Designing polymeric particles for antigen delivery
Stefaan
De Koker
ab,
Bart N.
Lambrecht
c,
Monique A.
Willart
c,
Yvette
van Kooyk
d,
Johan
Grooten
b,
Chris
Vervaet
a,
Jean Paul
Remon
a and
Bruno G.
De Geest
*a
aLaboratory of Pharmaceutical Technology, Department of Pharmaceutics, Ghent University, Ghent, Belgium
bDepartment of Molecular Biomedical Research, Ghent University, Ghent, Belgium
cLaboratory of Immunoregulation and Mucosal Immunology, Department of Pulmonary Medicine, Ghent University, Ghent, Belgium
dDepartment of Molecular Cell Biology and Immunology, Medical Centre, Vrije Universiteit Amsterdam, The Netherlands
First published on 9th November 2010
Abstract
By targeting dendritic cells, polymeric carriers in the nano to lower micron range constitute very interesting tools for antigen delivery. In this critical review, we review how new immunological insights can be exploited to design new carriers allowing one to tune immune responses and to further increase vaccine potency (137 references).
 Stefaan De Koker | Stefaan De Koker graduated as a bio-engineer from Ghent University in 2001. He started his PhD at the VIB, at the Department for Molecular Biomedical Research, which he obtained in 2009. Currently he is working as a post-doctoral associate affiliated to the Laboratory of Pharmaceutical Technology as well as the Laboratory of Molecular Immunology, both at Ghent University. The main focus of his work is to evaluate novel microparticulate systems for vaccine delivery. |
 Bruno G. De Geest | Bruno De Geest graduated as a chemical engineer in 2003 from Ghent University in Belgium, where he obtained his PhD in 2006. Following two years of post doctoral research at the University of Utrecht in The Netherlands he obtained a post doctoral fellowship at the Laboratory of Pharmaceutical Technology at Ghent University. His main interests are situated in the field of materials chemistry and immunology. |
1. Vaccine design: challenges of the 21st century
In an environment dominated by micro-organisms, vertebrates have developed multiple defence mechanisms to protect themselves against microbial attack. When the epithelial barrier gets breached by a micro-organism, the innate immune defence gets activated and induces a relatively non-specific antimicrobial and inflammatory defence. Besides this ancient and conserved innate immune defence, vertebrates have evolved a complex system of clonally expanding B-cell and T-cells that form the adaptive immune system, adding antigen specificity and immunological memory to the immune defence. The principal function of B cells is to produce antibodies upon antigen recognition via their B cell receptor. T cells recognize antigen-derived peptides presented by antigen-presenting cells via their T cell receptor. When activated by the appropriate signals, naïve T cells differentiate into effector T cells that subsequently exert their immunological role. CD4 T cells differentiate into T-helper (Th) cells which assist other white blood cells in their immune functions (e.g. activation of macrophages and cytotoxic T cells, promoting B cells to secrete antibodies, etc.) mainly by secreting cytokines. CD8 T cells in contrast differentiate into cytotoxic T cells (CTLs), which have the capacity to recognize and kill virally infected cells. Importantly, B-cells and T-cells also have the capacity to remember an encounter with an antigen, allowing them to react faster and more vigorous to re-exposure to the same antigen. This fundamental property of the adaptive immune system is called immunological memory and underlies the success of vaccination. By pre-exposing the immune system to either complete but weakened or killed pathogens, or (partially) purified immunogenic components of the pathogen, the immune system mounts a fast and strong response upon exposure to the native pathogen, ideally preventing illness. Since the pioneering work of Jenner and Pasteur over 200 years ago, vaccines have dramatically improved human health by preventing numerous infectious diseases and are now estimated to save 3 million lives annually.1 Nevertheless, many challenges still remain. First, there is an urgent need to develop effective but safe preventive vaccines against insidious pathogens such as HIV, Plasmodium (the causative agent of malaria), Dengue and Mycobacterium tuberculosis that affect millions of people. Second, given the high virulence of these pathogens, vaccines composed of live attenuated strains impose serious safety issues and are unlikely to become approved by the regulatory authorities. This is now enforcing the vaccination field to move towards entirely synthetic vaccines composed of recombinant antigens. Although much safer, such recombinant vaccines are far less immunogenic and will require the development of new adjuvants that can increase or modulate the adaptive immune response elicited to become effective. Third, the design of therapeutic vaccines that can induce strong cytotoxic T cell responses might lead to a successful immunotherapeutic treatments of cancer and chronic viral infections such as HIV and HCV.2Despite their tremendous impact on public health, vaccines have been developed largely on a trial and error basis. Only recently, it has become more and more clear how pathogens are recognized by the innate immune system, and perhaps even more importantly, how these initial interactions between pathogen and innate immune system shape the subsequent adaptive immune response.3 These insights have led to the realisation that although designing a vaccine starts with the choice of an appropriate antigen, the selection of the immune potentiator to activate the innate immune system and the way both antigen and immune potentiator are delivered to the immune system are equally important in determining the ultimate success of the vaccine. In this review, current knowledge on how dendritic cells (DCs) prime and modulate effector T cell responses is summarized. Then, we explore how this knowledge can be exploited to design better vaccines, with the focus on new materials and delivery strategies allowing us to present the antigen to the immune system in an optimal immunogenic way.
2. Dendritic cells: potent inducers of effector T cell responses
To prime a naïve T cell to become an effector T cell (Th or CTL) three signals are needed (Fig. 1). First, the antigen needs to be processed and presented as a peptide to the cognate T-cell receptor (TCR) by major histocompatibility complex (MHC) molecules at the cellular surface of professional antigen presenting cells (APCs). Two generally distinct pathways are used for presentation of antigens via MHCI and MHCII to respectively CD8 and CD4 T cells. The MHCI presenting pathway is present in almost all cell types, and is responsible for the processing and presentation of cytosolic proteins, which are cleaved by the proteasome, transported to the endoplasmatic reticulum (ER) and subsequently loaded onto MHCI molecules. By this, the internal proteome of the cell is made accessible for surveillance by cytolytic CD8 T cells, allowing them to recognize and kill virally infected and transformed cells. The MHCII processing pathway in contrast is restricted to professional APCs, including B cells, macrophages, DCs and probably also basophils. Endocytosed proteins are degraded in endo-lysosomal compartments, loaded onto MHCII and subsequently presented at the cell surface.4 Although B cells and macrophages can present antigens, they are far less efficient compared to DCs, which have the unique capacity of priming naïve T cells.5 As will be elaborated later on, DCs also have the capacity to present endocytosed antigens in combination with MHCI instead of MHCII, a feature called cross-presentation, which is essential for the induction of CTL responses against viruses and intracellular bacteria that do not infect DCs. In addition, cross-presentation is also crucial for inducing CTL responses against tumour cells.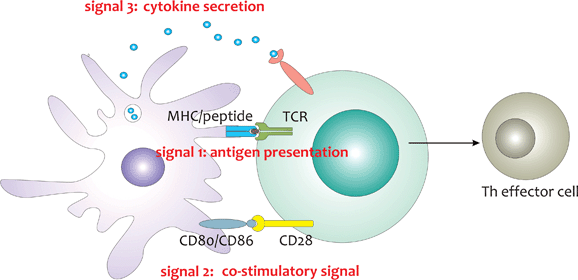 | ||
| Fig. 1 Initiation of effector T cell responses requires three signals. Stimulation of the TCR by MHC/peptide complexes delivers signal 1, interactions between co-stimulatory ligands on the APC and CD28 on the T cell provide signal 2 and the secretion of inflammatory cytokines that polarise T cell responses delivers signal 3. | ||
Besides antigen being presented by MHC molecules, priming of naïve T cells requires a co-stimulatory signal being delivered by the APC, which is mediated by interactions between the co-stimulatory ligands CD80 and CD86 on the DC and the receptor CD28 on the T cell. Expression of these co-stimulatory ligands is typically low on immature DCs present in peripheral tissues, making them weak APCs. Nevertheless, even in the absence of inflammation or infection, peripheral tissue DCs appear to undergo a functional maturation program, causing them to migrate to the lymph nodes and to express co-stimulatory ligands. Antigen presentation by these matured DCs to naïve T cells however leads to tolerance rather than immunity, by causing T cell anergy, clonal deletion or T cell differentiation towards immunosuppressive regulatory T cells, and constitutes an important mechanism for the maintenance of tolerance to self-antigens.6 Induction of effector T cell responses indeed requires the secretion of inflammatory and polarising cytokines.7 In case of microbial encounter, DCs become rapidly activated to upregulate co-stimulatory ligands and to secrete inflammatory cytokines via triggering of a group of germ-line encoded pattern recognition receptors (PRRs). These PRRs typically recognize conserved microbial associated molecular patterns (MAMPs) that are essential for microbial survival and thus difficult to alter.8–11 An overview of the most important PRRs and their microbial triggers is given in Fig. 2.
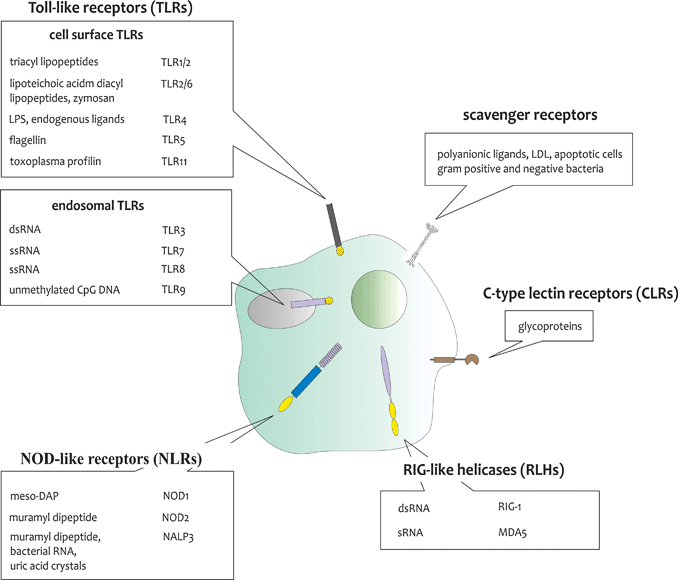 | ||
| Fig. 2 Overview of the most important classes of PRRs, their cellular localisation and microbial ligands. TLRs can be localized at the plasma membrane or the endosomal membrane, while NLRs and RLHs are located in the cytosol. Triggering of TLRs, NLRs and RLHs results in the initiation of potent pro-inflammatory and antimicrobial responses. CLRs and scavenger receptors are mainly involved in host–host interactions, but also recognize a wide variety of micro-organisms and have important roles in phagocytosis and antigen presentation. DCs have the capacity to integrate the signals received from triggering of different sets of PRRs and to translate them to induce the right type of response. | ||
Depending on the set of PRRs triggered, DCs secrete different cytokine profiles which will in turn largely determine the nature of the induced immune response. As a result, DCs link the recognition of a certain pathogen with the induction of the appropriate adaptive response by integrating the signals received from PRR triggering.12,13
The capacity of DCs to initiate different types of immune responses depending on the set of PRRs triggered is of crucial importance, as totally different types of immune defence are needed to combat distinct pathogen spectra. These different types of immunity are mainly regulated by different subsets of CD4 T helper cells. Th1 cells secrete IFN-γ and provide help to macrophages and cytotoxic T cells to kill intracellular pathogens. Th2 cells, in contrast, secrete IL-4 and IL-5 and combat helminth infections by recruiting mast cells and eosinophils.14 More recently, IL-17 secreting Th17 cells have been identified as a new subset of T-helper cells that mediate protection against extracellular bacteria and potentially fungi by recruiting neutrophils and stimulating the release of antimicrobial peptides.15–17 In response to a DC presenting the antigen as a MHCII/peptide complex, a naïve CD4 T cell can differentiate in either of the aforementioned T helper subsets depending on the cytokine microenvironment present. An overview on how different Th responses are elicited, and their roles in the immune defence are given in Fig. 3.
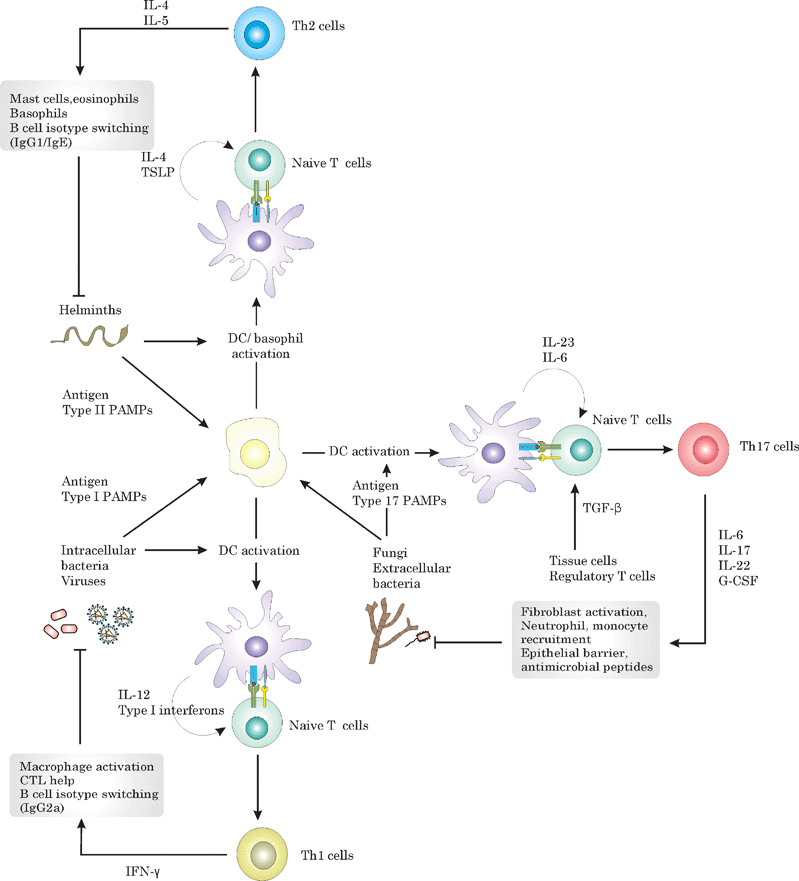 | ||
| Fig. 3 Overview on how APCs translate pathogen recognition into the induction of the appropriate Thelper (Th) response and of the role of different Th subsets in the immune defence against different pathogen spectra. Recognition of viruses and intracellular bacteria by DCs results in the secretion of IL-12 and type I interferons, which stimulate Th1 differentiation. By secreting IFN-γ, Th1 activate macrophages, provide help to CTLs and promote the secretion of neutralizing antibodies by B cells, which are all important to combat viruses and intracellular bacteria. Fungi and extracellular bacteria in contrast activate DCs to secrete IL-23, thereby promoting Th17 differentiation. Th17 cells have been demonstrated to enhance epithelial barrier function, and to recruit and activate neutrophils and monocytes. Helminth infections stimulate the generation of Th2 responses, which activate mast cells, basophils, eosinophils and provoke smooth muscle cell contraction in order to expel the helminth. Th2 differentiation is thought to require the cytokines IL-4 and TSLP, which might be basophile derived. | ||
3. Why vaccines need adjuvants
Adjuvants are generally defined as components that can enhance or modulate the intrinsic immunogenicity of an antigen in vivo. Most adjuvants have been derived empirically, based on their capacity to increase adaptive immune responses to co-delivered antigens but their mode of action has remained for a long time largely elusive.18 This empirical approach has given us only one FDA approved adjuvant for human use, aluminium hydroxide,19 and a second adjuvant approved by the EU, the oil-in-water emulsion MF59. Both these adjuvants are very useful for eliciting antibody responses, but largely fail to activate the cellular arm of the immune response, making them ineffective against many intracellular pathogens including Mycobacterium tuberculosis, HIV and malaria. Consequently, there is an urgent need to develop new adjuvants that also allow the induction of Th1, Th17 and CTL responses.Only during the last decade, due to our increased knowledge on how DCs initiate immune responses, we have begun to unfold the cellular and molecular mechanisms underlying the immune potentiating functions of adjuvants. Simplified, adjuvants can work by two modes of action: or they directly activate DCs or other innate immune cells, or they enhance antigen uptake and presentation by DCs.20 The identification of the crucial role of PRR triggering in DC activation and in the subsequent induction and steering of effector T cell responses has strongly boosted research in developing PRR agonists that mimic the immune stimulatory properties of natural PAMPs but display reduced toxicity during the last decade.21–25 Most of the PAMP mimics currently tested are agonists for diverse TLRs, and one of them, monophosphoryl lipid A, is now included in the human papillomavirus vaccine Cervarix (against cervical cancer)26 and in the improved hepatitis B vaccine Fendrix,27 demonstrating the tremendous potential of this approach. In contrast to aluminium salts, TLR agonists have the capacity to stimulate DCs to secrete IL-12 and type I interferons, thereby allowing the induction of Th1 and CTL responses, which might finally bring also certain intracellular pathogens in range.28–31
In addition to DC activation, strategies that enhance antigen targeting towards DCs and increase or modulate subsequent antigen presentation also bear the capacity to increase adaptive immune responses. Antigen targeting towards DCs can be obtained by coupling the antigens to antibodies or ligands specific for DC surface receptors,32 or alternatively by delivering antigens associated with particles in the 0.1–10 μm range.33,34 Particulates in this size range indeed mimic the dimensions of bacteria and viruses, to which DCs have evolved to react. Examples of particulate adjuvants are emulsions, liposomes, mineral salts, saponins, virus-like particles and polymeric carriers, which will be the main focus of this review.
4. Microparticulate antigen delivery
The new insights gathered in immunology have challenged drug delivery scientists to develop a myriad of delivery systems with the purpose of enhancing or modulating the induced immune response. As discussed in more detail in the sections below, there are several rationales to the formulation of antigen into a delivery system. Single shot formulations aim to replace the multiple booster injections often required to generate adequate immunity by a single administration.35 Other formulation strategies intend to enhance antigen targeting to DCs and to alter the way in which the processed antigen is subsequently presented to T-cells.36,37 In addition, systems are being designed to activate DCs by delivering immune potentiators together with antigens. Such strategies allow one to modulate the induced immune response through a rational design of one or multiple components of the delivery system. Predominantly, these systems involve microparticles which offer stable antigen encapsulation and subsequently gradually release the antigen, or allow the antigen to be processed by intracellular proteases following internalization by DCs.36 Although it is virtually impossible to go into detail on all different types of carrier systems that have been developed for vaccine delivery, we present here an overview of the main antigen encapsulation strategies that have been reported so far in literature.4.1 Generating microparticles for antigen delivery
Solvent evaporation is typically used in those cases where hydrophobic polymers are used as matrix particles.38,39 In a first step, an aqueous phase containing the antigen is emulsified in an immiscible organic solvent in which a non-water soluble polymer is dissolved. The obtained liquid is then emulsified a second time in an external aqueous phase containing a stabilizer. In this way a so-called double or water-in-oil-in-water emulsion is obtained which is subsequently stirred at a temperature above the boiling point of the organic solvent, allowing the solvent to evaporate and solid microparticles to form. The antigen that was in the innermost aqueous phase becomes in this way entrapped within a hydrophobic matrix. For such encapsulation procedures, different polymers have been used so far. One constant however is that they should be able to release their content upon administration to the body and, most preferably, readily release their payload upon internalization by antigen presenting cells. Therefore, polymers that are prone to erosion through hydrolytic degradation or that can respond to physico-chemical difference between the endolysosomal or cytoplasmatic compartments (i.e. slightly acidic and reductive medium) compared to the extracellular medium are highly desired.38 Further on in this review we will go more into detail on which type of polymers are the most suited for intracellular antigen release.
Although often applied, water-in-oil-in-water emulsification suffers from important drawbacks. The most significant one is the rather low encapsulation efficiency. Typically around 5% of the initial amount of antigen is retained within the microparticles due to leakage of the internal aqueous phase into the external aqueous phase upon emulsification. Moreover, the use of high shear forces, often produced by high energy ultrasound as well as the intimate contact between antigen and organic solvents might lead to protein denaturation which will evidently reduce the immune-activity of the antigen. Last but not least, an important issue which hampers large scale clinical applications is the difficulty to completely remove solvent traces from the microparticles as these are often retained within the hydrophobic polymer matrix.
An alternative approach to solvent evaporation which circumvents the use of organic solvents is spray-drying from an aqueous phase. Commonly, aqueous spray-drying involves spraying of an aqueous solution containing drug molecules in combination with one or more additional excipients which ensure stabilization of the drug molecules or which enhance the physico-chemical properties of the obtained dry powder. As mostly all the components are easily water soluble, a clear solution is obtained upon reconstitution in aqueous medium. Recently we have introduced a new concept involving spray-drying of stimuli-responsive polymers that are water soluble or which form tiny microscopic aggregates under the process conditions prior to atomization.40 After atomization and evaporation of the water, solid microparticles are obtained which retain their integrity upon reconstitution in water. Polymers well suited for such applications are, for example, thermosensitive polymers that are water soluble at low temperature but which precipitate at body temperature.41 Similar systems based on enzymatic degradable polymers could be anticipated as well.
The use of chemical synthesis is certainly a powerful tool to design new cross-linking methods and unique types of microparticles. It offers a tight control over the physico-chemical properties of the obtained microparticles, providing them with stimuli-responsive properties, tailored surface chemistry or well controlled antigen release rates. However, there are also serious disadvantages as side reactions between antigen and cross-linking moieties might occur as well. More specifically, amine and carboxylic acid moieties of the antigen arginine, glutamic acid, and aspartic acid residues might be affected by carbodiimide or aldehyde based cross-linking strategies. Also Michael addition between amines or thiols and (meth)acrylate moieties upon radical polymerisation are to be feared. To circumvent these issues the use of orthogonal chemistries such as ‘click’ chemistry might be a promising option.46
A milder approach to cross-link emulsion droplets is ionic cross-linking which commonly involves emulsification of the antigen together with a water-soluble polymer followed by the addition of a low or high molecular weight cross-linker. The most widespread example of ionic cross-linking is the calcium alginate system.47,48 Alginate is a polysaccharide consisting of alternating manuronic and glucuronic units, making the polymer abundantly substituted with carboxylic acid moieties. Addition of divalent calcium (Ca2+) ions induces instantaneous gelation through a so-called ‘zipper’-mechanisms involving the formation of ionic cross-links between the Ca2+ ions and carboxylic acid pairs, yielding a stable hydrogel network that can entrap proteins. These reaction conditions are very mild and therefore well suited for encapsulation of labile protein antigens. Decomposition of calcium alginate hydrogel microspheres takes place when they are transferred from a Ca2+-rich medium to a medium with physiological salt concentrations (e.g. 150 mM NaCl) by exchange of divalent Ca2+ ions by monovalent Na+ ions, which are not capable of retaining the network structure of the alginate gels. The simplicity of the calcium alginate system however has as major drawback that protein release rates are tough to be controlled. Release immediately starts upon contact with the physiological medium, thus prior to uptake of the microparticles by APCs, which means a considerable loss of antigen. Moreover, this remains an emulsion based encapsulation method involving the use of organic solvents as external phase as well as the use of high shear forces.
Besides vesicles which are self-assembled through hydrophobic interaction, electrostatic interaction is another strategy which has been intensively elaborated for antigen encapsulation. The most simple case is the inclusion of antigen in a polyelectrolyte complex through mixing the antigen with a polyelectrolyte bearing an opposite net charge. A well studied polymer for such application is chitosan which is a naturally occurring cationic polysaccharide that has been reported to open the tight junctions in the nasal-associated lymphoid tissue (NALT).53 Ovalbumin (OVA) is ubiquitous in numerous immunological studies and is due to its isoelectric point of 4 bearing a net anionic charge at physiological pH, which allows it to form electrostatic complexes, of several hundreds of nanometre in size, with the oppositely charged chitosan. This electrostatic self-assembly principle is not only restricted to anionic antigens as by carefully choosing the ratio between chitosan and polyphosphate molecules one is able to precipitate antigens regardless of their charge. However indisputedly attractive for its conceptual simplicity, such electrostatic precipitation procedure requires careful optimization for every case and definitely lacks a tight control over size and surface properties of the obtained microparticles.
A more recent, highly versatile strategy to design microparticles with an unmet degree of control over particle composition and physic-chemical properties is the Layer-by-Layer (LbL) technique.54 Sacrificial microparticles are coated with several polyelectrolyte bilayers of opposite charge, exploiting electrostatics as driving force for the deposition of each polyelectrolyte layer. Subsequently the microparticulate core templates are dissolved resulting in hollow capsules.55–57 As will be discussed in more detail further on in this review, the LbL technique is not only restricted to electrostatic interactions but also H-bonding has emerged as a highly promising strategy to design LbL capsules for drug delivery purposes.58,59 Both types of LbL capsules allow efficient antigen encapsulation using porous inorganic microparticles, such as calcium carbonate or silica, that are pre-filled with antigen prior to LbL coating. Subsequently the inorganic cores are extracted in an aqueous medium containing EDTA60 or HF,61 liberating the antigen into the hollow void of the capsules. These capsules are excellent in protecting their payload from degradation before reaching APCs as their target, while using polymers in the LbL coating that are prone to enzymatic or reductive degradation, release only after cellular uptake is assured.
4.2 Single shot vaccines
Current vaccines often require multiple injections to induce protective immunity. The need for multiple booster injections unfortunately often leads to patient non-compliance and logistic issues, especially in developing countries. As a result, there has been a lot of interest in developing antigen delivery systems that allow a controlled release of antigens in a pulsatile fashion to replace the classic prime-boost schedules. The polymer tested by far the most for such application is poly(lactic-co-glycolic) acid (PLGA). PLGA is a biodegradable and biocompatible polymer which has been used now for many years as resorbable suture material in humans. PLGA particles degrade by the non-enzymatic hydrolysis of the ester bonds in the backbone of the polymer, resulting in the release of lactic and glycolic acid, two acids that can be metabolized via the citric acid cycle. Early studies with PLGA as an encapsulation material focused mainly on the controlled long-term release of hormones and growth factors. These studies clearly demonstrated that the release of peptides/proteins from PLGA microspheres depends on several factors, such as the ratio lactic/glycolic acid, the molecular mass and hydrophobicity of the polymer, the type of emulsifier used and the size of the microspheres prepared. As a result, by carefully choosing the polymer's characteristics, one should be able to tailor the protein release as desired. In this view, a single injection with a mixture of hepatitis B surface antigen (HBsAg) containing PLGA microspheres with different degradation rates resulted in antibody titers comparable to three classical HBsAg/Alum injections.62 Similar results have been obtained with tetanus toxoid encapsulated in different PLGA microsphere preparations.63 Nevertheless, single shot PLGA vaccines suffer from significant drawbacks impeding their clinical application. First of all, release rates not only depend on particle intrinsic factors, but also on protein-specific factors such as molecular weight, hydrophobicity and charge.64 In addition, these large PLGA microspheres are generally prepared using a double emulsion process often resulting in low antigen encapsulation. Preparation of the microspheres also involves the use of chemical solvents such as dichloromethane or chloroform, which are not only highly toxic but also negatively affect protein stability as has been demonstrated for bacterial toxoids.65 Moreover, PLGA degradation results in an acidification of the injection spot, further contributing to protein denaturation. Even if protein stability can be improved by incorporating poorly soluble bases as Mg(OH)266,67 or protein stabilizers as trehalose,68 preparing antigen-loaded PLGA microspheres that release the antigens in the desired way remains a time-consuming and costly process that needs to be optimized for each antigen.69 Finally, also scaling-up has been proven difficult, further preventing industrial application.Besides PLGA, spherical hydrogels are also being explored for the controlled release of proteins. Such hydrogels might have significant value for the development of single shot vaccines, as their production does not involve the use of organic solvents and their degradation can be tightly controlled by varying the number of degradable cross-links. Whether such hydrogels can replace classical prime-boost schedules using Alum adsorbed antigens remains to be established. Recently, a novel approach to create single-shot vaccines, called self-exploding capsules,70–72 was explored by De Geest et al. These core–shell particles consisted of a degradable hydrogel core surrounded by a semi-permeable polyelectrolyte membrane. Upon degradation of the hydrogel core, an osmotic pressure builds up which finally ruptures the capsule membrane, resulting in a single release pulse. Fig. 4A shows a series of confocal snapshots taken at different time points during degradation. The capsules’ hydrogel core consists of polymerized methacrylated dextran whose methacrylate groups are connected to the dextran backbone through a hydrolysable carbonate ester. By varying the degree of cross-linking of the hydrogel core, one is able to alter the onset of capsule explosion and thus the time point of the release pulse. This is illustrated in Fig. 4B showing pulsed release of 50 nm latex beads, used as model antigen, from two types of exploding capsules with a different cross-link density and thus degradation kinetics. Theoretically, by injecting a mixture of coated gels with different degradation rates, classical prime-boost schedules could be mimicked with a single injection. Nevertheless, following subcutaneous injection of self-exploding capsules, the polyelectrolyte shell of the particles was prone to infiltration and degradation by recruited inflammatory cells, which probably prohibits a controlled release of their payload in a pulsatile manner.73 Consequently, although promising, much progress concerning shell stability and resistance to cellular infiltration needs to be made before such approach can fulfil its potential in vivo.
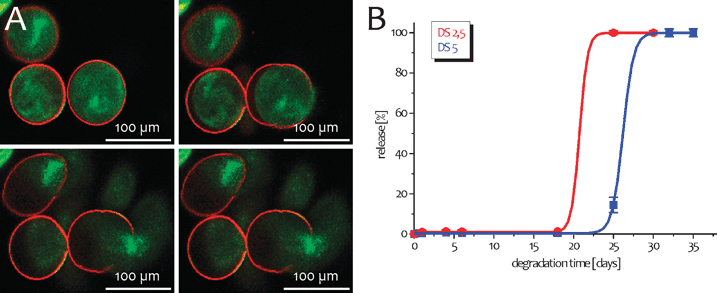 | ||
| Fig. 4 (A) Confocal microscopy images taken at different time intervals of self-exploding capsules. The red fluorescent membrane consists of 4 bilayers dextran sulfate /poly-L-arginine. The interior is a degradable hydrogel bead loaded with green fluorescent 50 nm latex beads. (B) Cumulative release curves of 50 nm latex beads from self-exploding capsules, using dex-HEMA hydrogel beads with different degradation kinetics. (Reprinted with permission from ref. 73. Copyright 2009, Elsevier.) | ||
Finally, it still remains to be established whether antigens really need to be released in a pulsatile manner for optimal induction of antibody responses. Several studies indicate that both continuous and discontinuous antigen release can induce similar antibody responses in small-animal models.74 In addition, due to the slow-decay kinetics of antibody responses in rodents, it has been suggested that such models might not be ideal to address the real value of single shot vaccine formulations.34
4.3 Micro-and nanoparticles: targeting antigens towards different APCs
A plethora of studies now have demonstrated that particles in the 50 nm to 5 μm range are efficiently taken up by DCs in vitro and in vivo. Nevertheless, DCs form a heterogeneous population, with multiple subtypes showing different functional properties.75 Resident lymphoid DCs directly differentiate in the lymphoid tissue after their emigration from the blood, without first entering peripheral tissues. Resident lymphoid DCs can be divided into CD8α− and CD8α+ subsets, which differ in their expression pattern of endocytosis receptors and in their capacity to present antigens, the CD8α+ subset being far more efficient in cross-presentation and the induction of CTL responses.76 CD8α− DCs in contrast have been reported to be more potent in MHCII mediated antigen presentation, and thus the priming of CD4 T cells.77 Besides resident lymphoid DCs, lymph nodes also contain migratory DCs. Migratory DCs differentiate in peripheral tissues where they reside in an immature status and continuously sample antigens. Such antigen sampling is illustrated in Fig. 5 showing a transmission electron microscopy (TEM) image of a DC stretching its dendrites to catch a microparticle. Even in the absence of inflammation, migratory DCs appear to undergo a functional maturation program, making them migrate to the draining lymph nodes. Antigen presentation by these mature (but not activated) DCs is of crucial importance to maintain peripheral tolerance to self-antigens. In case of infection, peripheral DCs get however activated by PRR triggering and become potent APCs capable of priming effector T cell responses. In addition, in case of infection inflammatory monocytes are recruited to the site of inflammation, which subsequently differentiate into DCs that are capable of priming CD4 and CD8 T cell responses.78,79 Differential targeting of these different DC subpopulations with their distinct properties most likely will also strongly impact the type of immune response elicited.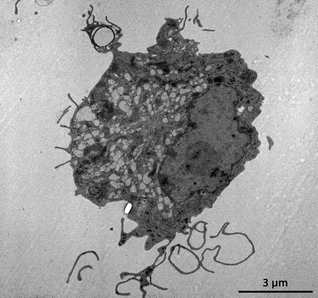 | ||
| Fig. 5 Transmission electron microscopy (TEM) image of a dendritic cell stretching out its dendritic to capture a hollow microparticle. | ||
Ultrasmall nanoparticles (20-50 nm) and particles in the lower μm range (0.5–5 μm) significantly differ in their in vivo fate following subcutaneous injection. Nanoparticles rapidly reach the lymph nodes through passive drainage via the lymphatics, making them interesting tools to directly target the lymphoid DC population. Targeting resident lymph node DCs to induce both cellular and humoral immune responses has been successfully applied by Reddy et al., who used 25 nm sized antigen-coupled polypropylene sulfide nanoparticles.80 In contrast to these ultrasmall nanoparticles, particles in the lower micron range are more tightly retained in the interstitial space and require active transport by migratory DCs to reach the lymph nodes. Recently, using polystyrene beads of different sizes, Manolova et al. have analyzed the effects of particle size on DC targeting following subcutaneous injection. Nanoparticles were shown to enter the lymph nodes via the subcapsular sinus, and to be subsequently taken up not only by resident lymphoid DCs but also by subcapsular macrophages and B cells. Approximately half of the DCs containing 20 nm particles were resident CD8α+ DCs, specialized in cross-presentation. Microparticles on the other hand were exclusively retrieved in CD8α− CD40low DCs, a phenotype consistent with DCs derived from phagocytic monocytes.81 These observations clearly demonstrate that micro- and nanoparticles target different APC populations in vivo. The functional repercussions of this differential targeting between micro- and nanoparticles remain currently unclear. Although microparticles do not target resident lymphoid CD8α+ DCs thought to be specialized in cross-presentation, many studies have now demonstrated their strong potential in inducing CD8 T cell responses in addition to CD4 T cell responses.82 This is likely due to their capacity to recruit inflammatory monocytes following injection. Following antigen uptake, these inflammatory monocytes differentiate into DCs that not only have the capacity to induce Th1 polarized CD4 T cell responses but are also highly efficient in cross-priming CD8 T cells. Consequently, micro- and nanoparticles can both elicit CD4 and CD8 T cell responses. Fig. 6 illustrates this for the particular case of antigen loaded polyelectrolyte capsules, demonstrating that antigen (ovalbumin; OVA) loaded capsules are far more potent inducers of T cell proliferation then merely soluble antigen. The particle size that produces the optimal immune response remains to be established, and might be even different depending on the pathogen one wants to target.
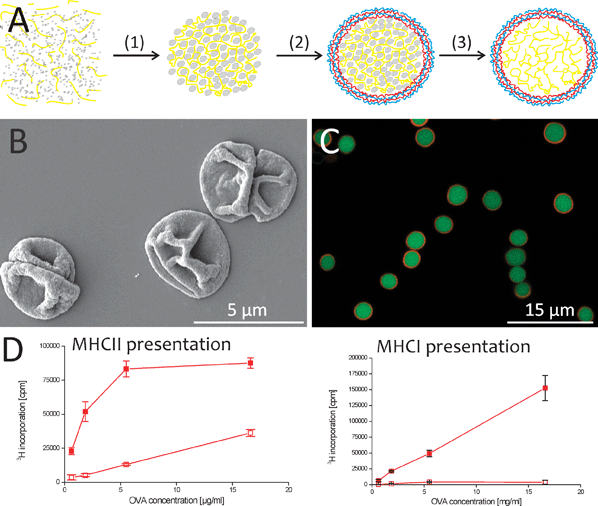 | ||
| Fig. 6 (A) Polyelectrolyte microcapsule synthesis. Antigen (yellow) is mixed with CaCl2 and Na2CO3, resulting in the generation of macromolecule-filled CaCO3 microparticles (gray), which are subsequently coated with alternating layers of dextran sulfate and poly-L-arginine (red, blue). Dissolution of the CaCO3 core by EDTA results in the generation of a hollow microcapsule composed of macromolecules surrounded by the polyelectrolyte shell. (B) Scanning electron and (C) confocal microscopy image of polyelectrolyte capsules. (D) Antigen presentation by BM-DCs after uptake of soluble and encapsulated ovalbumin. Proliferation of OT-I cells was used as a measure for MHC-I-mediated cross-presentation of ovalbumin (left graph), proliferation of OT-II cells as a measure for MHC-II mediated presentation (right). The open symbols represent soluble ovalbumin while the solid symbols represent encapsulated ovalbumin. | ||
Following subcutaneous injection, most of the microparticles injected are not transported by DCs to the lymph node but remain at the site of injection where they are taken up by other phagocytic cells (e.g. macrophages). As a result, microparticles generally target fewer DCs compared to nanoparticles that are passively drained to the lymph node. Strategies that increase microparticle targeting towards DCs following injection thereby might also enforce the strength of the induced immune response. Several approaches have been explored to enhance particle targeting to DCs or specific DC subsets. Particulate carriers can be modified with antibodies or ligands specific for DC surface markers and endocytosis receptors. In this view, functionalisation of liposomes with anti-CD11c antibody derivatives has been demonstrated to promote DC targeting and to provoke more potent immunity in a B16 melanoma model.83 Similarly, Kwon et al. have functionalized 1 μm sized pH sensitive particles with anti-DEC205 antibodies, resulting in an increased percentage of DEC205+ lymph node DCs containing the particles compared to non-functionalized particles. This increased DC targeting also resulted in an augmented CTL response, confirming the potential of this approach.84 Microparticulate uptake by peripheral DCs and inflammatory monocytes might also benefit from the incorporation of components that promote the selective recruitment of DCs and monocytes to the injection site. Carriers that have been used to gradually release DC attracting chemokines include polyester particles,85 and alginate based hydrogels.86,87 Alternatively, injection of the biomaterial itself might provoke the in situ production of chemokines and cytokines as has been demonstrated for aluminium hydroxide and the oil-in-water adjuvant MF59.88
4.4 Particulate carriers for increased cross-presentation
The most appealing part of using particulate carriers for antigen delivery is their capacity to promote cross-presentation. While endocytosed antigens generally enter the MHCII antigen processing pathway, DCs have the capacity to cross-present exogenous antigens via the MHCI route. For soluble antigens, this process appears to be dependent on their routing to stable early endosomes specialized for cross-presentation.89,90 Targeting of antigens to these specialized compartments is mediated by binding to specific endocytotic receptors, such as DEC205,32 the mannose receptor89 and langerin.91 Intriguingly, these receptors are mainly expressed on CD8α+ DCs, which have been reported to be specialized in cross-presentation.92 Nevertheless, although receptor-mediated endocytosis of soluble antigens can result in cross-presentation, the efficiency of this process is rather inefficient and requires high amounts of antigen. Antigens delivered to DCs in a particulate form in contrast, such as viruses and bacteria, are internalized via macropinocytosis or phagocytosis, and are far more efficiently cross-presented to CD8 T cells. The particulate nature of viruses and bacteria can be mimicked by encapsulating antigens in polymeric carriers with similar dimensions. Multiple studies have now demonstrated a strong increase in cross-presentation and the induction of CTL responses after antigen encapsulation in PLGA microparticles.93–95 However, the capacity to promote cross-presentation is certainly not restricted to PLGA, as many other particulate carriers including polystyrene beads,96 hydrogel particles97 and also polyelectrolyte microcapsules (Fig. 6)98 have been shown to promote cross-presentation, allowing cross-presentation at 100 to 1000-fold lower antigen doses. How particles mediate cross-presentation is still a matter of debate, and depending on the material tested multiple mechanisms have been proposed.As cross-presentation can be blocked in most cases by inhibitors of the proteasome, a mechanism has been proposed where antigen is exported from the phagosome or macropinosome, and subsequently enters the classical MHCI processing route similar to cytosolic proteins (Fig. 7A). Phagosomal escape has indeed been reported following uptake of PLGA nanoparticles, resulting in an increased presence of antigen in the cystosol and efficient cross-presentation.93,99 In contrast, Walter et al. failed to detect phagosomal escape using PLGA microparticles.100 These discrepancies might be due to differences in polymer hydrophobicity and charge, or in particle size, with the smaller nanoparticles being able to escape through membrane disruptions more easily than the bigger microparticles. The phagosomal escape hypothesis has driven researchers to develop microparticles capable of rupturing phagosomal membranes, in order to release the antigen directly into the cytosol. The main approach explored to achieve this goal has been the use of pH responsive particles that degrade or disassemble upon phagosomal acidification. Hydrazide or acetal containing microparticles are known to readily undergo acid hydrolysis and are well suited for this purpose as their single components exert an osmotic pressure on the phagosomal membrane leading to its rupture.97,101,102 On the other hand, amine-containing polymers often exhibit a so-called proton sponge effect which means that they buffer the phagosomal compartment, preventing the phagosomal acidification process. The subsequent influx of protons trying to force the acidification, induces an osmotic pressure which is also able to rupture the phagosomal membrane. Fig. 8 gives an excellent example of cytosolic antigen delivery mediated by pH responsive particles.103
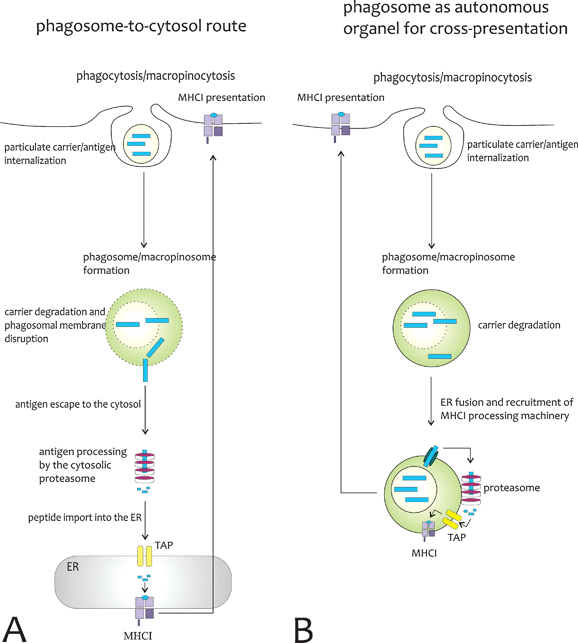 | ||
| Fig. 7 Proposed mechanisms for antigen cross-presentation mediated by particulate carriers (A) Phagosome-to-cytosol route for cross-presentation. Cross-presentation is dependent on the ability of the particulate carrier to disrupt phagosomal membranes and to release the antigen directly into the cytosol, where it is processed by the proteasome, imported via TAP transporters into the ER and subsequently loaded onto MHCI molecules. (B) Phagosomes as fully competent organelles for cross-presentation. Upon internalization, the antigen gets released from the particulate carrier into the phagosome, which contains all machinery for MHCI presentation, possibly by fusing with ER membranes. The antigen is exported from the phagosome, cleaved by immunoproteasomes associated to the phagosome, re-imported into the same phagasome via TAP transporters and loaded onto MHCI molecules in the phagosomal membrane. | ||
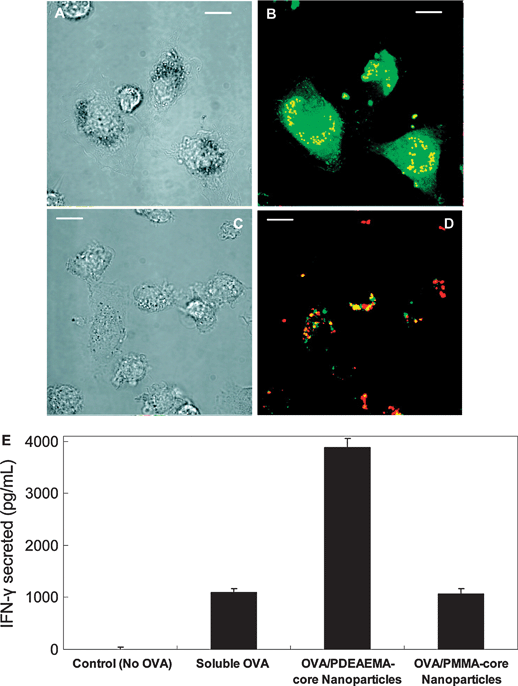 | ||
| Fig. 8 pH-sensitive core–shell nanoparticles deliver OVA to the cytosol of primary dendritic cells and promote CD8+ T cell priming. (A–D) CLSM images: (A, C) bright-field images; (B, D) fluorescence overlays of OVA (green) and nanoparticles (red). (A, B) BMDCs incubated with OVA adsorbed to PDEAEMA core–shell nanoparticles. (C, D) Cells incubated with OVA adsorbed to PMMA core–shell nanoparticles. Scale bars 10 μm. (E) BMDCs were incubated with medium alone (no OVA), soluble OVA, OVA-coated PDEAEMA-core nanoparticles, or OVA-coated PMMA-core nanoparticles, then washed and mixed with naïve OT-1 OVA-specific CD8+ T cells. IFN-γ secreted by the T cells in response to antigen presentation by the DCs was measured by ELISA after 72 h. Error bars represent standard deviation of triplicate samples. (Reprinted with permission from ref. 103. Copyright 2009, American Chemical Society.) | ||
Phagosomal disruption may however not be necessary to promote antigen cross-presentation. Recently, it has been proposed that phagosomes and macropinosomes are fully competent organelles for cross-presentation themselves and recruit all the necessary machinery for MHCI-mediated antigen presentation, possibly by fusing with ER membranes,104–106 although this is still a matter of ongoing debate. A mechanism has been suggested in which antigens are exported from the phagosomal lumen, processed by recruited immunoproteasomes, and re-imported into the same phagosome for loading onto MHCI molecules (Fig. 7B).107 Recent evidence indicates that antigen processing and loading onto MHCI and MHCII actually occur in the same phagosome, but at distinct time intervals following particle internalization. Active alkalization of the phagosome by recruitment of the NADPH oxidase NOX2 appears to be crucial for MHCI loading by preventing activation of lysosomal proteases and consequently rescuing antigens from fast degradation. This alkalization is however transient, and several hours after microparticle uptake NOX2 activity decreases causing the phagosomes to gradually acidify thus allowing the activation of lysosomal proteases that process the antigens into peptides for MHCII loading.108,109 These insights clearly have significant implications for the design of new particulate antigen carriers. If cross-presentation indeed occurs solely in the first hours after particle uptake, fast degrading particles should allow a more efficient cross-presentation compared to slow degrading ones such as PLGA. A recent study by Broaders et al. indicates that this indeed might be the case, as shown in Fig. 9. These authors encapsulated ovalbumin in acetylated dextran beads, which rapidly decompose upon phagosomal acidification. Degradation of the beads is depended on the degree of acetylation, with heavy acetylated particles degrading significantly slower. Fast degrading particles performed approximately ten times better in stimulating cross-presentation than slower ones, also including PLGA microparticles.110
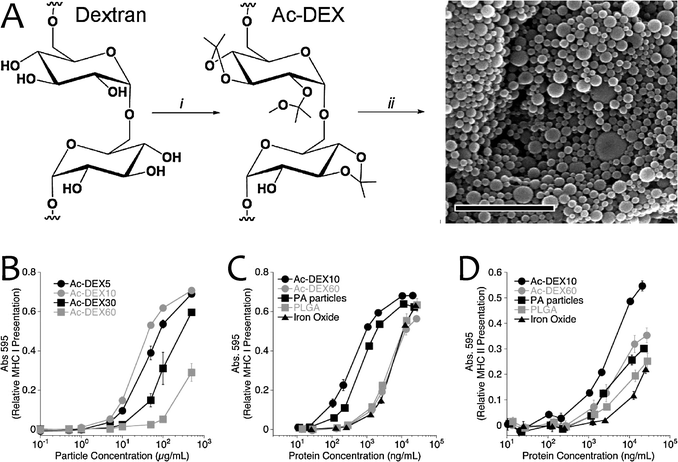 | ||
| Fig. 9 (A) Synthesis of acetal modified dextran (Ac-DEX) and particle formation: (i) 2-methoxypropene, pyridinium-p-toluenesulfonate, DMSO (ii) solvent evaporation based particle formation (scale bar is 2 μm). (B) Relative MHCI presentation from BMDCs for OVA-containing particles made from Ac-DEX5, Ac-DEX10, Ac-DEX30, and Ac-DEX60 (corresponding to degradation half-lives of 0.27, 1.7, 11, and 16 h) (n = 3, mean ± SD). (C) Relative MHCI presentation from BMDCs for OVA-containing particles made from Ac-DEX10, Ac-DEX60, PA particles, PLGA, and iron oxide. Quickly degrading materials (Ac-DEX10 and PA particles) show presentation at significantly lower protein concentrations (n = 3, mean ± SD). (D) Relative MHCII presentation from BMDCs for OVA-containing particles used in B (n = 3, mean ± SD). (Reprinted with permission from ref. 110. Copyright 2009, National Academy of Sciences.) | ||
Nevertheless, in these cases particle degradation still depends on a drop in pH, which might counteract cross-presentation and rather stimulate MHCII mediated presentation as it also results in the activation of lysosomal proteases. pH-independent strategies, in order to trigger rapid antigen release following particle uptake, might further improve MHCI loading. An elegant strategy to allow microcapsule decomposition in a pH-independent fashion might be the use of disulfide bonding stabilized particles. Zelikin et al. produced such biodeconstructible capsules by modifying poly(methacrylic acid) (PMA) with thiol groups (Fig. 10).111 In an oxidative environment, these capsules are stabilized by disulfide linkages between the polymer layers. However, in a more reductive environment, such as present in early endosomes and probably also in early phagosomes, the capsules are expected to decompose and to release their cargo. Use of thiolated PMA allowed the encapsulation of the cysteine-modified KP9 peptide into PMA particles by disulfide linkages. When placed in a reductive environment (5 mM GSH), over 80% of the peptide was released from the capsules within one hour.112 Proof of principle that such a strategy can indeed promote CD8 T cell responses was demonstrated in a non-human primate model of SIV infection.112,113 In a recent paper, Sexton et al. have extended this approach to protein antigens. Encapsulation of ovalbumin in disulfide stabilized PMA particles significantly enhanced antigen presentation to both CD4 and CD8 T cells in vitro when compared to soluble ovalbumin. Nevertheless, in vitro CD8 T cell proliferation was increased with merely a factor 3.8 to 7.9, which is at best moderate compared to other particles such as PLGA or acid degradable particles. CD4 T cell proliferation in contrast was enhanced 5.7–42 fold, indicating these particles mainly promoted the MHCII route of antigen presentation. Similar observations were made in vivo, with OVA loaded PMA particles increasing predominantly CD4 T cell responses (70-fold increase) and to a lesser extent CD8 T cell responses (6-fold increase).114 A possible reason why this approach only moderately stimulates CD8 T cell responses might be the subcellular localization of the microcapsules. Following uptake, microparticles tend to end up in phagosomal compartments, which have a far less reducing environment compared to the cytosol or the nucleus, resulting in inefficient disulfide reduction115 and slow microcapsule decomposition.
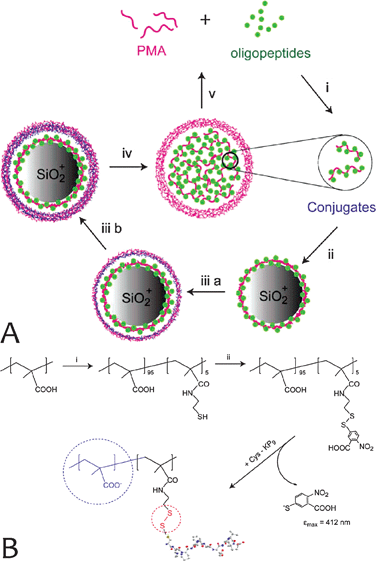 | ||
| Fig. 10 (A) Encapsulation of Cys-KP9 into degradable polymeric capsules: (i) conjugation of oligopeptides to an anchoring PMASH polymer; (ii) adsorption of conjugates onto an amine-functionalized silica particle; (iii) assembly of a thin polymer film prepared via the alternating deposition of PVPON and PMASH and oxidation of PMASH thiol groups into bridging disulfide linkages; (iv) removal of the core particle to result in a stable polymer capsule; (v) degradation of the capsule, releasing Cys-KP9. (B) Confinement of KP9 within polymer capsules through conjugating the oligopeptide to a carrier polymer. To achieve this, a sample of PMA was modified with thiol groups (i), which were subsequently activated for thiol-disulfide exchange using Ellman's reagent (ii). The resulting polymer was reacted with an N-terminal cysteine-modified KP9 to yield a PMA–KP9 conjugate wherein the PMA serves as an anchor for successful encapsulation. The disulfide linkage ensures a reversible nature of the linkage. (Reprinted with permission from ref. 112. Copyright 2009, Elsevier.) | ||
Similar to PMA capsules, a layer-by-layer technique can also be applied to produce polyelectrolyte microcapsules. In contrast to PMA particles such polyelectrolyte microcapsules are not stabilized by covalent disulfide bridges, but by electrostatic interaction.54,56,57,116 Recently, we have demonstrated that polyelectrolyte microcapsules composed of dextran-sulfate/poly-L-arginine strongly promoted antigen presentation of encapsulated ovalbumin to both CD4 and CD8 T cells.98 In contrast to the PMA particles, these microcapsules however stimulated predominantly CD8 T cell proliferation, an observation we have now also confirmed in vivo (unpublished data). These discrepancies might again be due to the kinetics of antigen release and/or availability for processing. Using DQ-OVA, a BODIPY labelled ovalbumin, which is self-quenched in its native state but becomes brightly fluorescent after enzymatic degradation, De Koker et al. showed that following uptake by DCs, dextran-sulfate/poly-L-arginine encapsulated ovalbumin becomes readily available for enzymatic processing, even before visual rupturing of the microcapsules’ shell 24 h after ingestion, as shown in Fig. 11.98 In this view, hollow microcapsules with the antigen only being surrounded by a thin shell might offer significant benefits compared to entire polymeric particles such as PLGA. In the case of hollow microcapsules, mere shell erosion or local rupturing is enough to make the entire microcapsule content available for processing, while in the case of ‘filled’ polymeric particles, polymer erosion starts at the border making only these proteins that are located near the particle border rapidly available for enzymatic processing. Using DQ-OVA, Heit et al. showed that degradation of ovalbumin encapsulated in PLGA microspheres only starts six hours after cellular uptake. Moreover, degradation was initiated at the border of the particle and then gradually proceeded towards the inner core of the PLGA microsphere.117
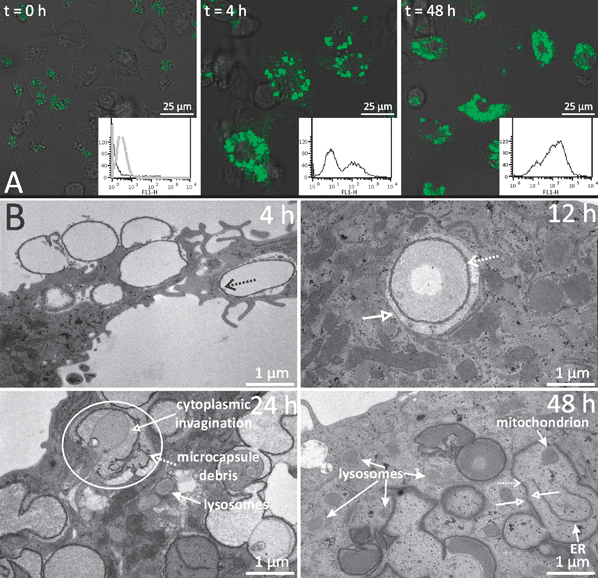 | ||
| Fig. 11 (A) Processing of dextran sulfate/poly-L-arginine microcapsule encapsulated OVA was analyzed using DQ-OVA. Confocal microscopy images of BM-DCs incubated with DQ-OVA-microcapsules for 0, 4 and 48 h (overlay of green fluorescence and DIC). Insets: flow cytometry analysis of green fluorescence intensity (FL1-H). An overlay of DC green autofluorescence (black line) and green fluorescence intensity of DQ-OVA microcapsules (gray line) is given in the first histogram. The second and third histograms show the green fluorescence intensity after incubating BM-DCs with DQ-OVA microcapsules for 4 and 48 h, respectively. (B) TEM images of BM-DCs that have internalized dextran sulfate/poly-L-arginine microcapsules at the indicated time intervals. Microcapsule shell: dotted arrows; membranes surrounding the microcapsules: open arrows. In the encircled area, microcapsule rupture and cytoplasmic invagination are clearly distinguishable. Lysosomes, endoplasmatic reticulu (ER), and a mitochondrion are indicated by the solid arrows. (Reprinted with permission from ref. 98. Copyright 2009, Wiley.) | ||
Another matter of intensive debate is whether there exists an optimal particle size to promote cross-presentation. In a recent study, Tran and Shen nicely demonstrated that, at least in vitro, DCs cross-present antigen bounded to 0.5–3 μm polystyrene beads far more efficient than antigen bounded to 50 nm beads.96 Importantly, as 50 nm nanoparticles were rapidly shuttled to acidic compartments while larger microparticles remained in a more neutral environment, these data are in accordance with the earlier mentioned observation that fast phagosomal acidification actually inhibits cross-presentation. Nevertheless, several other studies have claimed that nanoparticles are more potent in inducing CD8 T cell responses in vivo compared to microparticles.118 Differences in particle uptake and especially DC targeting may underlie this apparent paradox: while microparticles might be intrinsically superior in promoting antigen processing towards the MHCI route, they are less efficient in targeting DCs in vivo compared to nanoparticles, which can passively drain to the lymph nodes via the lymphatics. If so, strategies that can increase microparticle targeting to DCs or DC subpopulations might also result in superior cross-presentation, as has been demonstrated by Kwon et al. who used acid-labile 1 μm particles couple to the DC-specific antibody DEC205.84 Further studies are necessary to address these issues, and to resolve which particle size is indeed the most optimal.
4.5 Intrinsic immune activating properties of particulate carriers
To induce potent adaptive immune responses, mere antigen delivery to DCs is insufficient and can even lead to tolerance. Indeed, in order to induce effector T cell responses, DCs also need to become activated to upregulate co-stimulatory ligands and to secrete inflammatory cytokines. The most direct assay to evaluate the immunopotentiator properties of new biomaterials is assessing their capacity to activate DCs in vitro. Several studies have demonstrated a moderate upregulation of the co-stimulatory ligands CD83 and CD86 and an increased release of IL-12 and TNF-α by DCs following uptake of PLGA particles.119 Other groups however failed to detect any DC maturation following incubation with PLGA.120,121 These discrepancies might be attributed to differences in polymer and stabilizer used, but also to differences in bacterial contaminants such as LPS adsorbed to the microspheres’ surface. Similarly, incubation with poly-L-lactic acid (PLA),122 poly-(γ-glutamic acid)123 or poly-β-amino-ester containing particles124 has been reported to promote DC maturation. Recently, our group also observed a slight increase in the expression of CD40, CD86 and MHCII on bone marrow derived DCs after incubation with polyelectrolyte microcapsules composed of dextran-sulfate and poly-L-arginine (unpublished results). How particulate antigen carriers promote DC maturation has however largely remained a mystery, as they do not contain any known ligands for PRRs. Only very recently, a number of publications have shed a new light on how particulates might exert their adjuvant properties. First, it was recognized that at least part of the adjuvant properties of aluminium hydroxide components can be attributed to formation of the NALP3 inflammasome and the subsequent release of the potent pro-inflammatory cytokine IL-1β.19,125–128 NALP3 is a member of the nucleotide binding domain (NOD) family, a group of cytosolic PRRs that can be triggered by various endogenous and microbial danger signals. Inflammasome formation results in caspase-1 activation, pro-IL-1β cleavage and IL-1β release.129 Subsequently, these observations have been extended to particulate adjuvants including chitosan, QuilA,127 polystyrene and PLGA microparticles,130 which all appear to activate the NALP3 inflammasome. Sharp et al. demonstrated that following uptake of polystyrene and PLGA particles, NALP3 activation depended on phagosomal acidification and the lysosomal cysteine protease cathepsin B, indicating a possible role for phagosomal disruption in NALP activation.130 Although IL-1β release by DCs in response to particulates requires a pre-stimulation step of the DCs with LPS to produce pro-IL-1β in vitro, in vivo this appears not to be necessary as injection of PLGA particles in the absence of TLR agonists did induce local IL-1β at the injection site. Likely, injection of the particles resulted in the release of endogenous danger signals by inflicting tissue damage, which could replace for microbial PAMPs to trigger pro-IL-1β formation. Remarkably, although NALP3 activation was crucial for initiating cell-mediated immune responses, it was completely dispensable for initiating humoral immune responses, indicating that something else that remains to be unravelled must be involved in the adjuvant properties of particulates.4.6 Enhancing immune responses by co-delivery of PRR agonists
As most recombinant antigens fail to activate DCs, addition of microbial components or their synthetic derivatives (e.g. TLR ligands) can strongly enhance their immunogenicity by stimulating DC activation. Similarly, although the particulate nature of microparticles might be sufficient to be recognized by the immune system as intrinsically dangerous via inflammasome activation, most particulates are only poor activators of DCs. As a result, co-delivery of particulate antigen formulations with PRR agonists might well work synergistic in evoking potent immune responses by combining the antigen presentation promoting capacities of particulates with the DC activating properties of PRR agonists. Such co-administration of antigen carrier and DC activator can be achieved either by mere co-injection, or by physical linkage of the DC activation stimulus to the carrier via surface adsorption and encapsulation. Recent data clearly indicate that the latter strategy is superior in inducing strong effector T cell responses. Blander et al. have demonstrated that antigen processing and loading onto MHCII molecules is regulated at the level of the individual phagosome, and that antigen and TLR agonist need to be present in the same endocytic compartment to ensure optimal antigen presentation to CD4 T cells.131,132 Combining particles with TLR agonists might be of particular interest when using agonists for TLRs localized in phagolysosomal compartments such as TLR3, 7, 8 and 9. Schlosser et al. have used biodegradable PLGA microspheres to co-encapsulate ovalbumin and the TLR9 ligand CpG.133 Importantly, co-encapsulation of both antigen and CpG generated far superior CTL responses compared to a mixture of OVA containing microspheres with CpG containing microspheres, thus further extending the observations made by Blander et al. concerning the induction of CD4 T cell responses to cross-presentation and the induction of CD8 CTL responses. Similar results have been obtained by Heit et al., who demonstrated that co-encapsulating antigen and CpG in PLGA microspheres was far superior in inducing CD4 and CD8 T cell responses compared to a mixture of soluble CpG with soluble antigen. PLGA encapsulating both ovalbumin and CpG was able to confer protection against a lethal challenge with ovalbumin-expressing Listeria monocytogenes, while microspheres containing ovalbumin alone failed to protect. Unfortunately, in this study no comparison was made with a mixture of ovalbumin-containing PLGA and soluble CpG.117Increased humoral immune responses and levels of IFN-γ secreting T cells were also observed after co-encapsulation of ovalbumin and polyU, a synthetic TLR7/8 agonist mimicking ssRNA, inside polylactic particles.134 Enhancement of immune responses by co-delivery of TLR agonists is certainly not restricted to agonists that trigger endosomal TLRs. Kazzaz et al. made similar observations using PLGA and the TLR4 agonist monophosphoryl lipid A (MPL), a detoxified LPS analogue recently approved for human use in the EU.135 Encapsulation of MPL together with the naturally expressed tumor antigen tyrosinase related protein-2 (Trp-2) in PLGA nanoparticles could even induce therapeutic immunity against the highly aggressive B16 melanoma, further emphasizing the strength and potential of this approach.136 Although these initial experiments have clearly demonstrated the benefits of encapsulating antigen and immune potentiatior in PLGA microparticles, the encapsulation process itself is far from standard practice, and requires lots of optimization for each antigen and antigen–immune potentiator combination being applied. Given their capacity to efficiently encapsulate protein antigen and the high versatility of the layer-by-layer technique used to produce them, polyelectrolyte microcapsules might be an interesting alternative for PLGA microspheres. As these microcapsules are generated by the deposition of polyelectrolytes, their surface charge can be easily made cationic by depositing a positively charged polyelectrolyte as outer layer, which allows an easy binding of negatively charged TLR agonists such as CpG oligonucleotides and the dsRNA analogue polyI:C via mere electrostatic interaction (unpublished results).
Linking functional ligands to microparticles not only may enhance the general strength of the immune response, but might also allow us to steer the immune response towards the desired direction. For example, adding CpG to particulates skews the immune response towards a Th1 type. By using other ligands, for example β-glucans that promote IL-23 secretion by DCs, one might be able to induce a more Th17 skewed response.
In addition to potentiating immune responses by co-delivering antigen and DC activation stimulus, particulate delivery vehicles also have the significant benefit of reducing the inflammatory toxicity generally associated with the use of an immune potentiator by limiting its free diffusion and focusing its effects on the target cell, being the DC. In this respect, combining CpG oligonucleotides with poly-L-arginine, a polycationic amino acid, has been demonstrated to enhance CpG uptake by DCs in vivo, while inhibiting the systemic release of inflammatory cytokines observed after injection of free CpG.137
Finally, although co-delivery of antigens and immune potentiators such as TLR agonists within or associated to the same particle clearly has tremendous benefits in augmenting the strength of the induced immune response, co-encapsulating them remains a complex and challenging task. Developing polymeric particles with strong intrinsic immune activating properties could alleviate these complex procedures and might constitute a major breakthrough to pave the way for a broader clinical applicability of polymeric carriers in vaccines. One way to achieve this goal might be the use of biomaterials that activate the complement system. Complement activation not only constitutes a direct biochemical defense mechanism to kill and opsonize microorganisms, but also has been shown to modulate the DC activation status and to promote antigen-specific immune responses. Biomaterials containing high levels of free hydroxyls and amine nucleophiles strongly activate the complement cascade by binding to the exposed thioester of the complement factor C3b. Recently, Reddy et al. have developed antigen coupled polyhydroxylated nanoparticles composed of Pluronic stabilized polypropylene sulfide to exploit complement activation as activating stimulus, as shown in Fig. 12.80 Injection of these particles resulted in a fast and strong activation of APCs, followed by the generation of humoral and cellular immune responses, including the induction of IFN-γ secreting CD8 T cells, clearly showing the potential of this approach.
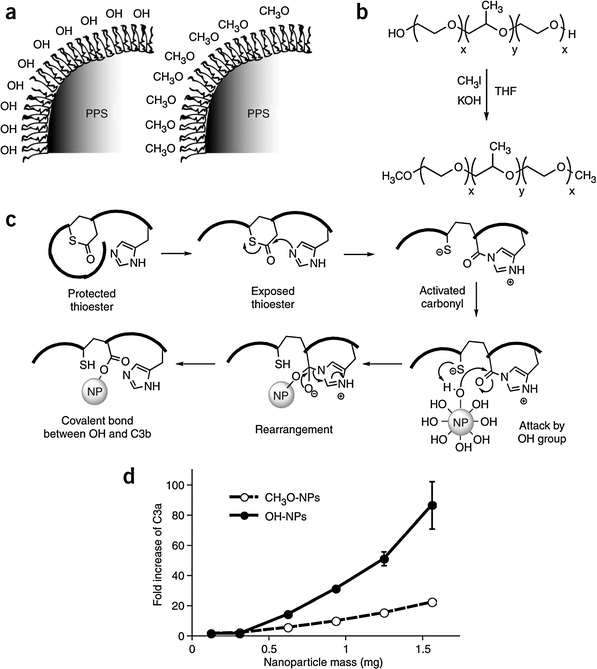 | ||
| Fig. 12 Polyhydroxylated nanoparticle surfaces activate complement. (a) Synthesis and stabilization with two different forms of Pluronic allowed the generation of polyhydroxylated- or polymethoxylatednanoparticles. (b) The α,ω-terminal OH groups on Pluronic could be converted to OCH3 groups. (c) The proposed mechanism where OH groups on the polyhydroxylated nanoparticles can bind to the exposed thioester of C3b to activate complement by the alternative pathway. (d) Nanoparticle-induced complement activation, as measured through C3a presence in human serum after incubation with nanoparticles, was demonstrated to be high with polyhydroxylated nanoparticles but low with polymethoxylated nanoparticles (OH– and CH3O–NPs, respectively). Results are normalized to control of serum incubation with PBS. Values are means of three independent experiments; error bars correspond to standard error of mean, s.e.m. (Reprinted with permission from ref. 80. Copyright 2007, Nature Publishing Group.) | ||
5. Conclusions and future perspectives
During the last decade, particulate antigen delivery using polymeric carriers has clearly demonstrated its strong benefits in enhancing antigen immunogenicity in vaccination. Importantly, particulates strongly promote cross-presentation, thereby allowing the induction of CTL responses, a feature hardly achievable when using soluble antigens. In addition, particulate carriers can also be used to target PRR agonists to DCs, which not only works synergistic in enforcing immunity, but also should allow one to steer immune responses to a certain direction and to reduce inflammatory side effects. Nevertheless, much progress remains to be made. Novel insights in the mechanisms underlying cross-presentation can provide drug delivery scientists with clues regarding the optimal size and composition of particles for cross-presentation. For example, if cross-presentation indeed occurs exclusively in the first hours after particle uptake and before the phagosome acidifies, particles that rapidly decompose and release their antigen following internalization should offer a significant benefit. In addition, modifying particles with certain ligands that target them to specific cellular compartments following receptor mediated internalization or to specific DC subsets, will also affect the way the antigen is presented and should allow one to further tune the immune response induced.Also many practical hurdles remain to be taken before polymeric carriers will become widespread antigen delivery vehicles in human vaccines. For most of the experimental carriers developed to date, antigen encapsulation is a complex and challenging process, involving multi-steps often resulting in low antigen encapsulation. Given the limited availability and cost of many recombinant antigens, there is consequently a strong need to develop new and more efficient encapsulation strategies. Nevertheless, cost might not be an insurmountable issue if particulate delivery can yield effective therapeutic vaccines against incurable diseases as cancer or HIV. Designing particle-based vaccines for large-scale preventive immunization schedules in contrast will certainly require the development of new strategies enabling an efficient, cheap and easy encapsulation of antigens and immune potentiators, preferably involving single step processes yielding a dry powder such as spray-drying. Achieving this will allow us to develop a next generation of vaccines, tailored towards 21st century needs.
Acknowledgements
S.D.K. thanks Ghent University (BOF-GOA) for funding. B.D.G. acknowledges the FWO for a postdoctoral scholarship.Notes and references
- R. Rappuoli, H. I. Miller and S. Falkow, Science, 2002, 297, 937–939 CrossRef CAS.
- R. Rappuoli, Nat. Biotechnol., 2007, 25, 1361–1366 CrossRef CAS.
- B. Pulendran and R. Ahmed, Cell, 2006, 124, 849–863 CrossRef CAS.
- P. E. Jensen, Nat. Immunol., 2007, 8, 1041–1048 CrossRef CAS.
- J. Banchereau, F. Briere, C. Caux, J. Davoust, S. Lebecque, Y. J. Liu, B. Pulendran and K. Palucka, Annu. Rev. Immunol., 2000, 18, 767–811 CrossRef CAS.
- N. Luckashenak, S. Schroeder, K. Endt, D. Schmidt, K. Mahnke, M. F. Bachmann, P. Marconi, C. A. Deeg and T. Brocker, Immunity, 2008, 28, 521–532 CrossRef CAS.
- J. K. Tan and H. C. O'Neill, J. Leukocyte Biol., 2005, 78, 319–324 Search PubMed.
- D. Kabelitz and R. Medzhitov, Curr. Opin. Immunol., 2007, 19, 1–3 CrossRef CAS.
- R. Medzhitov, Nature, 2007, 449, 819–826 CrossRef CAS.
- R. Medzhitov and C. Janeway, Jr., Immunol. Rev., 2000, 173, 89–97 CrossRef CAS.
- C. Pasare and R. Medzhitov, Semin. Immunol., 2004, 16, 23–26 CrossRef CAS.
- B. Pulendran, Immunol. Rev., 2004, 199, 227–250 CrossRef CAS.
- B. Pulendran, K. Palucka and J. Banchereau, Science, 2001, 293, 253–256 CrossRef CAS.
- K. Bottomly, Immunol. Today, 1988, 9, 268–274 CrossRef CAS.
- W. Huang, L. Na, P. L. Fidel and P. Schwarzenberger, J. Infect. Dis., 2004, 190, 624–631 CrossRef CAS.
- L. E. Harrington, P. R. Mangan and C. T. Weaver, Curr. Opin. Immunol., 2006, 18, 349–356 CrossRef CAS.
- F. L. van de Veerdonk, M. S. Gresnigt, B. J. Kullberg, J. W. van der Meer, L. A. Joosten and M. G. Netea, BMB Rep., 2009, 42, 776–787 Search PubMed.
- E. De Gregorio, E. Tritto and R. Rappuoli, Eur. J. Immunol., 2008, 38, 2068–2071 CrossRef CAS.
- B. N. Lambrecht, M. Kool, M. A. M. Willart and H. Hammad, Curr. Opin. Immunol., 2009, 21, 23–29 CrossRef CAS.
- B. Guy, Nat. Rev. Microbiol., 2007, 5, 505–517 Search PubMed.
- J. R. Baldridge, P. McGowan, J. T. Evans, C. Cluff, S. Mossman, D. Johnson and D. Persing, Expert Opin. Biol. Ther., 2004, 4, 1129–1138 Search PubMed.
- E. Celis, Cancer Res., 2007, 67, 7945–7947 CrossRef CAS.
- D. H. Persing, R. N. Coler, M. J. Lacy, D. A. Johnson, J. R. Baldridge, R. M. Hershberg and S. G. Reed, Trends Microbiol., 2002, 10, s32–37 CrossRef CAS.
- V. E. Schijns and W. G. Degen, Clin. Pharmacol. Ther., 2007, 82, 750–755 CrossRef CAS.
- B. S. Thompson, P. M. Chilton, J. R. Ward, J. T. Evans and T. C. Mitchell, J. Leukocyte Biol., 2005, 78, 1273–1280 Search PubMed.
- D. M. Harper, E. L. Franco, C. M. Wheeler, A. B. Moscicki, B. Romanowski, C. M. Roteli-Martins, D. Jenkins, A. Schuind, S. A. Costa Clemens and G. Dubin, Lancet, 2006, 367, 1247–1255 CrossRef CAS.
- M. Kundi, Expert Rev. Vaccines, 2007, 6, 133–140 CrossRef CAS.
- A. Pashine, N. M. Valiante and J. B. Ulmer, Nat. Med., 2005, 11, S63–68 CrossRef CAS.
- R. S. Chu, O. S. Targoni, A. M. Krieg, P. V. Lehmann and C. V. Harding, J. Exp. Med., 1997, 186, 1623–1631 CrossRef CAS.
- A. Heit, F. Schmitz, M. O'Keeffe, C. Staib, D. H. Busch, H. Wagner and K. M. Huster, J. Immunol., 2005, 174, 4373–4380 CAS.
- G. Napolitani, A. Rinaldi, F. Bertoni, F. Sallusto and A. Lanzavecchia, Nat. Immunol., 2005, 6, 769–776 CrossRef CAS.
- L. Bonifaz, D. Bonnyay, K. Mahnke, M. Rivera, M. C. Nussenzweig and R. M. Steinman, J. Exp. Med., 2002, 196, 1627–1638 CrossRef CAS.
- D. T. O'Hagan and M. Singh, Expert Rev. Vaccines, 2003, 2, 269–283 CrossRef CAS.
- D. T. O'Hagan, M. Singh and J. B. Ulmer, Methods, 2006, 40, 10–19 CrossRef CAS.
- J. Hanes, J. L. Cleland and R. Langer, Adv. Drug Delivery Rev., 1997, 28, 97–119 CrossRef CAS.
- S. T. Reddy, M. A. Swartz and J. A. Hubbell, Trends Immunol., 2006, 27, 573–579 CrossRef CAS.
- D. T. O'Hagan and N. M. Valiante, Nat. Rev. Drug Discovery, 2003, 2, 727–735 CrossRef CAS.
- C. Wang, Q. Ge, D. Ting, D. Nguyen, H. R. Shen, J. Z. Chen, H. N. Eisen, J. Heller, R. Langer and D. Putnam, Nat. Mater., 2004, 3, 190–196 CrossRef CAS.
- W. N. Haining, D. G. Anderson, S. R. Little, M. S. von Berwelt-Baildon, A. A. Cardoso, P. Alves, K. Kosmatopoulos, L. M. Nadler, R. Langer and D. S. Kohane, J. Immunol., 2004, 173, 2578–2585 CAS.
- B. G. De Geest, S. De Koker, Y. Gonnissen, L. J. De Cock, J. Grooten, J. P. Remon and C. Vervaet, Soft Matter, 2010, 6, 305–310 RSC.
- O. Soga, C. F. van Nostrum and W. E. Hennink, Biomacromolecules, 2004, 5, 818–821 CrossRef CAS.
- J. Ochoa, J. M. Irache, I. Tamayo, A. Walz, V. G. DelVecchio and C. Gamazo, Vaccine, 2007, 25, 4410–4419 CrossRef CAS.
- S. Jain, W. T. Yap and D. J. Irvine, Biomacromolecules, 2005, 6, 2590–2600 CrossRef CAS.
- N. Murthy, M. C. Xu, S. Schuck, J. Kunisawa, N. Shastri and J. M. J. Frechet, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4995–5000 CrossRef CAS.
- J. K. Oh, D. J. Siegwart, H. I. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5945 CrossRef CAS.
- B. G. De Geest, W. Van Camp, F. E. Du Prez, J. Demeester, S. C. De Smedt and W. E. Hennink, Chem. Commun., 2008, 190–192 RSC.
- D. Lemoine, F. Wauters, S. Bouchend'homme and V. Preat, Int. J. Pharm., 1998, 176, 9–19 CrossRef CAS.
- W. R. Gombotz and S. F. Wee, Adv. Drug Delivery Rev., 1998, 31, 267–285 CrossRef CAS.
- D. Christensen, K. S. Korsholm, I. Rosenkrands, T. Lindenstrom, P. Andersen and E. M. Agger, Expert Rev. Vaccines, 2007, 6, 785–796 CrossRef CAS.
- J. Kunisawa, S. Nakagawa and T. Mayumi, Adv. Drug Delivery Rev., 2001, 52, 177–186 CrossRef CAS.
- B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef.
- H. Lomas, I. Canton, S. MacNeil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Adv. Mater., 2007, 19, 4238 CrossRef CAS.
- L. Illum, I. Jabbal-Gill, M. Hinchcliffe, A. N. Fisher and S. S. Davis, Adv. Drug Delivery Rev., 2001, 51, 81–96 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- F. Caruso, R. A. Caruso and H. Mohwald, Science, 1998, 282, 1111–1114 CrossRef CAS.
- E. Donath, G. B. Sukhorukov, F. Caruso, S. A. Davis and H. Mohwald, Angew. Chem., Int. Ed., 1998, 37, 2201–2205 CrossRef.
- G. B. Sukhorukov, E. Donath, S. Davis, H. Lichtenfeld, F. Caruso, V. I. Popov and H. Mohwald, Polym. Adv. Technol., 1998, 9, 759–767 CrossRef CAS.
- J. F. Quinn, A. P. R. Johnston, G. K. Such, A. N. Zelikin and F. Caruso, Chem. Soc. Rev., 2007, 36, 707–718 RSC.
- V. Kozlovskaya, E. Kharlampieva, I. Erel and S. A. Sukhishvili, Soft Matter, 2009, 5, 4077–4087 RSC.
- D. V. Volodkin, A. I. Petrov, M. Prevot and G. B. Sukhorukov, Langmuir, 2004, 20, 3398–3406 CrossRef CAS.
- A. M. Yu, Y. J. Wang, E. Barlow and F. Caruso, Adv. Mater., 2005, 17, 1737 CrossRef CAS.
- L. Feng, X. R. Qi, X. J. Zhou, Y. Maitani, S. C. Wang, Y. Jiang and T. Nagai, J. Controlled Release, 2006, 112, 35–42 CrossRef CAS.
- P. Johansen, F. Estevez, R. Zurbriggen, H. P. Merkle, R. Gluck, G. Corradin and B. Gander, Vaccine, 2000, 19, 1047–1054 CrossRef CAS.
- M. Sandor, D. Enscore, P. Weston and E. Mathiowitz, J. Controlled Release, 2001, 76, 297–311 CrossRef CAS.
- S. P. Schwendeman, H. R. Costantino, R. K. Gupta, G. R. Siber, A. M. Klibanov and R. Langer, Proc. Natl. Acad. Sci.U. S. A., 1995, 92, 11234–11238 CrossRef CAS.
- G. Zhu, S. R. Mallery and S. P. Schwendeman, Nat. Biotechnol., 2000, 18, 52–57 CrossRef CAS.
- G. Zhu and S. P. Schwendeman, Pharm. Res., 2000, 17, 351–357 CrossRef CAS.
- H. Tamber, P. Johansen, H. P. Merkle and B. Gander, Adv. Drug Delivery Rev., 2005, 57, 357–376 CrossRef CAS.
- W. Jiang, R. K. Gupta, M. C. Deshpande and S. P. Schwendeman, Adv. Drug Delivery Rev., 2005, 57, 391–410 CrossRef CAS.
- B. G. De Geest, C. Dejugnat, M. Prevot, G. B. Sukhorukov, J. Demeester and S. C. De Smedt, Adv. Funct. Mater., 2007, 17, 531–537 CrossRef CAS.
- B. G. De Geest, C. Dejugnat, G. B. Sukhorukov, K. Braeckmans, S. C. De Smedt and J. Demeester, Adv. Mater., 2005, 17, 2357 CrossRef CAS.
- B. G. De Geest, C. Dejugnat, E. Verhoeven, G. B. Sukhorukov, A. M. Jonas, J. Plain, J. Demeester and S. C. De Smedt, J. Controlled Release, 2006, 116, 159–169 CrossRef CAS.
- B. G. De Geest, S. De Koker, J. Demeester, S. C. De Smedt and W. E. Hennink, J. Controlled Release, 2009, 135, 268–273 CrossRef CAS.
- R. K. Gupta, M. Singh and D. T. O'Hagan, Adv. Drug Delivery Rev., 1998, 32, 225–246 CrossRef CAS.
- W. R. Heath, G. T. Belz, G. M. Behrens, C. M. Smith, S. P. Forehan, I. A. Parish, G. M. Davey, N. S. Wilson, F. R. Carbone and J. A. Villadangos, Immunol. Rev., 2004, 199, 9–26 CrossRef CAS.
- J. M. den Haan, S. M. Lehar and M. J. Bevan, J. Exp. Med., 2000, 192, 1685–1696 CrossRef CAS.
- M. L. Lin, Y. Zhan, J. A. Villadangos and A. M. Lew, Immunol. Cell Biol., 2008, 86, 353–362 CrossRef CAS.
- E. Segura and J. A. Villadangos, Curr. Opin. Immunol., 2009, 21, 105–110 CrossRef CAS.
- B. Leon and C. Ardavin, Immunol. Cell Biol., 2008, 86, 320–324 CrossRef CAS.
- S. T. Reddy, A. J. van der Vlies, E. Simeoni, V. Angeli, G. J. Randolph, C. P. O'Neil, L. K. Lee, M. A. Swartz and J. A. Hubbell, Nat. Biotechnol., 2007, 25, 1159–1164 CrossRef CAS.
- V. Manolova, A. Flace, M. Bauer, K. Schwarz, P. Saudan and M. F. Bachmann, Eur. J. Immunol., 2008, 38, 1404–1413 CrossRef CAS.
- Y. Waeckerle-Men, E. U. Allmen, B. Gander, E. Scandella, E. Schlosser, G. Schmidtke, H. P. Merkle and M. Groettrup, Vaccine, 2006, 24, 1847–1857 CrossRef CAS.
- C. L. van Broekhoven, C. R. Parish, C. Demangel, W. J. Britton and J. G. Altin, Cancer Res., 2004, 64, 4357–4365 CrossRef CAS.
- Y. J. Kwon, E. James, N. Shastri and J. M. Frechet, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18264–18268 CrossRef CAS.
- X. Zhao, S. Jain, H. Benjamin Larman, S. Gonzalez and D. J. Irvine, Biomaterials, 2005, 26, 5048–5063 CrossRef CAS.
- Y. Hori, A. M. Winans, C. C. Huang, E. M. Horrigan and D. J. Irvine, Biomaterials, 2008, 29, 3671–3682 CrossRef CAS.
- Y. Hori, A. M. Winans and D. J. Irvine, Acta Biomater., 2009, 5, 969–982 CrossRef CAS.
- A. Seubert, E. Monaci, M. Pizza, D. T. O'Hagan and A. Wack, J. Immunol., 2008, 180, 5402–5412 CAS.
- S. Burgdorf, A. Kautz, V. Bohnert, P. A. Knolle and C. Kurts, Science, 2007, 316, 612–616 CrossRef CAS.
- S. Burgdorf, C. Scholz, A. Kautz, R. Tampe and C. Kurts, Nat. Immunol., 2008, 9, 558–566 CrossRef CAS.
- J. Idoyaga, C. Cheong, K. Suda, N. Suda, J. Y. Kim, H. Lee, C. G. Park and R. M. Steinman, J. Immunol., 2008, 180, 3647–3650 CAS.
- D. Dudziak, A. O. Kamphorst, G. F. Heidkamp, V. R. Buchholz, C. Trumpfheller, S. Yamazaki, C. Cheong, K. Liu, H. W. Lee, C. G. Park, R. M. Steinman and M. C. Nussenzweig, Science, 2007, 315, 107–111 CrossRef CAS.
- H. Shen, A. L. Ackerman, V. Cody, A. Giodini, E. R. Hinson, P. Cresswell, R. L. Edelson, W. M. Saltzman and D. J. Hanlon, Immunology, 2006, 117, 78–88 CrossRef CAS.
- C. D. Partidos, P. Vohra, D. H. Jones, G. Farrar and M. W. Steward, J. Controlled Release, 1999, 62, 325–332 CrossRef CAS.
- Y. Ataman-Onal, S. Munier, A. Ganee, C. Terrat, P. Y. Durand, N. Battail, F. Martinon, R. Le Grand, M. H. Charles, T. Delair and B. Verrier, J. Controlled Release, 2006, 112, 175–185 CrossRef.
- K. K. Tran and H. Shen, Biomaterials, 2009, 30, 1356–1362 CrossRef CAS.
- N. Murthy, M. Xu, S. Schuck, J. Kunisawa, N. Shastri and J. M. Frechet, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4995–5000 CrossRef CAS.
- S. De Koker, B. G. De Geest, S. K. Singh, R. De Rycke, T. Naessens, Y. Van Kooyk, J. Demeester, S. C. De Smedt and J. Grooten, Angew. Chem., Int. Ed., 2009, 48, 8485–8489 CrossRef CAS.
- J. Panyam, W. Z. Zhou, S. Prabha, S. K. Sahoo and V. Labhasetwar, FASEB J., 2002, 16, 1217–1226 CrossRef CAS.
- E. Walter, D. Dreher, M. Kok, L. Thiele, S. G. Kiama, P. Gehr and H. P. Merkle, J. Controlled Release, 2001, 76, 149–168 CrossRef CAS.
- W. N. Haining, D. G. Anderson, S. R. Little, M. S. von Bergwelt-Baildon, A. A. Cardoso, P. Alves, K. Kosmatopoulos, L. M. Nadler, R. Langer and D. S. Kohane, J. Immunol., 2004, 173, 2578–2585 CAS.
- N. Murthy, Y. X. Thng, S. Schuck, M. C. Xu and J. M. Frechet, J. Am. Chem. Soc., 2002, 124, 12398–12399 CrossRef CAS.
- Y. Hu, T. Litwin, A. R. Nagaraja, B. Kwong, J. Katz, N. Watson and D. J. Irvine, Nano Lett., 2007, 7, 3056–3064 CrossRef CAS.
- A. L. Ackerman, C. Kyritsis, R. Tampe and P. Cresswell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12889–12894 CrossRef CAS.
- P. Guermonprez, L. Saveanu, M. Kleijmeer, J. Davoust, P. Van Endert and S. Amigorena, Nature, 2003, 425, 397–402 CrossRef CAS.
- M. Houde, S. Bertholet, E. Gagnon, S. Brunet, G. Goyette, A. Laplante, M. F. Princiotta, P. Thibault, D. Sacks and M. Desjardins, Nature, 2003, 425, 402–406 CrossRef CAS.
- S. Burgdorf and C. Kurts, Curr. Opin. Immunol., 2008, 20, 89–95 CrossRef CAS.
- A. Savina, C. Jancic, S. Hugues, P. Guermonprez, P. Vargas, I. C. Moura, A. M. Lennon-Dumenil, M. C. Seabra, G. Raposo and S. Amigorena, Cell, 2006, 126, 205–218 CrossRef CAS.
- C. Jancic, A. Savina, C. Wasmeier, T. Tolmachova, J. El-Benna, P. M. Dang, S. Pascolo, M. A. Gougerot-Pocidalo, G. Raposo, M. C. Seabra and S. Amigorena, Nat. Cell Biol., 2007, 9, 367–378 CrossRef CAS.
- K. E. Broaders, J. A. Cohen, T. T. Beaudette, E. M. Bachelder and J. M. Frechet, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5497–5502 CrossRef CAS.
- A. N. Zelikin, J. F. Quinn and F. Caruso, Biomacromolecules, 2006, 7, 27–30 CrossRef CAS.
- S. F. Chong, A. Sexton, R. De Rose, S. J. Kent, A. N. Zelikin and F. Caruso, Biomaterials, 2009, 30, 5178–5186 CrossRef CAS.
- R. De Rose, A. N. Zelikin, A. P. R. Johnston, A. Sexton, S. F. Chong, C. Cortez, W. Mulholland, F. Caruso and S. J. Kent, Adv. Mater., 2008, 20, 4698 CrossRef CAS.
- A. Sexton, P. G. Whitney, S. F. Chong, A. N. Zelikin, A. P. Johnston, R. De Rose, A. G. Brooks, F. Caruso and S. J. Kent, ACS Nano, 2009, 3, 3391–3400 CrossRef CAS.
- C. D. Austin, X. Wen, L. Gazzard, C. Nelson, R. H. Scheller and S. J. Scales, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17987–17992 CrossRef CAS.
- D. V. Volodkin, N. I. Larionova and G. B. Sukhorukov, Biomacromolecules, 2004, 5, 1962–1972 CrossRef CAS.
- A. Heit, F. Schmitz, T. Haas, D. H. Busch and H. Wagner, Eur. J. Immunol., 2007, 37, 2063–2074 CrossRef CAS.
- T. Fifis, A. Gamvrellis, B. Crimeen-Irwin, G. A. Pietersz, J. Li, P. L. Mottram, I. F. McKenzie and M. Plebanski, J. Immunol., 2004, 173, 3148–3154 CAS.
- M. Yoshida and J. E. Babensee, J. Biomed. Mater. Res., 2004, 71a, 45–54 Search PubMed.
- S. Fischer, E. Uetz-von Allmen, Y. Waeckerle-Men, M. Groettrup, H. P. Merkle and B. Gander, Biomaterials, 2007, 28, 994–1004 CrossRef CAS.
- Y. Waeckerle-Men, E. Scandella, E. Uetz-Von Allmen, B. Ludewig, S. Gillessen, H. P. Merkle, B. Gander and M. Groettrup, J. Immunol. Methods, 2004, 287, 109–124 CrossRef CAS.
- A. Westwood, G. D. Healey, E. D. Williamson and J. E. Eyles, Vaccine, 2005, 23, 3857–3863 CrossRef CAS.
- T. Uto, X. Wang, K. Sato, M. Haraguchi, T. Akagi, M. Akashi and M. Baba, J. Immunol., 2007, 178, 2979–2986 CAS.
- S. R. Little, D. M. Lynn, Q. Ge, D. G. Anderson, S. V. Puram, J. Chen, H. N. Eisen and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9534–9539 CrossRef CAS.
- M. Kool, T. Soullie, M. van Nimwegen, M. A. M. Willart, F. Muskens, S. Jung, H. C. Hoogsteden, H. Hammad and B. N. Lambrecht, J. Exp. Med., 2008, 205, 869–882 CrossRef CAS.
- H. Li, S. Nookala and F. Re, J. Immunol., 2007, 178, 5271–5276 CAS.
- H. Li, S. B. Willingham, J. P. Ting and F. Re, J. Immunol., 2008, 181, 17–21 CAS.
- S. C. Eisenbarth, O. R. Colegio, W. O'Connor, F. S. Sutterwala and R. A. Flavell, Nature, 2008, 453, 1122–1126 CrossRef CAS.
- M. Kool, V. Petrilli, T. De Smedt, A. Rolaz, H. Hammad, M. van Nimwegen, I. M. Bergen, R. Castillo, B. N. Lambrecht and J. Tschopp, J. Immunol., 2008, 181, 3755–3759 CAS.
- F. A. Sharp, D. Ruane, B. Claass, E. Creagh, J. Harris, P. Malyala, M. Singh, D. T. O'Hagan, V. Petrilli, J. Tschopp, L. A. O'Neill and E. C. Lavelle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 870–875 CrossRef CAS.
- J. M. Blander, Trends Immunol., 2007, 28, 19–25 CrossRef CAS.
- J. M. Blander and R. Medzhitov, Nature, 2006, 440, 808–812 CrossRef CAS.
- E. Schlosser, M. Mueller, S. Fischer, S. Basta, D. H. Busch, B. Gander and M. Groettrup, Vaccine, 2008, 26, 1626–1637 CrossRef CAS.
- A. Westwood, S. J. Elvin, G. D. Healey, E. D. Williamson and J. E. Eyles, Vaccine, 2006, 24, 1736–1743 CrossRef CAS.
- J. Kazzaz, M. Singh, M. Ugozzoli, J. Chesko, E. Soenawan and D. T. O'Hagan, J. Controlled Release, 2006, 110, 566–573 CrossRef CAS.
- S. Hamdy, O. Molavi, Z. Ma, A. Haddadi, A. Alshamsan, Z. Gobti, S. Elhasi, J. Samuel and A. Lavasanifar, Vaccine, 2008, 26, 5046–5057 CrossRef CAS.
- K. Lingnau, A. Egyed, C. Schellack, F. Mattner, M. Buschle and W. Schmidt, Vaccine, 2002, 20, 3498–3508 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |