kent, ACS Nano, 2009, 3, 3391–3400 CrossRef CAS.
- Y. Wang and F. Caruso, Chem. Mater., 2005, 17, 953–961 CrossRef CAS.
- F. Caruso, D. Trau, H. Möhwald and R. Renneberg, Langmuir, 2000, 16, 1485–1488 CrossRef CAS.
- S. S. Ono and G. Decher, Nano Lett., 2006, 6, 592–598 CrossRef CAS.
- A. Zhuk, S. Pavlukhina and S. A. Sukhishvili, Langmuir, 2009, 25, 14025–14029 CrossRef CAS.
- S. A. Sukhishvili and S. Granick, J. Am. Chem. Soc., 2000, 122, 9550–9551 CrossRef CAS.
- I. Erel-Unal and S. A. Sukhishvili, Macromolecules, 2008, 41, 3962–3970 CrossRef CAS.
- I. Erel-Unal and S. A. Sukhishvili, Macromolecules, 2008, 41, 8737–8744 CrossRef CAS.
- A. L. Becker, A. N. Zelikin, A. P. R. Johnston and F. Caruso, Langmuir, 2009, 25, 14079–14085 CrossRef CAS.
- A. P. R. Johnston, L. Lee, Y. Wang and F. Caruso, Small, 2009, 5, 1418–1421 CrossRef CAS.
- Y. Yan, A. P. R. Johnston, S. J. Dodds, M. M. J. Kamphuis, C. Ferguson, R. G. Parton, E. C. Nice, J. K. Heath and F. Caruso, ACS Nano, 2010, 4, 2928–2936 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |