Biomedical applications of dendrimers: a tutorial
Meredith A.
Mintzer
and
Mark W.
Grinstaff
*
Boston University, Departments of Biomedical Engineering and Chemistry, Metcalf Center for Science and Engineering, 590 Commonwealth Ave, Boston, MA 02215, USA. E-mail: mgrin@bu.edu; Fax: +1 617 358-3186; Tel: +1 617 358-3429
First published on 28th September 2010
Abstract
Since their development in the mid-80s, dendrimers have become prominent synthetic macromolecules in the field of biomedical science. This tutorial review begins by discussing pertinent background information about dendrimers, focusing on their behavior in solution, how they are synthesized and what advantages they have over linear polymers. Then the focus of the review shifts to the biomedical applications of dendrimers, including their use in drug delivery, tissue engineering, gene transfection, and contrast enhancement for magnetic resonance imaging. This tutorial review is written for first-year graduate students or senior undergraduates and “asks” and “answers” many of the questions that arise in our first discussions of dendrimers.
 Meredith A. Mintzer | Meredith Mintzer obtained a BA in Chemistry from Franklin and Marshall College in 2005. She began her graduate studies that same year under the direction of Eric Simanek at Texas A&M University. Her research focused on the synthesis and evaluation of triazine dendrimers for both gene and siRNA transfection. After receiving her PhD in December 2009, she began working as a postdoctoral fellow at Boston University and Brigham and Women's Hospital under the direction of Professors Mark Grinstaff and Yolanda Colson. Her current research focuses on designing dendrimers and polymeric nanoparticles for biomedical applications. |
 Mark W. Grinstaff | Mark W. Grinstaff is a Professor of Biomedical Engineering and Chemistry at Boston University, a College of Engineering Distinguished Faculty Fellow, and Director of the NIH T32 grant program on Biomaterials. Mark received his PhD from the University of Illinois under the mentorship of Professor Kenneth S. Suslick and was an NIH postdoctoral fellow at the California Institute of Technology with Professor Harry B. Gray. Mark's awards include the ACS Nobel Laureate Signature Award, NSF Career Award, Alfred P. Sloan Research Fellowship, Pew Scholar in the Biomedical Sciences, Camille Dreyfus Teacher-Scholar, and Edward M. Kennedy Award for Health Care Innovation. He has published more than 120 peer-reviewed manuscripts and given more than 200 oral presentations. He is co-founder of three companies that are commercializing his ideas. His research interests cover a broad range of topics but always with an emphasis on new compositions, synthetic methods, and evaluation of properties. |
1. What are dendrimers and how do they behave?
Dendrimers are a class of macromolecules with a highly branched three-dimensional architecture whose structural elements can be tuned to affect both surface and internal properties of the macromolecule. The term dendrimer is derived from the Greek words dendri- meaning “tree-like” and meros meaning “part of”,1 but these structures have also been referred to as arborols2 or cascade molecules.3 The first dendrimer-like compound, polypropylenimine (PPI), was synthesized by Vögtle et al. in 1978.3 However, difficulties encountered with the synthetic approach resulted in the formation of only low generation compounds. It was not until the mid-80s that Newkome et al.2 and Tomalia1 synthesized dendrimers at higher generations with well-defined structures. Since then over 100 different dendrimer structures have been realized.4 Several of the most commonly referenced dendrimers are shown in Fig. 1 and include Tomalia's polyamidoamine (PAMAM),1 Denkewalter's poly(L-lysine) (PLL),5 Newkome's polyamide,2 Grinstaff's polyester (PGLSA-OH),6 Vögtle's polypropylenimine (PPI),3 and Hult's poly(2,2-bis(hydroxymethyl)propionic acid (bis-MPA)7 structures. Many of these dendrimers are commercially available from providers including Dendritech (PAMAM), Frontier Scientific (Newkome's polyamides—i.e. Ntrons—and Simanek's triazines), Colcom (poly(L-lysine)—i.e. DGL), Polymer Factory (bis-MPA), and DSM (PPI—i.e. Astromol).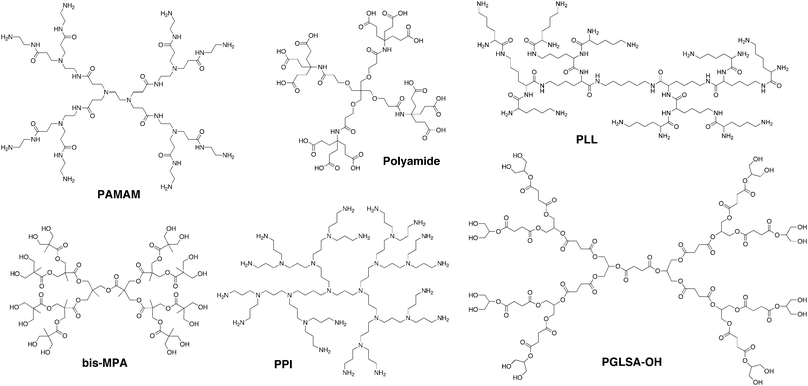 | ||
| Fig. 1 Chemical structures of several commonly used, commercially available dendrimer structures. | ||
Dendrimer structures can be divided into three main components: the core, the interior, and the shell. The core affects the 3D shape of the dendrimer (i.e., spheric, ellipsoidic, or cylindric scaffolds). The interior affects the host–guest properties of the dendrimer. The surface of the dendrimer can be further polymerized or modified with functional peripheral groups. Both the core and the number/type of interior branching units affect the overall dendrimer morphology. Because dendrimer diameters increase linearly while the number of surface groups increases exponentially for each generation, steric crowding at the surface occurs.8 Hence, dendrimers at low generations are usually flexible and open, while dendrimers at higher generations form more dense, three-dimensional shapes. A dendrimer's generation number also has an effect on the rigidity of the overall structure.9 Fréchet-type polyether dendrimers shift to higher rigidity (i.e. more globular structure) on going from generation three to four. PAMAM shifts to higher rigidity at generation 4.5. Finally, for PPI dendrimers a globular nature is observed for the fourth generation analogue. In general, the globular nature of the higher generation dendrimers results in differences in solubility and reactivity of the endgroups as compared to similar linear polymer analogues. Consequently, a majority of dendrimers used in biomedical applications are fourth generation or higher to take advantage of this unique physicochemical behavior.
However, describing dendrimer branches as simply extending outward from the core to create a “dense shell” of end groups at the periphery oversimplifies the behavior of most of these structures. Backfolding leads to the formation of structures with dense cores rather than dense shells and occurs for a number of different dendrimer analogues (Fig. 2).10
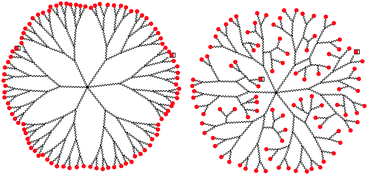 | ||
| Fig. 2 Representation of dendrimer backfolding. | ||
In aqueous conditions, dendrimer conformations are affected by ionic strength and pH, with changes depending on the type of charged group at the dendrimer surface. Molecular dynamic studies of amine-terminated PAMAM showed that globular, loosely compact structures are observed at high pH while the extended conformation dominates at low pH (<5) due to electrostatic repulsions of the protonated tertiary amines (pKa ≈ 5) at the interior of the dendrimer and the primary amines (pKa ≈ 9–11) at the surface.11 This conformational change affects the endosomal escape of dendrimers following cellular uptake. At physiological pH (7.4) only the primary amines are protonated but after exposure to the endosome environment (pH ≈ 5), the tertiary amines are protonated and the dendrimer conformation change causes endosome rupture. Conversely, for carboxylate-terminated PPI dendrimers, small angle neutron scattering studies have shown that at both low (<4) and high (>11) pH, the dendrimer displays an extended conformation due to electrostatic repulsions of either the protonated internal amines at low pH or the deprotonated carboxylic acids on the periphery at high pH. At a pH ≈ 6, however, the PPI dendrimer displays a condensed, backfolded structure due to intramolecular hydrogen bonding of the zwitterionic structure.12
2. How are dendrimers synthesized?
Most dendrimers are synthesized following either a convergent or divergent route, as depicted in Fig. 3. Each method has its own advantages and disadvantages. The initial syntheses of dendrimers pioneered by Tomalia et al.,1 Newkome et al.,2 and Vögtle et al.3 proceeded by divergent routes, whereby polyfunctional cores react with monomer units that have one reactive site and multiple protected or unreactive groups. Following reaction with the cores, the unreactive or protected groups are activated for further reactions with additional monomer units. Divergent route synthesis typically affords dendrimers that display repeated AB2 or AB3 branching motif, with AB2 branching being the most common.13 The divergent route can be used for the synthesis of a broad spectrum of dendrimer structures but can be limited by incomplete reaction of the groups leading to the defects in the branching. To overcome this limitation, the monomer unit is often added in excess, thus requiring purification after each step. However, such purification cannot eliminate all incomplete byproducts. For instance, PPI dendrimer growth can be limited by retro-Michael reactions or intramolecular amine cyclizations. Even if the desired reaction selectivity approaches 99.5%, by the fifth generation only 29% of the dendrimer will be defect-free, and above the seventh generation virtually no defect-free structures can exist. Similarly, for PAMAM, due to defects caused by retro-Michael additions and intramolecular lactam formation, the fourth generation dendrimer has only 8% defect-free product. Consequently, while calculations for the polydispersity of dendrimers synthesized divergently show values nearing monodispersity, the purity of these dendrimers is governed by statistics and defects will always be present. Still, several strategies have been pursued to improve this limitation.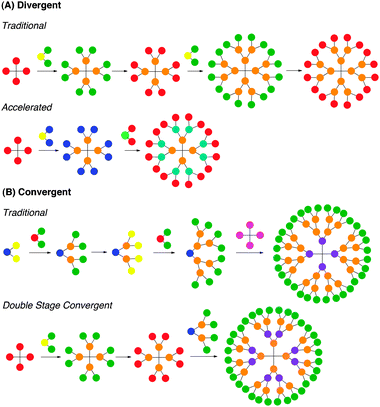 | ||
| Fig. 3 Pictorial representations of dendrimer synthesis by (A) divergent and (B) convergent strategies. | ||
To avoid purification procedures, user-friendly reactions that proceed in high yields have been pursued. In 2005 Hawker and Wooley et al. used the Huisgen 1,3-dipolar cycloaddition reaction to divergently grow dendrimers up to generation three.14 The synthesis involved iterative reaction between an azide dendrimer and alkyne monomer unit followed by halogenation and azido nucleophilic substitution of the newly formed periphery. The dendrimers could be obtained in decent yields with little purification required. In 2008 the Hawker group used thiol–ene chemistry to construct dendrimers up to generation four, as well as to functionalize the periphery.15 In addition to reducing the structural defects that can occur with the typical divergent approaches, the thiol–ene chemistry could be run in the absence of solvent without the use of metal catalyst, making it quite environmentally friendly.
In addition to improving the yields of the divergent strategy using Click chemistry techniques, accelerated synthetic strategies have been developed to reduce the number of steps. In this strategy, two different monomer units, AB2 and CD2, that have complementary functionalities can react spontaneously without the use of protecting groups or activating agents. In 2001 Caminade and Majoral et al. used this technique to synthesize their phosphorus-containing dendrimers through interactive condensation reactions between phosphorhydrazides and aldehydes followed by Staudinger reactions between phosphines and azides.16 Similarly, in 2007 Malkoch et al. used this technique to synthesize derivatives of both the Fréchet-type polyether dendrimers and 2,2-bis(methylol)propionic acid dendrimers using Click chemistry and etherification/esterification reactions, respectively.17
The second commonly used method for dendrimer synthesis is the convergent approach, pioneered by Fréchet et al.18 The method proceeds from the surface of the dendrimer inward to form a dendron that reacts with a suitable core to complete the synthesis. The convergent approach is advantageous because only a limited number of active sites are present per reaction, reducing structural defects in the product. As a result, higher percentages of defect-free product can be obtained per generation and can be isolated from the byproducts. However, the convergent approach is generally used to form only lower generation structures because steric hindrance is encountered when large dendrons are reacted with a small core to form a larger generation dendrimer. Still, because most dendrimers used for biomedical applications are fourth generation analogues, this has not been a limitation.
To increase coupling yields for the convergent approach, Hawker, Sharpless, and Fokin et al. used 1,3-dipolar cycloaddition reactions.19 Third generation dendrons were formed by iterative reactions of AB2 alkyl halide/alkyne monomer units with azidomethyl dendrons. The third generation dendron was coupled with a polyacetylene core to form a G4 dendrimer. Due to the high reactivity of the cycloaddition, the reaction between the dendrons and core could be run at low concentrations to avoid steric congestion and proceed to completion at room temperature.
The final, albeit significantly less utilized, route to dendrimer synthesis involves a double stage convergent method first reported in the early 90's by Fréchet et al.20 This method allows for the formation of monodisperse, higher generation dendrimers in fewer linear steps through a divergent approach that uses macromonomer units synthesized convergently. While this method is exploited less often than the convergent or divergent approaches, it has been used to synthesize polyester,21 polyamide,22 and triazine23 dendrimers.
3. What's so great about a dendrimer?
A common question is what makes dendrimers appealing over easily synthesized linear or branched polymers? The answer lies in the fact that the stepwise growth of dendrimers affords nearly monodisperse products, whereas polymerization involves chain growth procedures that afford statistical mixtures of products. By controlling details of the core, interior and periphery, it is possible to design macromolecules with nearly perfectly defined structures and compositions. Such macromolecules can function as synthetic analogues of peptides or polynucleotides, and the ability to control the details of the structure exactly can vary the physical, chemical, biological or rheological properties.It must be mentioned at this point in the tutorial that the term “monodisperse” is generally used to describe the unmodified dendrimer structure. However, for some of the dendrimers described in the remainder of this review, most notably PAMAM, the term “monodisperse” may be somewhat misleading. Commercially-available PAMAM actually has a polydispersity index (PDI) of ∼1.01, indicating that the well-defined branched structure has some defects (though significantly less than hyperbranched polymers). Furthermore, for biomedical purposes nearly all dendrimers are modified by conjugation with at least one type of ligand. Typically, the number of ligands attached per particle is reported as a mean value following stochastic synthesis techniques. Recently, Holl et al. have determined that using the mean value underestimates the heterogeneity in a ligand-conjugated dendrimer sample.24 For instance, when a G5 PAMAM dendrimer is conjugated with an alkyne ligand to achieve a mean value of 12.9 ligands per dendrimer, the actual sample contains 27 different species ranging from no ligands per dendrimer to 26 ligands per dendrimer, and the mode value is actually 16 ligands per dendrimer. Additionally, appending multiple ligands onto the same dendrimer following sequential reaction steps further increases the heterogeneity of the sample.
In addition to monodispersity, another key feature of dendrimers and probably their most important attribute is their multivalency, with the number of surface groups increasing exponentially with each generation. Appending ligands for a particular receptor onto the surface of a dendrimer creates a targeting molecule that exhibits increased avidity as compared to the monovalent ligand due to a “chelate” effect.25 In 2005, Finn, Fokin, Sharpless, and Hawker et al. probed this effect by synthesizing a bis-MPA dendrimer with mannose functionality on the periphery, and then evaluating the binding affinity of this dendrimer to concanavalin A (Con A) using a hemagglutination assay.26 The group observed that the dendrimer displayed a 240-fold greater binding affinity than monomeric mannose, correlating to a relative activity of 15 per sugar molecule. Additionally, it has been observed that the multivalency displayed by dendrimers exceeds that of traditional polymers. In 2009 Gillies et al. synthesized poly(butadiene-block-ethylene oxide) (PBD-PEO) with either hydroxyl or azide terminal groups.27 The polymers were used to form vesicles with dextran-coated iron oxide. The azide terminal groups were then functionalized with either alkyne-terminated dendrons displaying mannose surface groups or with alkyne functionalized mannose. The overall density of mannose groups on the surface of both dendritic and non-dendritic vesicles was kept constant. When evaluated for binding with Con A it was observed that the dendritic vesicles displayed up to 2-fold higher binding affinity than the non-dendritic analogues. This result was attributed to a “proximity” effect caused by the dendrimer structures, whereby the same density of mannose was displayed in localized clusters without steric inhibition caused by surrounding surface polymer chains. This multivalent effect of dendrimers has significant implications in designing high-affinity cell targeting agents using even ligands that normally have only weak binding interactions.
3.1 But do we really need monodispersity?
The answer to this question is yes, and this is not just because our research focuses on these structures. Monodispersity has been one of the main factors to catapult dendrimers to the forefront of biomedical research. For biological applications, monodispersity that can be consistently reproduced is key, as it allows for the investigation of structure activity relationships. By knowing the composition of macromolecules, we can relate the biological activity to specific aspects of the structure. This is a powerful tool for drug and medical device development. However, the question remains whether clinical applications always require monodisperse agents or materials. For clinical utility of dendrimers, the answer is not necessarily. Based on the fact that a number of linear polymers have made it to clinical trials for drug and gene delivery, polydispersity cannot be considered a limiting factor. N-(2-hydroxypropyl) methacrylamide (HMPA) polymers have shown promise in clinical trials for the delivery of doxorubicin, taxol, camptothecin, and diammineplatinum (anticancer agents). Polyethyleneglycol (PEG) has shown promise for the delivery of camptothecin, while polyglutamic acid (PGA) has shown potential in clinical trials for delivering both camptothecin and methotrexate. Cyclodextrin-based polymers are currently undergoing clinical trials for the delivery of both camptothecin and siRNA, and linear polyetheylenimine (PEI) conjugated to siRNA is being investigated in clinical trials for the treatment of bladder cancer as well as HIV.If monodispersity controls the reliability and precision of macromolecules for biomedical purposes, why have linear polymeric delivery agents continued to enter into clinical trials over dendrimers? The main factor hindering the use of dendrimers is the costly and time-consuming multistep synthesis required for the high generation structures. However, this hurdle will most likely be overcome with continued research into new and better synthetic routes and procedures.
4. Where can I learn more about dendrimers?
For individuals in the early stages of their scientific career, this tutorial reviews dendrimers and their relevance to biomedical applications. To keep the review concise, the scope of each topic was limited. However, a number of reviews that cover sections of this tutorial in more detail have been published and can be referred to for more information. The divergent synthesis of dendrimers has been reviewed by Newkome and Shreiner13 Also, Malkoch et al. described both traditional routes and recent advances in strategies for synthesizing dendrimers.28 Advances have been described by Franc and Kakkar, who reviewed the use of Diels–Alder click chemistry for dendrimer synthesis.29 Furthermore, Percec et al. have reviewed the behavior of dendrimers in detail, describing both self-assembly and self-organization of these structures.30The remainder of this review describes biomedical applications of dendrimers, but several recent publications have described each topic in more detail. The use of dendrimers for drug delivery has been reviewed by several groups.31,32 Several groups have also specifically reviewed the relevance of dendrimers with regards to transfection,33 tissue engineering,34 and MRI contrast enhancement,35 as well as vaccines,36 a topic which is not addressed by this review. Finally, it must be mentioned that dendrimers have been applied to many nonbiomedical applications as reviewed by Astruc et al.37
5. What are the biomedical applications of dendrimers?
It is clear that the dendrimer generation and the composition of the core and peripheral groups will have a significant effect on physical and chemical properties of these structures and thus their resulting utility in specific biomedical applications. In the following examples, we give the generation number and composition of the dendrimer under study and ask the reader to note the trends that are described, such as the use of cationic peripheral groups for the delivery of nucleic acids, the use of asymmetric dendrimers possessing a covalently linked drug for improved anti-tumor activity, and the increased evaluation of polyester-based dendrimers compared to amine or amide-based constructs for drug delivery and tissue engineering.5.1. Drug delivery
5.1.1.1 Antimicrobial. The emergence of a number of drug resistant strains of bacteria in the last few decades has increased interest in novel antimicrobial agents. Cationic dendrimers with amphiphilic properties are one potential option. In 2000 Cooper et al. synthesized PPI dendrimers with quaternary ammonium groups on the periphery.38 While the highest generation PPI dendrimer showed the highest antimicrobial activity, the activity did not correlate linearly from generation one to five. This was attributed to the interplay between multiple factors. While the higher generation structure should be more potent due to more surface groups, the permeability of the structures through the cell membrane is preferred for low generation structures. Also, the alkyl chain length of the quaternary ammonium group affected antimicrobial activity. Highest activity was observed for C10 chains, followed by C8 and C12. Similar trends have been observed for antimicrobial surfactants and are attributed to either (1) different cell binding affinities or (2) different aggregation behaviors. In this study the antimicrobial activity was observed below the critical aggregation concentration (CAC), suggesting that the difference in activity results from different binding affinities.
While the previously described cationic agents all show notable antimicrobial activity, they all suffer drawbacks due to their cationic nature. Cytotoxicity towards eukaryotic cells has been observed for nearly all positively charged macromolecular structures. Therefore, studies have investigated the effect of reducing the positive charge on the surface of the dendrimers. Cai et al. investigated the antimicrobial activity of a G5 PAMAM dendrimer in which ∼43% of the terminal amines were PEGylated.39 When exposed to Gram-negative bacteria strains (PA19660 and the clinical strain PA2219), the EC50 value of these dendrimers ranged from 0.9 to 1.5 μg mL−1, while only a slight decrease in eukaryotic cell survival was noted up to a concentration of 10 μg mL−1. Recently, the Grinstaff group has shown that cationic charge on the surface of a dendrimer can be completely eliminated while still maintaining antimicrobial activity. The group synthesized anionic amphiphilic dendrimers that self-assembled into supramolecular structures, and then evaluated these structures as antimicrobial agents.40 When exposed to cells at concentrations above the CAC, these structures were shown to be up to 36-fold more cytotoxic to B. subtilis cells than eukaryotic HUVEC cells. In general it was observed that the EC50 of cytotoxicity with respect to bacterial cells of the dendrimer assemblies was similar to the CAC. The reason for the reduced cytotoxicity of the dendrimer towards eukaryotic cells is unclear, and further studies are underway.
5.1.1.2 Antiviral. Dendrimers have also been investigated for their antiviral properties, particularly against HIV-1. At the early stage of HIV replication, the virus must target and enter the host cells. CD4+T cells are one of the most common targets for the virus, and cellular entry is promoted by the formation of a ternary complex between gp120, a glycoprotein presented on the surface of the HIV envelope, and CD4 and co-receptors CCR5 or CXCR4. The V3 loop of gp120 is a positively charged region of the protein, and by exposing the protein to polyanions, the ternary complex formation can be inhibited. One of the most successful anionic dendrimers used for antiviral purposes is VivaGel, a sulfonated polylysine dendrimer. This compound is currently undergoing phase I/II clinical trials, and will be discussed in detail in Section 6. However, other dendrimer structures have shown success as antiviral agents. Blanzat and Turrin et al. synthesized poly(phosphor-hydrazone) dendrimers with terminal phosphonic acid and alkyl chain groups (Fig. 4) and evaluated these agents as HIV antivirals for CEM-SS and MT-4 cells.41 Thirty minutes after exposure to the virus (HIV-1 LAI and HIV-1 IIIB, respectively) the cells were treated with various concentrations of the dendrimers and evaluated for inhibition of HIV infection after 5 days. The highest antiviral activity (IC50) was achieved using the dendrimer with a C3 alkyl chain (IC50 1.5 × 10−6 mol L−1). The dendrimers with no alkyl chain and with a C10 alkyl chain showed similar activity (IC50 1.0 × 10−5 and 3.5 × 10−5 mol L−1, respectively). This was attributed to the ability of the short alkyl chain to interact with the lipophilic portion of the V3 loop. The dendrimer containing the C10 alkyl group did not exhibit this effect due to favourable interactions of the long alkyl chain with the core of the dendrimer over external groups.
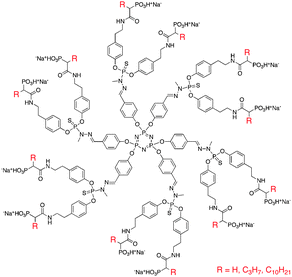 | ||
| Fig. 4 Structure of poly(phosphorhydrazone) dendrimers evaluated as antiviral agents. | ||
Alternative pathways of HIV inhibition using dendrimers have recently been investigated. The high infection of CD4+T cells has been attributed, in part, to interaction between HIV-1 and dendritic cells. These interactions occur between a mannose-binding C-type lectin (DC-SIGN) on the surface of dendritic cells, and the high density of mannose glycans on the surface of HIV-1. Wang, Liang and Wong et al. recently synthesized a series of dendrons with 3, 9, and 27 Man4 and Man9 surface moieties (Fig. 5).42 The second generation analogues, with nine Man4 or Man9 groups displayed on the surface, were investigated as inhibitors of the binding of DC-SIGN to gp120 using an ELISA. Plates were coated with gp120JR–FL and then treated with mixtures of the dendrons and Fc-DC-SIGN, a fusion protein of DC-SIGN. Bound Fc-DC-SIGN was detected with Cy3-labeled anti-human IgG antibody. The results showed that 50% of the binding of DC-SIGN to gp120 could be inhibited using nanomolar concentrations (IC50 20 nM and 8 nM for second generation Man4 and Man9 dendron, respectively).
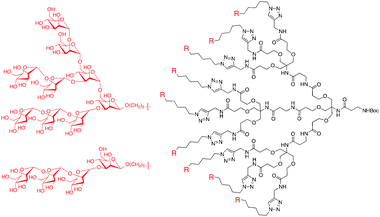 | ||
| Fig. 5 Dendrimers synthesized with Man4 and Man9 groups on the periphery to function as HIV-1 inhibitors. | ||
5.1.2.1 Encapsulation. Due to the hydrophobicity of most chemotherapeutic drugs, delivering these agents by intravenous (i.v.) or intraperitoneal (i.p.) injections is limited by poor water solubility. Furthermore, once free drug is incorporated into the bloodstream, it is quickly filtered and removed by the kidneys, thus requiring patients to undergo multiple rounds of treatments. Finally, because the free drug can circulate to most tissues in the body, there are always negative side effects associated with chemotherapy. Dendrimers have the potential to overcome these obstacles. First, dendrimers can function as unimolecular micelles capable of encapsulating and solubilizing drugs into the void spaces within the dendrimer's interior. Second, if a drug is carried by a dendrimer larger than 5 nm, the size of the carrier exceeds the renal threshold and is less able to be filtered out of the bloodstream by the kidneys. Therefore the drug remains in circulation for longer. Finally, due to the rapid growth and corresponding angiogenesis of tumors, tumor vasculature has larger pore sizes and ineffective lymphatic drainage as compared to healthy tissue. This results in increased uptake of large macromolecules by tumor tissue as compared to normal tissue, an effect known as enhanced permeation and retention (EPR effect). Hence, incorporating chemotherapeutic drugs with dendrimer carriers results in passive targeting of tumor tissue. These characteristics of dendrimer carriers have been previously reviewed by Fréchet and Szoka et al.32 This section focuses on dendrimer encapsulation used to deliver the most widely used chemotherapeutic agents (i.e. camptothecin, doxorubicin, methotrexate, and paclitaxel) and discusses modifications made to the dendrimers to improve this process.
Using dendrimers as drug carriers allows for the delivery of a high payload of drug with reduced cytotoxic side effect. Recently a dendrimer based on glycerol and succinic acid was used to deliver 10-hydroxy-camptothecin to various cell lines.43 When measured for cytotoxicity, the dendrimer/camptothecin complex showed low IC50 values (ng L−1) in human breast adenocarcinoma (MCF-7), colorectal adenocarcinoma (HT-29), non-small cell lung carcinoma (NCI-H460), and glioblastoma (SF-268) cells. Furthermore, cellular uptake was increased 16-fold for the dendrimer/camptothecin conjugate as compared to free drug, and cellular retention was increased from 35 to 50% after 32 h.44 Similarly, a triazine dendrimer–methotrexate complex has shown promising results in vivo. Forty-eight hours after i.p. injections of either dendrimer–methotrexate or methotrexate solution (2 mg MTX kg−1), C3H mice were sacrificed, and liver damage was quantified by measuring levels of alanine transaminase (ALT). The mice that received the dendrimer–methotrexate complexes showed 27% lower levels of ALT, indicating lower toxic side effects caused by the encapsulated drug.45
Despite successful drug encapsulation using dendrimers, the biocompatibility and rapid clearance of these structures warranted further modifications. The attachment of poly(ethylene glycol) (PEG) to the surface of dendrimers has been one such modification that proved successful, since PEG is a nontoxic, nonimmunogenic, water soluble polymer. Third and fourth generation PAMAM structures that have been conjugated at the surface with PEG-monomethyl ether chains of various lengths have been evaluated for drug encapsulation.46 Both generation number and PEG chain length affected encapsulation efficiency of doxorubicin, with the highest efficiency seen for PAMAM G4–PEG2000 (∼6.5 doxorubicin molecules per dendrimer). This same PAMAM–PEG construct also showed the highest capacity for encapsulating methotrexate (26 methotrexate per dendrimer). Higher efficiency for methotrexate encapsulation was attributed to electrostatic interactions between negatively charged methotrexate and the cationic PAMAM interior.
Even with the aid of dendrimer carriers, the delivery of drugs across the blood–brain barrier (BBB) is difficult. Appending glucosyl groups onto the surface of the dendrimer can help overcome this hurdle. Recently, polyether–copolyester dendrimers with either dihydroxy benzoic acid (dendrimer 1) or gallic acid ester (dendrimer 2) units were conjugated to 3 and 5 molecules of D-glucosamine per dendrimer, respectively, and used for methotrexate encapsulation.47 Dendrimer 1 was shown to encapsulate 17–22% w/w methotrexate, while dendrimer 2 showed encapsulation efficiency ranging from ∼20–25% w/w methotrexate. These compounds were then evaluated for their potential in crossing the BBB. Cellular uptake of glucosylated and nonglucosylated derivatives of dendrimer 1 was evaluated in two glioma cell lines (U 87 MG and U 343 MGa). Cellular uptake was higher for the glucosylated derivative (8-fold increase for U 87 MG cells, and 2-fold increase for U 343 MGa). When tested for transport across polycarbonate Transwell inserts seeded with a co-culture of brain capillary endothelial (bend.3) and U 373 MG cells, the glucosylated dendrimers were able to permeate 1.2- to 3.5-fold better than dendrimer alone. As compared to free methotrexate, the dendrimer–methotrexate complexes permeated 3- to 4-fold higher concentrations of drug. The compounds were then evaluated against an avascular human glioma tumor spheroid. The glioma tumor spheroid model, a multicellular 3-dimensional growth of cells, has advantages over typical monolayers of glioma cells for in vitro analysis because the spheroid model more accurately displays the cell–extracellular matrix adhesions and cell–cell adhesions that are present in vivo. While non-treated spheroids continued to grow and free methotrexate inhibited tumor growth for only the first few days, the dendrimer–methotrexate encapsulated complexes all showed reduced tumor volume when administered at 0.2 mM methotrexate and 0.4 mM methotrexate concentrations over the 7 day study. Furthermore, accumulation of ethidium bromide in the lysate of the tumor spheroids was used to monitor cell death. The glucosylated derivatives of both dendrimers induced statistically significant increase in cell death as compared to the non-glucosylated derivatives. The results indicate that the dendrimer–methotrexate inclusion complex have potential for delivering anticancer agents across the BBB. In vivo results are pending.
While successful in vitro drug delivery has been obtained via encapsulation with dendrimers as described above, limitations to the strategy exist. The main disadvantage of the encapsulation strategy is the rapid and uncontrollable release of drug molecules from the dendrimer core. In 2005 Baker et al. used fifth generation PAMAM with various surface groups to deliver methotrexate either through encapsulation or covalent attachment.48 The dendrimer–drug encapsulation complex showed minimal methotrexate release when dialyzed against water, but when dialyzed against phosphate buffered saline, >70% of the drug was released within 2.5 h regardless of functional groups on the surface of the dendrimer. Conversely, the covalently bound conjugates remained stable in both water and buffered solutions. This same trend has been observed for other non-covalent dendrimer–drug delivery systems.46,49 As such, the inability to control the rate of drug release from dendrimer encapsulates is a severely limiting issue for the use of these systems.
In addition, the drug loading capacity of dendrimers via encapsulation is a limiting factor for clinical translation. For instance, the previously-described third generation triazine dendrimer was shown to encapsulate approximately 3 molecules of methotrexate per dendrimer. For such structures to achieve clinical relevance, the dendrimer–drug complex would need to have a maximum tolerated dose above 490 mg m−2 in order to achieve a methotrexate regimen of 80 mg MTX m−2 (the maximum tolerated dose of methotrexate in humans ranges from 80 to 900 mg m−2). Furthermore, for the PAMAM G4–PEG2000 construct that encapsulated 6.5 doxorubicin molecules per dendrimer, the structure would need to be administered at a dosing regimen of 1.2 g m−2 to achieve the standard doxorubicin dose of 90 mg m−2. Such high doses of dendrimer–drug encapsulate are a potential concern. As a result, the development of alternative strategies for drug conjugation that afford higher drug loadings is necessary.
5.1.2.2 Covalent attachment. Due to the above limitations encountered with drug encapsulation, significant research has focused on covalent drug attachment. The types of linker groups used to covalently attach drugs to the surface of dendrimers can affect the activity of the dendrimer–drug conjugates and can be used to control the rate of drug release. Typically the linkers used to covalently attach drugs to dendrimers include (1) acid labile cis-aconityl or acyl hydrazone groups, which are readily cleaved in the acidic lysosomal environment following cellular uptake; (2) ester groups, which can be cleaved by a variety of esterase enzymes within the cell; or (3) disulfide groups, which can be reduced by glutathione within the cytosol. Recently a fourth generation PAMAM was conjugated with various amounts of PEG5000 chains (4, 16, and 32 chains per dendrimer), and doxorubicin was attached by either acid sensitive cis-aconityl (PPCD) or insensitive succinic amide linkages (PPSD).50 Based on intracellular localization studies evaluated by confocal laser scanning microscopy, it was determined that cis-aconityl-linked doxorubicin displayed doxorubicin-related fluorescence in both the lysosome and nucleus of ovarian cancer cells (SKOV-3), suggesting acid-triggered release of doxorubicin following cellular uptake. Furthermore, if the pH of the lysosomal environment was basified with chloroquine, doxorubicin release was inhibited, further strengthening this observation. No nuclear fluorescence was observed for the succinic amide-linked construct, indicating no doxorubicin release. Furthermore, in vivo studies of these constructs showed that the highest tumor accumulation occurred for the PPCD conjugate with the highest number of PEG chains, an effect attributed to enhanced permeation and retention (EPR) as well as the ability of the PEG chains to inhibit the adhesion of blood serum opsonins, thus allowing the particles to remain untargeted by phagocytic cells.
Similarly, doxorubicin (DOX) has been covalently conjugated through acyl hydrazone linkages to polyester–PEG dendrimers to improve bioavailability. In 2006 doxorubicin was conjugated to an asymmetric (i.e. bowtie), biodegradable G3 polyester dendrimer (Fig. 6).51 The construct consisted of eight PEG chains and sixteen pH-sensitive acyl hydrazone linker sites for drug conjugation. In vitro assays showed that the dendrimer–doxorubicin conjugate was 10-fold less toxic against colon carcinoma (C-26) cells than free drug, a feature attributed to slower cellular uptake and gradual drug release. When BALB/c mice with subcutaneous C-26 tumors were given a single i.v. injection of the dendrimer–doxorubicin complex (20 mg DOX kg−1) 8 days after tumor implantation, complete tumor regression and 100% survival were observed after the 60-day experiment. Furthermore, the dendrimer–DOX complex exhibited similar anti-tumor activity to Doxil, a clinically-approved, liposomal doxorubicin complex. However, the dendrimer–DOX can be stored in dry conditions, making it a more stable pharmaceutical agent, and has the potential for delivering a wider variety of drug agents due to the covalent attachment.
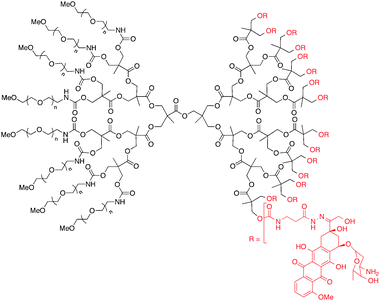 | ||
| Fig. 6 Chemical structure of asymmetric polyester bowtie dendrimer with doxorubicin conjugated by acyl hydrazone linkers. | ||
In addition to acid-labile linker groups, various efforts have investigated the attachment of drugs through enzyme-cleavable ester groups; with the length of the ester linker has been observed to affect the rate of drug release. In 2007 a trifunctional PAMAM dendrimer conjugate containing succinic acid ester-linked paclitaxel, folic acid, and FITC was compared to an analogue in which the drug was linked to the dendrimer using glutaric acid.52 Based on quantitative thin layer chromatography, it was observed that the succinic acid derivative hydrolyzed in PBS buffer with a half-life of approximately 10 h. Conversely, the glutaric acid conjugate showed no hydrolysis in the same conditions after 7 days. This study underscores the importance of the linking group used to conjugate drugs to dendrimers.
However, it must be stressed that the trends of linker groups have not been consistent for all dendrimer types. While the glutaric acid-linked PAMAM–paclitaxel conjugate showed no drug release in buffer conditions, 1,3,5-triazine based dendrimer–drug conjugates investigated by Simanek et al. have shown promising data.53 In 2008 a G2 triazine dendrimer was conjugated with a Bolton–Hunter-type moiety for radiolabeling, and 16 paclitaxel groups. The remaining amines were PEGylated with either PEG2000 or PEG5000 to create two dendrimer–drug conjugates with 18 wt% or 30 wt% drug, respectively. This degree of loading indicated that these conjugates could achieve drug concentrations that rivaled those of the clinically relevant Cremophor EL. More recently, biological studies were conducted on a set of similar triazine dendrimer–paclitaxel constructs, but these contained different linkers for the drug and PEG chains. Construct one was composed of ∼9 succinic acid ester-linked PEG2000 chains and 12 glutaric acid ester-linked paxlitaxel molecules. Construct two was composed of ∼8 succinic acid ester-linked PEG2000 chains and 12 glutaric acid ester/disulfide-linked paclitaxel molecules. Finally, construct three was composed of ∼7 ether-linked PEG2000 chains and 12 glutaric acid ester/disulfide-linked paclitaxel molecules. Because of the different drug linker combinations, it was expected that the cytotoxicities of these macromolecules would vary in the presence or absence of a disulfide reducing agent (i.e. glutathione or dithiothreitol). While constructs two and three showed increased cytotoxicities in the presence of glutathione (26 nM) and dithiothreitol (13 nM) as compared to the cytotoxicities observed without reductant (74 nM) as expected, the cytotoxicity of construct one was ∼29 nM in either condition. The reason why construct one was more cytotoxic than two and three without the reducing agent is unclear. Based on the biodistribution data of all of these constructs, it was observed that the elimination half-life was shorter than expected based on previously published predictions of PEGylated, non-drug loaded triazine dendrimer constructs. This indicates that drug loading may affect the biodistribution of the drug carriers. Tumor accumulation of these compounds following i.v. injections into SCID mice with PC-3 tumors was rapid and sustained over 48 h. In addition, the constructs could be administered at doses up to twice the maximum tolerated dose of free drug, which is relevant for clinical applications.
In addition to linking groups, targeting agents can affect the activity of dendrimer–drug constructs. Appending targeting agents onto the surface of dendrimer–drug conjugates has improved delivery to specific tissue as shown by studies conducted by Baker et al.54 In 2005 a G5 PAMAM dendrimer was partially acetylated to reduce the overall cationic charge, and the remaining amines were surface functionalized with fluorescein isothiocyanate (FITC) using a thiourea linkage, folic acid using an amide linkage, and methotrexate using an ester linkage. In vitro studies of this compound in folic acid receptor-expressing KB cancer cells showed a time- and dose-dependent cytotoxicity for the folate receptor-targeting dendrimer cojugate, whereas no toxicity was observed for the untargeted derivative. Furthermore, the results suggested that the dendrimer–drug conjugate was incorporated into the cell by folic acid receptors instead of reduced folate carriers. Because folic acid receptors are not involved in methotraxate-induced drug resistance, the PAMAM–methotrexate conjugate may be able to overcome drug resistant strains. In vivo studies of the conjugate involving 15 biweekly tail-vein injections into nude mice bearing human KB tumor xenografts showed that the complex had 10-fold higher efficacy and reduced toxicity as compared to equivalent doses of free methotrexate. Recently it was shown that a very similar methotrexate–folic acid–(G5)-PAMAM conjugate could be synthesized in a notably easier one-pot reaction. While slightly different structurally from the originally synthesized derivative, this new conjugate showed a nearly identical cytotoxic potency against KB cells.
The functional groups at the surface of a dendrimer can also affect drug delivery. In 2006 methotrexate was conjugated to both anionic (G2.5) and cationic (G3) PAMAM dendrimers.55 While the cationic dendrimer showed limited drug activity when compared to equimolar amount of free drug against both methotrexate sensitive (CCRF-CEM) and resistant (CEM/MTX) human lymphoblastoid leukemia cell lines, the anionic derivative showed higher drug activity in both cell lines (8- and 24-fold increases, respectively). It was hypothesized that the increased reactivity resulted from longer lysosomal residence time, allowing for better interactions with lysosomal proteases responsible for drug release from the dendrimer.
One novel dendrimer for drug delivery that avoids issues related to drug release and possibly tissue targeting came from the Shabat group. In 2006 a second generation, enzymatically-activated, self-immolative dendrimer with four camptothecin molecules and two PEG chains per dendrimer (Fig. 7) was tested for toxicity against two human leukemia (MOLT-3 and JURKAT) and one human embryonic kidney (HEK 293) cell lines.56 The prodrug alone was between 100- and 1000-fold less toxic than free camptothecin in all cell lines. However, when the cells were treated with penicillin-G-amidase, an enzyme that cleaves the phenylacetamide group of the self-immolative dendrimer causing disassembly of the molecule though cyclization of N,N′-dimethylethylendiamine, the toxicity of the prodrug rivaled that of free camptothecin. These data suggest that if a substrate could be included in the dendrimer structure that would be cleaved by a protease overexpressed by tumor cells, the camptothecin drug could be cancer-cell specific.
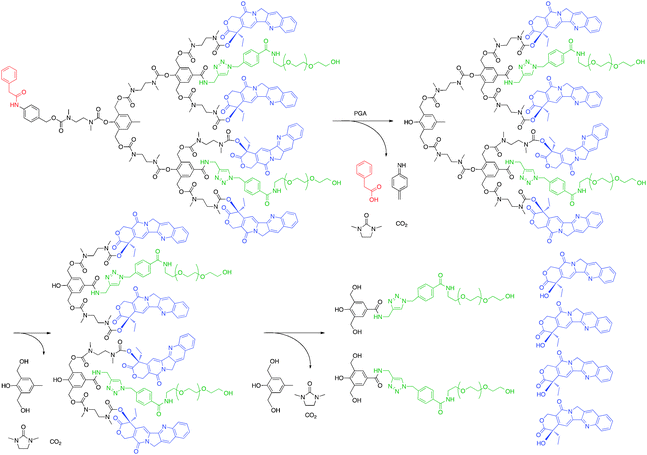 | ||
| Fig. 7 Degradation of self-immolative dendrimer following enzymatic activation to release camptothecin. | ||
5.2 Tissue engineering
5.2.1.1 Cell proliferation. Collagen is widely used as a scaffold for tissue engineering, but crosslinking of the structure is often necessary due to its weak mechanical properties and its rapid biodegradation. The most common crosslinking methods involve either glutaraldehyde (GTA) or 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide (EDC) agents. However, GTA crosslinking is limited by the cytotoxicity induced by unreacted aldehyde groups while EDC crosslinking is limited by reduced biostability and structural integrity as compared to the GTA crosslinked analogues. In 2008 Pandit et al. used EDC and N-hydroxysuccinimide (NHA) to activate the carboxylic acids of a cholecyst-derived extracellular matrix (CEM) containing ∼80% collagen.57 The activated construct was then treated with a first generation PAMAM dendrimer. As compared to untreated or EDC/NHS crosslinked scaffolds, the dendrimer construct displayed higher shrink temperatures, indicating higher crosslinking. Furthermore, the dendrimer crosslinked scaffolds showed swelling behavior that mimicked that for the EDC/NHS crosslinked structures. Using a collagenase assay, it was observed that the dendrimer crosslinked scaffolds exhibited higher resistance to degradation and lower cytotoxicity towards murine 3T3 fibroblast cells without negatively affecting cell proliferation.
More recently Yung et al. used larger, second generation PAMAM dendrimers to crosslink collagen scaffolds and improve their mechanical properties.58 The group crosslinked the scaffolds using either PAMAM and EDC or PAMAM and GTA. Both dendrimer-crosslinked structures exhibited higher thermal stability than the unmodified or EDC-crosslinked scaffolds. Also, the dendrimers displayed reduced degradation patterns following a 1-day incubation in collagenase solution. While all of the constructs displayed similar cytotoxicity behavior, the cell proliferation of the dendrimer constructs varied. The EDC–PAMAM scaffold showed notably higher proliferation of human conjunctival fibroblasts as compared to the EDC crosslinked scaffold. This effect was attributed to the increased density of crosslinking units, resulting in enhanced biostability of the scaffold. Conversely, the cell proliferation of the GTA–PAMAM construct was not improved over the GTA crosslinked analogues and neither construct showed enhanced cellular proliferation as compared to unmodified collagen. The authors attribute this to the cytotoxic effects of unreacted GTA, which nullifies the enhanced biostability gained through the crosslinking.
5.2.1.2 Corneal tissue engineering. Tissue-engineered corneal equivalents (TECE) are synthetic cornea implants composed of extracellular matrices and immortalized corneal cells designed to increase cellular repair of the cornea following tissue damage. Typically, TECE matrices are collagen-based as the cornea is predominantly type-I collagen. In 2005 Sheardown et al. investigated the effect of using an amine-terminated dendrimer to improve the crosslinking of a collagen matrix.59 While incorporating a G1-PPI dendrimer did little to improve matrix stability, G2 and G3 analogues afforded matrices with suitable properties. The G2- and G3-PPI cross-linked matrices showed high denaturation temperatures (80–90 °C) by DSC and reduced collagenase-induced degradation for matrices with a 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 collagen to dendrimer ratio. Additionally, the dendrimer cross-linked matrices showed up to 70% reduction in swelling. Later in 2007, the Sheardown group expanded upon this study by using G2 PPI dendrimers to both crosslink as well as incorporate additional biological functionality into the collagen scaffolds.60 A YIGSR peptide, a sequence derived from laminin that has been shown to promote nerve growth, was covalently attached to G2 PPI prior to crosslinking. DSC measurements of the dendrimer–YIGSR crosslinked matrices showed slightly lower maximum denaturation temperatures, suggestive of slight interference to crosslinking caused by the peptide. This was also supported by a slightly lower modulus. However, the dendrimer–peptide conjugation did not drastically affect the maximum load nor displacement at maximum load. Furthermore, human corneal epithelial cells showed more rapid growth and confluence on YIGSR-modified collagen surfaces, and both the nerve density and length of extended neurites grown from dorsal root ganglia increased 1.5- to 2-fold as compared to those grown on an unmodified dendrimer–collagen matrix.
∶1 collagen to dendrimer ratio. Additionally, the dendrimer cross-linked matrices showed up to 70% reduction in swelling. Later in 2007, the Sheardown group expanded upon this study by using G2 PPI dendrimers to both crosslink as well as incorporate additional biological functionality into the collagen scaffolds.60 A YIGSR peptide, a sequence derived from laminin that has been shown to promote nerve growth, was covalently attached to G2 PPI prior to crosslinking. DSC measurements of the dendrimer–YIGSR crosslinked matrices showed slightly lower maximum denaturation temperatures, suggestive of slight interference to crosslinking caused by the peptide. This was also supported by a slightly lower modulus. However, the dendrimer–peptide conjugation did not drastically affect the maximum load nor displacement at maximum load. Furthermore, human corneal epithelial cells showed more rapid growth and confluence on YIGSR-modified collagen surfaces, and both the nerve density and length of extended neurites grown from dorsal root ganglia increased 1.5- to 2-fold as compared to those grown on an unmodified dendrimer–collagen matrix.
5.2.2.1 Ophthalmic applications. Damage to corneal tissue can arise from a number of factors, including trauma, infections, and surgical procedures. While nylon sutures are currently available to repair such damages, these devices are limited by (1) the infliction of additional tissue damage from suture placement, (2) the potential for infection or inflammation caused by the sutures, (3) uneven healing that results in astigmatisms, (4) the potential for postoperative suture loosening or breakage, (5) the required removal following the healing process, and (6) a variation in the success of operation due to disparity in the surgical skill of the attending physician. Consequently, synthetic polymers that can function as molecular glues and sealants are of clinical relevance. For such sealants to be suitable, they must function within the mechanical and optical constraints of the corneal tissue (i.e. withstand high intraocular pressure, display higher elasticity than the corneal tissue, and have a refractive index that mimics the native tissue).
The Grinstaff group has attained significant success using the photochemical or chemical crosslinking of dendrimers to form ocular sealants. This section will focus only on the use of these dendrimers as a sealant for corneal lacerations and for corneal transplants or penetrating keratoplasty (PKP). In early studies photocrosslinkable first generation dendrimers designed using dendrons based on succinic acid and glycerol linked by various PEG chains lengths and terminated with methacrylate were investigated as sealants for 4.1 mm full-thickness corneal lacerations and PKP autografts to an enucleated porcine eye (Fig. 8).61 Based on evaluations for laceration repair using hydrogels formed using various weight % dendrimer (10, 20 and 40), it was observed that as dendrimer concentration increased, the corresponding hydrogel's viscosity and its ability to withstand high leaking pressures increased. Furthermore, as the PEG chain length increased from 3.4 kDa to 10 kDa, the ability to withstand high leaking pressures increased for the 10 and 20 weight % dendrimers. However, this trend was reversed as the PEG chain length was increased further to 20 kDa. When these same dendrimers were used in addition to either 8 or 16 sutures to seal a PKP, it was observed that the hydrogels increased the maximum leaking pressure that could be endured by the wound. India ink studies, which can be used to observe the flow of ocular fluid across the wound, showed that a hydrogel formed with 20 weight % dendrimer linked with 10 kDa PEG prevented fluid penetration at the wound interface, suggesting it could help prevent post-operative infection.
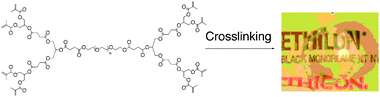 | ||
| Fig. 8 First generation dendrimer with PEG core and methacrylate end groups that can be photocrosslinked to form a clear gel with both ophthalmic and orthopaedic applications. | ||
More recently, this dendrimer was investigated for full thickness corneal laceration repair in vivo.62 The right eyes of 60 white leghorn chickens were given central full-thickness 4.1 mm lacerations. Half of the animals were treated with hydrogels formed using the photocrosslinkable dendrimers and the other half were treated with 3 interrupted 10–0 nylon sutures. Slitlamp examinations were performed to evaluate wound integrity, corneal clarity, Seidel positivity, and inflammation at 6 h and then once daily for 7 days followed by weekly evaluations for 21 days. The corneal repair using the dendrimer-based hydrogel could be accomplished more rapidly than suturing, and the eyes showed less scarring through day 28 following treatment. Futhermore, histologic evaluation at the same time points showed promising results for the dendrimer adhesives. Based on the ease of application and the equally suitable sealing properties of the dendrimer-based hydrogels, these materials may offer a superior alternative to current procedures.
Despite the significant success of these glycerol–succinic acid based dendrimers as ocular adhesives, these structures are still limited by the required argon-laser initiation step for cross-linking. To circumvent this barrier, the Grinstaff group has also investigated “self-gelling” biodendrimers. In 2004 the group synthesized lysine-based peptide dendrimers with either 4 or 8 N-terminal cysteine groups.63 These dendrimers could form hydrogels within minutes following mixing with PEG dialdehyde chains due to the formation of thiazolidine linkages. The complex modulus measured for these hydrogels was shown to increase with increased number of terminal cysteines as a result of a higher crosslinking density. However, the dendrimer with the lower number of cysteine groups was shown to form a suitable and more easily synthesized hydrogel, and thus was used for evaluating the leaking pressures of enucleated eyes. It was observed that the dendrimer-based sealant could withstand higher pressures than one interrupted 10–0 nylon suture. More recently, the group has shown that these dendrimers with either 4 or 8 terminal cysteine groups can increase the leaking pressure for autografts.64 Although the hydrogels alone could not secure the autograft at suitable pressures, it increased the leaking pressure of autografts with 8 sutures from 5 mm Hg to 77 mm Hg. Because intraocular pressure is ∼15 mm Hg, 8 sutures alone would not be suitable to secure an autograph in vivo, and additional sutures would be required. Therefore, the dendrimer-based hydrogels are clinically relevant as they offer the potential to reduce the number of sutures required for corneal autograft repair.
5.2.2.2 Orthopedic applications. In addition to their use as molecular glues, dendrimers have been applied in the treatment of osteoarthritis, a degenerative tissue disease in which proteoglycans and collagen-based proteins in the joints degrade, resulting in eventual exposure of subchondral bone. Tissue engineering treatment strategies for the disease typically involve the introduction of a synthetic scaffold, cells, and growth factors to repair the cartilage loss. The synthetic scaffolds, usually polymer-based, must (1) form an appropriate porous, three dimensional network that is resorbable in vivo, (2) mimic the mechanical properties of the native cartilage, (3) allow for the growth of necessary cells in the surrounding joint area, (4) be biocompatible and avoid eliciting an immune response in vivo, and (5) integrate with the remaining cartilage in the joint area and withstand the physiological loads until the tissue repair is complete. Dendrimers offer an advantage over polymer-based scaffolds because their increased number of endgroups can provide a more densely crosslinked matrix that resists excess swelling.
The Grinstaff group has used its previously described dendrons based on succinic acid and glycerol linked via ester units with a PEG core for cartilage repair.65 The first generation construct was modified to contain terminal methacrylate units for eosin Y-based photocrosslinking. In vitro evaluation of the mechanical properties of the hydrogels made with 7.5, 10, and 15 weight % dendrimer showed only a 10% weight gain due to swelling over 30 days in phosphate buffered saline (PBS), as compared to an unfavourable 100% weight gain observed for PEG dimethacrylate polymer hydrogels. The equilibrium compressive modulus E for these hydrogels increased with dendrimer weight %, from 3 kPa to 600 kPa. The 7.5 and 15 weight % dendrimer solutions were mixed with porcine chondrocytes prior to crosslinking, and the resulting hydrogels were processed for histology at 4 and 12 weeks. The constructs were stained using a marker for proteoglycans (Safranin-O) and a marker for collagen (Masson's Trichrome) as well as immunostained for Types I and II collagen. It was observed that, at the lower dendrimer concentration, the cells encapsulated in the hydrogel produced significant amount of extracellular matrix containing both proteoglycans and collagen, indicative of its potential use for cartilage repair. However, over 4 weeks the scaffold prepared with the low dendrimer concentration had degraded too quickly, limiting its use in vivo.
To overcome the degradation of the original dendrimer, new dendrimers that contained both ester and carbamate linkages were synthesized by incorporating β-alanine units into the structure.66 Based on evaluation of the mechanical properties of these structures, it was observed that the hydrogel formed with the highest dendrimer concentration was the most resilient as indicated by a higher compressive and complex shear modulus and a lower loss angle (supporting a more elastic material). However, this hydrogel also showed up to 25% swelling over 30 days. Therefore, in vivo studies were conducted using hydrogels formed with 10 wt% dendrimer. New Zealand white rabbits (n = 3) with osteochondral defects were treated with an injection of the dendrimer solution followed by in situ photocrosslinking to yield a hydrogel. After 24 weeks the rabbits were sacrificed and their knees were subject to histological evaluation. It was observed that the dendrimer hydrogels were tolerated by the animals and remained intact at the site of application. Furthermore, staining of the knees revealed high concentrations of Type II collagen and GAG, indicative of the appropriate healing response. It is important to note that this response was observed in the absence of cell encapsulation by the hydrogel, indicating that the dendrimer scaffold alone provided the positive outcome.
∶1 ratio and enzymatically crosslinked, the collagen-like matrix formed was evaluated for cytotoxicity and cell adhesion. The collagen mimetic dendrimer matrix showed nearly no cytotoxicity towards L929 mouse fibroblast cells, and Hep3B cell adhesion that rivaled that of natural collagen. Staining assays showed that the cells adhered to the dendrimer scaffold by strong focal adhesion contacts that resembled the cell affinity of calf-skin collagen.
5.3 Transfection
Significant interest in gene therapy stems from its potential for targeting numerous diseases. While free DNA or siRNA can be injected intravenously into a patient, serum nucleases rapidly bind to and degrade polynucleotides. As a result, vectors that can compact and protect the nucleotides are vital. This section will review dendrimers used for transfection, focusing on the structural modifications used to overcome gene delivery barriers.The main barriers for in vitro gene delivery stem from problems related to (1) toxicity, (2) cell targeting and membrane permeation, (3) complex stability under physiological conditions, and (4) release of the gene from the complex following cellular uptake. Cellular uptake of dendriplexes, a term used to describe the complex formed from the electrostatic interactions between cationic dendrimers and anionic DNA/RNA, varies depending on whether the construct is targeted or non-targeted. While targeted constructs are incorporated into the cell based on clatharin-dependent pathways, non-targeted constructs can be taken up by alternative endocytic routes. Both phagocytosis and macropinocytosis have been shown to occur, but neither of these additional routes result in successful gene expression. Regardless of the route of endocytosis, the cellular uptake of dendriplexes is usually affected by the size of the complex, with smaller complexes more readily incorporated than larger analogues. Higher generation number of the dendrimer usually correlates with higher uptake. For PAMAM it has been observed that efficient transfection is generally limited to generations >4.68 However, variations from this trend still exist. For PPI (G1–G5) it was determined that the most stable, positively charged complexes are formed for the highest generation, suggesting that these complexes would result in high transfection efficiency.69 However, when these complexes were tested in vitro, it was observed that the lower generation structures afforded higher gene transfer.70 Therefore, while size of the complex can affect the in vitro transfection efficiency of dendriplexes, there are clearly other influential factors.
Often high generation dendrimers are cytotoxic due to the cationic endgroups. Some surface functionalities have been appended onto these molecules to mask the cationic groups. Across the board, the most commonly used of these surface groups is poly(ethylene glycol). When G5 PAMAM dendrimer with 128 surface amines was conjugated with ∼14 PEG3400 chains (10% conversion), the corresponding dendriplex showed up to 20-fold increase in gene delivery in Chinese hamster ovarian (CHO) cells as compared to SuperFect, possibly due to the notably lower cytotoxicity.71 Similarly, all 32 terminal amines of G4 PPI have been conjugated with tri-glycol gallyl chloride to create a PEG-like exterior. This PEG–G4–PLL construct showed approximately 10% reduction in cytotoxicity and approximately 20% increase in transfection efficiency of a DNAzyme in ovarian carcinoma (A2780) cells.72
Other neutral groups have also been incorporated onto the surface of dendrimers to reduce cytotoxicity. In 2003 an internally quaternized G4 PAMAM dendrimer with a hydroxyl periphery was tested for cytotoxicity and transfection efficiency in 293T cells. While the hydroxyl periphery of the dendrimer resulted in reduced cytotoxicity of the dendriplex, the transfection efficiency was reduced as well due to decreased electrostatic interactions with the cell membrane.73 However, it must not be concluded that dendrimers with hydroxyl peripheries are ineffective transfection vectors. In 2009 a G4 PAMAM-OH dendrimer was conjugated to a synthetic analogue of luteinizing hormone-releasing hormone (LHRH) followed by quaternization of amines to afford a dendrimer that targets cancer cells.74 The targeted dendriplexes showed increased cellular uptake as compared to the non-targeted analogues. Additionally, the degree of quaternization was observed to affect siRNA expression, with the highest gene knockdown seen for dendrimers having 85% quaternization of amines. This effect was attributed to a high number of cationic amines capable of interacting electrostatically with DNA and a sufficient number of unquaternized amines that could function as a “proton sponge” following cellular uptake.
To improve the cell targeting and membrane permeation of dendriplexes, dendrimers are modified to mimic cell-penetrating peptides. The most common method for achieving this goal is modifying the amino groups by appending arginine moieties that mimic the TAT peptide, as has been investigated by Park et al.75 In 2004 arginine was conjugated onto the surface of G4 PAMAM dendrimers through an amide linker, and the corresponding dendriplexes showed up to two orders of magnitude higher DNA transfection efficiency than the unmodified PAMAM analogue in HepG2, Neuro2A, and primary rat aorta smooth muscle (SMC) cells. Later these arginine-conjugated PAMAM dendrimers were improved by replacing the amide linker with an ester group. The one order of magnitude higher transfection efficiency observed for the ester-linked analogues in both SMC and HUVEC cells as compared to the amide-linked derivative was attributed to reduced cytotoxicity. In fact, confocal microscopy images showed that the ester-linked analogues were cleared from the cells following ester hydrolysis because the hydroxyl-terminated PAMAM byproduct did not interact with anionic proteins within the cell. These data indicate that dendrimers with arginine peripheries can be more effective DNA carriers than amine-terminated analogues due to their membrane penetrating capacity, but the higher cationic charge density of the arginine derivatives can cause cytotoxicity. The density of arginine groups on the periphery of dendrimers can affect the intracellular trafficking of the corresponding dendriplexes. When the gene delivery of arginine and di-arginine conjugated G3 PAMAM dendriplexes was compared, the efficiency of the di-arginine derivate was higher, regardless of cell line or the presence of serum. Studies showed that the higher transfection efficiency resulted from differences in intracellular trafficking patterns. Following cellular uptake, the arginine-conjugated derivatives were located primarily in the cytoplasm, while the di-arginine analogues could be found inside the nucleus. The same trafficking pattern was observed for G4 analogues. However, the G4 di-arginine dendriplex showed enhanced transfection efficiency only in the presence of serum. The reduced transfection efficiency of the G4 di-arginine derivative in the absence of serum was attributed to the high cytotoxicity of the dendriplex. Thus, the improvements in cellular trafficking of densely-packed arginine groups on the dendrimer surface must be weighed against the higher cytotoxicity.
Finally, following cellular uptake, the dendrimer must be released from the endosome. Fortunately, two of the most commonly used dendrimers, PAMAM and PPI, have an innate capacity to rupture endosomes by a process referred to as the proton sponge effect.76 At physiological pH (7.4), only the primary amines of these dendrimers are protonated; the internal, tertiary amines remain unprotonated. Following endocytosis the dendriplex is exposed to the acidic endosomal environment (pH ≈ 5), and these titratable tertiary amines can function as a buffer, causing chloride accumulation and eventual endosome lysis. However, the buffering capacity of PAMAM has been further enhanced via degradation of the dendrimer structure. By heating a G6 PAMAM in water/butanol solvent, solvolysis of the amide bonds occurs.77 Despite having a lower charge density of amines, the corresponding dendriplex of the “degraded” dendrimer promotes more efficient gene transfer. This improved transfection was attributed to the increased flexibility of the dendrimer, which allowed for tight complexation with DNA prior to cellular uptake and enhanced swelling of the dendrimer within the endosome as a result of protonation.
When used for in vivo studies, dendriplexes face additional barriers that hinder gene delivery. To be an effective agent, the carrier must (1) readily circulate in the bloodstream without being cleared by the reticuloendothelial system (RES); (2) avoid causing non-specific toxic effect; and (3) extravasate beyond the endothelial lining of the blood vessels into the proper target tissue. The combination of high complex stability in the bloodstream with high availability of the DNA or siRNA following cellular uptake is a difficult task to achieve. As such, only limited successful in vivo gene delivery using dendrimers has been observed, and it is important to point out how these results were achieved.
To deliver the complex to specific tissue, targeting agents have been appended onto dendrimer surfaces. In 2007 Liu et al. was able to achieve targeted hepatic gene delivery by conjugating G3 PPI with 3 galactose residues linked by a branched alkyl spacer unit.78 When this asialoglycoprotein receptor-targeting construct was complexed with DNA and injected into CD-1 mice and organs were harvested after 8 h, luciferase expression occurred preferentially in the liver over the heart, lungs, spleen, and kidneys. This liver-specific gene delivery was confirmed by injecting asialofetuin, a ligand for asialoglycoprotein, prior to injecting the dendrimer–DNA complex. A dose-dependent decrease in luciferase activity was observed, confirming hepatic gene targeting in vivo. However, it must be pointed out that usually targeted delivery agents aim to avoid targeting the liver and other organs that make up the RES.
One recent in vivo study using PPI as a delivery agent has investigated the synergistic effect of both particle stabilizer and targeting agent. Minko et al. complexed siRNA with G5 PPI, and following complexation, cross-linked the individual complexes using a dithiobispropionimidate (DTBP) to create a caging effect with reducible disulfides.79 The caged dendriplex was then PEGylated with NHS–PEG5000–MAL to improve stability and then finally targeted with LHRH peptide via the maleimide functionality. These targeted dendriplexes showed BCL-2 gene knockdown preferentially in LHRH positive (A549 and A2780) cells in vitro. When investigated in vivo, these targeted dendriplexes showed predominate uptake of labeled siRNA and labeled dendrimers in tumor tissue over other organs. However, it must be noted that in vivo gene silencing activity of the delivered siRNA is still pending.
5.4 Magnetic resonance imaging (MRI)
Magnetic resonance imaging has become a widely used technique for disease diagnosis. However, due to the low sensitivity of MRI, contrast agents, typically gadolinium-based, are necessary (Fig. 9). Generally, the longitudinal relaxivity (r1) of the contrast agents is used to describe its efficiency, where a higher r1 value correlates with improved MRI signal (eq. 1). While contrast agents enhance the signal of MRI, they usually suffer from low contrast efficiency, no tissue specificity, and rapid excretion. To avoid needing to administer high doses of the agents to overcome these limitations, research has focused on appending multiple contrast ligands to a single core scaffold (i.e. dendrimers).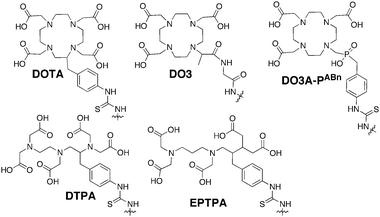 | ||
| Fig. 9 Commonly used gadolinium chelating agents. | ||
Much of the early work using dendrimers as contrast agents focused on PAMAM as investigated by Wiener et al.80 G2 and G6 PAMAM dendrimers were conjugated with diethylene triamine pentaacetic acid (DTPA), a commonly used gadolinium chelating agent, to create structures with 11 and 170 surface DTPA groups and molecular weights of ∼8.5 and 139 kDa, respectively. Based on NMRD studies, the second and sixth generation dendrimer chelates increased r1 to 21 and 34 mM−1 s−1, respectively. These values correspond to 4 and 6-fold higher ion relaxivities as compared to free Gd(III)DTPA (5.4 mM−1 s−1). The increase in r1 was attributed to increased rotational correlation times for the dendrimer constructs. In vivo analysis showed that these dendrimers increased enhancement half-life from 24 min for Gd(III)DTPA to 40 and 200 min for the G2 and G6 analogues, respectively. Bryant et al. probed the relationship between r1 and generation number of PAMAM–DOTA scaffolds and determined that r1 increases up to G7, but then plateaus. This plateau was attributed to slow water exchange.81
Changing the gadolinium chelator appended to the dendrimer can increase the water exchange rate. Ethylenepropylenetriamine pentaacetic acid (EPTPA) has a 10-fold higher exchange rate as compared to Gd(III)DTPA due to steric crowding of gadolinium. When Merbach et al. conjugated G5–G9 PAMAM with EPTPA, it was observed that relaxivities increased from G5 to G7. However, this trend was reversed for the G9 structure.82 It was observed that decreasing the pH of the solution results in higher relaxivity due to protonation of the tertiary amines of PAMAM, which induces scaffold rigidity.
Several studies have further investigated the effect of scaffold rigidity on relaxivity enhancement. Botta and Hermann et al. synthesized PAMAM chelating scaffolds using 1,4,7,10-tetraazacyclo-dodecane-4,7,10-triacetic-(methyl-(4-aminophenylmethyl) phosphinic acid) (DO3A-PABn) and then created adducts between negatively charged PAMAM–DO3A-PABn and polyarginine or polylysine.83 The adduct formation increased relaxivity by reducing the internal motion of the dendrimer. Additionally, it has been realized that the rigidity of the dendrimer–chelate linking group affects relaxivity. Kobayashi et al. synthesized a series of PPI–Gd(III)DTPA constructs with a short, rigid linker group.84 For the G5 analogue, a relaxivity of 29 mM−1 s−1 was observed. Later, Meijer et al. synthesized G5 PPI–Gd(III)DTPA construct linked with a more flexible group.85 It was observed that the relaxivity was reduced (19 mM−1 s−1) due to more Gd3+ movement allowed by the flexible linker.
While the macromolecular size of dendrimer–Gd(III) chelate helps improve enhancement half-life, these constructs are still limited with respect to tissue specificity. Several groups have investigated target-specific MRI contrast agents. For instance, a tumor-targeting dendrimer–chelate agent has been synthesized by appending folate on the surface of a G4 PAMAM dendrimer. When injected into athymic mice with either hFR-positive or negative ovarian tumors, it was observed that the hFR-positive tumors accumulated 3.6% injected dose g−1, while the negative tumors showed no notable accumulation.86 Also, Kobayashi et al. conjugated G4 PAMAM with a DTPA derivative (1B4M) followed by the addition of OST7, a murine monoclonal IgG1 antibody.87 The Gd(III)–OST7–G4–(1B4M)43 constructs accumulated more readily in KT005 tumors (16.1% injected dose g−1) as compared to the untargeted analogue (5.1% injected dose g−1). However, the OST7 antibody–1B4M Gd(III) constructs showed the highest tumor accumulations (27.7% injected dose g−1).
Recently, Tsien et al. developed a dendrimer-based chelating agent that functions as both a contrast agent and a fluorescent probe.88 The synthesis proceeds by conjugating a G5 PAMAM dendrimer to activatable cell penetrating peptides (ACPPs) masked by PEG. The PEG chain can be cleaved from the ACPPs by proteases, and the cell penetrating peptides can then adhere to and be taken up by surrounding cells. Because the proteases MMP-2 and MMP-9 are linked with tumor growth and metastasis, the PAMAM–ACPP conjugates afford tumor-targeting constructs. Following PAMAM–ACPP conjugation, the macromolecule was further labeled with Cy5 fluorescing agent and DOTA by NHS linkers. Finally, the construct was PEGylated and complexed with Gd(III) (Fig. 10). When administered via tail-vein injections into mice with HT-1080 tumors 48 h prior to scanning, it was observed that the ACPP–dendrimer constructs increased r1 and signal intensity by 32% and 21%, respectively. An uncleavable dendrimer control showed much lower relaxivity and signal intensity (8% and 11%, respectively). Furthermore, for HT-1080 tumors injected intramuscularly, it was observed that following ACPP–dendrimer administration, the Cy5 ligand showed brightest fluorescence at the tumor edges. As a result, these constructs have potential for aiding in the complete surgical removal of tumor tissue. This was confirmed by the same group in an in vivo investigation. When the ACPP–dendrimer constructs were injected into mice isografted with a melanoma (B16F10) or breast cancer (8199) cell line, tumor-free survival was increased 1.5- and 5-fold, respectively, after 24 weeks. These results indicate that this construct has potential use for tumor detection using MRI and intraoperative guidance using fluorescence.
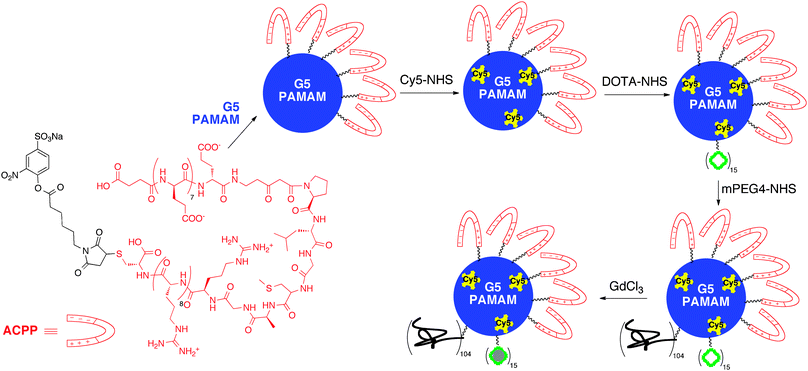 | ||
| Fig. 10 PAMAM dendrimer functionalized with activatable cell penetrating peptides and gadolinium contrast agents for targeted cancer imaging. | ||
It must be mentioned at the end of this section that researchers have recently observed a correlation between gadolinium-based contrast agents and nephrogenic systemic fibrosis (NSF). Briefly, when gadolinium detaches from the chelating agent in circulation, it can cause fibrosing and scar tissue in a variety of organs, resulting in NSF. Although this disease is typically observed in only patients previously suffering from kidney or renal failure, the trend is quite concerning. A number of clinically used gadolinium agents aim to overcome this dilemma by adding excess ligand to the formula.89 However, because the ligand itself can cause adverse affect, this solution is not ideal and is clearly a problem that the dendrimer community must take into consideration when designing a delivery agents. Brechbiel et al. have determined methods to potentially avoid this problem when using dendrimer-based contrast agents.90 First, it has been observed that macrocyclic ligands (e.g. DOTA) are more stable than open chain analogues (e.g. DTPA). Therefore, conjugating the more stable, macrocyclic analogue to the dendrimer may be more ideal for preventing the release of gadolinium in vivo. Additionally the group has introduced a metal preligation technique, whereby the gadolinium is chelated by a ligand prior to attachment to a G4 PAMAM dendrimer. In vivo data showed that the preligation technique resulted in the formation of structures whose rate of clearance was nearly 3-fold faster than a similar analogue in which the gadolinium was chelated in the final step. This increased rate of clearance has the potential to reduce the amount of gadolinium that leaches out from the complex while in circulation and correspondingly reduce the occurrence of NSF.
6. Are dendrimers ready for clinical use?
The current need to develop better diagnostic systems, methods to deliver drugs, and ways to engineer new tissue or promote tissue repair has resulted in dendrimers becoming prominent agents for a number of biomedical applications. Just like any new drug candidates or device formulation, dendrimers must possess certain basic characteristics before they have potential to be used clinically.91 First there is clearly a danger in administering non-biodegradable dendrimers into cells and tissue, as this may result in cellular accumulation leading to lysosomal storage disease in a patient. Therefore, while a number of different dendrimer systems have been described in the previous sections, it is important to note that those dendrimers that are biodegradable, such bis-MPA, PLL, or PGLSA-OH, may offer advantages over the non-biodegradable analogues. In addition, translating toxicity results from in vitro and in vivo studies to clinical applications is often difficult. For instance, in most in vitro studies of chemotherapeutic agents, cytotoxicity is determined using immortalized cancer cell lines. While these cell types are easy to obtain and use, they are limited by behavior that does not exactly mimic the phenotype and cellular mechanisms of normal cells. Primary cells lines can be used for in vitro studies to overcome these limitations, but such cells are more difficult to obtain and may produce less repeatable results. Furthermore, the specific treatment for which the dendrimer will be used is quite important. If the dendrimer will be used in a formulation in which repeated administrations will be required (e.g. chemotherapy), it is possible that the macromolecular drug can induce immune response in a patient, resulting in anaphylactic shock. Furthermore, even if the carrier does not elicit an immune response on its own, it is possible that a drug–dendrimer system can result in a hapten effect. Thus, determining the immunogenicity of a dendrimer complex is vital and may eliminate successful in vitro and in vivo studies from becoming clinically relevant. However, the immune response can be an asset if the dendrimer is being designed for vaccination purposes.Although the number of dendritic polymers that have advanced to the clinic is small, the success of several of these compounds for both treatment and diagnostic purposes suggests that current dendrimer research has the potential to make a significant clinical impact. For instance, PAMAM dendrimers have been used as a component of a clinical diagnostic tool for patients presenting with suspected myocardial ischemia, a heart condition caused by lack of blood flow to the heart.92 The Stratus CS (Dade Behring) detection kit contains monoclonal antibodies immobilized on a glass fiber matrix using fifth generation PAMAM dendrimers as the linking agent.93 The instrument can analyze 6 key analytes, including cardiac troponin I and N-terminal pro-brain natriuretic peptide, two important diagnostic markers for cardiac disorders or damage.
In addition to being used in clinical settings as diagnostic tools, dendrimers have also found use in clinical settings as antiviral agents. VivaGel® (SPL7013, Starpharma), a topical vaginal microbicide with a PLL dendrimer as the active ingredient, is currently undergoing phase II clinical trials. The PLL dendrimer in VivaGel is a fourth-generation analogue with thirty-two naphthalene disulfonate groups conjugated to the surface. The compound inhibits HIV-1 and HSV-2 infections by binding to gp120 glycoprotein receptors on the surface of these viruses, thus preventing the viruses from binding to CD4+ receptors on human T-cells. Data from recently completed Phase I dose-ranging studies of SPL7013 in patients have shown positive safety, tolerability, and pharmacokinetic properties. When applied vaginally to sexually abstinent women, once daily for seven days at concentrations of 0.5–3.0% dendrimer, no evidence of toxicity or absorption was observed. The adverse affects were mild and included abdominal pain or discomfort.94 Additionally, when the 3% SPL7013 gel was applied to the penis of either circumcised or uncircumcised men once daily for seven days, no toxicity or absorption was observed. The adverse affects generally involved itching and redness at the application site.95
Dendrimers have also made it into clinical trials as MRI contrast agents. Gadomer-17 (SH L 643A, Schering) is a polylysine dendrimer with a trimesic acid core and 24 gadolinium chelate groups conjugated to the surface.96 The compound has shown success as a contrast agent for coronary magnetic resonance angiography. Therefore, this dendrimer-based contrast agent offers an alternative to the typical invasive coronary artery angiography used to diagnose coronary artery disease. The compound is currently in phase II clinical trials.
Lastly, based on the wound repair and tissue engineering applications of dendrimers, HyperBranch Medical Technology, Inc. has developed and commercialized several sealants for tissue repair. The first product is a sealant for the closure of corneal wounds—OcuSeal™—which has received a CE mark enabling the product to be sold and used in Europe. This new sealant is superior to the use of sutures to close such wounds. Building upon this success, HyperBranch Medical Technology has developed and commercialized specialized sealants for securing a hernia mesh as well as for closing the dura and preventing CSF leaks. Both of these products are also approved and being sold and used in Europe. In addition, the pilot US clinical trial for the dura sealant has been successfully completed.
Conclusions
Since the discovery of dendrimers approximately 25 years ago, significant progress has been made in the synthesis and use of these macromolecules for biomedical applications. The unique ability to control size, structural properties, and polydispersity has made these compounds ideal candidates for a number of clinically relevant applications. The early use of dendrimers for drug delivery exploited the host–guest properties of these molecules and their ability to encapsulate a variety of hydrophobic drugs. More recent studies have investigated the covalent attachment of drugs to the surface of dendrimers to help control drug release. While clinical trials of dendrimer–drug conjugates as anticancer agents are still pending, these macromolecules have been successfully introduced into the clinical setting as antiviral agents. Similarly, the ability to conjugate gadolinium chelating agents onto the surface of dendrimers has allowed for their use as MRI contrast agents. The success of such research studies is reflected by the clinical use of Gadomer-17, a polylysine-based dendrimer, which is currently in phase II trials.Additionally, because of the multivalent nature of dendrimers these compounds are ideal materials for tissue engineering applications. Finally, the ability to functionalize the surface of dendrimers with exquisit control has allowed for their use as vectors for either DNA or siRNA transfection. The cationic nature of such carriers typically results in cytotoxic effects, but the ability to mask these cations on the surface of the dendrimers by conjugating neutral or hydrophobic groups and then appending a targeting agent onto the structure has made dendrimers valuable non-viral carriers for transfection. However, the use of these compounds in clinical trials is still awaiting.
At this point in the tutorial review, we are taking the liberty to give an opinion. This opinion is unavoidably biased. The dendrimer community is at a unique stage in the research and development of these unique structures where there are still opportunities for young scientists to make significant contributions provided he/she considers several key factors. First, if you are interested in biomedical applications, you must be cognizant of the potential intended clinical indication in the design of your experiments and the composition of your dendrimer. The dendrimer community as a whole has learned a great deal from work on PAMAM and polyamide dendrimers, for example, but these are not our favorite candidates for in vivo use. We favor the polyester dendrimers like bis-MPA and PGLSA-OH, where known degradation routes combined with all the advantages for post-functionalization exist for optimization of the composition. Second, the development of structure–activity relationships is key. We encourage you to synthesize a dendrimer, make discrete structural changes, and assay the resulting effects on a particular physical property or biological response. These studies will then guide you in preparation of a specific dendrimer as well as provide motivation for new synthetic approaches towards a particular structure, a scalable production, or even a new method for preparing dendrimers.
In summary, dendrimers have achieved significant success in a variety of biomedical applications. While the use of dendrimers in the clinic has still not reached the success of linear polymers, this fact is attributed to the relative newness of this field and the difficulties encountered with the synthesis of these materials at the kilogram and larger scales under GMP conditions. It is clear that with improved syntheses, further understandings of their properties, clever solutions to existing problems, and recognition of new applications, dendrimers will become prominent materials in a number of clinical applications. As scientist at the beginning of their career, we challenge you to ask questions, to critically analyze data, to do the hard experiments, to be cognizant of the next step in your research, and, if appropriate, apply your results and knowledge to the many interesting medical applications that exist today.
Conflict of interest. MWG is a co-founder of HyperBranch Medical Technology, Inc.
Notes and references
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J. (Tokyo), 1985, 17, 117–132 Search PubMed.
- G. R. Newkome, Z. Yao, G. R. Baker and V. K. Gupta, J. Org. Chem., 1985, 50, 2003–2004 CrossRef CAS.
- E. Buhleier, W. Wehner and F. Vogtle, Synthesis, 1978, 2, 155–158 CrossRef.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS.
- R. G. Denkewalter, J. Kolc and W. J. Lukasavage, US Pat., 4289872, 1981 Search PubMed.
- M. A. Carnahan and M. W. Grinstaff, Macromolecules, 2006, 39, 609–616 CrossRef CAS.
- H. Ihre, A. Hult and E. Söderlind, J. Am. Chem. Soc., 1996, 118, 6388–6395 CrossRef CAS.
- P. G. de Gennes and H. Hervet, J. Phys., Lett., 1983, 44, 351–360 CrossRef CAS.
- A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665–1688 CrossRef CAS.
- A. D. Meltzer, D. A. Tirrell, A. A. Jones and P. T. Inglefield, Macromolecules, 1992, 25, 4549–4552 CrossRef CAS; M. Chai, Y. Niu, W. J. Youngs and P. L. Rinaldi, J. Am. Chem. Soc., 2001, 123, 4670–4678 CrossRef CAS; K. X. Moreno and E. E. Simanek, Macromolecules, 2008, 41, 4108–4114 CrossRef CAS.
- I. Lee, B. D. Athey, A. W. Wetzel, W. Meixner and J. R. Baker Jr., Macromolecules, 2002, 35, 4510–4520 CrossRef CAS.
- I. B. Rietveld, W. G. Bouwman, M. W. P. L. Baars and R. K. Heenan, Macromolecules, 2001, 34, 8380–8383 CrossRef CAS.
- G. R. Newkome and C. D. Shreiner, Polymer, 2008, 49, 1–173 CrossRef CAS.
- M. J. Joralemon, R. K. O'Reilley, J. B. Matson, A. K. Nugent, C. J. Hawker and K. L. Wooley, Macromolecules, 2005, 38, 5436–5443 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- L. Brauge, G. Magro, A.-M. Caminade and J.-P. Majoral, J. Am. Chem. Soc., 2001, 123, 6698–6699 CrossRef CAS.
- P. Antoni, D. Nyström, C. J. Hawker, A. Hult and M. Malkoch, Chem. Commun., 2007, 2249–2251 RSC.
- C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef CAS.
- P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3932 CrossRef CAS.
- K. L. Wooley, C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 113, 4252–4261 CrossRef CAS.
- H. Ihre, A. Hult, J. M. J. Fréchet and I. Gitsov, Macromolecules, 1998, 31, 4061–4068 CrossRef CAS.
- Y. Ishida, M. Jikei and M. Kakimoto, Macromolecules, 2000, 33, 3202–3211 CrossRef CAS.
- J. Lim, M. A. Mintzer, L. M. Perez and E. E. Simanek, Org. Lett., 2010, 12, 1148–1151 CrossRef CAS.
- D. G. Mullen, M. Fang, A. Desai, J. R. Baker Jr., B. G. Orr and M. M. B. Holl, ACS Nano, 2010, 4, 657–670 CrossRef CAS.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef.
- P. Wu, M. Malkoch, J. N. Hunt, R. Vestberg, E. Kaltgrad, M. G. Finn, V. V. Fokin, K. B. Sharpless and C. J. Hawker, Chem. Commun., 2005, 5775–5777 RSC.
- A. L. Martin, B. Li and E. R. Gillies, J. Am. Chem. Soc., 2009, 131, 734–741 CrossRef CAS.
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- G. Franc and A. K. Kakkar, Chem.–Eur. J., 2009, 15, 5630–5639 CrossRef CAS.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. Imam and V. Percec, Chem. Rev., 2009, 109, 6275–6540 CrossRef CAS.
- O. Rolland, C.-O. Turrin, A.-M. Caminade and J.-P. Majoral, New J. Chem., 2009, 33, 1809–1824 RSC; S. Svenson, Eur. J. Pharm. Biopharm., 2009, 71, 445–462 CrossRef CAS; S. H. Medina and M. E. H. El-Sayed, Chem. Rev., 2009, 109, 3141–3157 CrossRef CAS.
- M. E. Fox, F. C. Szoka and J. M. J. Fréchet, Acc. Chem. Res., 2009, 42, 1141–1151 CrossRef CAS.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS; D. K. Smith, Curr. Top. Med. Chem., 2008, 8, 1187–1203 CrossRef; D. G. Shcharbin, B. Klajnert and M. Bryszewska, Biochemistry, 2009, 74, 1070–1079 CAS.
- N. Joshi and M. Grinstaff, Curr. Top. Med. Chem., 2008, 8, 1225–1236 CrossRef CAS.
- M. Longmire, P. L. Choyke and H. Kobayashi, Curr. Top. Med. Chem., 2008, 8, 1180–1186 CrossRef CAS; A. J. L. Villaraza, A. Bumb and M. W. Brechbiel, Chem. Rev., 2010, 110, 2921–2959 CrossRef CAS.
- P. M. H. Heegaard, U. Boas and N. S. Sorensen, Bioconjugate Chem., 2010, 21, 405–418 CrossRef CAS.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS.
- C. Z. Chen, N. C. Beck-Tan, P. Dhurjati, T. K. van Dyk, R. A. LaRossa and S. L. Cooper, Biomacromolecules, 2000, 1, 473–480 CrossRef CAS.
- M. K. Calabretta, A. Kumar, A. M. McDermott and C. Cai, Biomacromolecules, 2007, 8, 1807–1811 CrossRef CAS.
- S. R. Meyers, F. S. Juhn, A. P. Griset, N. R. Luman and M. W. Grinstaff, J. Am. Chem. Soc., 2008, 130, 14444–14445 CrossRef CAS.
- A. Pérez-Anes, G. Spataro, Y. Coppel, C. Moog, M. Blanzat, C.-O. Turrin, A.-M. Caminade, I. Rico-Lattes and J.-P. Majoral, Org. Biomol. Chem., 2009, 7, 3491–3498 RSC.
- S.-K. Wang, P.-H. Liang, R. D. Astronomo, T.-L. Hsu, S.-L. Hsieh, D. R. Burton and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3690–3695 CrossRef CAS.
- M. T. Morgan, M. A. Carnahan, C. E. Immoos, A. A. Ribeiro, S. Finkelstein, S. J. Lee and M. W. Grinstaff, J. Am. Chem. Soc., 2003, 125, 15485–15489 CrossRef CAS.
- M. T. Morgan, Y. Nakanishi, D. J. Kroll, A. P. Griset, M. A. Carnahan, M. Wathier, N. H. Oberlies, G. Manikumar, M. C. Wani and M. W. Grinstaff, Cancer Res., 2006, 66, 11913–11921 CrossRef CAS.
- M. F. Neerman, H.-T. Chen, A. R. Parrish and E. E. Simanek, Mol. Pharmaceutics, 2004, 1, 390–393 CrossRef CAS.
- C. Kojima, K. Kono, K. Maruyama and T. Takagishi, Bioconjugate Chem., 2000, 11, 910–917 CrossRef CAS.
- R. S. Dhanikula, A. Argaw, J.-F. Bouchard and P. Hildgen, Mol. Pharmaceutics, 2008, 5, 105–116 CrossRef CAS.
- A. K. Patri, J. F. Kukowska-Latallo and J. R. Baker Jr., Adv. Drug Delivery Rev., 2005, 57, 2203–2214 CrossRef CAS.
- M. Liu, K. Kono and J. M. J. Fréchet, J. Controlled Release, 2000, 65, 121–131 CrossRef.
- S. Zhu, M. Hong, L. Zhang, G. Tang, Y. Jiang and Y. Pei, Pharm. Res., 2010, 27, 161–174 CrossRef CAS.
- C. C. Lee, E. R. Gillies, M. E. Fox, S. J. Guillaudeu, J. M. J. Fréchet, E. E. Dy and F. C. Szoka, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16649–16654 CrossRef CAS.
- X. Bi, X. Shi, I. J. Majoros, S. Rameshwer and J. R. Baker Jr., J. Comput. Theor. Nanosci., 2007, 4, 1179–1187 CAS.
- J. Lim and E. E. Simanek, Org. Lett., 2008, 10, 201–204 CrossRef CAS; J. Lim, A. Chouai, S.-T. Lo, W. Liu, X. Sun and E. E. Simanek, Bioconjugate Chem., 2009, 20, 2154–2161 CrossRef CAS; J. Lim, Y. Guo, C. L. Rostollan, J. Stanfield, J.-T. Hsieh, X. Sun and E. E. Simanek, Mol. Pharmaceutics, 2008, 5, 540–547 CrossRef CAS.
- I. J. Majoros, T. P. Thomas, C. B. Mehta and J. R. Baker Jr., J. Med. Chem., 2005, 48, 5892–5899 CrossRef CAS; T. P. Thomas, I. J. Majoros, A. Kotlyar, J. F. Kukowska-Latallo, A. Bielinska, A. Myc and J. R. Baker Jr., J. Med. Chem., 2005, 48, 3729–3735 CrossRef CAS; J. F. Kukowska-Latallo, K. A. Candido, Z. Y. Cao, S. S. Nigavekar, I. J. Majoros, T. P. Thomas, L. P. Balogh, M. K. Khan and J. R. Baker Jr., Cancer Res., 2005, 65, 5317–5324 CrossRef CAS; Y. Zhang, T. P. Thomas, A. Desai, H. Zong, P. R. Leroueil, I. J. Majoros and J. R. Baker Jr., Bioconjugate Chem., 2010, 21, 489–495 CrossRef CAS.
- S. Gurdag, J. Khandare, S. Stapels, L. H. Matherly and R. M. Kannan, Bioconjugate Chem., 2006, 17, 275–283 CrossRef CAS.
- A. Gopin, S. Ebner, B. Attali and D. Shabat, Bioconjugate Chem., 2006, 17, 1432–1440 CrossRef CAS.
- J. C. Y. Chan, K. Burugapalli, H. Naik, J. L. Kelly and A. Pandit, Biomacromolecules, 2008, 9, 528–536 CrossRef CAS.
- S. Zhong and L. Y. L. Yung, J. Biomed. Mater. Res., Part A, 2009, 91, 114–122 CrossRef.
- X. Duan and H. Sheardown, J. Biomed. Mater. Res., Part A, 2005, 75, 510–518 CrossRef CAS.
- X. Duan, C. McLaughlin, M. Griffith and H. Sheardown, Biomaterials, 2007, 28, 78–88 CrossRef CAS.
- M. A. Carnahan, C. Middleton, J. Kim, T. Kim and M. W. Grinstaff, J. Am. Chem. Soc., 2002, 124, 5291–5293 CrossRef CAS; L. Degoricija, C. S. Johnson, M. Wathier, T. Kim and M. W. Grinstaff, Invest. Ophthalmol. Visual Sci., 2007, 48, 2037–2042 CrossRef.
- J. P. Berdahl, S. Johnson, A. D. Proia, M. W. Grinstaff and T. Kim, AMA Arch. Ophthalmol., 2009, 127, 442–447 Search PubMed.
- M. Wathier, P. J. Jung, M. A. Carnahan, T. Kim and M. W. Grinstaff, J. Am. Chem. Soc., 2004, 126, 12744–12745 CrossRef CAS.
- M. Wathier, C. S. Johnson, T. Kim and M. W. Grinstaff, Bioconjugate Chem., 2006, 17, 873–876 CrossRef CAS.
- S. Söntjens, D. L. Nettles, M. A. Carnahan, L. A. Setton and M. W. Grinstaff, Biomacromolecules, 2006, 7, 310–316 CrossRef CAS.
- L. Degoricija, P. N. Bansal, S. H. M. Söntjens, H. M. Serge, N. Joshi, M. Takahashi, B. Snyder and M. W. Grinstaff, Biomacromolecules, 2008, 9, 2863–2872 CrossRef CAS.
- S. T. Khew, Q. J. Yang and Y. W. Tong, Biomaterials, 2008, 29, 3034–3045 CrossRef CAS.
- J. Haensler and F. C. Szoka, Bioconjugate Chem., 1993, 4, 372–379 CrossRef CAS.
- V. A. Kabanov, V. G. Sergeyev, O. A. Pyshkina, A. A. Zinchenko, A. B. Zezin, J. G. H. Joosten, J. Brackman and K. Yoshikawa, Macromolecules, 2000, 33, 9587–9593 CrossRef CAS.
- B. H. Zinselmeyer, S. P. Mackay, A. G. Schatzlein and I. F. Uchegbu, Pharm. Res., 2002, 19, 960–967 CrossRef CAS.
- D. Luo, K. Haverstick, N. Belcheva, E. Han and W. M. Saltzman, Macromolecules, 2002, 35, 3456–3462 CrossRef CAS.
- F. Tack, A. Bakker, S. Maes, N. Dekeyser, M. Bruining, C. Elissen-Roman, M. Janicot, M. Brewster, H. M. Janssen, B. F. M. De Waal, P. M. Fransen, X. Lou and E. W. Meijer, J. Drug Targeting, 2006, 14, 69–86 Search PubMed.
- J. H. Lee, Y.-B. Lim, J. S. Choi, Y. Lee, T.-I. Kim, H. J. Kim, J. K. Yoon, K. Kim and J.-S. Park, Bioconjugate Chem., 2003, 14, 1214–1221 CrossRef CAS.
- M. L. Patil, M. Zhang, O. Taratula, O. B. Garbuzenko, H. He and T. Minko, Biomacromolecules, 2009, 10, 258–266 CrossRef CAS.
- J. S. Choi, K. Nam, J.-Y. Park, J.-B. Kim, J.-K. Lee and J.-S. Park, J. Controlled Release, 2004, 99, 445–456 CrossRef CAS; H. Y. Nam, K. Nam, H. J. Hahn, B. H. Kim, H. J. Lim, H. J. Kim, J. S. Choi and J.-S. Park, Biomaterials, 2009, 30, 665–673 CrossRef CAS; T.-I. Kim, C. Z. Bai, K. Nam and J.-S. Park, J. Controlled Release, 2009, 136, 132–139 CrossRef CAS.
- N. D. Sonawane, F. C. Szoka Jr. and A. S. Verkman, J. Biol. Chem., 2003, 278, 44826–44831 CrossRef CAS.
- M. X. Tang, C. T. Redemann and F. C. Szoka Jr., Bioconjugate Chem., 1996, 7, 703–714 CrossRef CAS.
- K. S. Kim, Y. Lei, D. B. Stolz and D. Liu, Gene Ther., 2007, 14, 704–708 CrossRef CAS.
- O. Taratula, O. B. Garbuzenko, P. Kirkpatrick, I. Pandya, R. Savla, V. P. Pozharov, H. He and T. Minko, J. Controlled Release, 2009, 140, 284–293 CrossRef CAS.
- E. Wiener, M. W. Brechbiel, H. Brothers, R. L. Magin, O. A. Gansow, D. A. Tomalia and P. C. Lauterbur, Magn. Reson. Med., 1994, 31, 1–8 CrossRef CAS; E. C. Wiener, F. P. Auteri, J. W. Chen, M. W. Brechbiel, O. A. Gansow, D. S. Schneider, R. L. Belford, R. B. Clarkson and P. C. Lauterbur, J. Am. Chem. Soc., 1996, 118, 7774–7782 CrossRef CAS.
- L. H. Bryant Jr., M. W. Brechbiel, C. Wu, J. W. Bulte, V. Herynek and J. A. Frank, J. Magn. Reson. Imaging, 1999, 9, 348–352 CrossRef CAS.
- S. Laus, A. Sour, R. Ruloff, E. Toth and A. E. Merbach, Chem.–Eur. J., 2005, 11, 3064–3076 CrossRef CAS.
- J. Rudovsky, M. Botta, P. Hermann, K. I. Hardcastle, I. Lukes and S. Aime, Bioconjugate Chem., 2006, 17, 975–987 CrossRef CAS.
- H. Kobayashi, S. Kawamoto, S.-K. Jo, H. L. Bryant Jr., M. W. Brechbiel and R. A. Star, Bioconjugate Chem., 2003, 14, 388–394 CrossRef CAS.
- S. Langereis, Q. G. de Lussanet, M. H. P. van Genderen, W. H. Backes and E. W. Meijer, Macromolecules, 2004, 37, 3084–3091 CrossRef CAS.
- S. D. Konda, S. Wang, M. Brechbiel and E. C. Wiener, Invest. Radiol., 2002, 37, 199–204 CrossRef CAS.
- H. Kobayashi, N. Sato, T. Saga, Y. Nakamoto, T. Ishimori, S. Toyama, K. Togashi, J. Konishi and M. W. Brechbiel, Eur. J. Nucl. Med., 2000, 27, 1334–1339 CrossRef CAS.
- E. S. Olson, T. Jiang, T. A. Aguilera, Q. T. Nguyen, L. G. Ellies, M. Scadeng and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4311–4316 CrossRef CAS; Q. T. Nguyen, E. S. Olson, T. A. Aguilera, T. Jiang, M. Scadeng, L. G. Ellies and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4317–4322 CrossRef CAS.
- M. A. Sieber, P. Lengsfeld, J. Walter, H. Schirmer, T. Frenzel, F. Siegmund, H.-J. Weinmann and H. Pietsch, J. Magn. Reson. Imaging, 2008, 27, 955–962 CrossRef.
- K. Nwe, H. Bryant Jr. and M. W. Brechbiel, Bioconjugate Chem., 2010, 21, 1014–1017 CrossRef CAS.
- R. Duncan and L. Izzo, Adv. Drug Delivery Rev., 2005, 57, 2215–2237 CrossRef CAS.
- S. Altinier, M. Mion, A. Cappelletti, M. Zaninotto and M. Plebani, Clin. Chem. (Washington, DC), 2000, 46, 991–993 CAS.
- P. Singh, F. Moll III, S. H. Lin, C. Ferzli, K. S. Yu, R. K. Koski, R. G. Saul and P. Cronin, Clin. Chem. (Washington, DC), 1994, 40, 1845–1849 CAS.
- J. O. O'Loughlin, I. Y. Millwood, H. M. McDonald, C. F. Price, J. M. Kaldor and J. R. A. Paull, Sex. Transm. Dis., 2010, 37, 100–104 Search PubMed.
- M. Y. Chen, I. Y. Millwood, H. Wand, M. Poynten, M. Law, J. M. Kaldor, S. Wesselingh, C. F. Price, L. J. Clark, J. R. A. Paull and C. K. Fairley, J. Acquired Immune Defic. Syndr., 2009, 50, 375–380 CrossRef CAS.
- C. U. Herborn, J. Barkhausen, I. Paetsch, P. Hunold, M. Mahler, K. Shamsi and E. Nagel, Radiology, 2003, 229, 217–223 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |