Asymmetric Cu(II) catalyses for cycloaddition reactions based on π–cation or n–cation interactions
Akira
Sakakura
a and
Kazuaki
Ishihara
*b
aEcoTopia Science Institute, Nagoya University, Japan
bGraduate School of Engineering and JST, CREST, Nagoya University, Furo-cho, Chikusa, Nagoya, 464-8603, Japan. E-mail: ishihara@cc.nagoya-u.ac.jp; Fax: +81 52 789 3222; Tel: +81 52 789 3331
First published on 6th September 2010
Abstract
The rational design of small but highly functional artificial catalysts is very important for practical organic synthesis. Asymmetric Lewis acid catalyses with non-covalent secondary interactions have been developed for enantioselective reactions. This tutorial review describes the concept, design and examples of asymmetric Cu(II) catalyses for cycloaddition reactions based on intramolecular π–cation or n–cation interactions between the copper(II) cation and auxiliary Lewis basic sites of the chiral ligands.
 Akira Sakakura | Akira Sakakura received his Bachelor's degree and PhD from Nagoya University under Professor Kiyoyuki Yamada and Professor Yoshihiro Hayakawa, respectively. In 2001, he became Assistant Professor at University of Tsukuba. In 2003, he became Associate Professor at Graduate School of Engineering, Nagoya University. From 2007, he has been Associate Professor at EcoTopia Science Institute, Nagoya University. He has received Banyu Pharmaceutical Award in Synthetic Organic Chemistry, Japan (2005), Lectureship Award of Young Generation Special Forum from the Chemical Society of Japan (2006), Thieme Chemistry Journal Award (2006), and Incentive Award in Synthetic Organic Chemistry, Japan (2007), Banyu Chemist Award (BCA) (2009). |
 Kazuaki Ishihara | Kazuaki Ishihara received his PhD from Nagoya University in 1991 under the direction of Professor Hisashi Yamamoto. He had the opportunity to work under the direction of Professor Clayton H. Heathcock at the University of California, Berkeley, as a visiting graduate student in 1988. After completing his postdoctoral studies with Professor E. J. Corey at Harvard University, he joined Professor H. Yamamoto's group at Nagoya University as an assistant professor in 1992, and became associate professor in 1997. In 2002, he was appointed to his current position as a full professor at Nagoya University. He has received the Chemical Society of Japan Award for Young Chemists (1996), the Green & Sustainable Chemistry Award (2003), the JSPS Prize (2005), the Japan IBM Science Prize (2007), and the Mukaiyama Award (2009). |
1. Introduction
Enzyme-induced reactions are environmentally benign, but the range of substrates is generally narrow. To act as highly functional catalystsin vivo, enzymes have become very large and complicated. However, the active sites of enzymes are generally small, and some large parts of enzymes are not required for catalytic functions in vitro. In this context, the design of small but highly functional artificial catalysts is very important for practical organic synthesis. A great deal of research has been focused on designing minimal artificial enzymes from natural L-amino acids which enantioselectively catalyze synthetically useful organic reactions.1–3For the construction of an efficient active site in enzymes, weak non-covalent interaction between a metal cation and the functional groups of organic molecules plays an important role. For example, a natural Diels–Alderase, fungal macrophomate synthase (MPS) is a Mg2+-dependent enzyme with 339 amino acid residues (relative molecular mass Mr = 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 244).4–6 In MPS, both the coordination of two carboxyl oxygens (Glu185 and Asp211) and two water molecules with Mg2+ and hydrogen bonding between the water molecules and some amino acid residues create an efficient active site. π–Cation interactions between metal cations and the aromatic arms of natural amino acids, such as L-phenylalanine, L-tyrosine and L-tryptophan, are also known to play important roles in biological systems.7,8
244).4–6 In MPS, both the coordination of two carboxyl oxygens (Glu185 and Asp211) and two water molecules with Mg2+ and hydrogen bonding between the water molecules and some amino acid residues create an efficient active site. π–Cation interactions between metal cations and the aromatic arms of natural amino acids, such as L-phenylalanine, L-tyrosine and L-tryptophan, are also known to play important roles in biological systems.7,8
Based on these biological systems, the use of weak non-covalent interactions should be key for the rational design of small but highly functional artificial catalysts. The weak secondary coordination of rationally designed auxiliary Lewis basic sites of the chiral ligand to the metal center can create an efficient asymmetric environment around the metal center through high-level control of the conformation of the catalyst. In addition, the secondary interaction can increase the Lewis acidity of the catalyst by displacing counteranions to generate cationic active species.9 This tutorial review summarizes our recent contributions directed toward the rational design of chiral copper(II) catalysts10,11 based on π–cation12–14 and n–cation15 interactions for the enantioselective Diels–Alder reaction16–20 and [2+2] cycloaddition reaction.21
2. Asymmetric Cu(II) catalyses based on π–cation interactions
2.1 Asymmetric Diels–Alder reactions using chiral ligands bearing electron-rich aromatic groups
Previously, some chiral α-amino acid and α-hydroxy acid derivatives bearing electron-rich aromatic groups have been developed as chiral ligands for asymmetric Diels–Alder catalyses. In these asymmetric catalysts, the electron-rich aromatic groups play an important role in the construction of an efficient asymmetric environment around the metal center. For example, in 1988, Yamamoto's group reported chiral acyloxyborane (CAB) catalyst 1 derived from 2,6-di(isopropoxy)benzoyl L-tartaric acid and borane·THF for the enantioselective Diels–Alder reaction with acrylic acids, α,β-alkenals and α,β-alkynals (Scheme 1).22–25 Effective shielding of the si face of the 1-coordinated α,β-alkenal arises from π–π stacking between the 2,6-diisopropoxybenzene ring and the coordinated aldehyde.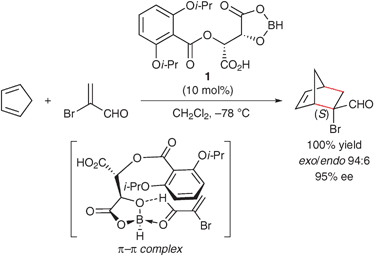 | ||
| Scheme 1 Enantioselective Diels–Alder reaction catalyzed by CAB catalyst 1. | ||
In 1991, Corey's group reported chiral oxazaborolidine catalyst 2 derived from N-(p-toluenesulfonyl)-L-tryptophan and borane·THF or butylboronic acid for the Diels–Alder reaction with α-bromoacrolein (Scheme 2).26–28 The high enantioselectivity is induced by attractive interaction between the π-donor indole ring and the coordinated aldehyde. Based on the bright orange–red color of the complex of 2 with methacrolein, this attractive interaction may be due to charge-transfer complexation, which is stronger than π–π attractive interaction.
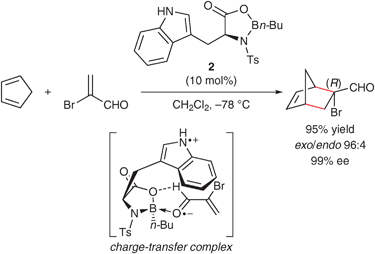 | ||
| Scheme 2 Enantioselective Diels–Alder reaction catalyzed by chiral oxazaborolidine catalyst 2. | ||
In 1998, Engberts and coworkers reported the asymmetric Diels–Alder reaction of cyclopentadiene with 3-phenyl-1-(2-pyridinyl)-2-propen-1-one in water catalyzed by copper(II) complexes of N-methyl-L-amino acids. The use of amino acids featuring an aromatic group allows the isolation of the corresponding endo-adducts with good enantioselectivities. In particular, copper(II) complexes of L-abrine (N-methyl-L-tryptophan) or N-methyl-L-tyrosine show high enantioselectivities (Scheme 3).29,30 In this reaction, water enhances the enantioselectivity up to 74% ee. On the other hand, the enantioselectivity is reduced to 17–44% ee in organic solvents, such as acetonitrile, THF, ethanol and chloroform. Engberts and coworkers proposed that the enantioselectivity could be attributed to the folding back of the aromatic side-chain of the amino acids and π–π attractive interactions between the aromatic group and the dienophile.
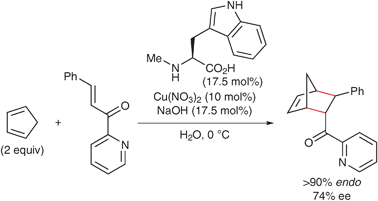 | ||
| Scheme 3 Enantioselective Diels–Alder reaction in water catalyzed by the copper(II)·L-abrine complex. | ||
The crystal structure of the bis(L-tyrosinato)copper(II) complex was determined by van der Helm's group (Fig. 1).31 They observed intramolecular weak π–cation attractive interaction between the copper(II) cation and one of the phenolic rings of tyrosinates. The intramolecular π–cation interaction of the L-(tryptophyl-glycinato)copper(II) complex has also been reported.32,33 Based on these studies, it is conceivable that the origin of the asymmetric induction in the Engberts' Diels–Alder reaction is the intramolecular π–cation interaction between the copper(II) cation and the indole ring.
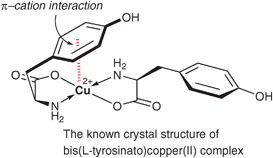 | ||
| Fig. 1 Crystal structure of the bis(L-tyrosinato)copper(II) complex. | ||
2.2 Concept for the design of π–cation catalyses
L-Amino acids, which are natural chiral sources, are useful as readily available chiral ligands. In contrast to C2-symmetric ligands, which basically require more than one chiral center, we designed a chiral amino acid-derived ligand which has a single chiral center, with “chiral economy” in mind. In practice, the side arm of an amino acid derivative would be flexible and it might be difficult to create an effective asymmetric environment for enantioselective reactions. In the above-mentioned chiral borane catalysts, the side arms are controlled by the π–π attractive interaction between the aromatic ring of the side arm and the electron-deficient π system of the substrates.Based on the pioneering works by Engberts' group,29,30 we envisioned that the π–cation interaction between a metal cation and the aromatic ring of the side arm could control the side arm for the construction of an effective asymmetric environment around the metal cation. Engberts' monocationic catalyst [3·CuII]X is prepared from CuX2 and the sodium salt of L-amino acid H-3. In contrast, we envisioned that the dicationic catalyst [4·CuII]X2, prepared from CuX2 and L-amino esters or L-amino amides 4, should be more enhanced than Engberts' monocationic catalyst with regard to catalytic activity and π–cation interaction, due to the greater cationic nature of the copper(II) center in the chelate-type catalyst (Scheme 4).
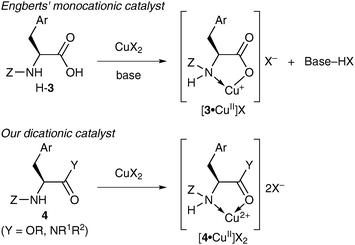 | ||
| Scheme 4 Rational design of copper(II)·L-amino acid derivative catalysts for the asymmetric Diels–Alder reaction. | ||
2.3 Enantioselective Diels–Alder reaction with 5
The catalytic activities of copper(II)·L-DOPA derivative complexes [3·CuII]OTf, [H-3·CuII](OTf)2 and [4·CuII](OTf)2 (Fig. 2) were examined for the enantioselective Diels–Alder reaction of cyclopentadiene with α,β-unsaturated 1-acyl-3,5-dimethylpyrazoles 5 in acetonitrile. Monocationic catalyst [3·CuII]OTf was prepared from H-3 (15 mol%) and Cu(OTf)2 (10 mol%) in the presence of Et3N (15 mol%), while dicationic catalysts [H-3·CuII](OTf)2 and [4·CuII](OTf)2 were prepared from H-3 or 4 (11 mol%) and Cu(OTf)2 (10 mol%) in the absence of Et3N. As expected, [H-3a·CuII](OTf)2 was more active than [3a·CuII]OTf and gave the endo-(2S)-6a with 87% ee (Scheme 5). Thus, Y of 4 was further screened to attain higher enantioselectivity. Isopropyl ester 4a was less effective than the corresponding acid H-3a with regard to enantioselectivity and catalytic activity. On the other hand, pyrrolidine amide 4b was extremely effective and gave endo-(2S)-6a with 97% ee.![Copper(ii)·l-DOPA derivative complexes [3·CuII]OTf, [H-3·CuII](OTf)2 and [4·CuII](OTf)2.](/image/article/2011/CS/b924478f/b924478f-f2.gif) | ||
| Fig. 2 Copper(II)·L-DOPA derivative complexes [3·CuII]OTf, [H-3·CuII](OTf)2 and [4·CuII](OTf)2. | ||
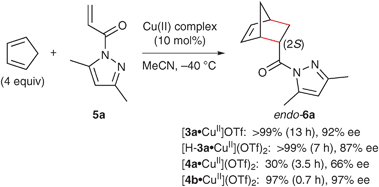 | ||
| Scheme 5 Catalytic activities of copper(II)·L-DOPA derivative complexes for the Diels–Alder reaction of cyclopentadiene with 5a. | ||
The generality and scope of the enantioselective Diels–Alder reaction with 5 catalyzed by [4b·CuII](OTf)2 or more active [4b·CuII](NTf2)2 were examined in acetonitrile (Scheme 6). The reaction with not only simple dienophiles 5a–c (R1 = H, Me, Ph) but also β-functionalized dienophiles 5d–f (R1 = CO2Et, OCOPh, Cl) gave the corresponding Diels–Alder adducts 6–10 with high enantioselectivities. Highly electron-deficient 5d (R1 = CO2Et) reacted with not only cyclic dienes but also acyclic dienes such as 2,3-dimethylbutadiene, isoprene, 2-phenylbutadiene and 2-methoxybutadiene with high enantioselectivities.
![Enantioselective Diels–Alder reaction catalyzed by [4b·CuII]X2.](/image/article/2011/CS/b924478f/b924478f-s6.gif) | ||
| Scheme 6 Enantioselective Diels–Alder reaction catalyzed by [4b·CuII]X2. | ||
The pyrazolyl amide moiety in Diels–Alder adducts 6–10 may be transformed into a range of carboxylic acid derivatives by treatment with appropriate nucleophiles: hydrolysis,34alcoholysis,35aminolysis,36,37 reductive cleavage to aldehydes,38 and alkylative cleavage to ketones39 or β-ketoesters.40
2.4 Mechanistic analysis
The crystal structure of bis(L-tyrosinato)copper(II) complex has been determined by van der Helm and coworkers (Fig. 1).31 On the basis of the π–cation interaction between the copper(II) cation and one of the phenolic rings of tyrosinates, the absolute stereochemical outcome in the [4b·CuII](OTf)2-catalyzed Diels–Alder reaction can be understood through the proposed transition-state assembly, trans-s-cis-TS 11, shown in Fig. 3. In addition, the N-cyclopentyl and pyrrolidinyl groups in 4b would sterically assist the π–cation interaction. The 3- and 5-methyl groups of 5 would sterically control the coordination environment around the copper(II) [trans (favored) or cis (disfavored)] and the conformation of 5 [s-cis (favored) or s-trans (disfavored)], respectively.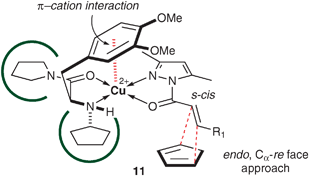 | ||
| Fig. 3 Proposed transition-state assembly 11 based on the π–cation interaction. | ||
To experimentally ascertain the significance of the aromatic moiety in 4b for asymmetric induction, the enantioselective Diels–Alder reaction of 2,3-dimethylbutadiene with 5a was performed using a pyrrolidine amide of N-cyclopentyl-3-cyclohexyl-L-alanine in place of 4b (Scheme 7). As expected, the enantioselectivity was significantly diminished (from 91 to 19% ee). These results also suggest that the aromatic π–copper(II) cation interaction plays an important role. Furthermore, the catalytic activity of [4b·CuII](OTf)2 was slightly greater than that of the control catalyst. The cationic character of copper(II) might be increased by the release of counteranions (−OTf) from the copper(II) center. When these experimental results are taken into consideration, the π–cation interaction may contribute to the stabilization of a transition-state assembly that includes a diene.
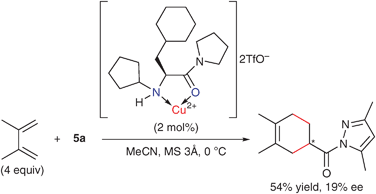 | ||
| Scheme 7 Enantioselective Diels–Alder reaction catalyzed by the copper(II)·L-aliphatic amide complex. | ||
Theoretical calculations for a 1∶1∶1 chelate complex of copper(II) cation, N,O-dimethyl-L-tyrosine N,N-dimethylamide, and N-formylpyrazole suggest that the folded geometry is more stable than the extended geometry (Fig. 4).13† The difference in free energy between the two geometries is 6.9 kJ mol−1. It is reasonable to assume that the stability of the folded geometry is due to intramolecular π–cation interaction, since the distance between CuII and C1 of the folded geometry and its Mulliken overlap population value are 2.860 Å and 0.114, respectively. These results strongly suggest that the enantioselective Diels–Alder reaction may proceed via a transition-state assembly that includes a diene, analogous to the folded geometry, as shown by theoretical calculations.
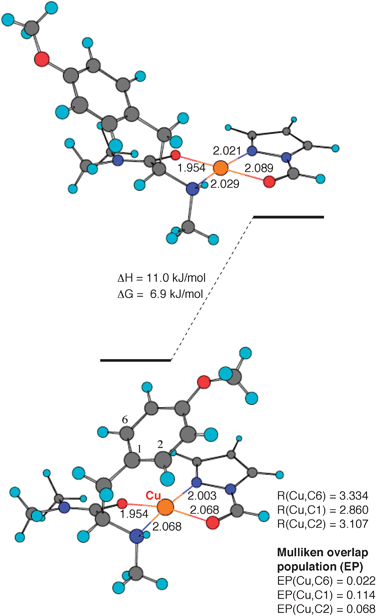 | ||
| Fig. 4 Theoretical calculations for a 1∶1∶1 chelate complex of copper(II) cation, N,O-dimethyl-L-tyrosine N,N-dimethylamide, and N-formylpyrazole. Distances are in angstroms. | ||
2.5 Enantioselective [2+4] and [2+2] cycloaddition reactions with propiolamides 12
A highly asymmetric induction of the π–cation catalysts [4·CuII](NTf2)2 was also observed in the enantioselective Diels–Alder reaction with acetylenic dienophiles. In general, acetylene derivatives are less reactive than the corresponding ethylene derivatives as electron-deficient dienophiles. Acetylene derivatives with a linear sp–sp bond make the asymmetric induction with chiral Lewis acid difficult because the alkynyl moiety is far from the acidic metal center. Therefore, only a few successful methods have been reported for the enantioselective Diels–Alder reaction of acetylenic dienophiles.41–44Investigation of the activities of π–cation catalysts revealed that copper(II) complex [4c·CuII](NTf2)2 bearing a 2-naphthyl group (Fig. 5) showed a higher enantioselectivity than [4b·CuII](NTf2)2 for the Diels–Alder reaction of cyclopentadiene with propiolamide 12a to give the corresponding (1R,4S)-adduct 13a with 88% ee (Scheme 8). The expanded asymmetric environment constructed by π–cation interaction between copper(II) cation and the 2-naphthyl group might be effective for high-level asymmetric induction. The addition of MS 4 Å activated [4b·CuII](NTf2)2 and prevented the hydrolysis of 12a. Under the influence of 10–20 mol% of [4b·CuII](NTf2)2, propiolamides 12 smoothly reacted with various cyclic dienes. In particular, more electron-deficient 3-iodopropiolamide 12c gave the corresponding 3-iodo adducts in high yields (82–89%) with high enantioselectivities (89–97% ee). The 3-iodo adducts can be transformed into 3-nonsubstituted derivatives and 3-alkyl derivatives by known methods.41 Several 2- and 1-substituted cyclohexadienes reacted with 12 and regioselectively gave the corresponding 5- and 1-substituted bicyclo[2.2.2]octadienyl-2-carboxamides 13–16 without the production of any 6- and 4-substituted regioisomers, respectively.
2.](/image/article/2011/CS/b924478f/b924478f-f5.gif) | ||
| Fig. 5 Copper(II)·3-(2-naphthyl)-L-alanine complex [4c·CuII](NTf2)2. | ||
2.](/image/article/2011/CS/b924478f/b924478f-s8.gif) | ||
| Scheme 8 Enantioselective Diels–Alder reaction of cyclic dienes with 12 using π–cation catalyst [4c·CuII](NTf2)2. | ||
Based on the experimental results for the Diels–Alder reaction of dienes with 5 catalyzed by [4b·CuII](OTf)2, it is conceivable that the carbonylre face of 12a would be preferentially shielded by the 2-naphthyl group of 4c, which would be conformationally folded through π–cation interaction with CuII. Cyclopentadiene would predominantly approach the si-face side of 12a to give (1R,4S)-13a through an exo-transition-state assembly. The frontier molecular orbital theory also explains the predominance of the exo-transition-state in terms of secondary antibonding interaction41,45 between the lobes at the C-2 position of cyclopentadiene and the carbonyl oxygen of 12a in the endo-transition state.
Recently, impressive progress has been made in the development of chiral diene ligands for the asymmetric catalysis of Rh and Ir.46 The Diels–Alder reaction of cyclic dienes with acetylenic dienophiles may provide a concise method for the preparation of chiral bicyclic 2,5-diene ligand candidates.
The π–cation catalyst [4c·CuII](NTf2)2 was also efficient for the [2+2] cycloaddition reaction21 of enol ethers with propiolamide 12a (Scheme 9). In the presence of [4c·CuII](NTf2)2 (10 mol%), the reaction of 2-methoxy-5,5-dimethylcyclohexadiene with 12a gave the [2+2] cycloadduct 17 in 65% yield with 80% ee. Interestingly, no corresponding Diels–Alder adducts were obtained, probably due to the steric hindrance of the 5,5-dimethyl substituents of the diene. 1-Trialkylsiloxycyclopentenes and its 2-methyl analogue also reacted with 12a to give cyclobutenecarboxamides 18–20 with 81–83% ee.
![Enantioselective [2+2] cycloaddition reaction with 12a using π–cation catalyst [4c·CuII](NTf2)2.](/image/article/2011/CS/b924478f/b924478f-s9.gif) | ||
| Scheme 9 Enantioselective [2+2] cycloaddition reaction with 12a using π–cation catalyst [4c·CuII](NTf2)2. | ||
The bicyclic [2+2] adducts are useful chiral intermediates for further synthetic elaboration. For instance, the adduct 20 was efficiently transformed into the known (S)-bicyclo[4.3.0]nonenone 2147 (Scheme 10).48 Thus, the absolute configuration of 20 was established to be (1S,5R).
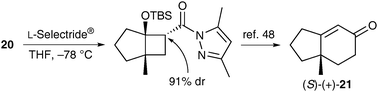 | ||
| Scheme 10 Conversion of (1S,5R)-20 to (S)-(+)-21. | ||
3. Asymmetric Cu(II) catalyses based on n–cation interactions
3.1 Asymmetric reactions using chiral bis(oxazoline) ligands bearing auxiliary Lewis basic sites
Bis(oxazoline)s are among the most important classes of chiral ligands that are widely used for asymmetric catalyses.49–53 For example, Evans and coworkers reported that copper(II) complexes of chiral bis(oxazoline) ligand 22a (R = t-Bu, Fig. 6) are efficient catalysts for the enantioselective Diels–Alder reaction of dienes with α,β-unsaturated N-acyloxazolidinones 24.54,55 Steric hindrance of bulky tert-butyl groups at the 4,4′-positions successfully controls the enantiofaceselectivity of 24 in a distorted square-planar catalyst–substrate complex.![Copper(ii)·bis(oxazoline) complexes [22·CuII]X2 and [23·CuII]X2.](/image/article/2011/CS/b924478f/b924478f-f6.gif) | ||
| Fig. 6 Copper(II)·bis(oxazoline) complexes [22·CuII]X2 and [23·CuII]X2. | ||
Recently, some chiral bis(oxazoline) ligands bearing auxiliary Lewis basic sites through a methylene linker have been developed for the construction of an efficient asymmetric environment around a metal center. For example, in 2004, Zhou and Tang reported the asymmetric Diels–Alder reaction of cyclopentadiene using a copper(II) complex of tridentate tris(oxazoline) 25 (Scheme 11).56 The additional oxazoline moiety interacts with the CuII center (n–cation interaction), which makes the complex air- and water-stable. Furthermore, the enantioselectivity of the reaction with α-ketoesters using the tris(oxazoline) ligand is higher than that in the reaction using the 4,4′-t-Bu-bis(oxazoline) ligand 22a. Another example of an asymmetric reaction using a bis(oxazoline) ligand bearing auxiliary Lewis basic sites has been reported by Reiser's group (Scheme 12).57 They developed bis(oxazoline) 26 bearing 4,4′-hydroxymethyl groups, which is an efficient ligand for the asymmetric 1,4-addition of diethylzinc to enones. It is proposed that the bis(oxazoline) moiety coordinates to CuII and the 4,4′-hydroxymethyl group coordinates to ZnII to form a bimetallic complex as an active species. In the transition state, an enone is locked in a two-point binding mode via the CuII and ZnII atoms.
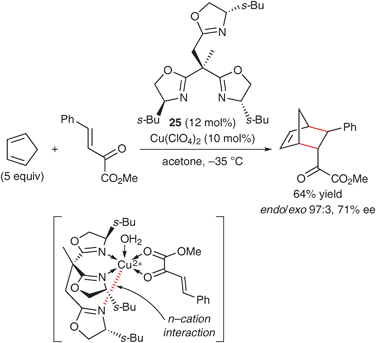 | ||
| Scheme 11 Asymmetric Diels–Alder reaction with α-ketoesters using a copper(II)·tris(oxazoline) complex. | ||
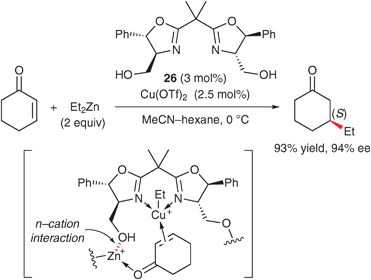 | ||
| Scheme 12 Asymmetric 1,4-addition of Et2Zn to enones using a copper(II)·bis(oxazoline) complex. | ||
3.2 Concept for the design of n–cation catalyses
In the above-mentioned catalyses, the auxiliary Lewis basic sites create an efficient asymmetric environment via interaction with metal cations. In addition, rationally designed chiral ligands bearing auxiliary Lewis basic sites are also expected to increase the Lewis acidity of the catalysts by displacing counteranions.9 Our working hypothesis for the design of n–cation catalysts is shown in Scheme 13. Lewis basic sites (:Y) in the 4,4′-substituents of a bis(oxazoline) ligand 23 would coordinate to the CuII center (Y: → CuII), which generates an equilibrium between complexes A and B. Complex B should be more acidic than complex A due to the cationic character of CuII. However, rather basic counteranions Tf2N− and TfO− may coordinate to the CuII center to predominantly form less-basic complex A. In contrast, when the ligand has protic Lewis basic sites (:Y = :ZH) in the 4,4′-substituents, released counteranions may successfully interact with :ZH through hydrogen bonding (X−⋯H–Z), to form cationic catalyst C which is more acidic and stable.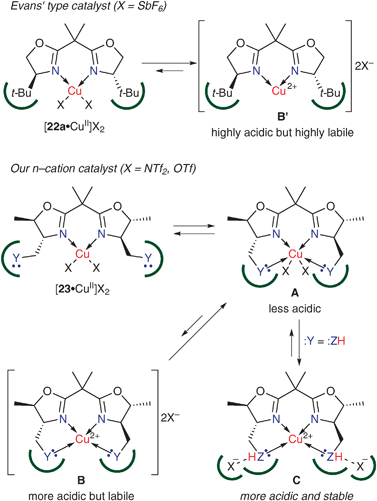 | ||
| Scheme 13 Rational design of copper(II)·bis(oxazoline) complexes based on n–cation interaction. | ||
Catalyst B′ (X = SbF6) is more Lewis acidic than catalyst C (X = Tf2N or TfO) because SbF6− is less basic than Tf2N−. However, highly Lewis acidic catalysts B′ are more labile than catalyst C. In our study, the catalyst B′ sometimes caused the polymerization of a significant amount of dienes probably due to a strong Brønsted acid generated by decomposition of the catalyst. This undesired side reaction would seriously limit the generality of the substrate.
To achieve high catalytic activity, the exchange between the product and the substrate on the catalyst should be fast. In the reaction catalyzed by C, the exchange on the catalyst should be assisted by the weak coordination of :ZH to copper(II). In contrast, the exchange on the highly acidic catalyst B′ should be relatively slow because of the tight chelation of the product with the catalyst (product inhibition).
3.3 Enantioselective Diels–Alder reaction with 24
Based on our working hypothesis, catalytic activities of Cu(OTf)2 complexes with bis(oxazoline)s 23 bearing Lewis basic sites (:Y) at the 4,4′-substituents were investigated in the Diels–Alder reaction of cyclopentadiene with α,β-unsaturated N-acyloxazolidinone 24a in CH2Cl2 (Scheme 14). Bis(oxazoline) ligands 23 were prepared from L-threonine methyl ester using molybdenum(VI) oxide-catalyzed dehydrative cyclization as a key reaction.58–60 As a result of investigation, when the reaction was conducted with 23d (:ZH = NHMs) and 23e (:ZH = NHTf) bearing protic Lewis basic sites at the 4,4′-substituents, the endo-(2R)-adduct 27a was obtained with excellent enantioselectivities (93 and 80% ees) and yields (97 and 99%), as expected. These results suggest that the sulfonamido groups of 23d and 23e may play a key role in the construction of an efficient asymmetric environment and the activation of 24a through the formation of more acidic complexes C. In contrast, the use of 23c (:Y = OMs) bearing aprotic Lewis basic sites showed poor reactivity (18%), although the enantioselectivity was high (84% ee). The predominant formation of complex A or B may cause the low catalytic activity of [23c·CuII](OTf)2,‡ although the efficient asymmetric environment created by the coordination of methanesulfonato groups of 23c may afford the high enantioselectivity. The poor reactivity and enantioselectivity with 23a and 23b may be attributed to the formation of complexes A with an inefficient asymmetric environment. The use of EtNO2 or MeNO2 instead of CH2Cl2 significantly increased the reactivity,61 while Evans and coworkers reported that the use of MeNO2 slightly decreased the reactivity and the enantioselectivity of [22a·CuII]X2-catalyzed Diels–Alder reaction.54 In addition, the use of Cu(NTf2)2 instead of Cu(OTf)2 successfully improved the enantioselectivity. Only 1 mol% of [23·CuII](NTf2)2 efficiently promoted the reaction to give 27a in 94% isolated yield with 98% ee.2 in CH2Cl2.](/image/article/2011/CS/b924478f/b924478f-s14.gif) | ||
| Scheme 14 Enantioselective Diels–Alder reaction of cyclopentadiene with 24a using n–cation catalysts [23·CuII](OTf)2 in CH2Cl2. | ||
To explore the generality and scope of the present enantioselective Diels–Alder reaction using n–cation catalysts, the reactions of various dienes were examined under the optimized conditions (Scheme 15). For the reaction at −20 °C or higher, the use of MeNO2 instead of EtNO2 improved the reactivity. The reaction of cyclic dienes such as cyclohexadiene and furan successfully gave the corresponding endo-adducts 28 and 29 with high enantioselectivities.
2 in nitroalkane.](/image/article/2011/CS/b924478f/b924478f-s15.gif) | ||
| Scheme 15 Enantioselective Diels–Alder reaction of various dienes with 24 catalyzed by [23·CuII](NTf2)2 in nitroalkane. | ||
Conventional chiral ligands which create rather rigid asymmetric environments using sterically bulky substituents sometimes decrease the substrate generality. For example, Evans and coworkers reported that the [22a·CuII](SbF6)2-catalyzed Diels–Alder reaction of isoprene with 24a gave 30a with 59% ee.55 In contrast, the present Diels–Alder reaction using [23d·CuII](NTf2)2 and [23e·CuII](NTf2)2 was highly effective for a series of 2-substituted butadienes and gave the corresponding adducts 30–34 with excellent regio- and enantioselectivities (>99∶1 dr, >90% ee). 1-Substituted butadienes such as 1,3-pentadiene and 1-acetoxy-3-methylbutadiene were also suitable substrates for the present Diels–Alder reaction and gave the corresponding adducts 35 and 36 with high enantioselectivities, albeit the diastereoselectivities were moderate.
The present n–cation catalysis was also effective for the Diels–Alder reaction with β-substituted dienophiles 24b (R1 = Me), 24c (R1 = CO2Et) and 24d (R1 = CH2OTBS). The reaction of cyclopentadiene with 24b and 24c proceeded smoothly to give the endo-adducts 27b and 27c with high enantioselectivities. The Diels–Alder reaction of acyclic dienes with β-substituted dienophiles is challenging. The n–cation catalyst [23d·CuII](NTf2)2 successfully promoted the reaction of isoprene with 24c and 24d to give the corresponding adducts 30c and 30d with high enantioselectivities. The absolute configuration of 30d was determined to be (1S,6R) after conversion to the corresponding benzyl ester (BnOLi, THF), which is a key intermediate for the synthesis of the decahydrofluorene core of hirsutellones.62
3.4 Mechanistic analysis
In general, the coordination of a Lewis basic ligand to a metal cation decreases its Lewis acidity. Actually, the catalytic activity of [22f·CuII](NTf2)2 (Fig. 6) was much lower than that of Cu(NTf2)2 itself for the reaction of cyclopentadiene with 24a (Scheme 16). In contrast, the use of 22d, 23d and 23e bearing 4,4′-sulfonamide groups significantly increased the catalytic activities and enantioselectivities. In particular, the catalytic activities of [23d·CuII](NTf2)2 and [23e·CuII](NTf2)2 were higher than that of Cu(NTf2)2 itself. These results strongly suggest that 4,4′-sulfonamide groups successfully increase the Lewis acidity of the copper(II) complexes by displacing counteranions.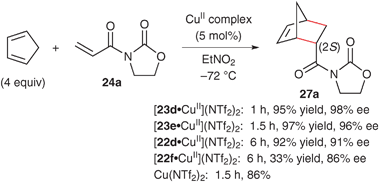 | ||
| Scheme 16 Effect of the 4,4′-substituents of bis(oxazoline)s 22 and 23. | ||
The postulated intramolecular secondary n–cation interaction of sulfonyl oxygens was considered based on theoretical calculations for a [23d·CuII](NTf2)2–24a complex.† As shown in Fig. 7, the sulfonyl oxygens coordinate with the copper(II) cation (n–cation interaction), whereas counteranions (Tf2N−) do not coordinate to the copper(II) cation but rather to protons of sulfonamido groups, as expected. It is conceivable that dienes predominantly approach the si-face of s-cis acrylimides, since a sulfonamide group and a Tf2N− preferentially shielded the re-face of the acrylimide moiety.
2–24a complex (left) and proposed transition-state assembly (right). Hydrogen atoms, except for protons of sulfonamido groups, are omitted for clarity. Distances are in angstroms.](/image/article/2011/CS/b924478f/b924478f-f7.gif) | ||
| Fig. 7 Theoretical calculations for a [23d·CuII](NTf2)2–24a complex (left) and proposed transition-state assembly (right). Hydrogen atoms, except for protons of sulfonamido groups, are omitted for clarity. Distances are in angstroms. | ||
4. Conclusions
In this tutorial review, we have described the rational design of asymmetric copper(II) catalyses based on π–cation and n–cation interactions for the enantioselective Diels–Alder reaction and [2+2] cycloaddition reaction. These non-covalent secondary interactions between the copper(II) cation and the auxiliary Lewis basic sites on the chiral ligands successfully created an efficient asymmetric environment around the metal center to afford the high-level induction of enantioselectivity. Furthermore, these secondary interactions activated the catalysts by displacing counteranions. The π–cation and n–cation catalysts are expected to promote various asymmetric reactions. Currently, investigations into the applications of π–cation and n–cation catalysts to other asymmetric reactions are underway.Acknowledgements
Financial support for this project was provided by JSPS.KAKENHI (20245022), the Toray Science Foundation, and the Global COE Program of MEXT. Some of the calculations were performed at the Research Center for Computational Science (RCCS), Okazaki Research Facilities, National Institute of Natural Science (NINS).Notes and references
- K. Ishihara, A. Sakakura and M. Hatano, Synlett, 2007, 686 CrossRef CAS.
- E. J. Corey, Pure Appl. Chem., 1990, 62, 1209 CrossRef CAS.
- S. J. Miller, Acc. Chem. Res., 2004, 37, 601 CrossRef CAS.
- T. Ose, K. Watanabe, T. Mie, M. Honma, H. Watanabe, M. Yao, H. Oikawa and I. Tanaka, Nature, 2003, 422, 185 CrossRef CAS.
- T. Ose, K. Watanabe, M. Yao, M. Honma, H. Oikawa and I. Tanaka, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1187 CrossRef.
- C. R. W. Guimarães, M. Udier-Blagovic and W. L. Jorgensen, J. Am. Chem. Soc., 2005, 127, 3577 CrossRef CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS.
- D. A. Dougherty, Science, 1996, 271, 163 CrossRef CAS.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS.
- S. Reymond and J. Cossy, Chem. Rev., 2008, 108, 5359 CrossRef CAS.
- L. M. Stanley and M. P. Sibi, in Acid Catalysis in Modern Organic Synthesis, ed. H. Yamamoto and K. Ishihara, Wiley-VCH, Weinheim, 2008, vol. 2, pp. 903–985 Search PubMed.
- K. Ishihara and M. Fushimi, Org. Lett., 2006, 8, 1921 CrossRef CAS.
- K. Ishihara, M. Fushimi and M. Akakura, Acc. Chem. Res., 2007, 40, 1049 CrossRef CAS.
- K. Ishihara and M. Fushimi, J. Am. Chem. Soc., 2008, 130, 7532 CrossRef CAS.
- A. Sakakura, R. Kondo, Y. Matsumura, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2009, 131, 17762 CrossRef CAS.
- P. Merino, E. Marqués-López, T. Tejero and R. P. Herrera, Synthesis, 2010, 1 CrossRef CAS.
- E. J. Corey, Angew. Chem., Int. Ed., 2009, 48, 2100 CrossRef CAS.
- E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef CAS.
- J. Shen and C.-H. Tan, Org. Biomol. Chem., 2008, 6, 3229 RSC.
- E. Lee-Ruff and G. Mladenova, Chem. Rev., 2003, 103, 1449 CrossRef CAS.
- K. Furuta, Y. Miwa, K. Iwanaga and H. Yamamoto, J. Am. Chem. Soc., 1988, 110, 6254 CrossRef CAS.
- K. Furuta, S. Shimizu, Y. Miwa and H. Yamamoto, J. Org. Chem., 1989, 54, 1481 CrossRef CAS.
- K. Ishihara, Q. Gao and H. Yamamoto, J. Org. Chem., 1993, 58, 6917 CrossRef CAS.
- K. Ishihara, Q. Gao and H. Yamamoto, J. Am. Chem. Soc., 1993, 115, 10412 CrossRef CAS.
- E. J. Corey and T.-P. Loh, J. Am. Chem. Soc., 1991, 113, 8966 CrossRef CAS.
- E. J. Corey, A. Guzman-Perez and T.-P. Loh, J. Am. Chem. Soc., 1994, 116, 3611 CrossRef CAS.
- E. J. Corey, T.-P. Loh, T. D. Roper, M. D. Azimioara and M. C. Noe, J. Am. Chem. Soc., 1992, 114, 8290 CrossRef.
- S. Otto, G. Boccaletti and J. B. F. N. Engberts, J. Am. Chem. Soc., 1998, 120, 4238 CrossRef CAS.
- S. Otto and J. B. F. N. Engberts, J. Am. Chem. Soc., 1999, 121, 6798 CrossRef CAS.
- D. van der Helm, M. B. Lawson and E. L. Enwall, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 2307 CrossRef CAS.
- O. R. Nascimento, A. J. Costa-Filho, D. I. DeMorais, J. Ellena and L. F. Delboni, Inorg. Chim. Acta, 2001, 312, 133 CrossRef CAS.
- A. J. Costa-Filho, O. R. Nascimento and R. Calvo, J. Phys. Chem. B, 2004, 108, 9549 CrossRef CAS.
- C. Kashima, K. Fukusaka and K. Takahashi, J. Heterocycl. Chem., 1997, 34, 1559 CrossRef CAS.
- C. Kashima, H. Harada, I. Kita, I. Fukuchi and A. Hosomi, Synthesis, 1994, 61 CrossRef CAS.
- C. Kashima, I. Fukuchi, K. Takahashi and A. Hosomi, Heterocycles, 1994, 38, 1407 CrossRef CAS.
- C. Kashima, I. Fukuchi, K. Takahashi and A. Hosomi, Tetrahedron, 1996, 52, 10335 CrossRef CAS.
- W. Ried and F.-J. Königstein, Justus Liebigs Ann. Chem., 1959, 622, 37 CrossRef CAS.
- C. Kashima, I. Kita, K. Takahashi and A. Hosomi, J. Heterocycl. Chem., 1995, 32, 25 CrossRef CAS.
- C. Kashima, I. Kita, K. Takahashi and A. Hosomi, J. Heterocycl. Chem., 1995, 32, 723 CAS.
- K. Ishihara, S. Kondo, H. Yamamoto, H. Ohashi and S. Inagaki, J. Org. Chem., 1997, 62, 3026 CrossRef CAS.
- E. J. Corey and T. W. Lee, Tetrahedron Lett., 1997, 38, 5755 CrossRef CAS.
- G. Hilt, W. Hess and K. Harms, Org. Lett., 2006, 8, 3287 CrossRef CAS.
- J. N. Payette and H. Yamamoto, Angew. Chem., Int. Ed., 2009, 48, 8060 CrossRef CAS.
- I. Fleming, Frontier Orbitals and Organic Chemical Reactions, John Wiley & Sons, New York, 1976, pp. 106–109 Search PubMed.
- F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 3364 CrossRef CAS.
- M. Pfau, G. Revial, A. Guigant and J. Angelo, J. Am. Chem. Soc., 1985, 107, 273 CrossRef CAS.
- E. Canales and E. J. Corey, J. Am. Chem. Soc., 2007, 129, 12686 CrossRef CAS.
- G. C. Hargaden and P. J. Guiry, Chem. Rev., 2009, 109, 2505 CrossRef CAS.
- R. Rasappan, D. Laventine and O. Reiser, Coord. Chem. Rev., 2008, 252, 702 CAS.
- G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561 CrossRef CAS.
- J. S. Johnson and D. A. Evans, Acc. Chem. Res., 2000, 33, 325 CrossRef CAS.
- A. K. Ghosh, P. Mathivanan and J. Cappiello, Tetrahedron: Asymmetry, 1998, 9, 1 CrossRef CAS.
- D. A. Evans, S. J. Miller, T. Lectka and P. von Matt, J. Am. Chem. Soc., 1999, 121, 7559 CrossRef CAS.
- D. A. Evans, D. M. Barnes, J. S. Johnson, T. Lectka, P. von Matt, S. J. Miller, J. A. Murry, R. D. Norcross, E. A. Shaughnessy and K. R. Campos, J. Am. Chem. Soc., 1999, 121, 7582 CrossRef CAS.
- J. Zhou and Y. Tang, Org. Biomol. Chem., 2004, 2, 429 RSC.
- M. Schinnerl, M. Seits, A. Kaiser and O. Reiser, Org. Lett., 2001, 3, 4259 CrossRef CAS.
- A. Sakakura, R. Kondo, S. Umemura and K. Ishihara, Tetrahedron, 2009, 65, 2102 CrossRef CAS.
- A. Sakakura, S. Umemura, R. Kondo and K. Ishihara, Adv. Synth. Catal., 2007, 349, 1641 CrossRef CAS.
- A. Sakakura, R. Kondo and K. Ishihara, Org. Lett., 2005, 7, 1971 CrossRef CAS.
- M. Johannsen and K. A. Jørgensen, J. Chem. Soc., Perkin Trans. 2, 1997, 1183 RSC.
- S. D. Tilley, K. P. Reber and E. J. Sorensen, Org. Lett., 2009, 11, 701 CrossRef CAS.
Footnotes |
| † Theoretical calculations were performed using Gaussian 98 and 03 programs. The geometries of the complex were optimized using gradient-corrected density functional theory (DFT) calculations with Becke's three-parameter exchange with the Lee, Yang and Parr correlation functional (B3LYP). The basis set was chosen as follows. For Cu, Wachter's primitive set (14s9p5d), supplemented with a three f polarization function (Wachters+f), was used, and the final basis set was (14s9p5d3f)/[8s6p4d1f]. For C, N, O, F, S and H, the standard 6-31G(d) basis set was used. |
| ‡ A theoretical calculation of the geometry optimization of [23c·CuII](NTf2)2 did not give a local minimum structure, since the counteranions were caught by 23c and completely liberated. |
| This journal is © The Royal Society of Chemistry 2011 |