Stimuli-responsive gels as reaction vessels and reusable catalysts†
David
Díaz Díaz
*ab,
Dennis
Kühbeck
a and
Rudy J.
Koopmans
c
aInstitut für Organische Chemie, Universität Regensburg, Universitätsstr. 31, 93040 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de; Fax: +49-941-943-4121
bICMA, CSIC-Universidad de Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain
cDow Europe GmbH, Bachtobelstrasse 3, 8810 Horgen, Switzerland
First published on 28th September 2010
Abstract
As part of a continuing scientific challenge, a substantial effort during the past few decades has been devoted towards altering the selectivity of chemical transformations by arranging the potential reactants in a number of organized and confining media. Such systems, having features significantly different from those of isotropic solutions, include, for example, micelles, microemulsions, molecular aggregates, liquid crystals, and zeolites. Among these materials, stimuli-response gels constitute another important class of nanostructured and dynamic systems with high active surface areas and remarkable diffusion properties. Within this group, polymer gels have been traditionally used to obtain catalytic and reactive soft materials. Moreover, gels made of low-molecular-weight compounds represent a major novelty in this area as potential soft-vessels to carry out chemical reactions with control on product selectivity. In addition, the possibility of integrating switchable catalytic functions in both organo- and hydrogels shall accelerate the development of robust platforms for the ‘bottom–up’ tailor-fabrication of more sophisticated functional materials. The present critical review reports on the most important results published during the last decade regarding the use of ‘smart’ gels that has displayed promising properties as selective soft-nanoreactors and/or heterogeneous recyclable catalysts (152 references).
 David Díaz Díaz | David Díaz Díaz was born in 1974 in Tenerife (Spain). He received both BSc (1997) and PhD (2002) degrees in Chemistry (University of La Laguna, Spain) under the supervision of Prof. Martín. In 2002, he joined Prof. Finn's group at TSRI (USA) for a post-doctoral stay. In 2006, he was appointed as a ‘RyC’ research fellow at The Autonomous University of Madrid. In 2007, he joined The Dow Chemical Company (Switzerland) as Sr. Chemist. He is currently a Tenured Scientist at the ICMA-CSIC and an AvH Fellow at the University of Regensburg (Germany). His research interests lie on the synthesis of soft materials for catalysis and biomedicine. |
 Dennis Kühbeck | Dennis Kühbeck was born in 1984 in Passau (Germany). In 2008, he completed his Bachelor Thesis “Synthese fluoreszenter Cyclen-Komplexe” in the group of Prof. König (University of Regensburg, Germany) and received his BSc degree in 2008. In 2009, he completed, the Practical Research “Synthese von Cobalt(II) Schiff Basen Komplexen und deren Immobilisierung auf mit Graphit beschichteten Cobalt-Nanopartikel” in the group of Prof. Reiser, and “Synthese (M)ANT-ähnlicher Verbindungen”, in the group of Prof. König. Currently, he carries out his Master Thesis on the synthesis and applications of functional gels in the group of Dr Díaz Díaz. |
 Rudy J. Koopmans | Rudy J. Koopmans was born in 1956 in Kruibeke (Belgium). He obtained an MSc (1979) degree in Chemistry and a PhD (1983) degree in Physical & Macromolecular Chemistry from Antwerp University (Belgium) under the supervision of Prof. E. Vansant. In 1983, he joined The Dow Chemical Company, where he has held various R&D positions in Europe and USA. He is currently a Scientist in the Basic Plastics organization (Switzerland). He is also a visiting professor at Leeds University. His research aims at developing next generation of polymeric materials with specific interest in rheology of fluids and dynamics of self-assembling molecules. |
1. Introduction
Gels of both organic solvents (organogels) and water (hydrogels) have defined a significant research field for well over a century.1–3 These hierarchical, self-assembled, and viscoelastic materials may be considered to be either hard or soft based on their rheological characteristics. Among different classification criteria, their categorization according to the driving forces for molecular aggregation is probably the most familiar. Thus, it is usual to find literature dealing with chemical gels4,5—based on covalent bonds, usually cross-linked polymers-, or physical gels1,6,7—made of either low-molecular-weight (LMW) compounds or polymers, and based on non-covalent bonds, predominantly hydrogen-bonding, van der Waals, charge-transfer, dipole–dipole, π–π stacking, and coordination interactions, which usually lead to reversible gel-to-sol phase transitions. Moreover, systems based on both types of connections are also known.8,9The solid-like appearance of a gel is a result of the entrapment of the liquid (major component) in the interstices of a solid 3D-matrix of a large surface area (minor component), typically through surface tension and capillary forces.10 The formation of the solid matrix is a consequence of the entanglement of 1D-polymeric (or suprapolymeric) strands of macromolecules (or LMW molecules), -so called gelators-, typically of micrometer scale lengths and nanometer scale diameters, through the above mentioned forces. Hence, in many cases, gels may immobilize up to 105 liquid molecules per gelator through a metastable dissolution-crystallization equilibrium, and increase the viscosity of the medium by a factor of 1010, with the potential to respond to a variety of external stimuli.11,12
It is true that the limited understanding of the complex molecular gelation phenomena still prevents the development of a quantitative set of rules for the design of new gelators.13 However, as it has frequently happened for example with many industrial products, the development of high-tech applications has taken place much faster than the acquisition of a full knowledge on the mechanisms governing the key physical and/or chemical processes. In this regard, ‘smart’ gels are now widely used in diverse applied fields such as regenerative medicine, drug-delivery, sensors, actuators, cosmetic, foods, environmental remediation, and nanoelectronic (‘bottom–up’ approaches), among others.14 Moreover, development of new synthetic methods, together with the substantial progress made in the control of the properties of porous materials on the nano-, meso-, and macroscale,15 and the possibility of carrying out analytical measurements in the dynamic gel medium using, for instance, fluorescence spectroscopy or cyclic voltammetry, have stimulated the study of a number of diffusion-controlled processes within the constrained environment of these materials, which could have an impact on their potential catalytic properties and the control of product selectivity.16,17 Indeed, the variety of possible compositions and the porous nature of many of these supramolecular gels, made of self-assembled fibrillar domains easily dispersed in a liquid phase of micrometer size, may bear a resemblance to several kinds of macromolecular nanoreactors, such as organic polymers, micelles or zeolites.18,19 Most importantly, supramolecular aggregation has been also declared to play a crucial role in several catalytic processes connected with chirality amplification,20,21 which could enhance the potential of chiral gels as medium for asymmetric reactions.
Within this challenging topic in materials science, we review here the most important results published during the last decade dealing with gels that have showed promising properties as selective nanoreactors and/or heterogeneous recyclable catalysts. Although the results regarding practical applications are still in a very preliminary stage, making the scope of the field still limited, we believe that the efforts carried out during the last years well deserve a first overview of examples to highlight timely the new horizons of these materials in catalysis. To the best of our knowledge, this is the first review that focuses entirely on reactions carried out within gel-based materials. On the other hand, the design, synthesis, physical properties, and other applications of stimuli-responsive self-assembling gels such as their use as templates in the synthesis of well-defined nanostructured materials, which have been already discussed in numerous review articles,6,7,22 are hence beyond the scope of this manuscript. Nevertheless, we highly recommend reading these comprehensive overviews to help for a better understanding of the structural properties of these systems. Although the presented review concentrates on gels in which one of their constitutive phases is a liquid, other related materials in which the liquid phase has been replaced by a gas phase—usually via supercritical drying—(i.e. aerogels), are also representatively cited taking into consideration the existing number of reviews that deal with their application in heterogeneous catalysis.23–26
It is well known that polymer gels constitute the traditional approach to obtain catalytic and reactive soft materials.27 Therefore, in order to achieve a systematic structure during the following chapters a broader classification system of the gels than the mentioned above has been adopted. Thus, this overview presents in first place a selection of the most representative examples of “polymer gels” used in catalysis during the last years. Herein, the term “polymer gels” refers to gels made of polymers, which may include systems where either covalent –chemical gels– or non-covalent forces –polymer physical gels28– are the driving force for molecular aggregation. For convenience, the examples collected in this section are discussed within the context of the following renowned type of materials: electroactive-, acrylamide-, polyvinyl alcohol (PVA)-, and biopolymer-based hydrogels (Fig. 1). Next, a section focused on “low-molecular-weight gels” (gels made of self-assembled low-molecular-weight compounds) is conferred as one of the most recent and innovative area of research dealing with gel-type soft materials. Hydrogels, organogels, and metallogels constitute the three major pillars of this section (Fig. 1). In addition, subheadings have been built-in to distinguee examples where the gel acts as a catalyst and examples where the gel is a reactive framework.
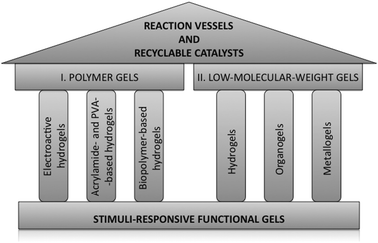 | ||
| Fig. 1 Temple-like structure of the present overview. | ||
2. Polymer gels as catalytic systems
There is an exceptional long history associated to the attachment of catalysts to gel-type materials (i.e. cross-linked resins, silica gel), and the development of technologies to precisely control their surface area, pore volume, and average pore diameter of such materials.29 If we focus on the recent achievements within this context, the work from Wagner, Mioskowski and co-workers30 could serve as a representative example for the reader. This group demonstrated that polyionic gel beads 1 (Scheme 1), prepared from a Merrifield-type resin, constitute a highly polar microenvironment for both efficient metal scavenging and active heterogeneous metal-based catalyst preparation. Such insoluble polyionic gels promote the formation of stabilized Pd nanoparticles of 3–10 nm in diameter during the Suzuki reaction. A number of experiments based on hot-filtration and three-phase tests confirmed that the heterogeneous gel-embedded colloidal Pd nanoparticles are indeed the catalytically actives species. The soaking ability and catalytic activity of these recyclable gel-embedded colloidal catalysts was found to depend on both the strength of the ionic environment and the non-covalent metal–ion interactions. As a major interest from a practical standpoint, the catalytic activity of the system was found almost constant after five runs, and ICP-AES analyses of the supernatant showed only less than 2% of metal leaching. The same concept was later applied to the fabrication of Rh-soaked catalysts, which allowed, for instance, the regio- and stereoselective hydrosilylation of phenylacetylene affording the corresponding (E)-1-silyl-1-alkyne in good yields.31,32 Similarly, an useful combination of catalytic abilities could be achieved by a bimetallic polyionic-gel Rh–Pd catalyst, which showed a notable effectiveness in a sequential one-pot hydrosilylation/Hiyama coupling protocol affording stereoselectively (E)-disubstituted olefins, thus exemplifying the potential wide scope and modularity of this approach.33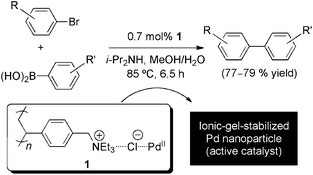 | ||
| Scheme 1 Suzuki cross-coupling reaction of aryl bromides catalyzed by ionic-gel-stabilized Pd nanoparticles formed from 1.30 | ||
On the other hand, on the extensive subject of silica gel supported catalysts, Marr and co-workers have recently described the preparation of molecular hydrogenation catalysts co-entrapped into ionic liquids inside a silica matrix by the sol–gel method. In this case, the combination of an ionic liquid and a solid oxide matrix forms stable pores that contain the active species, allowing a rapid diffusion of the substrate and product to and from the catalyst in the gel state.34 However, the exact role of the catalyst in templating the matrix is still unknown in these systems. In another recent work, Vioux and co-workers35 reported the preparation of highly processable Pd(II)-containing ionogels through the incorporation of Pd(OAc)2 into a solution of an ionic liquid. Although the materials were successfully used as catalysts in Heck–Mizoroki coupling reactions, the reaction rates were compared to homogeneous systems showing no significant difference. Leaching tests showed that catalysis actually took place in the ionic liquid phase, and salts by-products were trapped in the ionogels, offering practical advantages over other supported ionic liquids as media for organic reactions.
Despite being a very active area of research, the rigidity associated to these materials has moved the interest of many research groups towards softer gel-based systems with high active surface area and excellent diffusion properties through a viscoelastic matrix. The advances made in this direction point out that the confinement effects in these materials could be responsible for the enhancement of substrate-catalyst interactions in comparison with reactions carried out in homogeneous phase. As it is classified below, most of the examples of viscoelastic polymer gels can be integrated in the following major categories within the general context of catalysis: electroactive-, acrylamide-, PVA, and biopolymer-based hydrogels.
2.1 Electroactive hydrogels
It is well-documented that redox hydrogels constitute the only electron-conducting phases in which the permeation of water-soluble biological reactants and products, including ions and electrons, are all rapid, facilitating the transport of electrons between electrodes and multiple layers of redox enzymes without a particular orientation.36,37 In addition, the volume change of hydrogels has showed a great potential for application as biomaterials that quickly can be reduced by an applied electric field. This volume change of the hydrogel is similar to natural muscle, and can be utilized as biosensors and as artificial muscle when employing an electrical stimulus. An enormous body of contributions on this topic has appeared during the last decades and has been reviewed in many publications and books.38,39Rescuing some recent examples on the use of electroactive hydrogels in catalysis we should highlight the work of Mano and co-workers40,41 who described a 0.55 V (Ag/AgCl) laccase-wiring redox hydrogel as a more efficient O2 electroreduction catalyst than the classical platinum catalyst (Fig. 2).
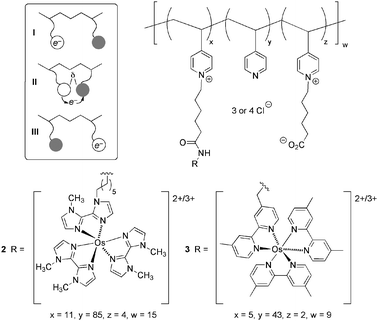 | ||
| Fig. 2 Left: Three-steps electrons diffusion by collisions between mobile redox centres tethered to backbone of the cross-linked polymer gel. Right: Structure of hydrogelators 2–3 designed to electrically connect the reaction centres of glucose oxidase and laccase to electrodes respectively.40 | ||
The cyclic voltammograms of hydrogelators-bound osmium complexes 2 and 3 (Fig. 3, top) having the same redox potential (+550 mV vs. Ag/AgCl) indicated that a long eight-atoms flexible spacer arm results in a higher apparent electron diffusion coefficient (Dapp) (7.6 × 10−7 cm2 s−1) in comparison with a shorter tether, which make easier the collisional electron transfer between proximal redox centres. On the other hand, the coordination of the Os2/3 centres with three substituted bipyridines provided the required stability against ligand substitution. Optimization studies of the catalysts composition for these bioelectrocatalysts (Fig. 3, bottom) using redox hydrogelators 2 and 3 resulted in the following practical recipe: 45.6 wt% laccase, 47.2 wt% 2, 7.2 wt% poly-(ethylene glycol) (400) diglycidyl ether (PEGDGE), and 40 wt% laccase, 52.8 wt% 3, 7.2 wt% PEGDGE.
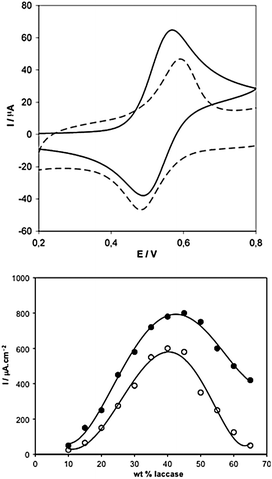 | ||
| Fig. 3 Top: Cyclic voltammograms of polymers hydrogelators 2 (solid line) and 3 (dashed line) on 3 mm diameter glassy carbon electrodes under argon (pH 5, 37 °C, 20 mV/s). Anodic/cathodic peaks separation: 65 mV for 2, and >120 mV for 3. Bottom: Dependence of the current density on the laccase weight percentage for the bioelectrocatalysts made with polymer 2 (solid circles) and with polymer 3 (open circles). Adapted with permission from ref. 40, ® 2006, The American Chemical Society. | ||
It is easy to realize that studies towards the mathematical modelling of hydrogel-based reaction-diffusion systems could help for a better understanding of the effect of self-assembled fibrillar networks (SAFINs) on product selectivity when used as reaction vessels or catalysts. In this context, Merkin and co-workers42 validated a simplified model that accurately describes a hydrogel-based nanoreactor that behaves like a semiconductor diode. Therein, a chemically cross-linked PVA hydrogel cylinder was submitted to a pH gradient by having an acid and a base maintained at constant concentrations in reservoirs at each end of the 1D-hydrogel reactor, across of which a potential difference of a given strength was also applied. The stationary concentration profiles for the different ions were successfully described by a system of equations involving migration caused by the electric field.42
Moreover, the possibility of controlling the diffusion of macromolecular systems through a polymer gel for catalytic purposes (e.g. immobilization of enzymes) has been addressed in many historical papers and continues being a highly active area of research (see biocatalysis below -subchapters 2.2 and 2.3-).43,44 A few examples dealing with electroactive enzyme-containing hydrogels are discussed in this subchapter for convenience. Thus, Banta and co-workers45 reported a protein-engineering approach to advanced functional hydrogels for biocatalytic, biosensing, and tissue-engineering applications. Specifically, the authors described two bifunctional protein building blocks that co-assembled to form a bioelectrocatalytic supramolecular hydrogel, that, when polarized, generated a catalytic current in the presence of molecular oxygen with the consequent reduction to water (Fig. 4, bottom).
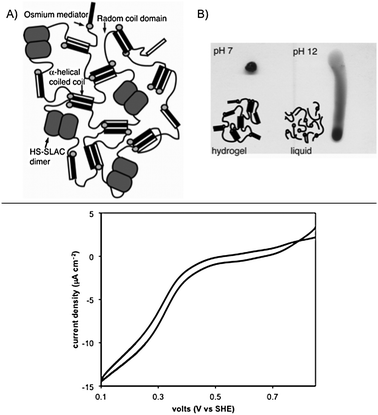 | ||
| Fig. 4 Top: (A) Schematic diagram of OsHSH-1 and HS-SLAC hydrogel. (B) Function of the H domains confirmed by the formation of hydrogel through a change in pH from 12 (loss of secondary structure) to 7 (regain of secondary structure and physical cross-linking functionality). The photographs were taken using 7.5 wt% of OsHSH-1 deposited on vertical glass slides. Bottom: Bioelectrocatalysis of a mixed hydrogel of 7.5 wt% OsHSH-1 and 2.5 wt% HS-SLAC on a 3 mm glassy carbon rotating disk electrode (pH = 7.0, 25 °C, 1 mmol L−1 O2). Protein sequences: a-helix = SGDLENE VAQLENE VRSLEDE AAELEQK VSRLKNE IEDLKAE; random coil = (AGAGAGPEG)10. Reprinted with permission from ref. 45, ® 2008, National Academy of Sciences, USA. | ||
The building blocks used to prepare this biohybrid consisted of (1) a chimera of artificial α-helical leucine zipper and random coiled domain fused to a polyphenol oxidase, small laccase Streptomyces coelicolor (SLAC); and (2) an electron-conducting metallopolypeptide (HSH) having two α-helical domains (H domain) separated by a randomly coiled domain (S domain), which formed a hydrogel via tetrameric coiled coils. The helical and random domains were identical to those fused to the polyphenol oxidase. Electron-conducting functionality was derived from the divalent attachment of an osmium bis-bipyridine complex to histidine residues within the peptide (OsHSH-1), whereas the active dimers of HS-SLAC were incorporated within the OsHSH-1 network by means of physical cross-linking at the coiled-coil bundles (Fig. 4A). The bioelectrocatalytic results demonstrated that HS-SLAC maintained enzymatic function while bound within the hydrogel network, requiring less osmium redox mediator than previous hybrid systems reported in the literature for similar purposes. Nonetheless, further studies are still needed in order to demonstrate the scope and applicability of such system in a manufactured biofuel cell or biosensor.
Xian and co-workers46 also described the fabrication of a biohybrid hydrogel by integrating Horseradish peroxidase (HRP) into a porous cross-linked polyhydroxyl cellulose (PHC)-based hydrogel, which provided a biocompatible microenvironment for the effective immobilization of the enzyme, and subsequent development of a third generation H2O2 biosensor. UV-vis spectroscopy showed that the native secondary structure of the HRP-loaded hydrogel was well retained in comparison to free HRP. In addition, the direct electrochemistry of HRP could be observed using this strategy because PHC could afford a native platform for a proper conformation of the enzyme for efficient electron transfer. Under optimized conditions, the enzyme electrode showed a good stability and a reproducible linear response over the range from 1.0 μmol L−1 to 1.0 mmol L−1 with a detection limit (S/N = 3) of 0.5 mmol L−1.
2.2 Acrylamide- and PVA-based hydrogels
Tanaka and co-workers47 prepared a polymer gel with a remarkably catalytic activity that could be switched on and off by an infinitesimal change in solvent composition. This gel consisted of two species of monomers (Scheme 2): (1) the major component, N-isopropylacrylamide (NIPA), that made the gel swell and shrink in response to a change in composition of ethanol/water mixtures; and (2) the minor component, 4(5)-vinylimidazole (Im), which was capable of catalysis, was copolymerized into the gel network.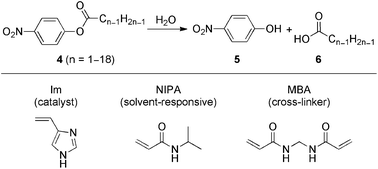 | ||
| Scheme 2 Hydrolysis of p-nitrophenyl esters 4 (top), which is catalyzed by the gels made of NIPA, Im, and N,N′-methylenebisacrylamide (MBA) (bottom).47 | ||
The reaction rate for catalytic hydrolysis of p-nitrophenyl caprylate 4 to afford 5 + 6 was small when the gel was swollen (Fig. 5, left). In contrast, when the gel was shrunken, the reaction rate increased dramatically to 5 times higher than that catalyzed by the monomer solution (Fig. 5, right). These results indicated that the absorption of the substrate by the hydrophobic environment created by the N-isopropylacrylamide polymer could well be a primary factor determining the kinetics in the shrunken gel but not in the swollen state.
 | ||
| Fig. 5 Concentration of p-nitrophenol (5) as a function of time. Open circles: catalysis by the gel in in the swollen state at 10 vol% ethanol (left), and in the shrunken state at 12.5 vol% ethanol (right). Solid circles: catalysis by an imidazole monomer solution (control experiment). Reprinted with permission from ref. 47, ® 2000, National Academy of Sciences, USA. | ||
On the other hand, a quick look to the bibliography on nanosciences confirms that the development of new procedures for the synthesis of well-defined and stable metallic nanoparticles has been an intensive research field during the last decades. Such interest comes mainly because of their remarkable electronic, optical, and catalytic properties in comparison to their bulk metal counterparts.48,49 In this regard, and just within the scope of this review, Zhang and co-workers50 reported the use of a thermoresponsive hydrogel based on cross-linked poly(glycidyl methacrylate-co-N-isopropylacrylamide) (PGMA-co-PNIPA) as a thermotunable and recyclable nanoreactor of gold nanoparticles (∅ = 15 nm) due to the effective coordination of nitrogen atoms of the crosslinker (diethylenetriamine) with gold ions.
The hydrogel was characterized as uniform spheres (∅ = 280 nm) at a temperature (T) < lower critical solution temperature (LCST = 50 °C), which gradually shrunken at T > LCST until a constant diameter of 113 nm at 75 °C. The reduction of 4-nitrophenol catalyzed by the thermoresponsive hydrogel/gold nanocomposite at different temperatures was studied as a proof of concept (Fig. 6). The conversion at T < LCST homogenous catalyst was found to be relatively fast (i.e. conversion(25 °C, 200 s) = 65%), whereas at T > LCST heterogeneous catalyst reduction rate significantly decreased (i.e. conversion(75 °C, 600 s) = 10%). Slowly reduction of part of the reactant was also observed at T > LCST, presumably due to its diffusion through the hydrophobic shell of the soft-nanoreactor.
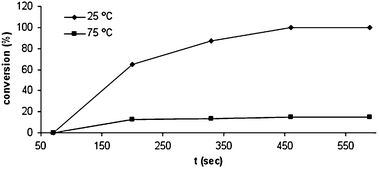 | ||
| Fig. 6 Conversion of 4-nitrophenol catalyzed by the thermoresponsive hydrogel/gold nanocomposite at different temperatures. Reaction conditions: 4-nitrophenol, 0.10 mmol L−1; NaBH4, 3.3 mmol L−1; hydrogel/gold nanocomposite containing 8.3 × 10−5 mmol of gold. Adapted with permission from ref. 50, ® 2007, Wiley-VCH Verlag GMBH & Co. | ||
Independently, Geckeler and co-workers51 reported the synthesis of hydrogel networks based on NIPA and sodium acrylate (SA) by redox-polymerization in the presence of MBA, which were used as a carrier for the fabrication of highly stable and uniformly distributed silver nanoparticles via in situ reduction of silver nitrate in the presence of sodium borohydride as a reducing agent. Preliminary experiments also indicated that the hydrogel hybrid-stabilized silver nanoparticles (∅ = 2.67 nm) could be efficiently employed as antibacterial material towards E. Coli in comparison with larger non-stabilized silver nanoparticles. Within this context, highly stable and uniformly distributed silver nanoparticles (∅ = 20–30 nm) were prepared more recently using dextran-based hydrogel networks as a carrier via in situ reduction of silver nitrate.52 In principle, the same approach could easily be applied to synthesize other metal nanoparticles.
Zhang's group published last year an illustrative example of catalytic metallic species in acrylamide-based gel materials.53,54 Therein, a porous, thermoresponsive (sol-to-gel phase-transition temperature (Tgel) = 32 °C) and pH-responsive, and chelating hydrogel of poly(N-isopropylacrylamide)-co-poly2-methacrylic acid 3-(bis-carboxymethylamino)-2-hydroxypropyl ester (PNIPAM-co-PMACHE) was proposed as both a reaction medium and a recyclable—via swelling/deswelling—Pd catalyst support for organic reactions. As shown in Scheme 3, Suzuki and Heck reactions performed within the PNIPAM-co-PMACHE hydrogel demonstrated that the hydrogel is indeed a suitable reaction medium where organic synthesis can be accelerated in comparison to the processes in water (Fig. 7), since the hydrophobic reactants and Pd catalyst can indeed become highly enriched within the hydrogel.
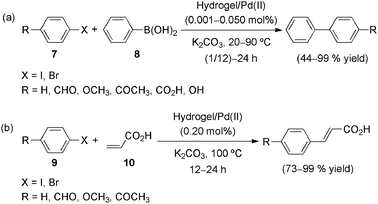 | ||
| Scheme 3 Suzuki (a) and Heck (b) reactions catalyzed by PNIPAM-co-PMACHE hydrogel/Pd(II) composite. Reaction conditions: aryl halide 7, 9 (1.0 mmol), phenylboronic acid (8) or acrylic acid (10) (1.5 mmol), K2CO3 (3.0 mmol).53 | ||
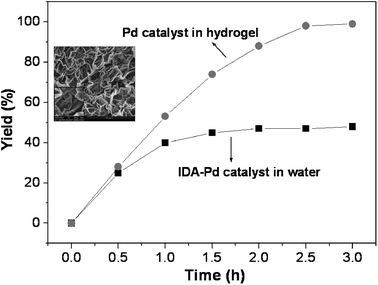 | ||
| Fig. 7 Comparison of the kinetic profiles of Suzuki reaction within the hydrogel (circle) and in water (squares). Reaction conditions: 4-bromoacetophenone (1.0 mmol), benzenboronic acid (1.5 mmol), K2CO3 (3.0 mmol), 0.050 mol% of Pd(II) catalyst, 40 °C. Inset: SEM image of the porous structure of the PNIPAM-co-PMACHE hydrogel. Adapted with permission from ref. 54, ® 2009, The American Chemical Society. | ||
Very recently, the same group also reported the preparation of size-controlled Au nanoparticles within the PNIPAM-co-PMACHE hydrogel.55 The Au-loaded thermoresponsive hydrogel obtained in this case was found to be an effective and reusable catalyst for aerobic alcohol oxidations, where the catalytic activity resulted highly dependent on the size of the encapsulated metal nanoparticles.
Sabitha, Shailaja and co-workers56 reported the in situ synthesis of ligand-free, palladium-supported 13, poly(N-isopropylacrylamide-co-potassium methacrylate) poly(NIPA-co-PMA) hydrogel nanocomposites with good swelling properties and high thermal-stability, using different co-monomer ratios and MBA as cross-linker (Scheme 4).
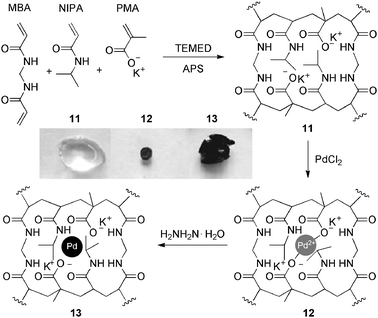 | ||
| Scheme 4 Synthesis of poly(NIPA-co-PMA)-Pd hydrogel nanocomposites. Inset: photographs of the core hydrogel 11 (swollen) and hydrogel-Pd nanocomposites 12–13 (after complexation and reduction respectively). Pd particles in all the gels showed a narrow size distribution with an average size of 5 nm. TEMED = N,N,N′,N′′-tetramethylethylenediamine, APS = ammonium persulfate. Adapted with permission from ref. 56, ® 2008, Elsevier B.V. | ||
The catalytic performance of these Pd-hydrogel nanocomposites was examined for Suzuki–Miyaura cross-coupling reaction of aryl halides (1.0 mmol) with arylboronic acids (1.5 mmol) in the presence of Na2CO3 (3.0 mmol) in an aqueous medium (0.125 M) under inert refluxing conditions. The results showed that the use of 1% w/w hydrogel (9.84 mmol Pd) with comonomer ratio of 8.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6 mmol of NIPA:PMA exhibited optimum catalytic activity, affording the corresponding biaryl and heteroaryl products in very good yields (86–98%) within 1.5–3 h, which represent a good mark in this area. The Pd-hydrogel catalyst could be effectively reused 5–6 times after simple filtration without significantly loss of activity.
:1.6 mmol of NIPA:PMA exhibited optimum catalytic activity, affording the corresponding biaryl and heteroaryl products in very good yields (86–98%) within 1.5–3 h, which represent a good mark in this area. The Pd-hydrogel catalyst could be effectively reused 5–6 times after simple filtration without significantly loss of activity.
Acrylamide-based hydrogels have also found interesting applications as artificial enzymes. In this regard, Liu and his group57 recently reported the preparation of a model microgel catalyst with modulatory glutathione peroxidase (GPx) activity by introducing GPx active sites into temperature responsive PNIPAM scaffolds. In this case a tellurium-containing compound, bis-(3-acryloyloxypropyl)-telluride, was used as the catalytic centre. Compared with diphenyl diselenide (PhSeSePh), a well-studied GPx mimic, the hydrogel-based catalyst was about 339000-fold more efficient than that of PhSeSePh for catalyzing the model reduction of cumene hydroperoxide by 3-carboxyl-4-nitrobenzenethiol. More importantly, the catalytic activity of the microgel-based artificial enzyme could be reversibly turned on and off by changing the temperature, which induced dramatic changes of both pore size and hydrophobic character of the core.
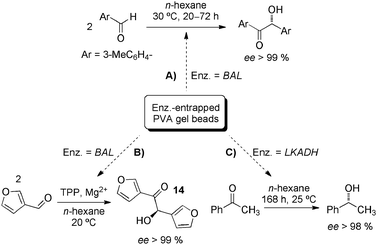
BAL-containing hybrid gel system was also used later for the same group for the continuous synthesis of enantiopure (R)-3,3′-furoin (14) in a fluidised bed reactor (Scheme 5B).60 The results showed an enhanced stability with half-life times under operation conditions of more than 100 h, as well as superior enzyme utilisation in terms of productivity. PVA-gel beads (∅ = 500–1000 μm) have been also employed to encapsulate alcohol dehydrogenase from Lactobacillus kefir LKADH and used as a catalyst for the conversion of prochiral ketones to chiral alcohols in n-hexane (Scheme 5C).61 As a practical feature it is worth to remark that the general compatibility of these enzyme-containing hydrogel-beads with organic solvents allows the transformation of poor water-soluble substrates.
More recently, Aida and co-workers62 reported the encapsulation of myoglobin in self-healing hydrogels made of <0.4 wt% of organic components (sodium polyacrylate and a G3-dendritic macromolecule –Fig. 8–) and 2–3 wt% of clay nanosheets. Such hybrid hydrogel was tested for the oxidation of o-phenylenediamine to 2,3-diamino-5,10-dihydrophenazine (λmax = 420 nm) in H2O2 at 20 °C. The results showed retention of ca. 71% of the protein activity relative to the free form, which highlighted the potential utility of this hydrogel as an effective biological carrier.
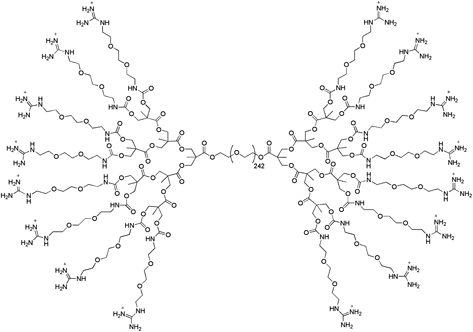 | ||
| Fig. 8 G3-binder used by Aida and co-workers to prepare self-healing hydrogels with potential as biological carriers.62 | ||
2.3 Biopolymer-based hydrogels
It is well known that during the last decades the emphasis of science and technology has shifted more towards environmentally friendly and sustainable resources and processes. Under this standpoint, the direct utilization of natural materials such as biopolymers (e.g. chitosan, alginate, carrageenan, etc.) for catalytic applications represents a very attractive strategy to allow the production and ready separation of large quantities of products with the use of a small amount of catalyst. In this direction, early studies conducted by Paradossi and co-workers63 demonstrated the feasibility of using biobased-metal nanoreactors for oxidative processes. They reported the preparation of an Cu(II)-loaded chitosan-based hydrogel using a polyfunctionalized β-cyclodextrin as cross-linking agent. The copper-loaded hydrogel acted as an efficient heterogeneous catalyst for the oxidation of adrenaline (15) to adrenochrome (16) by molecular oxygen in aqueous media (Scheme 6). Kinetic analyses indicated that both the reaction rate and mechanism were preserved working in the presence of the catalyst anchored to the gel phase, but with the consequent advantage of being recyclable. In principle, the host-biocavities using this approach might be modulated according to the geometry of the guest molecules and/or multivalent ions inside the hydrogel.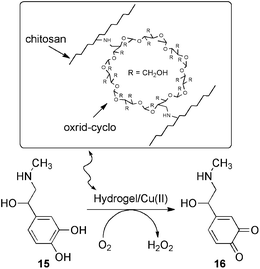 | ||
| Scheme 6 Oxidation of adrenaline catalyzed by Cu(II)-loaded hydrogel based on chitosan/oxrid-cyclo. Reaction conditions: 0.17 g of swollen gel per mL of reaction mixture, 0.1 M Hepes buffer at pH 6.78, 25 °C. Turnover number of the catalyst kmax = 10−4 s−1.63 | ||
Reddy and co-workers64 reported the use of chitosan-based hydrogel beads as an environmentally benign and recyclable base organocatalyst for non-stereoselective aldol and Knoevenagel reactions of arylaldehydes 17, providing the products 18 and 19 respectively, in good yields and chemoselectivity (aldol/dehydration) under biphasic conditions (Scheme 7). The higher activity observed of the hydrogel in comparison to commercial chitosan and dried chitosan beads may have the origin in the higher accessibility of the amine functional groups in the former (55–65%, 2.5%, and 1.65%, respectively, based on ca. 80% deacetylated commercial chitosan).
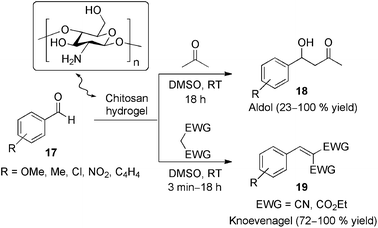 | ||
| Scheme 7 Chitosan hydrogel-catalyzed aldol and Knoevenagel reactions without formation of self-condensation or Cannizaro products. The catalyst could be recovered by simple filtration and reused efficiently in further experiments.64 | ||
As discussed in the previous section, a different approach to hydrogel-based catalysts consists in incorporating metallic species directly into the viscoelastic matrices. In this direction, Quignard and co-workers65 reported the preparation of hybrid aerogels based on palladium doped alginate gel and their use as efficient heterogeneous catalysts for Suzuki C–C coupling reactions. On the other hand, Reddy's group66 also reported the immobilization of copper(II) ions onto alginate via interactions with their carboxylate groups. Using copper acetate monohydrate as source of Cu(II) ions, uniform and high-temperature stable copper-alginate gel beads were prepared with a metal content of ca. 0.0042 mmol per gel bead.67 These materials were used as effective heterogeneous and recyclable catalysts for regioselective 1,3-dipolar cycloaddition of alkynes 20 with azides 21 (AAC), as well as for the oxidative coupling of 2-naphthols and phenols 23 in water to afford the 1,4-disubstituted 1,2,3-triazoles 22 and biaryl compounds 24 respectively (Scheme 8).
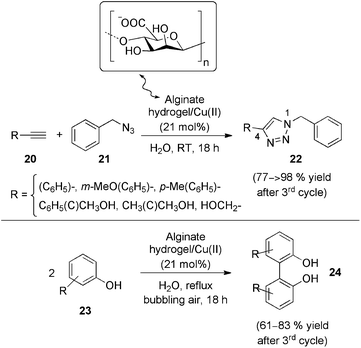 | ||
| Scheme 8 Copper-alginates catalyzed 1,3-cycloaddition reactions between terminal alkynes (20 mol% excess) and azides (top); and oxidative coupling of 2-naphthols/phenols (bottom).66 | ||
Despite the striking results reported with this metallogel, especially from a practical point of view, the exact origin of their catalytic activity towards AAC has not been carefully studied yet. Cu(II) salts has been previously claimed as a highly active catalyst for the AAC,68 which apparently contradict the extensive mechanistic evidences reported so far regarding the metal-catalyzed version of this reaction.69–71 Such studies suggest that any non-background AAC that occurs in the presence of oxidant Cu(II) salts comes from reasonable reduction to Cu(I) under most experimental conditions (e.g. via Glazer coupling of acetylenes), which is indeed very difficult to avoid and detect in trace amounts, which on the other hand can be enough to catalyze the AAC. Therefore, additional investigations have to be performed before the modularity and scope of different metal-catalyzed reactions can be established. On the other hand, the removal of the liquid phase in this type of materials has been recently employed to prepare alginate-based metalloxerogels that can act as heterogeneous catalysts in the reduction of 4-nitrophenol.72,73
:1 mixture bromohexane/cyclohexane mixture (1:1) afforded a slightly higher relative rate (up to 10%) in a fluidized-bed reactor, which essentially invites to a further necessary optimization for practical purposes. More recently, Lin and co-workers75 investigated the activity of γ-glutamyltranspeptidase from Escherichia coli (EcGGT) entrapped in Ca-alginate-κ-carrageenan beads and their use in the synthesis of L-theanine (25), a neurologically-active aminoacid (Scheme 9B). The authors determined an optimum alginate concentration of 2% (w/v), and a maximum enzyme activity using a loading enzyme concentration of 1.5 mg/g alginate. The optimum bead size for the enzyme immobilization was found to be 1.9 mm. In this case, the catalytic system was found to be stable on repeated reuse for five cycles without any significant loss in the enzyme activity, which dropped to 45% of the initial value after the sixth cycle. Gotor's76,77 and Qader's78 groups reported also the immobilization of Manihot esculenta (ME) and Bacillus subtilis, respectively, in Ca-alginate hydrogel beads. The former used the hybrid hydrogel for the stereoselective acetylation of a wide range of racemic alcohols (Scheme 9C), and the latter tested the protease activity using casein as a substrate to quantify different factors that influence the immobilization of the enzyme (i.e. temperature, pH, calcium chloride, salts and substrate concentrations, incubation time).
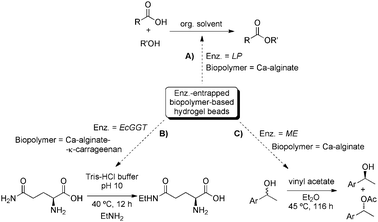 | ||
| Scheme 9 Representative biocatalytic processes carried out using enzymes encapsulated in biopolymer-based hydrogels.74–76 | ||
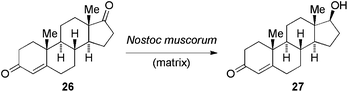
In spite of the numerous efforts reported in this area, it is surprising the lack of more comparative studies to determine the influence of the polymer nature and gel-beads properties (e.g. morphology, cross-linking density, size, diffusion coefficient, etc.) on the outcome of the studied reaction. The use of Ca-alginate hydrogel beads as matrices and concentrations up to 2% for optimal enzyme activity seem to be a constant in most of the reported examples. Nevertheless, such mentioned comparative research would facilitate drawing conclusive statements regarding the factors to consider when preparing enzyme-containing hydrogel beads for a specific biotransformation. In this sense, Faramarzi and co-workers79 very recently reported a valuable study on the influence of whole microalgal cell immobilization and organic solvent on the bioconversion of androst-4-en-3,17-dione (26) to testosterone (27) by Nostoc muscorum. These cells were immobilized by entrapment methods in different gelling media (i.e. agar, agarose, κ-carrageenan, polyacrylamide, polyvinyl alcohol, and sodium alginate). As outlined in Table 1, hydrogel matrices made of sodium alginate with a concentration of 2% (w/v) were found to be the most favourable for the immobilization and activity of the cells, affording a conversion three times higher than the free form (Table 1).
| Type of matrix | 27 Concentration (g/L) | Residual 26 (g/L) | Conversion (%) |
|---|---|---|---|
| a Reaction conditions: 20 mL hexadecane, 30 °C, 100 rpm, 5 days. Cell volume = 5 mL. Substrate concentration = 10 mg/20 mL. | |||
| Whole free cell | 0.12 | 0.38 | 24 ± 1.3 |
| Agar | 0.125 | 0.375 | 25 ± 1.8 |
| Agarose | 0.099 | 0.401 | 19.8 ± 1.2 |
| κ-Carrageenan | 0.155 | 0.345 | 31 ± 3.1 |
| Polyacrylamide | 0.0 | 0.5 | 0.0 |
| PVA | 0.018 | 0.482 | 3.6 ± 0.6 |
| Na-alginate 2% (w/v) | 0.36 | 0.14 | 72 ± 2.3 |
A number of suitable methods have been developed during the last decades to prepare stable dry materials with high surface area, which retain the dispersion of polymer hydrogels while encapsulating different biocatalysts80 or drugs.81 Quignard and co-workers82 have also recently published a comprehensive review on polysaccharide aerogels and their use in heterogeneous catalysis or adsorbents.
3. LMW gels as reaction vessels and catalytic systems
As outlined in the previous section, polymer gels have been conventionally used to prepare recyclable catalysts. The present chapter is focused in the recent achievements using LMW gel-based soft materials as confined media for organic reactions or as new heterogeneous catalysts. Undoubtedly, this section exemplifies the highest novelty in this research area. Along several differences mentioned in the introduction between polymer gels and low-molecular-weight gels, the often well-defined mode of self-assembly in the latter could be consider as a major one regarding their potential function in catalysis. Miravet, Escuder and co-workers83 recently illustrated this important concept by tuning the pKa of stimuli-responsive LMW gels through the sol-to-gel phase transition. Such highly organized self-assembling process could control the orientation of existing reactive or catalytic groups and bring them into a critical proximity, which in principle could have an impact on the reactivity in comparison with homogeneous phase or less structured polymer-based gels. The next subchapters categorize the bibliography within two major types of LMW gels according to the liquid phase: hydrogels and organogels. One last subchapter dealing with coordination LMW gels is discussed separately in order to facilitate the drawing of a general blueprint of available materials within this new area of research.3.1 Hydrogels based on low-molecular-weight compounds
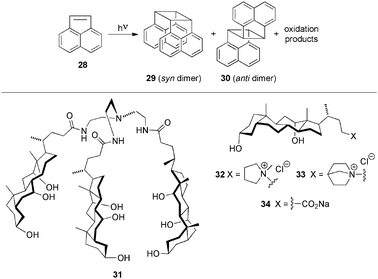 | ||
| Scheme 10 Top: Typical photodimerization reaction of ACN (28). Bottom: Low-molecular-weight gelators (LMWGs) 31–34 used for the preparation of hydrogel-based nanoreactors.85 | ||
The photodimer ratio for ACN photodimerization reactions performed in the gel-bound state was 3–10 times higher in magnitude compared to reactions carried out in micellar solutions, where the dimer ratios are similar to those observed in water (Table 2). Very interestingly, the product selectivity was found to be highly dependent on the rheological properties of the hydrogels, obtaining the higher values in the most rigid gels (higher storage modulus, G′: 33 > 32 > 34 > 3187). This represents one of the first studies where the outcome of a reaction is directly correlated with the mechanical strength of the viscoelastic matrix.
| Reaction medium | Gelator concentration (% w/v) | anti/syn dimer ratioa (error ≤ 10%) |
|---|---|---|
| a The isomeric product ratios was calculated from the 1H-NMR spectra using the cyclobutane-H signal as reference: δsyn = 4.837; δanti = 4.095, b In 20% AcOH/H2O, c In 1 M NaCl, d In 0.5 M NaCl, e In phosphate buffer (pH 8.9). | ||
| 20% AcOH/H2O | — | 0.7 |
| Hydrogel made of 31b | 1.2 | 2.0 |
| 1 M NaCl | — | 0.9 |
| Hydrogel made of 32c | 0.5 | 5.0 |
| 0.5 M NaCl | — | 1.0 |
| Hydrogel made of 33d | 0.4 | 11.0 |
| Phosphate buffer (pH 6.9) | — | 1.3 |
| Hydrogel made of 34e | 1.2 | 3.5 |
In comparison with the classical alkyl surfactant systems, bile salt-based micellar aggregates possess (1) higher rigidity, and (2) stronger hydrophobic microenvironments, where polyaromatic hydrocarbons are known to lie down.88 Although these properties could favour the role of the hydrogels as reaction vessels vs. micellar systems, the basic origin of their effect on the dimer ratio of ACN photodimerization has not been rationalized yet. Indeed, preliminary fluorescence experiments85 showed different intensity of the ACN-excimer band in the gel-bound state and in micellar solution, thus pointing out the government of different molecular interactions patterns, which have not been elucidated yet. In addition, the scope of this approach can be only evaluated after investigating other model reactions where the chiral environment of the confined media could also influence the stereoselectivity of the process. In general, only small amounts of reactants have been used in the examples dealing with LMW gels as reaction media. Hence, further detailed studies on the effect of the concentration of reactants would help to perceive the scope of these materials, something that is difficult to discern with the existing data.
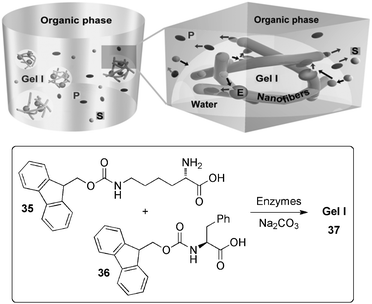 | ||
| Scheme 11 Illustration of hydrogel-immobilized enzymes 37 made of LMW 35+36 for catalyzing reactions in an organic solvent (E = enzymes; S = substrates; P = products). Reprinted with permission from ref. 90, ® 2007, The Royal Society of Chemistry. | ||
This approach represents an elegant solution to the low activity of enzymes in organic solvents mainly due to unfavourable substrate desolvation, suboptimal pH, and reduced conformational mobility. Indeed, using the oxidation of pyrogallol by H2O2 as a model reaction (Table 3), it was found that the enzymes enclosed in this hydrogel exhibited higher activity (e.g. up to eight times) in the organic media than that of unconfined enzymes in water, being as well reusable without losing activity up to three runs.
| Enzyme | Substrate/Product | Activity (half-life/min) of E(I) in toluene | Activity (half-life/min) of E(U) in H2O |
|---|---|---|---|
| a Methemoglobin. b Horseradish peroxidase. c Laccase from trameters versicolor. d Alpha-cymotrypsin from bovine pancreas. e Unit = mmol min−1 mg−1. f Unit = nmol min−1 U−1. | |||
| Hba | Pyrogallol/purpurgallin | 7.98e (7.0) | 0.92e (1.9) |
| HRPb | Pyrogallol/purpurgallin | 2165f (5.3) | 802f (2.8) |
| Laccasec | o-Phenyldiamine/phenazine-2,3-diamine | 229.3f (13.6) | 54.6f (4.2) |
| Alpha-CTd | N-Benzoyl-L-tyrosine-p-nitroanilide/p-nitroanilide | 5.83f (28.4) | 1.06f (5.9) |
In addition, the hydrogel acts as a more effective carrier of enzymes in organic solvents than a polymeric hydrogel, suggesting a unique role of the self-assembled nanofibers in the former. This observation is particularly significant because it implies that tailoring the nanofibers via the control of the structure of hydrogelators may provide an optimal microenvironment for enzyme-catalyzed biotransformations. This is an important statement, which might easily be applicable to many other chemical processes. The authors pointed out two plausible reasons to explain the observed high stabilities of the enzymes: (1) an aqueous microenvironment provided by the hydrogel that protects the enzyme from deactivation in the organic solvent, and (2) an easier transport of the product back to the organic phase, thus reducing inhibition of the catalyst, due to the relatively large pore size and amphiphilic nature of the hydrogel. Nevertheless, the general scope of this novel approach still remains to be established.
Although aerogels do not fall within the scope of this review (see introduction), their use in areas like biocatalysis has been particularly raised in the last years. Some recent representative examples involve the encapsulation of different lipases in silica aerogels base on tetramethoxysilane (TMSO) and methyltrimethoxysilane (MTMS) for their use in biodiesel synthesis by transesterification of sunflower seed oil with methyl acetate. The activities of these systems have been found to be comparable with other well-known commercial lipases immobilized on macroporous acrylic resin.91 Nonetheless, the performance of such TMSO-based bioaerogels are limited by substrate diffusion through the aerogels, which may denote a major limitation in comparison with the wet counterpart gels.92 Similar aerogels have been also used to encapsulate superstructures formed by cytochrome c and metal nanoparticles (i.e. Au, Ag). In these cases, the viability of the encapsulated proteins depends on the size of the colloidal metal nanoparticles that results in stable aerogels.93
3.2 Organogels based on low-molecular-weight compounds
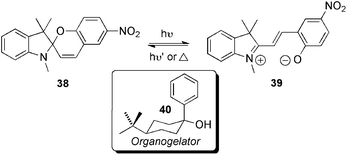
A number of reactions have been also reported in the presence of LMW gels networks made in organic solvents. In this subchapter, the examples have been classified in function of the role of the viscoelastic matrix (i.e. as reaction medium, as reactive framework and/or as catalytic system).In this field, Shumburo and Biewer94 described the photochromic behavior of the spiropyran dye 38 in an organogel medium made from 4-tert-butyl-1-phenylcyclohexanol (40) (Scheme 12). Interestingly, control experiments and decay rate data showed that in the gel phase, the succinyl functionality of 38 seemed to interact with the gelator to retard remarkably the thermal 38-to-39 isomerization process (the lifetime of the colored photomerocyanine 39 was increased by over 300-fold in the organogel vs. solution!). Nonetheless, more detailed experiments are still necessary to unequivocally establish the apparent important spatial orientation of the photochromic compound relative to the gel fibers.
Shortly after this report, Koshima and co-workers95 reported the pinacol homocoupling of a series of benzophenone derivatives. However, in this case the kinetic of the reaction was found to be slower in the gel state, which was attributed to a restricted molecular mobility within the gel and to the opaqueness of the medium leading to diffuse reflection of light. These results envisage the strong influence that the nature of the gels (e.g. rheology, morphology, chirality, etc.) might have on the outcome of a reaction if they are used as media. We could finally include in this section also a remarkable number of photo-responsive organogels, in which the chromophore is covalently linked to a gelator or micellar structure.96 Most of them are based on the sol–gel phase transition induced by UV-(trans-to-cis)-photoisomerization of azobenzene units, and are usually hypersensitive to the nanostructure of the gel.97–101 Other examples include also photo-induce ring opening of chromene102 and spiropyran103,104 units, photo-cyclization of diarylethenes,105 and photo-dimerization of anthracene moieties.106
Hitherto, also a number of diacetylene-containing gelators with different molecular geometries (Fig. 9: (A)115–120, (B)121,122, (C)123, (D)124, (E)125,126) have been successfully polymerized upon photoirradiation to yield conjugate poly(diacetylene)s.115 The post-polymerization of preorganized assemblies made of these structures has allowed the fabrication of shape-controlled and mechanically robust functional materials in different organized media including both organic and aqueous environments.117,123,126–128 The general concept has been recently reviewed.129,130
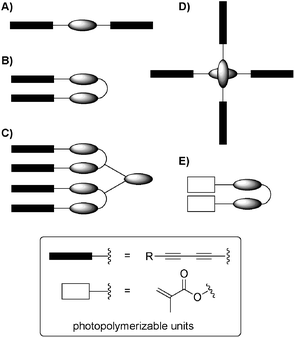 | ||
| Fig. 9 Principal molecular geometries used for post-polymerization of gels materials.115–123 | ||
Shinkai and co-workers131 reported also the capture of super-structures and the strength enhancement of the material by in situ post-polymerization of an external diisocyanate reagent (e.g. toluene-2,4-diisocyanate = TDI) and the hydroxyl groups presented in a glucosamine-based organogelator 41 (Fig. 10). Remarkably, under optimized conditions (molar ratio gelator:TDI = 1:1, reaction time = 14 days) the Tgel could be enhanced up to 60 °C higher than the starting material affording a cross-linked gel with an estimated average molecular weight Mn = 2–6 × 103. It is worth to highlight that the reaction with TDI in the sol state only afforded an insoluble and amorphous powder. This finding indicates that the stabilization of such self-assembled structures could be indeed fine-tuned by controlling the polymerization chemistry within the gel.
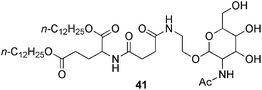 | ||
| Fig. 10 Sugar-integrated organogelator used for the cross-linking at room temperature in the gel phase by reaction with TDI.131 | ||
Seminal work by Miravet and Escuder132,133 opened a new doorway for the use of organogels (made by low-molecular-weight organogelators) bearing reactive groups as a platform for the gel-phase synthesis of new functional and stable gels. In particular, compound 42 having reactive p-nitrophenyl carbamoyl groups can react with nucleophiles such as a variety of amines to yield amino acyl bisureas 43a–e (Scheme 13). Herein, the gel was assembled before the reaction by post-diffusion of the reagents, acting as a scaffold for the formation of the new material with higher thermostability (Tgel > 100 °C) as compared to the starting gel (Tgel > 50–55 °C). The results showed that reactions took place quantitatively within few hours within the gel and that supramolecular aggregation modified the product distribution in those cases where several compounds could be obtained such as macrocyclic 44–45 and/or acyclic products 46–47.
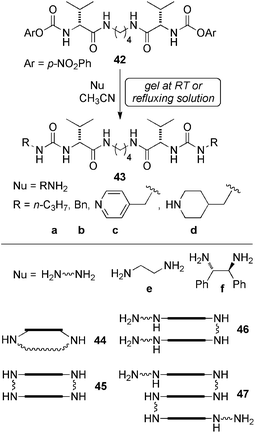 | ||
| Scheme 13 Reactive organogel 42 and its reaction with a variety of primary amines. Rectangles in the products 44–47 represent peptidomimetic backbone.132,133 | ||
Thus, in the case of the reaction between 42 and ethylenediamine, the formation of supramolecular aggregates preorganizes the system in such a way that cyclization is favored. The kinetic profiles obtained by NMR experiments indicate that reaction is taking place either simultaneously in both gel and sol phases or as fast as the disassembly of the gel into monomer units.132,133 An extensive collection of experimental data suggested that the reversible nature of a supramolecular gel together with the high active surface of its fibrillar network allow it to act as an on-demand supplier of reagents to the solution phase, as pointed out by the authors. The promising results reported by this group might serve as a basis to extend the scope of functional gels as catalytic systems to other fields of chemistry by introducing other reactive groups within the gel networks.
In no doubt, post-polymerization is in most cases a powerful strategy for creating robust materials by allowing intermolecular reactions without introducing reagents that might interfere with the self-assembled supramolecular nanostructure. Nevertheless, especially from an industrial perspective, it is worth to keep in mind that the inherent difficulties to control many polymerization reactions lead the conversion of LMW gels into cross-linked polymer gels with a consequent loss of some of their important functional properties such as thermoreversibility and/or swelling capacity. In this direction, Smith and co-workers134 reported the synthesis of robust swellable organogels from soft gels made of self-assembled dendrimers 48–51 (Fig. 11).
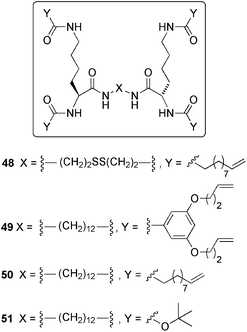 | ||
| Fig. 11 Structures of gelators studied by Smith and co-workers for the metathesis within gels.114,130,134 | ||
The strategy of Smith's group implies the decoration of the dendritic building blocks with multiple terminal alkenes and their subsequent Grubbs ring-closing metathesis within the self-assembly that acts as reaction vessel. In this case, the metathesis reaction generated an insoluble material (75 wt%) in which the individual gel fibers of the cross-linkable gelator were captured, whereas the non-reactive gelator could be washed away. This example clearly illustrates how a soft gel is converted into a robust gel via a chemical instead a photochemical reaction in the gel phase. The experimental data suggested that the second-generation Grubbs’ catalyst was able to diffuse into the gel and react over a 24 h period, during which time the catalyst changed colour from dull red to gray/black, probably because of decomposition.
The kinetic profiles indicated that the metathesis reaction was effectively complete in general up to 7 h. It is important to highlight that the covalently fixed material obtained by this approach presented a degree of ‘memory’ of its previous properties like solvent compatibility, morphology, and swelling ability (Fig. 12).130 Thus, the solid material obtained after the cross-linking reaction could be reswollen in solvents that were originally compatible with the gel but not in others. Furthermore, the drying/resolvation process could be repeated multiple times without a major disruption of the swelling capacity.
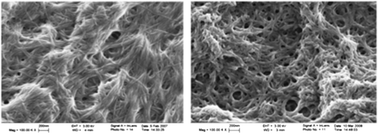 | ||
| Fig. 12 Field emission gun scanning electron microscopy (FEGSEM) images of xerogel (left) formed by gelator 50 dried from toluene under ambient conditions, and insoluble material (right) produced by the metathesis reaction after reswelling in toluene and ambient drying. Scale bars = 200 nm. Reprinted with permission from ref. 130, ® 2009, The American Chemical Society. | ||
The broad concept was also used for the incorporation of reactive nanoscale self-assemblies within polymeric materials to yield ‘nano-imprinted’ materials.114 Therein, the ingrained of the nanostructured fibrillar network (3 wt% of gelator) could increase the Tg value of the polymer by ca. 20 °C, with a ca. 18% reduction in the degree of damping (tan δ) at the Tg value and an increase of the storage modulus (G′) by almost an order of magnitude. Furthermore, the toughening effect by the embedded network was accompanied by the maintenance of the optical properties of the material.
In a similar direction, the first practical use of the Cu(I)-catalyzed cycloaddition reaction of alkynes and azides (CuAAC)69 for the stabilization of gel super-structures with the minimum disruption of their functional properties was reported by Finn and co-workers in 2006.135 In this approach, small and nonprotic azides and alkynes groups were placed at the end of the hydrophobic chains of the gelator molecule to avoid a major disruption of the intermolecular interactions that lead to gelation, followed by in situ CuAAC cross-linking.
LMWG based on the undecylamide of trans-1,2-diaminocyclohexane, 52, its “clickable” analogs 53 and 54 (Scheme 14) were here used as a proof of concept. In general, gels made from 53 or 54 were strengthened by the incorporation of an equimolar amount of 52 into the mixture. The introduction of copper iodide and linear cross-linkers (e.g.55–56, optimized ratio gelator:cross-linker = 10:1) into these samples afforded brittle gels with even much greater thermostability, as confirmed by oscillatory rheological measurements.
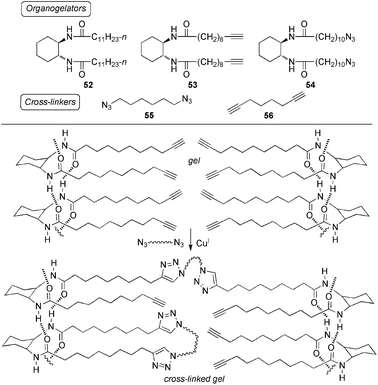 | ||
| Scheme 14 Top: Organogelators 52–54 and linear divalent cross-linkers 34–35 used for the CuAAC reaction. Bottom: Proposed hydrogen-bond pattern for gelation by 53 and Cu(I)-catalyzed cross-linking with a linear diazide. The presence of triazole moieties in cross-linked materials could be confirmed by NMR analysis.135 | ||
The results of click-mediated gel stabilization were very similar when the reactions were allowed to proceed for only a short time in the isotropic solution (followed by incubation in the gel state at room temperature) or exclusively in the gel phase at room temperature by layering on copper iodide after gel formation and diffusion of the catalyst into the viscoelastic matrix, which acts as reaction vessel.135 Control experiments demonstrated that enhancement of gel thermostability upon cross-linking is dependent upon the simultaneous presence of the metal in the active Cu(I) oxidation state and the appropriate bivalent additive. Nevertheless, spectroscopic studies indicated that neither gross morphological changes nor the expected Cu-triazole interactions upon successful reaction are imperative to the stabilization effect of the materials. Remarkably, the low concentration of click connectivity in the cross-linked gels allowed the preservation of the thermoreversible function.135
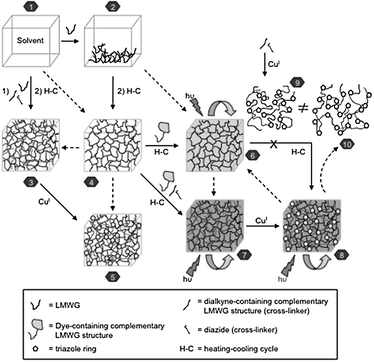
The above strategy could be also applied to more complex systems such as photoactive organogels.107 It is important to note that, in this case, the CuAAC represents a much more challenging process, since the cycloaddition must occur in an already multicomponent pre-formed organic gel, in which the appropriate “clickable” acetylene monomer should be at least partially associated with the gel fibres by virtue of multiple non-covalent interactions.136 Indeed, the gel network acts here as an efficient reaction vessel to carry out the coupling reaction between low-molecular-weight additives that are unable to form stable gels by themselves. Fig. 13 provides an overview of the global procedure developed for the preparation of standard functional organogels and their further stabilization by in situ cross-linking through CuAAC. System 4 represents a standard organogel formed upon cooling of the isotropic solution of the appropriate LMWG (system 2). If further strength enhancement of the material is required, a well-defined concentration of a suitable diacetylene-containing complementary LMWG structure, linear diazide and Cu(I)-catalyst can be either swollen into the organogel network or premixed with the LMWG prior to gel formation (system 3) in the heating–cooling process. The subsequent controllable cross-linking reaction could enhance the thermostability of the organogel while preserving its thermoreversibility (system 5). On the other hand, the organogel 4 may be prepared in the presence, for instance, of the appropriate dye-containing complementary organogelator structures to afford a multicomponent photoactive viscoelastic material (system 6). Also in this case, suitably designed clickable monomers (diacetylene-containing complementary LMWG structure and linear diazide) and the Cu(I)-catalyst could be incorporated into the gel network (system 7) in order to achieve further cross-linking and generate a new photoactive organogel with enhanced thermo- and mechanical stability. Nevertheless, the structural differences between a preformed 1,3,5-triazole-based polymer (system 9) -made by CuAAC of the appropriate monomers in solution- and the cross-linked material (system 10) -obtained by in situ CuAAC of the clickable monomers-, usually induce a fast-phase separation process when the former is incorporated into the photoactive organogels.
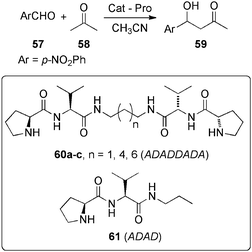
It was suggested that the close proximity of pyrrolidinic residues on the surface of the gel fibres could provoke the cooperative assistance of several basic groups allowing for the observed enhancement of the basicity. As a result they behave as enantioselective catalysts for the diffusion-controlled aldol reaction between acetone (58) and 4-nitrobenzaldehyde (57) in solution to generate the corresponding aldol-adduct 59, but they produce a base-catalyzed aldol racemisation in the gel state over time (Fig. 14).
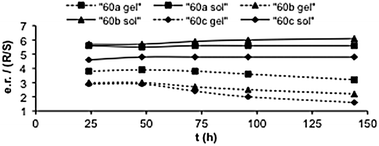 | ||
| Fig. 14 Enantiomeric ratio (e.r., R/S) of the aldol reaction between 57 (60 mmol L−1) and 58 (1.2 M) in CH3CN catalyzed by 60a–c in solution and in gel phase. Adapted with permission from ref. 83, ® 2009, The Royal Society of Chemistry. | ||
This work has been further extended to the use of L-proline-derived low-molecular-weight gelators as efficient basic catalysts for the non-stereoselective Henry nitroaldol reaction, albeit unfortunately neither stereoselectivity nor enantioselectivity were achieved.137,138Scheme 16 illustrates the proposed mechanism for the reaction of nitroalkanes 62 with aromatic aldehydes 64 in the gel phase and in solution. In the presence of a gel, the L-proline fragment would act as a basic catalyst promoting the nitroaldol reaction through the so-called ionic pair type mechanism, which is initiated by nitroalkane deprotonation. However, in solution an iminium-based mechanism would be operative and responsible for the formation of dehydrated product 66 that could suffer a further conjugated addition of nitroalkane.
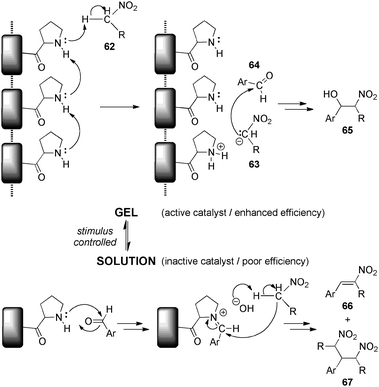 | ||
| Scheme 16 Proposed reaction mechanisms for the reaction of nitroalkanes with aromatic aldehydes in the gel phase and in solution.137 | ||
Control experiments showed that the quantitative conversion of the aldehyde to the corresponding nitroaldol 65 was obtained only in the case where a gel was present (Table 4). In addition to the low conversion obtained in solution by deactivation of the catalyst, side products corresponding to dehydratation and conjugated addition to the nitroalkene were detected (products 66 and 67 respectively). On the other hand, the use of the non-gelling molecule 61, which is formally a half of 60c, yielded nitroaldol in poor yields both in solution and in the gel state accompanied by similar amounts of side products.
| Cat.a | Reagentb | Reaction time (days) | T (° C) | Yield 65 (%) | Yield 66+67 (%) |
|---|---|---|---|---|---|
| a 13 mmol L−1 in 60c, 26 mmol L−1 in 61. b 130 mmol L−1 in aldehyde. Total volume = 1.3 mL. BA = benzaldehyde. | |||||
| 60c | 4-NitroBA | 2 | 5 (gel) | 38 | 2 |
| 60c | 4-NitroBA | 6 | 5 (gel) | 84 | 3 |
| 60c | 4-NitroBA | 2 | 50 (sol.) | 46 | 33 |
| 61 | 4-NitroBA | 2 | 5 (sol.) | 7 | 7 |
| 61 | 4-NitroBA | 6 | 5 (sol.) | 13 | 25 |
| 61 | 4-NitroBA | 2 | 50 (sol.) | 24 | 54 |
| 60c | 4-ChloroBA | 2 | 5 (gel) | 11 | 4 |
| 60c | 4-ChloroBA | 2 | 50 (sol.) | — | — |
| 60c | 4-MethoxyBA | 2 | 5 (gel) | — | 99 |
| 60c | 4-MethoxyBA | 2 | 50 (sol.) | — | 100 |
3.3 Coordination organic gels
An alternative approach for the synthesis of highly processable, recyclable, and stimuli-responsive organic gels-based catalysts makes use of the tunable metal–ligand interactions in metal complexes.139 Indeed, the foremost catalytic potential of gel-based materials lies on two key advantages: (1) the two-phase nature of the system, which allows the easy separation of the catalysts, and (2) a much higher accessibility of small reagents to the highly solvated 3D-porous network in comparison to several standard heterogeneous catalysts. This strategy was reported by Xu and co-workers,140 who described a series of catalytic cross-linked metallogels prepared in DMSO by using transition-metal ions with multiple coordination sites and flexible multidentate ligands 68–73 (Fig. 15). These metallogels were stable in water and most organic solvents, and could act as efficient heterogeneous catalysts for the oxidation of benzyl alcohol to benzaldehyde. The best catalytic turnover number (about twice that of Pd(OAc)2) was observed for the metallogel B made of 70 + Pd(OAc)2 (Fig. 16, top). The lower catalytic activity of the solid coordination polymers (“dry gels”) in comparison to their gels and Pd(II) complexes, was attributed respectively to: (1) lack of the metallogel's solution in the DMSO phase to deliver the reaction substrates to the metal sites; (2) relatively smaller surface area of the insoluble coordination polymers.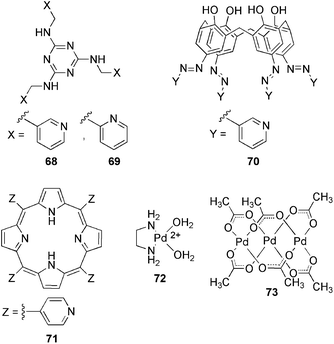 | ||
| Fig. 15 Multidentate ligands 68–71 and metal complexes 72–73.140 | ||
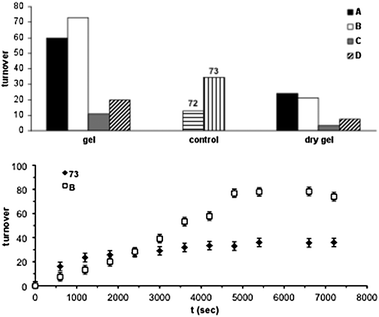 | ||
| Fig. 16 Top: Cumulative catalytic turnovers for the oxidation of benzyl alcohol by air using the metallogels (catalyst:substrate molar ratio ≈ 1:104), their corresponding dry gels, and their corresponding transition-metal complexes as catalysts. Bottom: Catalytic turnovers of gel B and compound 73vs. time, showing the higher stability of the former. The metallogels were prepared in DMSO as follow: Gel A = 68 + Pd(OAc)2; Gel B = 70 + Pd(OAc)2; Gel C = 71 + Pd(en)2+; Gel D = 70 + Pd(en)2+. Adapted with permission from ref. 140, ® 2002, Wiley-VCH Verlag GMBH & Co. | ||
The turnover profile of metallogel B planed off starting at about two hours (Fig. 16, bottom), suggesting that the catalytic reaction rate was controlled by a diffusion mechanism of substrates from the surrounding medium to the catalytic sites in the gel and products from the gel to the surroundings.141 Nonetheless, neither conclusive experimental data on this regard nor comparative studies with other transition metal-based gels have been reported until now.
Later, Miravet and Escuder142 reported also the efficient and general preparation of Pd-functionalised materials with catalytic activity based on pyridine-containing gelators (Fig. 17). They use as gelators compounds 74 and 75, which are able to form both organogels and pH-responsive hydrogels, apparently through a dimmer structure stabilized by four intermolecular H-bonds. To achieve catalytic function, the authors made a Pd(II)-doped gel by post-diffusion of palladium salts into the preformed organogels. Microscopic and spectroscopic evidences showed the presence of Pd in the gel fibers and the absence of substantial changes in the H-bonding network by the doping process. Very interestingly, the pre-assembly of the gelator seems to be a critical factor for the stability of Pd(II) complexes being a requirement for the formation of stable Pd(II)-doped gels. These metallogels showed a modest catalytic activity towards the aerobic oxidation of benzyl alcohol (maximum yield of ca. 50% after 48 h, presumably due to the inactivation of the catalyst over time).
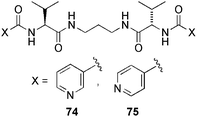 | ||
| Fig. 17 Pyridine-functionalized gelators used to prepare Pd-containing gels with catalytic activity.142 | ||
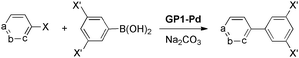
Recently, Zhang and co-workers143 reported the preparation of Fe-dicarboxylate based phosphine-functionalized polymer gels in alcohols or DMF from 5-diphenylphosphanylisophthalic acid (76) (Scheme 17), which were subsequently functionalized with Pd(II) to yield recyclable and highly active Pd(II)-immobilized catalytic gels for Suzuki C–C coupling of aryl halides/bromopyridines with arylboronic acids (Table 5). In this case, the catalytic activity of the gels was found to be, at least, comparable to unsupported complexes.
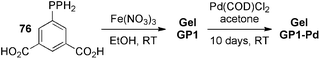 | ||
| Scheme 17 Synthesis of Pd(II)-loaded organogels for catalysis. Pd content = 1.0 wt%. Thermogravimetric analyses (TGA) showed that the coordination network resisted at least until 240 °C. COD = 1,5-cyclooctadiene.143 | ||
| Aryl halide | Aryl boronic acid | Time (h) | Yield (%) |
|---|---|---|---|
| a The reactions were carried out using 1 mmol% of catalyst, at 60 °C in methanol unless otherwise indicated. b Reaction carried out in water. c Reaction carried out at room temperature. | |||
| X = I; a = b = c = CH | X′ = H | 1.0 | 100 |
| X = I; a = b = c = CH | X′ = H | 2.0 | 81b |
| X = I; a = b = c = CH | X′ = H | 3.5 | 98c |
| X = Br; a = b = c = CH | X′ = H | 3.0 | 74 |
| X = Br; a = COMe; b = c = CH | X′ = H | 3.0 | 39 |
| X = Br; a = CCOMe; b = c = CH | X′ = H | 1.0 | 99 |
| X = Br; a = N; b = c = CH | X′ = F | 2.0 | 95 |
| X = Br; a = N; b = c = CH | X′ = H | 1.0 | >99 |
| X = Br; b = N; a = c = CH | X′ = F | 3.0 | 71 |
| X = Br; c = N; a = b = CH | X′ = F | 3.0 | 53 |
Recent studies144,145 demonstrated that pyridine-based fibrous networked metallogels showed higher catalytic activity in Suzuki-Miyaura coupling reaction than spherical analogous. In agreement with previous examples, these results point out the significance of controlling the morphology of metallogels, by adjusting the metal/ligand ratio as well as the crosslinking density, for catalytic purposes of these materials.146 The general concept of metal-bound to swellable polymer matrixes have been already extended to other reactions such as hydrogenations.147 Even though most of the examples discussed in this subheading deal with Pd-catalyzed coupling reactions, they have pioneered a completely new gateway for the study and use of other transition metals within the concept of catalysis using soft-materials. Therefore it seems reasonable to presume an increasing number of contributions in this direction during the upcoming years. Moreover, metal leaching to act as true active species has been critically analyzed only outwardly in most cases. Hence, this aspect still constitutes the major threat of using metallogels as heterogeneous catalysts if not appropriate tests are carried out to consistently quantify metal leaching during the reaction or recycling process.
Last but not least, other less renowned self-assembled gels such as those made from ionic liquids (ionogels) also represent promising materials for catalysis. In 2005, Deng and co-workers148 reported the enhanced catalytic performance of the carbonylation of aniline in the presence of ionogels doped with Rh(PPh3)3Cl and Pd(PPh3)2Cl2. Moreover, the immobilization of enzymes (e.g. HRP) in ionogels has been found to be an efficient approach to achieve higher activities in biocatalysis.149,150
4. Conclusions and outlook
During the last decade, a growing body of publications has showed the potentiality of functional gels (e.g. reactive gels, electron-conducting gels, metallogels, biohydrogels) as recyclable catalysts and/or media for chemical reactions. In comparison to traditional polymer gels, viscoelastic materials made of low-molecular-weight compounds denote the highest innovation in this area, by means of a better-defined mode of self-assembly than the former. Fig. 18 attempts to provide a general pattern of the gel-based soft materials presented in this review and their role and/or type of reactions for which they are most suited. Some of the described processes have been accomplished with even enhanced selectivity due specific interactions between the reactants. Such envisaged usefulness have been mainly possible due to some remarkable advantages of the gels-based soft materials including a high active surface area, highly solvated heterogeneous nature with excellent diffusion properties, variety of compositions, simple preparation from readily available building blocks, and, in many cases, their stimuli-response reversible nature.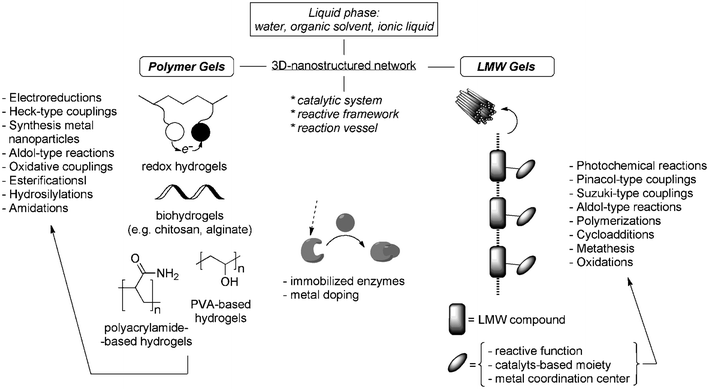 | ||
| Fig. 18 Schematic “toolbox” of available materials presented in this overview. | ||
Nonetheless, in order for these materials to compete with a number of reactions in solution, that offer high yields and conversions through the use of either artificial or natural catalytic systems, many scientific challenges still remain, such as: (1) the efficient chirality induction using chiral gel networks151,152 to achieve enantioselectivity; (2) the enhancement of the specificity and strength absorption of substrates, as well as the chemical reactivity of the catalytic centre; (3) the improvement in the loading capacity of the gels; (4) the development of new methodologies to increase the robustness of the gel networks for higher operational stability, while preserving the required functional properties; (5) the fundamental understanding of the mechanisms through which nanostructured gels favour specific reaction pathways (e.g. accretion of reactants into nanostructured subdomains creating hot-spots of catalytic activity, steric interactions that could control special orientation of the reactive groups and affect the stereoselectivity); and 6) the mapping of the critical relationships between the properties of the gels and the outcome of a reaction carried out within the viscoelastic matrix.
Undoubtedly, more detailed fundamental studies on the molecular organization within the gel fibers will still be necessary for a better understanding of the gelation mechanisms. In addition, further comparative studies directed to unequivocally establish the influence of the gel properties (e.g. morphology, cross-linking density, size, diffusion coefficient, thermodynamic properties, etc.) on the outcome of different chemical reaction is a current demand. The anticipated framework of research in the above directions will ultimately boost the challenged use of self-assembly gels as realistic user-friendly, efficient and selective nanoreactors and/or heterogeneous asymmetric catalysts, as well as robust platforms for the ‘bottom–up’ tailor-fabrication of more advanced and greener nanocomposite materials. Indeed, most of functional gels will become more than a scientific curiosity if the full potential of selectivity and efficiency is brought closer to the challenges of conventional catalyst research in for example the chemical and pharmaceutical industry.
Appendix
| Acronym | Term |
| AAC | Alkyne-azide cycloaddition |
| ACN | Acenaphthylene |
| APS | Ammonium persulfate |
| BAL | Benzaldehyde lyase |
| COD | 1,5-Cyclooctadiene |
| CTAC | Cetyltrimethylammonium chloride |
| CuAAC | Copper catalyzed alkyne-azide cycloaddition |
| DETA | Diethylenetriamine |
| DVB | Divinylbenzene |
| EcGGT | γ-Glutamyltranspeptidase from Escherichia coli |
| Enz. | Enzyme |
| EVB | Ethyl vinyl benzene |
| EWG | Electron Withdrawing Group |
| FEGSEM | Field emission gun scanning electron microscopy |
| GPx | Glutathione peroxidase |
| HRP | Horseradish peroxidase |
| ICP-AAS | Inductively-coupled plasma atomic absorption spectroscopy |
| Im | 4(5)-Vinylimidazole |
| LCST | Lower critical solution temperature |
| LKADH | Alcohol dehydrogenase from Lactobacillus kefir |
| LMW | Low-molecular-weight |
| LMWGs | Low-molecular-weight gelators |
| LP | Lipase |
| MBA | N,N′-Methylenebisacrylamide |
| ME | Manihot esculenta |
| MTMS | Methyltrimethoxysilane |
| NIPA | N-Isopropylacrylamide |
| Nu | Nucleophile |
| PEG 400 | Polyethylene glycol 400 |
| PEGDGE | Polyethylene glycol diglycidyl ether |
| PGMA-co-PNIPA | Poly(glycidylmethacrylate-co-N-isopropylacrylamide) |
| PHC | Polyhydroxyl cellulose |
| PNIPAM-co-PMACHE | Poly(N-isopropylacrylamide)-co-poly-2-methacrylic acid 3-(bis-carboxymethylamino)-2-hydroxypropyl ester |
| poly(NIPA-co-PMA) | Poly(N-isopropylacrylamide-co-potassium methacrylate) |
| PVA | Polyvinyl alcohol |
| RT | Room temperature |
| SA | Sodium acrylate |
| SAFINs | Self-assembled fibrillar networks |
| SLAC | Small laccase |
| TDI | Toluene-2,4-diisocyanate |
| TEMED | N,N,N′,N′′-Tetramethylenediamine |
| TGA | Thermogravimetric analyses |
| TMSO | Tetramethoxysilane |
| T gel | Sol-to-gel phase-transition temperature |
Acknowledgements
The authors thank all students and professors who have kindly provided some graphic material needed for this manuscript. The background-image of the graphical abstract was kindly taken by E. Morin. D.D.D. thanks Universität Regensburg and the Alexander von Humboldt Foundation for financial support.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef; R. G. Weiss and P. Terech, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, Springer, New York, 2006 Search PubMed; M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489 Search PubMed , and references therein.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263 CrossRef.
- O. Gronwald and S. Shinkai, Chem.–Eur. J., 2001, 7, 4328 CrossRef CAS.
- T. Tanaka, Sci. Am., 1981, 244, 110.
- D. Derossi, Y. Kajiwara and Y. Osada, Polymer Gels: Fundamentals and Biomedical Applications, Plenum Press, New York, 1991 Search PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS; N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC; A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109 RSC.
- P. Xie and R. Zhang, J. Mater. Chem., 2005, 15, 2529 RSC; M. George, R. Mathew and R. G. Weiss, Mol. Gels, 2006, 449 Search PubMed; D. K. Smith, Chem. Commun., 2006, 34 RSC.
- S. M. Aharoni, in Synthesis, Characterization, and Theory of Polymeric Networks and Gels, ed. S. M. Aharoni, Plenum, New York, 1992 Search PubMed.
- E. R. Zubarev, M. U. Pralle, E. D. Sone and S. I. Stupp, Adv. Mater., 2002, 14, 198 CrossRef CAS.
- X. Huang, P. Terech, S. R. Raghavan and R. G. Weiss, J. Am. Chem. Soc., 2005, 127, 4336 CrossRef CAS.
- F. Ilmain, T. Tanaka and E. Kokufuta, Nature, 1991, 349, 400 CrossRef , and references therein.
- Y. Osada and A. R. Khokhlov, Polymer Gels and Networks, Marcel Dekker, New York, 2002 Search PubMed.
- J. H. van Esch, Langmuir, 2009, 25, 8392 CrossRef CAS.
- N. Fujita, P. Mukhopadhyay and S. Shinkai, Annu. Rev. Nano Res., 2006, 1, 385 Search PubMed; A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS.
- F. Schueth, Annu. Rev. Mater. Res., 2005, 35, 209 CrossRef.
- E. G. Kovaleva, L. S. Molochnikov, V. G. Kharchuk, O. V. Kuznetsova, A. B. Shishmakov, M. Y. Yanchenko, L. Y. Buldakova, I. N. Zhdanov, Y. V. Mikushina and L. A. Petrov, Kinet. Catal., 2004, 45, 752 CrossRef CAS; M. I. Burguete, M. A. Izquierdo, F. Galindo and S. V. Luis, Chem. Phys. Lett., 2008, 460, 503 CrossRef CAS , and references therein; G. Feng, Y. Xiong, H. Wang and Y. Yang, Electrochim. Acta, 2008, 53, 8253 Search PubMed.
- For the early development of heterogeneous gel-derived catalysts, including aerogels and xerogels of silica materials, see: C. Colmenares, M. Connor, C. Evans and R. Gaver, Eur. J. Sol. State Inor., 1991, 28, 429 Search PubMed , and references therein; R. A. Kemp and C. T. Adams, Appl. Catal., A, 1996, 134, 299 Search PubMed; M. Buechler-Skoda, R. Gill, D. Vu, C. Nguyen and G. Larsen, Appl. Catal., A, 1999, 185, 301 Search PubMed , and references therein.
- S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195 CrossRef.
- A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665 CrossRef CAS.
- D. G. Blackmond and M. Klussmann, Chem. Commun., 2007, 3990 RSC.
- L. Schiaffino and G. Ercolani, Angew. Chem., Int. Ed., 2008, 47, 6832 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869 CrossRef; Y. Osada, J. P. Gong and Y. Tanaka, Polym. Rev., 2004, 44, 87 Search PubMed; J. H. Jung and S. Shinkai, Top. Curr. Chem., 2005, 248, 223 CAS; F. Zhao, M. L. Ma and B. Xu, Chem. Soc. Rev., 2009, 38, 883 RSC.
- M. A. Cauqui and J. M. Rodriguez-Izquierdo, J. Non-Cryst. Solids, 1992, 147–148, 724 CAS; E. I. Ko, ChemTech, 1993, 23, 31 CAS; L. W. Hrubesh, J. Non-Cryst. Solids, 1998, 225, 335 CrossRef.
- G. M. Pajonk, Catal. Today, 1997, 35, 319 CrossRef CAS; M. Schneider and A. Baiker, Catal. Today, 1997, 35, 339 CrossRef CAS; G. M. Pajonk, Catal. Today, 1999, 52, 3 CrossRef CAS.
- A. M. Orlovic, D. T. Janackovic, T. Djordje, Skala and Dejan U. Belgrade, in New Developments in Catalysis Research, ed. L. P. Bevy, Nova Science Publishers, Inc., 2005, pp. 39–84 Search PubMed; A. Vallribera and E. Molins, in Nanoparticles and Catalysis, ed. D. Astruc, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008, ch. 5, pp. 161–194 Search PubMed.
- Al. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243 CrossRef CAS.
- S. Ikegami and H. Hamamoto, Chem. Rev., 2009, 109, 583 CrossRef CAS , and references therein.
- M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2010, 39, 455 RSC.
- M. R. Buchmeiser, in Polymeric Materials in Organic Synthesis and Catalysis, Wiley-VCH, Weinheim, 2003 Search PubMed.
- C. Thiot, M. Schmutz, A. Wagner and C. Mioskowski, Angew. Chem., Int. Ed., 2006, 45, 2868 CrossRef CAS.
- C. Thiot, A. Wagner and C. Mioskowski, Org. Lett., 2006, 8, 5939 CrossRef CAS.
- C. Thiot, C. Mioskowski and A. Wagner, Eur. J. Org. Chem., 2009, 3219 CrossRef CAS.
- C. Thiot, M. Schmutz, A. Wagner and C. Mioskowski, Chem.–Eur. J., 2007, 13, 8971 CrossRef CAS.
- S. J. Craythorne, K. Anderson, F. Lorenzini, C. McCausland, E. F. Smith, P. Licence, A. C. Marr and P. C. Marr, Chem.–Eur. J., 2009, 15, 7094 CrossRef CAS.
- S. Volland, M. Gruit, T. Régnier, L. Viau, O. Lavastre and A. Vioux, New J. Chem., 2009, 33, 2015 RSC.
- A. Hellera, Curr. Opin. Chem. Biol., 2006, 10, 664 CrossRef CAS.
- H. F. Cui, J. S. Ye, W. D. Zhang and F. S. Sheu, Biosens. Bioelectron., 2009, 24, 1723 CrossRef CAS.
- M. T. Cortés and J. C. Moreno, e-Polymers, 2003, 1 Search PubMed; A. Eftekhari, Chem. Mater., 2010, 22, 2689 CrossRef CAS.
- M. Shahinpoor, K. J. Kim and M. Mojarrad, in Artificial Muscles. Applications of Advanced Polymeric Nanocomposites, Taylor & Francis, 2007 Search PubMed; H.-J. Schneider and R. M. Strongin, Acc. Chem. Res., 2009, 42, 1489 Search PubMed.
- N. Mano, V. Soukharev and A. Heller, J. Phys. Chem. B, 2006, 110, 11180 CrossRef CAS.
- A. Heller, Curr. Opin. Chem. Biol., 2006, 10, 664 CrossRef CAS.
- J. H. Merkin, P. L. Simon and Z. Noszticzius, J. Math. Chem., 2000, 28, 43 CrossRef CAS.
- I. Gill, Chem. Mater., 2001, 13, 3404 CrossRef CAS.
- A. Buthe, A. Kapitain, W. Hartmeier and M. B. Ansorge-Schumacher, J. Mol. Catal. B: Enzym., 2005, 35, 93 CrossRef CAS; P. Hoyos, A. Buthe, M. B. Ansorge-Schumacher, J. V. Sinisterra and A. R. Alcántara, J. Mol. Catal. B: Enzym., 2008, 52–53, 133 CrossRef CAS.
- I. R. Wheeldon, J. W. Gallaway, S. C. Barton and S. Banta, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15275 CrossRef CAS.
- L. Feng, L. Wang, Z. Hu, Y. Tian, Y. Xian and L. Jin, Microchim. Acta, 2009, 164, 49 CrossRef CAS.
- G. Wang, K. Kuroda, T. Enoki, A. Grosberg, S. Masamune, T. Oya, Y. Takeoka and T. Tanaka, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9861 CrossRef CAS.
- Y. Y. Li, F. Cunin, J. R. Link, T. Gao, R. E. Betts, S. H. Reiver, V. Chin, S. N. Bhatia and M. J. Sailor, Science, 2003, 299, 2045 CrossRef CAS.
- J. Zhang, S. Xu and E. Kumacheva, J. Am. Chem. Soc., 2004, 126, 7908 CrossRef CAS , and references therein.
- X. Jiang, D. Xiong, Y. An, P. Zheng, W. Zhang and L. Shi, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2812 CrossRef CAS.
- Y. M. Mohan, K. Lee, T. Premkumar and K. E. Geckeler, Polymer, 2007, 48, 158 CrossRef CAS.
- Y.-Q. Ma, J.-Z. Yi and L.-M. Zhang, J. Macromol. Sci. A, 2009, 46, 643 CrossRef CAS.
- Y. Wang, G. Wei, F. Wen, X. Zhang, W. Zhang and L. Shi, J. Mol. Catal. A: Chem., 2008, 280, 1 CrossRef CAS.
- Y. Wang, J. Zhang, W. Zhang and M. Zhang, J. Org. Chem., 2009, 74, 1923 CrossRef CAS.
- Y. Wang, R. Yan, J. Zhang and W. Zhang, J. Mol. Catal. A: Chem., 2010, 317, 81 CrossRef CAS.
- K. S. Sivudu, N. M. Reddy, M. N. Prasad, K. M. Raju, Y. M. Mohan, J. S. Yadav, G. Sabitha and D. Shailaja, J. Mol. Catal. A: Chem., 2008, 295, 10 CrossRef.
- X. Huang, Y. Yin, Y. Tang, X. Bai, Z. Zhang, J. Xu, J. Liu and J. Shen, Soft Matter, 2009, 5, 1905 RSC.
- M. B. Ansorge-Schumacher, H. Slusarczyk, J. Schümers and D. Hirtz, FEBS J., 2006, 273, 3938 CrossRef CAS.
- T. Hischer, D. Gocke, M. Fernández, P. Hoyos, A. R. Alcántara, J. V. Sinisterra, W. Hartmeier and M. B. Ansorge-Schumacher, Tetrahedron, 2005, 61, 7378 CrossRef CAS.
- M. B. Ansorge-Schumacher, L. Greiner, F. Schroeper, S. Mirtschin and T. Hischer, Biotechnol. J., 2006, 1, 564 Search PubMed.
- D. Metrangolo-Ruiz De Temiño, W. Hartmeier and M. B. Ansorge-Schumacher, Enzyme Microb. Technol., 2005, 36, 3 CrossRef.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339 CrossRef CAS.
- G. Paradossi, E. Chiessi, F. Cavalieri, D. Moscono and V. Crescenzi, Polym. Gels Networks, 1997, 5, 525 CAS.
- K. R. Reddy, K. Rajgopal, C. U. Maheswari and M. L. Kantam, New J. Chem., 2006, 30, 1549 RSC.
- A. Primo, M. Liebel and F. Quignard, Chem. Mater., 2009, 21, 621 CrossRef CAS.
- K. R. Reddy, K. Rajgopal and M. L. Kantam, Catal. Lett., 2007, 114, 36 CrossRef.
- For recent reviews on biopolymers-based heterogeneous catalysts, see: E. Guibal, Prog. Polym. Sci., 2005, 30, 71 Search PubMed; E. Guibal, T. Vincent and F. P. Blondet, Ion Ex. Solv., 2007, 18, 151 CrossRef CAS.
- K. R. Reddy, K. Rajgopal and M. L. Kantam, Synlett, 2006, 957 CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- V. O. Rodionov, V. V. Fokin and M. G. Finn, Angew. Chem., Int. Ed., 2005, 44, 2210 CrossRef CAS; V. O. Rodionov, S. I. Presolski, D. D. Díaz; V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705 CrossRef CAS.
- W. S. Brotherton, H. A. Michaels, J. T. Simmons, R. J. Clark, N. S. Dalal and Lei Zhu, Org. Lett., 2009, 11, 4954 CrossRef CAS.
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2010, 26, 2885 CrossRef CAS , and references therein.
- H. Yang, K. Nagai, T. Abe, H. Homma, T. Norimatsu and R. Ramaraj, Appl. Mater. Interfaces, 2009, 1, 1860 Search PubMed.
- B. Doumèche, M. Heinemann, J. Büchs, W. Hartmeier and M. B. Ansorge-Schumacher, J. Mol. Catal. B: Enzym., 2002, 18, 19 CrossRef CAS.
- C. P. Hung, H. F. Lo, W. H. Hsu, S. C. Chen and L. L. Lin, Appl. Biochem. Biotechnol., 2008, 150, 157 CrossRef CAS.
- L. L. Machado, T. L. G. Lemos, M. C. de Mattos, M. da Conceição, F. de Oliveira, G. de Gonzalo, V. Gotor-Fernández and V. Gotor, Tetrahedron: Asymmetry, 2008, 19, 1419 CrossRef CAS.
- L. L. Machado, G. de Gonzalo, T. L. G. Lemos, M. C. de Mattos, M. da Conceição, F. de Oliveira, V. Gotor-Fernández and V. Gotor, J. Mol. Catal. B: Enzym., 2009, 60, 157 CrossRef CAS.
- A. Anwar, S. A. U. Qader, A. Raiz. S. Iqbal and A. Azhar, World Appl. Sci. J., 2009, 10, 1281 Search PubMed.
- H. Arabi, M. T. Yazdi and M. A. Faramarzi, J. Mol. Catal. B: Enzym., 2010, 62, 213 CrossRef CAS.
- M. T. Reetz, Adv. Mater., 1997, 9, 943 CrossRef CAS.
- C. Shah, R. Patel, R. Mazumder, S. Bhattacharya, A. Mazumder and R. N. Gupta, Pharma Rev., 2009, 7, 164 Search PubMed.
- F. Quignard, R. Valentin and F. Di Renzo, Chem. Soc. Rev., 2008, 32, 1300 Search PubMed , and references therein.
- F. Rodríguez-Llansola, B. Escuder and J. F. Miravet, Org. Biomol. Chem., 2009, 7, 3091 RSC.
- L. J. M. Gaspar and G. Baskar, Chem. Commun., 2005, 3603 RSC.
- S. Bhat and U. Maitra, Molecules, 2007, 12, 2181 Search PubMed.
- D. O. Cowan and R. L. E. Drisko, J. Am. Chem. Soc., 1970, 92, 6286 CrossRef CAS.
- S. Bhat and U. Maitra, Tetrahedron, 2007, 63, 7309 CrossRef CAS.
- S. Mukhopadhyay and U. Maitra, Curr. Sci., 2004, 87, 1666.
- M. O. Guler and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 12082 CrossRef CAS.
- Q. Wang, Z. Yang, L. Wang, M. Ma and B. Xu, Chem. Commun., 2007, 1032 RSC.
- O. Orçaire, P. Buisson and A. C. Pierre, J. Mol. Catal. B: Enzym., 2006, 42, 106 CrossRef CAS.
- A. Karout, C. Chopard and A. C. Pierre, J. Mol. Catal. B: Enzym., 2007, 44, 117 CrossRef CAS.
- J. M. Wallace, R. M. Stroud, J. J. Pietron, J. W. Long and D. R. Rolison, J. Non-Cryst. Solids, 2004, 350, 31 CrossRef CAS; J. M. Wallace, B. M. Dening, K. B. Eden, R. M. Stroud, J. W. Long and D. R. Rolison, Langmuir, 2004, 20, 9276 CrossRef CAS.
- A. Shumburo and M. C. Biewer, Chem. Mater., 2002, 14, 3745 CrossRef CAS.
- H. Koshima, W. Matsusaka and H. Yu, J. Photochem. Photobiol., A, 2003, 156, 83 CrossRef CAS.
- T. Ishi-i and S. Shinkai, Top. Curr. Chem., 2005, 258, 119 CAS.
- X. Song, J. Perlstein and D. G. Whitten, J. Am. Chem. Soc., 1997, 119, 9144 CrossRef CAS.
- K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664 CrossRef CAS , and references therein.
- M. de Loos, J. van Esch, R. M. Kellogg and B. J. Feringa, Angew. Chem., Int. Ed., 2001, 40, 613 CrossRef CAS.
- M. Moriyama, N. Mizoshita, T. Yokota, K. Kishimoto and T. Kato, Adv. Mater., 2003, 15, 1335 CrossRef CAS.
- Y. Zhao and X. Tong, Adv. Mater., 2003, 15, 1431 CrossRef CAS.
- W. M. Müller, U. Müller, F. Vögtle and J. L. Pozzo, Langmuir, 2002, 18, 7096 CrossRef CAS.
- S. J. Lee, S. H. Jung, S.-H. Lee, W. S. Han and J. H. Jung, J. Nanosci. Nanotechnol., 2009, 9, 5990 CrossRef CAS.
- Q. Zhenjun, Y. Haitao, L. Jinbo, W. Yu and Z. Yan, Chem. Commun., 2009, 23, 3342 Search PubMed.
- J. J. D. de Jong, J. N. Lucas, R. M. Kellogg, J. H. van Esch and B. L. Feringa, Science, 2004, 304, 278 CrossRef CAS.
- M. Ayabe, T. Kishida, N. Fujita, K. Sada and S. Shinkai, Org. Biomol. Chem., 2003, 1, 2744 RSC.
- D. D. Díaz, J. J. Cid, P. Vázquez and T. Torres, Chem.–Eur. J., 2008, 14, 9261 CrossRef CAS , and references therein.
- R. J. H. Hafkamp, B. P. A. Kokke, I. M. Danke, H. P. M. Geurts, A. E. Rowan, M. C. Feiters and R. J. M. Nolte, Chem. Commun., 1997, 545 RSC.
- W. Gu, L. Lu, G. B. Chapman and R. G. Weiss, Chem. Commun., 1997, 543 RSC.
- G. Tan, M. Singh, J. He, V. T. John and G. L. McPherson, Langmuir, 2005, 21, 9322 CrossRef.
- F. X. Simon, N. S. Khelfallah, M. Schmutz, N. Díaz and P. J. Mésini, J. Am. Chem. Soc., 2007, 129, 3788 CrossRef CAS.
- U. Beginn, S. Sheiko and M. Möller, Macromol. Chem. Phys., 2000, 201, 1008 CrossRef CAS.
- E. R. Zubarev, M. U. Pralle, E. D. Sone and S. I. Stupp, Adv. Mater., 2002, 14, 198 CrossRef CAS; J. C. Stendahl, E. R. Zubarev, M. S. Arnold, M. C. Hersam, H. J. Sue and S. I. Stupp, Adv. Funct. Mater., 2005, 15, 487 CrossRef; J. R. Moffat, G. J. Seeley, J. T. Carter, A. Burgess and D. K. Smith, Chem. Commun., 2008, 4601 RSC.
- J. R. Moffat, G. J. Seeley, J. T. Carter, A. Burgess and D. K. Smith, Chem. Commun., 2008, 4601 RSC.
- M. George and R. G. Weiss, Chem. Mater., 2003, 15, 2879 CrossRef CAS.
- J. Nagasawa, M. Kuso, S. Hayashi and N. Tamaoki, Langmuir, 2004, 20, 7907 CrossRef CAS; K. Aoki, M. Kudo and N. Tamaoki, Org. Lett., 2004, 6, 4009 CrossRef CAS.
- S. H. Kang, B. M. Jung, W. J. Kim and J. Y. Chang, Chem. Mater., 2008, 20, 5532 CrossRef CAS , and references therein.
- E. Jahnke, N. Severin, P. Kreutzkamp, J. P. Rabe and H. Frauenrath, Adv. Mater., 2008, 20, 409 CrossRef CAS , and references therein.
- N. Fujita, Y. Sakamoto, M. Shirakawa, M. Ojima, A. Fujii, M. Ozaki and S. Shinkai, J. Am. Chem. Soc., 2007, 129, 4134 CrossRef CAS.
- O. J. Dautel, M. Robitzer, J.-P. Lère-Porte, F. Serein-Spirau and J. J. E. Moreau, J. Am. Chem. Soc., 2006, 128, 16213 CrossRef CAS.
- K. Inoue, Y. Ono, Y. Kanekiyo, K. Hanabusa and S. Shinkai, Chem. Lett., 1999, 429 CrossRef CAS.
- X. Nie and G. Wang, J. Org. Chem., 2006, 71, 4734 CrossRef CAS.
- C. Kim, S. J. Lee, I. H. Lee, K. Taek, K. H. H. Song and H.-J. Jeon, Chem. Mater., 2003, 15, 3638 CrossRef CAS.
- M. Shirakawa, N. Fuijita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 4164 CrossRef CAS.
- M. de Loos, J. Van Esch, I. Stokroos, R. M. Kellog and B. L. Feringa, J. Am. Chem. Soc., 1997, 119, 12675 CrossRef CAS.
- N. Mizoshita and T. Kato, Adv. Funct. Mater., 2006, 16, 2218 CrossRef CAS.
- Y. Hirai, N. Mizoshita, M. Moriyama and T. Kato, Langmuir, 2009, 25, 8423 CrossRef CAS.
- L. Zhong, T. Jiao and M. Liu, J. Phys. Chem. B, 2009, 113, 8867 CrossRef CAS.
- K. Sada, M. Takeuchi, N. Fujita, M. Numata and S. Shinkai, Chem. Soc. Rev., 2007, 36, 415 RSC.
- J. R. Moffat, I. A. Coates, F. J. Leng and D. K. Smith, Langmuir, 2009, 25, 8786 CrossRef CAS.
- K. Inoue, Y. Ono, Y. Kanekiyo, S. Kiyonaka, I. Hamachi and S. Shinkai, Chem. Lett., 1999, 225 CrossRef CAS.
- J. F. Miravet and B. Escuder, Org. Lett., 2005, 7, 4791 CrossRef CAS.
- J. F. Miravet and B. Escuder, Tetrahedron, 2007, 63, 7321 CrossRef CAS.
- C. S. Love, V. Chechik, D. K. Smith, I. Ashworth and C. Brennan, Chem. Commun., 2005, 5647 RSC.
- D. D. Díaz, K. Rajagopal, E. Strable, J. Schneider and M. G. Finn, J. Am. Chem. Soc., 2006, 128, 6056 CrossRef CAS.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 316 RSC.
- F. Rodríguez-Llansola, J. F. Miravet and B. Escuder, Chem. Commun., 2009, 7303 RSC.
- F. Rodríguez-Llansola, B. Escuder and J. F. Miravet, J. Am. Chem. Soc., 2009, 131, 11478 CrossRef CAS.
- T. Tao, A. Wilfried, P. Herwig, W. Ralf, N. Martin and D. K. Heinz, Angew. Chem., Int. Ed., 2007, 46, 6368 CrossRef CAS.
- B. Xing, M.-F. Choi and B. Xu, Chem.–Eur. J., 2002, 8, 5028 CrossRef CAS.
- F. Fergg and K. J. Keil, Chem. Eng. Sci., 2001, 56, 1305 CrossRef CAS.
- J. F. Miravet and B. Escuder, Chem. Commun., 2005, 5796 RSC.
- J. Zhang, X. Wang, L. He, L. Chen, C.-Y. Su and S. L. James, New J. Chem., 2009, 33, 1070 RSC.
- Y.-R. Liu, L. He, J. Zhang, X. Wang and C.-Y. Su, Chem. Mater., 2009, 21, 557 CrossRef CAS.
- J. Huang, L. He, J. Zhang, L. Chen and C.-Y. Su, J. Mol. Catal. A: Chem., 2010, 317, 97 CrossRef CAS.
- Y. Dong, Y. Ma, T. Zhai, Y. Zeng, H. Fu and J. Yao, J. Nanosci. Nanotechnol., 2008, 8, 6283 CrossRef CAS.
- Z. S. Liu and G. L. Rempel, J. Mol. Catal. A: Chem., 2007, 278, 228 CrossRef CAS.
- F. Shi, Q. Zhang, D. Li and Y. Deng, Chem.–Eur. J., 2005, 11, 5279 CrossRef CAS.
- Y. Liu, M. Wang, J. Li, Z. Li, P. He, H. Liu and J. Li, Chem. Commun., 2005, 1778 RSC; Y. Liu, L. Shi, M. Wang, Z. Li, H. Liu and J. Li, Green Chem., 2005, 7, 655 RSC.
- H. Y. Xiong, T. Chen, X. H. Zhang and S. F. Wang, Electrochem. Commun., 2007, 9, 2671 CrossRef CAS.
- A. Brizard, R. Oda and I. Huc, Top. Curr. Chem., 2005, 256, 167 CAS.
- D. K. Smith, Chem. Soc. Rev., 2009, 38, 684 RSC.
Footnote |
| † This work is dedicated to the memory of Prof. Toyoichi Tanaka. |
| This journal is © The Royal Society of Chemistry 2011 |