Mastering fundamentals of supramolecular design with carboxylic acids. Common lessons from X-ray crystallography and scanning tunneling microscopy†
Oleksandr
Ivasenko
and
Dmitrii F.
Perepichka
*
Department of Chemistry and Centre of Self-Assembled Chemical Structures, McGill University, 801 Sherbrooke Street West, Montreal, Quebec H3A 2K6, Canada. E-mail: dmitrii.perepichka@mcgill.ca; Fax: 1 514 398 3797; Tel: 1 514 398 6233
First published on 29th September 2010
Abstract
Hydrogen bonding is one of the most important non-covalent interactions in both biological (DNA, peptides, saccharides etc.) and artificial systems (various soft materials, host–guest architectures, molecular networks, etc.). Carboxylic acids are some of the most simple yet powerful hydrogen-bonding building blocks, that possess a particularly rich supramolecular chemistry. This tutorial review focuses on the structural diversity of supramolecular architectures accessible viahydrogen bonding of carboxylic acids, as observed both in single crystals using X-ray analysis and in monolayers on surfaces using scanning probe techniques. It provides a concise overview of the key concepts and principles of modern supramolecular design and is given in the form of case studies of finely selected literature examples, covering formation of macrocycles, chains, ladders, rotaxanes, catenanes, various 2D and 3D nets, host–guest systems and some applications thereof.
 Oleksandr Ivasenko | Oleksandr Ivasenko received his BSc and MSc degrees from Donetsk National University (Ukraine) and in 2004 he joined the group of Prof. Perepichka to do his PhD. The main focus of his research is supramolecular chemistry of carboxylic acids and their self-assembly at solid–liquid interfaces. |
 Dmitrii F. Perepichka | Dmitrii Perepichka is an Associate Professor in the Department of Chemistry, McGill University. He received his PhD in chemistry in 1999 from the Institute of National Academy of Science of Ukraine, followed by post-doctoral stays at Durham University and at UCLA. His first independent appointment was at INRS-University of Quebec (2003), from where he moved to McGill University in 2005. The research of his group is in organic electronic materials, which includes the synthesis of polyaromatic molecules and conjugated polymers, their self-assembly, and reactions on crystalline surfaces, and applications in molecular and thin film electronics. |
1. Introduction
In 1989 Desiraju defined crystal engineering as ‘the understanding of intermolecular interactions in the context of crystal packing and the utilization of such understanding in the design of new solids with desired physical and chemical properties’.1 Since then, its importance has spanned far beyond the design of tailored crystalline solids. The supramolecular interactions and motifs discovered within the field of crystal engineering have become design tools used in different areas of chemistry, biology, physics and materials engineering. We thus felt that there is a need for concise yet systematic introduction of the basic concepts and design principles of crystal engineering for an audience with a general interest in molecular science. In this review we focus on hydrogen-bonding interactions of carboxylic (COOH) groups, emphasizing the diversity of accessible supramolecular motifs and their relations to molecular structure of the building blocks.Hydrogen bonding (H-bonding) is a specific interaction between a positively polarized hydrogen that is covalently bonded to an electronegative atom of an H-bond donor molecule, and a negatively polarized atom of an H-bond acceptor molecule. H-bonding is often divided into three categories: very strong (∼15–40 kcal mol−1; typically involves a charged H-bond acceptor, e.g., COOH⋯COO−, so called “charge-assisted H-bonding”), moderate (∼5–15 kcal mol−1, most often observed for neutral H-bond donors and acceptors with N and O as electronegative atoms) and weak (∼1–5 kcal mol−1, e.g., CH⋯O). It is largely electrostatic in origin although orbital interactions as well as dispersion forces (van der Waals interactions, vdW) can have a significant contribution to the overall energy of some H-bonds. An important feature of H-bonding brought, at least in part, by orbital interactions, is a pronounced directionality. This directionality along with significant strength render H-bonding as one of the most important supramolecular interactions, both in nature (cf., DNA double-helix) and in the lab.
Carboxylic groups, containing strong hydrogen-bond donor (O–H) and acceptor (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) components, are naturally predisposed to self-associate through H-bonding. There are several possible modes of such intermolecular association: linear dimers (D), linear chains [also known as catemers, C(4), where the number 4 indicates that four atoms of a building block form the chain] or rings (R) (Scheme 1). In the crystals, these acts as connections (supramolecular synthons) linking together the nodes (molecules) in a network structure.2 Ring synthon, the most characteristic association modes of COOH groups, can be further specified by notation Rxy(Z), where x and y are the number of H-bond donor and acceptor centers and Z is a total number of atoms in the ring.
O) components, are naturally predisposed to self-associate through H-bonding. There are several possible modes of such intermolecular association: linear dimers (D), linear chains [also known as catemers, C(4), where the number 4 indicates that four atoms of a building block form the chain] or rings (R) (Scheme 1). In the crystals, these acts as connections (supramolecular synthons) linking together the nodes (molecules) in a network structure.2 Ring synthon, the most characteristic association modes of COOH groups, can be further specified by notation Rxy(Z), where x and y are the number of H-bond donor and acceptor centers and Z is a total number of atoms in the ring.
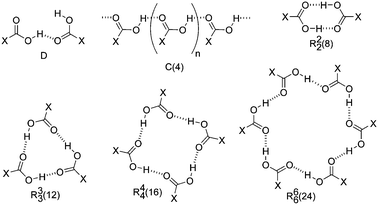 | ||
| Scheme 1 Homo-synthons of self-association of carboxylic groups. | ||
Hydrogen bonding has been extensively studied in the gas phase and in solutions by spectral methods and particularly in the solid state by X-ray crystallographic analysis. A significant part of this data is collected in Cambridge Structure Database (CSD) that contains over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 entries of exact crystal structures of carboxylic acids.3 More recently, Scanning Tunneling Microscopy (STM) has emerged as a convenient tool to analyze molecular assemblies on surfaces with submolecular resolution. Although STM does not generally provide direct atomic-precision information of the supramolecular organization, this can be often deduced through analysis of the STM images aided by molecular modeling. In contrast to X-ray analysis, STM allows for real-space/real-time imaging and thus can be applied to studies of aperiodic and dynamic structures. Also, restricting the supramolecular interactions within a two-dimensional space of a surface-adsorbed monolayer, greatly simplifies the structural analysis.
000 entries of exact crystal structures of carboxylic acids.3 More recently, Scanning Tunneling Microscopy (STM) has emerged as a convenient tool to analyze molecular assemblies on surfaces with submolecular resolution. Although STM does not generally provide direct atomic-precision information of the supramolecular organization, this can be often deduced through analysis of the STM images aided by molecular modeling. In contrast to X-ray analysis, STM allows for real-space/real-time imaging and thus can be applied to studies of aperiodic and dynamic structures. Also, restricting the supramolecular interactions within a two-dimensional space of a surface-adsorbed monolayer, greatly simplifies the structural analysis.
This review is created mostly from analysis of CSD records of COOH association in 3D crystals3 and original literature reporting STM studies of carboxylic acids in 2D monolayers. It is focused on relationships between the geometry of building blocks and resulting supramolecular structure. We have purposely avoided extensive discussions of other aspects of self-assembly such as the role of the solvent or the preparation conditions, and selected the examples highlighting the role of the building block. We note, however, that these “external” conditions can have a profound effect on the mode of self-assembly often controlling which supramolecular structure, among the multitude of possibilities, is formed in the given case.
The paper is divided in 5 sections. In the next section 2 we examine R22(8) homoassembly of carboxylic group in great details to illustrate general strategies of building block design for targeting various architectures. These strategies are generally applicable to other synthons, and thus we only outlined specifics of other homo- and hetero-synthons of the COOH group (in sections 3 and 4, respectively). Finally, in the section 5 we give selected examples of applications based on utilization of H-bonding capabilities of the carboxylic acids.
We note that self-assembly of carboxylic acids in crystals was summarized much earlier by Leiserowitz.4 Most recently, 2D assembly of aromatic carboxylic acids on surfaces, as studied by STM, was reviewed by Lackinger and Heckl.5 Various aspects of supramolecular design including H-bonding have been detailed out in eleven volumes of Comprehensive supramolecular chemistry.6 While there certainly is an overlap in the covered literature and concepts between these works and the present paper, our purpose was to create a tutorial review that summarizes the last ∼50 years of developing an understanding of self-assembly of carboxylic acids, and to show recent general examples of how this understanding is used in the rational design of complex and functional structures, not bound by a specific field, application or dimensionality.
2. Self-assembly via R22(8) homodimers
R22(8) synthon involves H-bond donor and acceptor centers of a COOH group in a cyclic manner and leads to dimerization. Thus, for monocarboxylic acids it can give only discrete, “zero-dimensional” (0D) structures—supramolecular dimers. For dicarboxylic acids either 0D (cycles) or 1D (chains) structures can be created, while building blocks with more than two COOH groups could potentially be used to form any architectures.2.1 Zero-dimensional structures viaR22(8) synthons
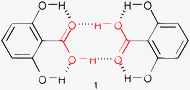
An ambiguity arises when two different carboxylic acids (A and B) co-crystallize together. One possibility is formation of segregated AA and BB homodimers (Fig. 1) as observed for the salt of p-aminobenzoic acid with 4-hydroxysulfonyl-2-hydroxybenzoic acid (FETWIL†). Such segregation is favorable when it increases packing efficiency and/or provides additional inter-molecular interactions.
 | ||
| Fig. 1 Segregated homodimers in cocrystals of different carboxylic acids. | ||
Often, however, carboxylic acids with different pKa's are predisposed to form mixed homodimers AB.7 This was effectively used in the construction of H-bonded aggregates using phenyl–pentafluorophenyl π-stacking interactions. Thus, co-crystallization of pentafluorobenzoic acid (2) with benzoic (3) or 2,4,6-trimethylbenzoic acid (4) afforded the corresponding mixed dimers assembled into face-to-face stacks with alternating phenyl and pentafluorophenyl rings (Fig. 2).8
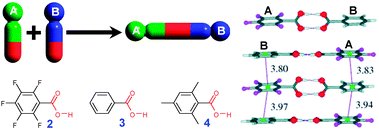 | ||
| Fig. 2 Formation of mixed carboxylic dimers for cocrystals 2·3 (UKOKIO†)and 2·4. (UKOKOU†). X-ray structure of 2·3 is depicted on the left. Adopted from ref. 8 with permission. | ||
Decreasing the structural difference between two components diminishes energy differentiation between alternative packing arrangements of segregated and mixed dimers leading to disorder, as is the case for 1:1 co-crystals of 2-methylbenzoic and 2-chlorobenzoic acids (TOJFUU†),9 or even to a fully random (isomorphic) substitution. Indeed co-crystallization of benzoic and 4-fluorobenzoic acid yields solid solution crystals (e.g.SATHOK† and SATJAY†) with a wide compositional range (10–70%).10
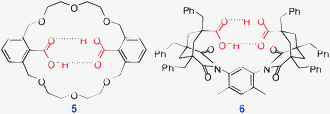
2.1.2.1 Intramolecular cyclic assemblies (n = 1). Simplest supramolecular macrocycles can be formed by intramolecular dimerization of carboxylic groups as illustrated by diacids 5 (COXYLC†) or 6 (RIXKIS†). Obviously, the structure of a building block controls the distance between the carboxylic groups and strongly affects the efficiency (and the existence) of such H-bonding. In turn, H-bonding can also control the conformation and rigidify flexible building blocks as exemplified by diacid 5.
2.1.2.2 Bimolecular monocyclic assemblies (n = 2). For simple aliphatic diacids, a preferential all-trans conformation of the hydrocarbon chains leads to infinite chains as a preferred association motif (7–9, Fig. 3). Substituting α-carbons with methyl groups induces rotation of the carboxylic groups to minimize the intramolecular strain. In diacids with odd number of carbons (10 and 12) this co-aligns the COOH groups resulting in cyclization.11 In diacids with even number of carbons (11) such substitution predictably leads to anti-alignment of the COOH groups thus giving rise to zig-zag chains (Fig. 3).
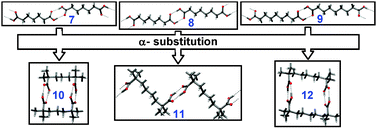 | ||
| Fig. 3 Effect of α-substitution on H-bonding in single crystals of aliphatic dicarboxylic acids. 7: PIMELA07†, 8: SUBRAC05†, 9: AZELAC05†, 10: AVOZIU†, 11: AVOZUG†, 12: AVOZOA†. Adopted from ref. 11 with permission. | ||
H-bonding macrocylization of flexible dicarboxylic acids can also be enforced by templating. Thus, complexation of ferullic acid derivative 13 with alkali metal cations (Na, K, Cs) stabilizes a U-shaped conformation (Fig. 4).12 In the solid state, these complexes readily dimerize into supramolecular macrocyles via H-bonding of COOH groups.
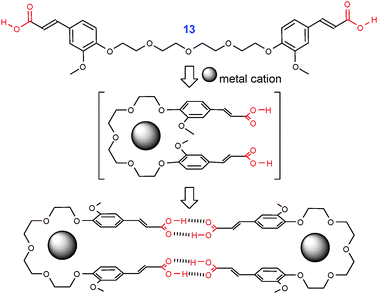 | ||
| Fig. 4 Formation of supramolecular macrocycles of 13viaalkali metal templating. Adopted from ref. 12 with permission | ||
We note that the co-alignment of two carboxylic groups does not always guarantee formation of macrocycles as illustrated by the crystal structure of anthracene-1,8-dicarboxylic acid 14 (WISJAJ†) which assembles into 1D chains (Fig. 5). In this case, cyclization can be enforced by decreasing a distance between the co-aligned carboxylic groups to the point where H-bonding with more than one molecule (polymerization) becomes sterically unfavorable, as shown for the naphthalene-1,8-dicarboxylic acid 15 (KOFJUK†, Fig. 5)
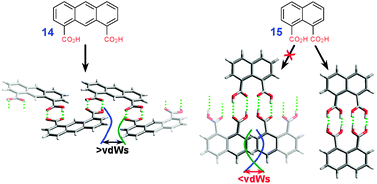 | ||
| Fig. 5 Forced supramolecular cyclization caused by preferential steric interactions as observed in single crystals of acenedicarboxylic acids. | ||
2.1.2.3 Oligomeric monocyclic assemblies (n > 2). In principle, formation of cyclic {R(COOH)2}n equilateral polygons could be expected for rigid diacid building blocks if the angle (θ) between the two carboxylic groups is equal to (180–360/n). Accordingly, phthalic (16, θ = 60°), isophthalic (17, θ = 120°) and furan-2,5-dicarboxylic acid (19, θ = 129°) should give triangular, hexagonal and heptagonal macrocycles, respectively (Fig. 6). In the solid state, however, all three crystallize into infinite chains (section 2.2.1) due to higher packing efficiency of the later.
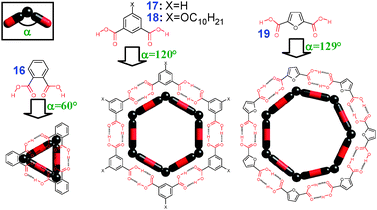 | ||
| Fig. 6 Effect of the orientation of the carboxylic groups on the expected structure of corresponding H-bonded macrocycles. | ||
A common practice in synthesis of covalent macrocycles is using high dilution conditions to kinetically favor an intramolecular cyclizationvs. intermolecular polymerization. Using this strategy in synthesis of supramolecular cycles was demonstrated in ultra-high vacuum (UHV) STM studies by regulating a dosage of the molecules on the surface. Thus, porphyrin diacid 20 self-assembles into cyclic tetramers at low surface coverage, while at coverage >0.5 monolayer the equilibrium is shifted towards infinite zig-zag chains (Fig. 7).13
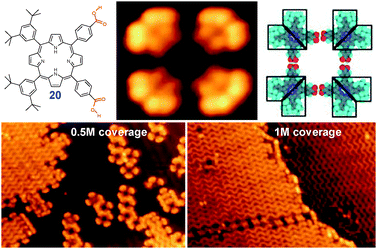 | ||
| Fig. 7 Effect of surface concentration (coverage) on self-assembly of 20 on Au(111) surface. Adopted from ref. 13 with permission. | ||
Under thermodynamic control, the choice between the cyclization and polymerization pathways is often defined by secondary interactions and relative packing efficiencies of the two possible structures. Thus, Valiyaveettil and Müllen have shown that introduction of the decyloxy substituent in the isophthalic acid (18) leads to preferential cyclization, both in the solid state and in solution (based on vapor pressure osmometry).14 Single crystal X-ray analysis of 18 shows that cavities of a hexagonal macrocycle are filled with alkyl chains (belonging to molecules of adjacent hexagons). This is not possible when alkyl chains become longer than C10 and higher homologues (C12–C20) form H-bonded zig–zag polymers wherein the alkyl chains are more tightly packed into lamellas.
As shown above for 13, the macrocyclization can also be enforced by templating. While homo-assembly of isophthalic acid 17 at solid/liquid interface leads to zigzag chains, co-assembly with coronene favors H-bonding cyclization where the resulting hexagonal cavities are occupied by coronene guest molecules (Fig. 8).15 This templating effect was recently studied by Tobe, De Feyter et al. in an elegant programmable self-assembly of four-component 2D crystal.16
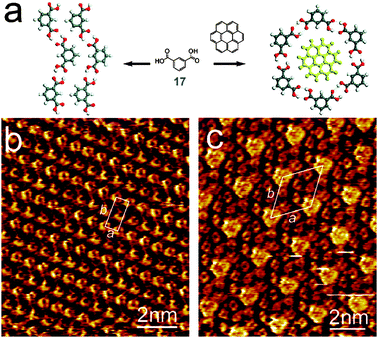 | ||
| Fig. 8 Molecular scheme (a) and STM images of: (b) the “zigzag” pattern of 17 and (c) the coronene-templated assembly of 17 hexamers. Reproduced from ref. 15 with permission. | ||
Non-planar cyclic trimers and tetramers were observed in crystals of isomeric diphenic acids 21 and 22, respectively (Fig. 9).17 The orientation of the carboxylic groups is dictated by the dihedral angle of the biphenyl core which is larger in 22 due to repulsion between o-methyl groups. Unlike 21 which can cyclize in its racemic form (forming both enantiomeric rings in its unit cell), 22 has to be enantiomerically pure in order to form the cycles. A racemic mixture of 22, on the other hand, assembles in zig–zag chains in the solid state. Notably, this fundamental difference in self-assembly of the homochiral and racemic diacid 22 can be also seen in solution. Vapor pressure osmometry of the homochiral diacid (R)-22 gives an apparent degree of association of ∼4 in CHCl3 solution indicating formation of the discrete tetrameric cycles. In contrast, much less soluble polymeric chains are formed by racemic form (R,S)-22 under comparable conditions.
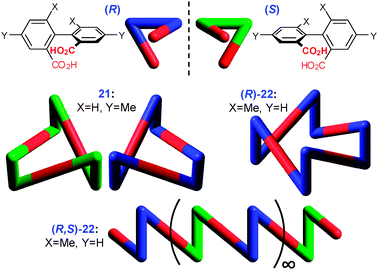 | ||
| Fig. 9 Self-assembly of substituted diphenic acids 21 (SAZQEP†) and 22 (PEQBIW†). | ||
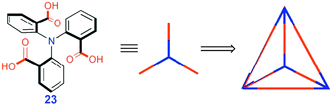 | ||
| Fig. 10 Formation of H-bonded tetrahedron. Reproduced from ref. 18 with permission. | ||
Hexameric prisms can be formed in solution from the tetradentate derivative of isophthalic acid (24), as shown by Zimmerman et al.19 The supramolecular cyclization in this case is directed by a large dendritic substituent R (Fig. 11).
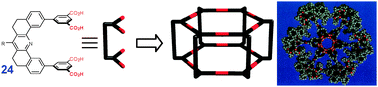 | ||
| Fig. 11 Design of H-bonded hexameric prism. Adopted from ref. 19 with permission. | ||
 | ||
| Fig. 12 Examples of crystallosolvates of polycarboxylic acids formed via mixed homo-R22(8) synthon. | ||
2.2 One-dimensional structures viaR22(8) synthons
 | ||
| Fig. 13 1D chains formed by (a) terephthalic acid (TEPHTH13), (b) phthalic acid (16: PHTHAC02†), (c) isophthalic acid (17: BENZDC01†), (d) furan-2,5-dicarboxylic acid (19: FURDCA†) and e) phenylene-1,3-diacetic acid (HOFBOU).3 | ||
This simple geometrical relationship between the orientation of the COOH groups in diacids and the structure of the formed 1D chains might not work for flexible building blocks. Thus, even short methylene bridges in 1,3-phenylenediacetic acid makes the structure of the supramolecular chain difficult to predict (Fig. 13e).
More complex 1D structures can be formed from non-planar bidentate building blocks. Thus, helical structures constructed with R22(8) synthons were observed in crystals of 2,2′,4,4′-tetranitrodiphenic acids 29 (FINMEU†, Fig. 14a). Unfortunately, predictability of such systems is far from straightforward: a closely related 2,2′-dinitrodiphenic acid 30 packs into zig–zag chains (SEFYEH†, Fig. 14b).
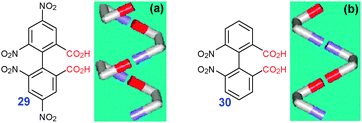 | ||
| Fig. 14 Chemical structures of 29 and 30 together with schematic models of (a) supramolecular helix and (b) zig–zag showing different enantiomeric composition necessary for their formation. Adopted from ref. 22 with permission. | ||
Generally for non-planar dicarboxylic acids, a zigzag assembly requires two atropisomers (rotational isomers) while association of one single atropisomer can be expected to produce a helical structure. This later condition could be achieved by inducing asymmetry (chirality) in the molecule through substitution. An interesting attempt at directing the assembly motif for symmetric molecules was reported by Field and Venkataraman.22 Varying the bridge X in bis(benzoic) acids 2-HOOC-Ph-X-Ph-2′-COOH, they were able to change the symmetry of the molecule by controlling intramolecular H-bonding of either one or both COOH groups with the bridge atom. This led to either symmetric or asymmetric orientation of the COOH groups in the molecule thereby directing the assembly into either zigzag or helical pathway, respectively.
A 1D cavity created in such H-bonded helixes potentially allows hosting other molecules of interest. Sanders et al. have used diastereomeric naphthalenediimide derivatives to create chiral helixes, both in solution and in the solid state (Fig. 15).23 An interior of these supramolecular structures can host different guest molecules (fullerenes, polyaromatic hydrocarbons, quaternary ammonium salts) which are additionally stabilized by π–π interactions with planar naphthalenediimides.
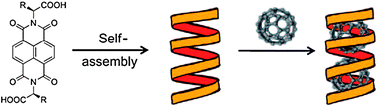 | ||
| Fig. 15 Schematic representation of the design of helical nanotubular host and its interaction with C60 guest molecules. Reproduced from ref. 23 with permission. | ||
 | ||
| Fig. 16 H-bonded polyrotaxanes in co-crystals of aminium-containing dicarboxylic acids and crown ethers. Reproduced from ref. 24 with permission. | ||
A related recent example is the design of “polycatenated” chains combining coordinational chemistry of copper and H-bonding of carboxylic groups (Fig. 17).26
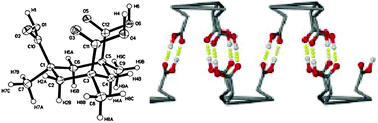 | ||
| Fig. 18 1D chains in crystals of 28. Reproduced from ref. 21 with permission. | ||
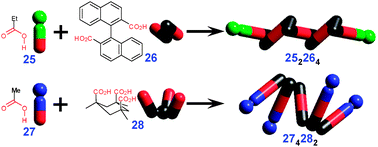
The 1D self-association can also be favorable for tricarboxylic acids containing long aliphatic chains (e.g.31 NISMEH†, Fig. 19). To achieve close packing, such molecules distort from the C3-symmetric conformation by bringing two of the three alkyl chains together. Such conformation co-aligns corresponding COOH groups and leads to the 1D supramolecular association.
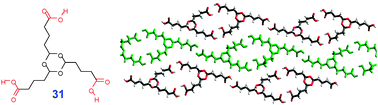 | ||
| Fig. 19 Crystal packing of 1D chains formed from flexible tricarboxylic acid 31. | ||
Same principle operates for triacid 32 which forms 1D chains at a solid–liquid interface. The internal pores in these structures can be tuned through incorporation of various guest molecules (Fig. 20).27 In its homologue 33, the shorter aliphatic chains are too stiff to allow co-alignment and so less strained dimerized polymer strands are formed (RENCUI†, Fig. 21).
 | ||
| Fig. 20 Template-induced inclusion structures of 32 with coronene as a guest (STM image at solid liquid interface). Reproduced from ref. 27 with permission. | ||
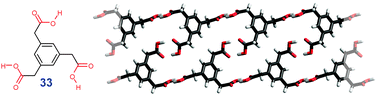 | ||
| Fig. 21 Supramolecular organization in crystals of 33. | ||
Removing the methylene bridges completely results in a very rigid trimesic acid (34) which can only self-associate into 2D nets (see below). Yet in the crystals of its acetic acid solvate interesting ladder structures have been observed (LERSAD†, Fig. 22).28 Here the acetic acid molecules cap the edges of TMA ribbon forming quasi-1D structures.
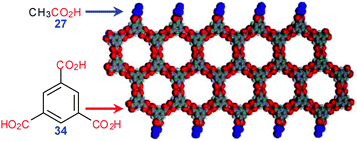 | ||
| Fig. 22 1D features found in acetic acid crystallosolvate of 34. Adopted from ref. 28 with permission. | ||
Finally, some tetracarboxylic acids can also be used to create porous 1D structures, as illustrated in Fig. 23.
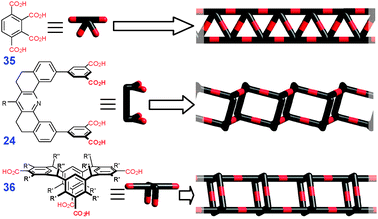
2.3 Two-dimensional structures viaR22(8) synthons
2D nets are structures with an infinite number of nodes that propagate infinitely in two directions. Depending on the valency and geometry of the building blocks, different 2D H-bonded networks can be formed. For association through R22(8) synthons, the 2D networks necessarily require at least three carboxylic groups in the building block, although dicarboxylic acids can also self-associate in the 2D structures via other synthons (see section 3.1).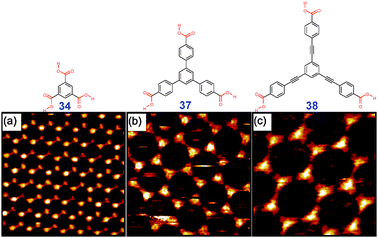 | ||
| Fig. 24 STM images of chicken wire polymorphs of (a) trimesic acid 34, (b) 37 and (c) 38 at solid–liquid interface Adopted from ref. 5 with permission. | ||
Formation of the chicken-wire net can be predicted for any rigid building block of C3-symmetry which self-associates through R22(8) synthons. Thus, changing the size of the building block allows to rationally control the diameter of the pores (Fig. 24).5
A family of bicomponent porous structures can be created by combining two building blocks. The diameter of the pore and the overall pattern in such systems can be tuned by varying the ratio of the components (Fig. 25).5
 | ||
| Fig. 25 STM images and structures of co-assemblies of 34 and 37 at solid–liquid interface. Adopted with permission from L. Kampschulte, T. L. Werblowsky, R. S. K. Kishore, M. Schmittel, W. M. Heckl and M. Lackinger, J. Am. Chem. Soc., 2008, 130, 8502. | ||
An appropriate substitution of the building-blocks can ‘decorate’ the chicken-wire cavities creating a chiral environment,31 introducing favorable interactions with guest molecules32 or even blocking them completely.33 Lowering the rigidity of the building block can cause either a random disorder, a uniform distortion or even form 1D structures as was seen in the cases of 31–33 (section 2.2.3).
Using tricarboxylic acids of lower symmetry makes the supramolecular architecture rather difficult to predict. Thus, 1,2,3-benzenetricarboxylic acid forms a very complex non-planar 2D H-bonded network (TANRIK†)34 while a 3D network was found for 1,2,4-benzenetricarboxylic acid (see below).
 | ||
| Fig. 26 Self-assembly of CoTCPP at solid liquid interface: (a) structure and (b) STM image. Reproduced from ref. 35 with permission. | ||
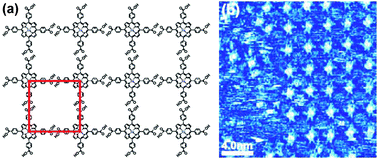
C2-symmetric tetracarboxylic building blocks self-assemble viaR22(8) synthons in either p2 symmetry “oblique” (Fig. 27a) or p6 symmetry “Kagome” (Fig. 27b) nets, depending on the size of the core. As shown by Wuest et al., a smaller biphenyltetracarboxylic acid 39 preferentially forms oblique nets while a larger building block 41 prefers Kagome nets.36 The intermediate size tolanetetracarboxylic acid 40 gave a randomly mixed network of both motifs. A similar behavior was found by Beton et al. in terphenyltetracarboxylic acid 42 which has been proposed as a model system for organic glasses.37
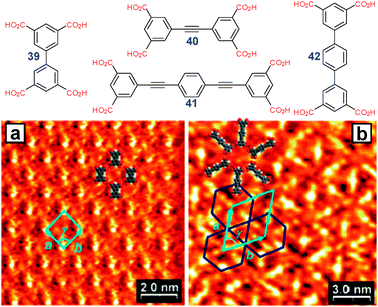 | ||
| Fig. 27 STM images of (a) oblique and (b) Kagome networks formed by 39 and 41, respectively, at solid–liquid interface. Adopted from ref. 36 with permission. | ||
Preferential inclusion of guest molecules (coronene) in a porous Kagome network was used to enforce/impose the formation of the Kagome polymorph from teteracid 43 and 44 which otherwise produce an oblique polymorph under these experimental conditions (Fig. 28).38
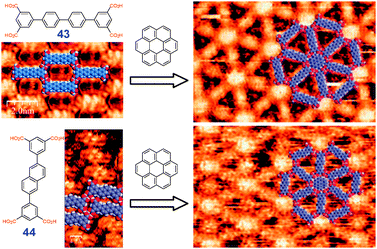 | ||
| Fig. 28 Templating Kagome polymorphs of 43 and 44 by coronene observed by STM at solid–liquid interface. Adopted from ref. 38 with permission. | ||
Finally, non-planar porous 2D square nets can be formed from non-planar tetraacids, as shown for biphenyltetracarboxylic acid 45 (Fig. 29).39
 | ||
| Fig. 29 Design of square nets from 45 (X = CO2H). Reproduced from ref. 39 with permission. | ||
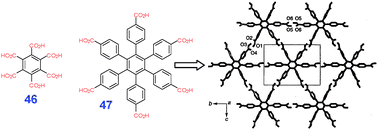 | ||
| Fig. 30 Examples of hexacarboxylic acids capable of formation of (3,6)-nets (X-ray analysis of the crystals). Adopted from ref. 40 with permission. | ||
Interestingly, in the case of benzenepentacarboxylic acid (JEXZUH†) four carboxylic groups in positions 1,2,4,5 form a 2D sheet somewhat similar to the oblique polymorph of 39, while the remaining carboxylic group dimerizes such sheets into a complex 2D net.
2.4 Three-dimensional structures viaR22(8) synthons
3D nets are usually built from tetradentate building blocks of tetrahedral symmetry, which naturally assemble in diamond-type structures, as exemplified by methanetetraacetic acid 48 and adamantanetetracarboxylic acid 49 (Fig. 31).41 Interpenetrations are very typical in these highly porous networks. More complex 3D structures (e.g., LECKAG†) have also been observed for conformationally flexible tetracarboxylic building blocks.42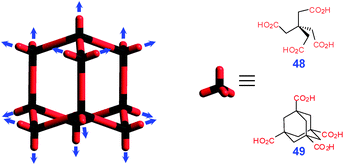
In principle, the 3D nets can also be formed by tridentate carboxylic acids. For example, in 1,2,4-benzenetricarboxylic acid (TRIMEL†) the COOH groups in 1- and 4- positions form infinite chains which are crosslinked into a 3D structure via the remaining 2-carboxylic groups.
A very unusual case of 3D network was observed in a hybrid germanium oxide/carboxylic acid structure 50 (CXEGEO†) synthesized by sol–gel chemistry from trichloro(β-cyanoethyl)germane (hydrolysis followed by annealing). In the resulting structure, the germanium oxide core forms 2D sheets substituted with carboxylic groups. Association of these sheets viaR22(8) synthons leads to a cross-linked 3D layered material (Fig. 32).43
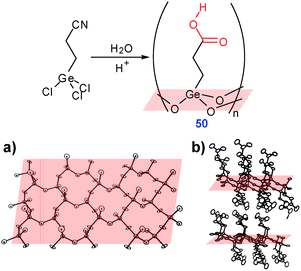 | ||
| Fig. 32 Synthesis of 50 and its crystal structure showing (a) GeOx layer and (b) inter-layer interactions viaR22(8) dimerization of the carboxylic groups. Adopted from ref. 43 with permission. | ||
In general, stabilization of 3D porous networks via simple H-bonding of carboxylic groups is difficult and such structures are relatively rare. We note, however, that the 3D supramolecular networks built by stronger interactions, e.g. metallorganic frameworks (MOFs), is a burgeoning area of research.44
3. Self-assembly via other homosynthons
Until now we have discussed the design of various supramolecular architectures viaR22(8) synthons, focusing on the relationship between the building block structure and resulting self-assembly motif. Many of the presented concepts are also applicable for other homosynthons (Scheme 1). Such a variety of homosynthons significantly expands the diversity of self-assembled architectures that can be created even with structurally primitive building blocks. On the other hand, polymorphism brought by possible associations through several synthons, complicates a rational design of the desired architecture.3.1 Cyclic homosynthons
The cyclic H-bonding of carboxylic acids other than viaR22(8) sythons, is rare and only a few tens of such structures have been reported. Stabilization energies of the trimeric (R33(12)), tetrameric (R44(16)) and hexameric (R66(24)) H-bonding synthons are comparable but usually somewhat lower than that of R22(8) synthon, due to a less favorable angle of the CO⋯H contact and/or steric repulsions between the building blocks. However, when realized, these homosynthons can expand dramatically the supramolecular capabilities of simple building blocks. In construction of 2D and 3D nets the R22(8) synthon can serve only as simple linear connections, while R33(12), R44(16) and R66(24) homosynthons can be used as tri-, tetra- and hexadentate nodes. Thus, Fasel et al. demonstrated a square, p4 symmetry 2D net formed from a biphenyl-4,4′-dicarboxylic acid is monolayers on Au(111) surface (Fig. 33a).45 In the case of symm-trithienobenzene-2,2′,2′′-tricarboxylic acid, association via an extremely rare R66(24) synthons leads to a high-density enantiomerically pure polymorph (Fig. 33b,c).32 Interestingly, a corresponding chicken-wire polymorph of this acid built viaR22(8) synthons is racemic with a completely random orientation of both enantiomers. This is due to a rapid decrease of distances between the building blocks in the range: R22(8) > R33(12) > R44(16) > R66(24), that accentuates secondary interactions.
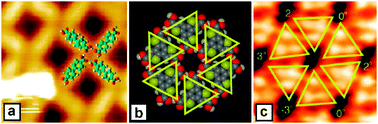 | ||
| Fig. 33 STM image and structural models of (a) biphenyldicarboxylic acid and (b, c) trithienobenzenetricarboxylic acid self-assembled on flat surfaces. Reproduced from ref. 45 and 32 with permission. | ||
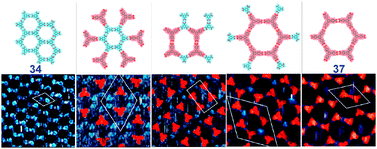
A remarkable diversity of self-assembly in topologically different nets can be achieved even with one of the simplest building blocks, trimesic acid 34. As discussed before, a “chicken-wire” network where all molecules are linked viaR22(8) synthons, is a naturally expected mode of self-assembly of trimesic acid. However, depending on the solvent (and likely the concentration), a different polymorph, nick-named ‘flower’, can be created through a spontaneous adsorption at the solid–liquid interface (Fig. 34).46 The molecules of trimesic acid in this flower polymorph are linked viaR22(8) synthons to form hexagonal macrocycles, which are then hold together viaR33(12) synthons. In the same work,46 Lackinger et al. have predicted existence of ‘superflower’ polymorph built solely with trimeric R33(12) synthons although they were not able to observe it. We have occasionally seen small domains of such superflower polymorph, also in STM images of trimesic acid at solid–liquid interface, and proposed an existence of a whole spectrum of 2D polymorphs of TMA built from R22(8) and R33(12) synthons.47 An elegant experimental evidence of this was provided by Yeet al. in a UHV STM investigation of self-assembly of trimesic acid on Au(111).48 It was found that formation of these polymorphs is coverage driven and can be controlled by adjusting the dosage of 34 (Fig. 34).
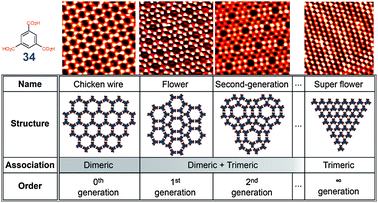 | ||
| Fig. 34 Set of polymorphic nets formed from trimesic acid 34via different combinations of R22(8) and R33(12) homosynthons (UHV-STM images and corresponding molecular models). Adopted from ref. 47 and 48. | ||
3.2 Catemers
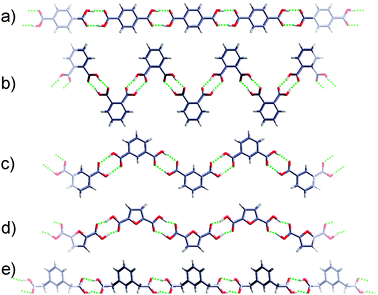
Finite acyclic associates of carboxylic group (such as dimers D, Scheme 1) are intrinsically less stable than cyclic homo-assemblies because they only partially utilize the H-bonding functionality of the COOH group. For example, acyclic dimer of acetic acid rapidly transforms into thermodynamically more stable R22(8) dimer even at 40 K.49 However, such dimers can be stabilized via additional H-bonding with solvent molecules (H2O, tetrahydrofuran, etc.) and as such are very common.51Infinite 1D homo-assemblies of carboxylic groups (catemers C(4), Scheme 1), are relatively more stable. All units of a catemer participate in H-bonding with stabilization energies comparable to that of R22(8). However much closer steric interactions in the former (between the groups X, Scheme 1) imposes a size limitation of the building block capable to self-associate in catemers. Indeed polymeric H-bonding is mainly observed for small building blocks such as formic and acetic acids, and a few derivatives of propiolic acid. Interestingly, it is also frequently found for 2,6-disubstituted benzoic acids50 which can be rationalized by the fact that in 2,6-disubstituted benzoic acids the COOH group is locked perpendicular to the aromatic ring and the resulting steric effect of aromatic core is much smaller than in the case of unsubstituted analogues.
When these steric requirements to the building block are met, the formation of a specific catemer on an R22(8) dimer is dictated by the considerations of packing efficiency. An elegant control of H-bonding of carboxylic group by means of secondary interactions (weak C–H⋯X and X⋯X interactions) was demonstrated in the design of helical catemers of substituted benzoic acids (Fig. 35).51
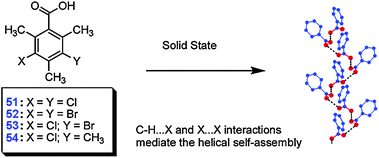 | ||
| Fig. 35 Helical catemers found in crystals of substituted benzoic acids 51–54. Reproduced from ref. 51 with permission. | ||
4. Self-assembly via heterosynthons
Since carboxylic groups can serve both as H-donors and H-acceptors, any sufficiently polar group can potentially form one or more types of H-bonding synthons with it. The main advantage of such hetero-synthons is that several geometrically different building blocks can be easily programmed into supramolecular structures, which are extremely difficult to achieve with homosynthons. In terms of energy, H-bonding interactions of carboxylic groups ranges from just 1–4 kcal mol−1 (e.g, for C(OH) = O⋯H–C interactions) to over 30 kcal mol−1 for charge-assisted H-bonding (e.g. COOH⋯(−)OOC). It is not possible to cover in this review in any details the diversity of heterosynthons and corresponding H-bonding interactions between carboxylic groups and other functionalities. However, within the scopes of presenting the supramolecular design strategies, it is important to classify these in two distinct cases: (i) very strong H-bonding without competitive alternatives, such as charge-assisted H-bonding52 and (ii) weaker H-bonding with two or more competitive interactions.An example of case (i) is a strong and selective H-bonding between amidine (–C(NR)–NHR) and carboxylic groups, that can be used to design multicomponent supramolecular structures, topology of which is tailored by changing one of the components (carboxylic acid) as illustrated in Fig. 36.53 Such complexity of the design is not generally achievable with homosynthons because of a large number of competitive interactions.
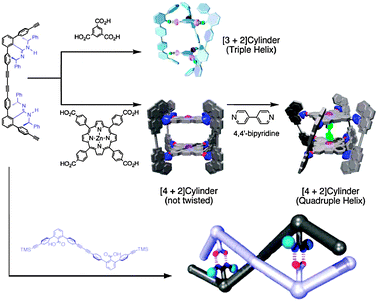 | ||
| Fig. 36 Complex supramolecular structures assembled in solutions viaR22(8) heterosynthon between amidine and carboxylic groups. Adopted from ref. 53 with permission. | ||
In the case of several competing synthons, the self-assembly pattern tends to follow an hierarchical principle: the best H-bond donor pairs with best H-bond acceptor, the second best donor with the second-best acceptor, etc. In some cases, it allows to predict the exact structure of a multicomponent assembly where each component contains several H-bonding functionalities (Fig. 37).54
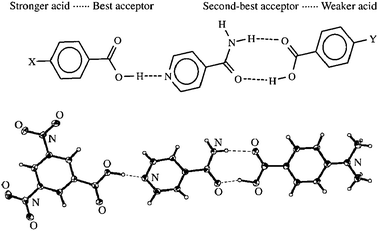 | ||
| Fig. 37 The hierarchy principle in crystal engineering of ternary H-bonded assembly. Reproduced from ref. 54 with permission. | ||
We also note that hybrid homo/heterosynthons can be formed when COOH groups simultaneously form links to one of its kind as well as to another H-bonding functionality, e.g. an alcohol group. We have recently used this mode of association in design of 2D molecular networks with tailored periodicity.55 When trimesic acid 34 is co-assembled with a long-chain alcohol at solid–liquid interface, the resulting supramolecular structure consists of homo-R22(8) synthons (which form dimerized tapes of trimesic acid) and hetero-R33(10) synthons that also binds the alcohol molecules (Fig. 38). This example shows how heterosynthons can be used to change dimensionality of assembly of carboxylic acids (from 2D for pure 34 to 1D for the 34:alcohol co-assembly) and rationally modulate the periodicity of the pattern (by adjusting the length of the alcohol).
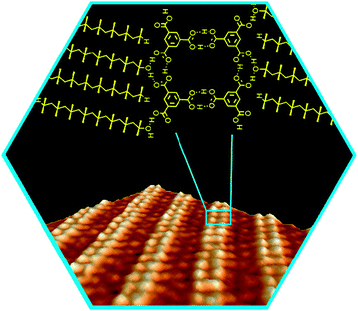 | ||
| Fig. 38 STM image and structural model of a molecular network formed at solid–liquid interface by association of trimesic acid 34 with aliphatic alcoholsviahomo-R22(8) and hetero-R33(10)-synthons. Reproduced with permission from K. G. Nath, O. Ivasenko, J. A. Miwa, H. Dang, J. D. Wuest, A. Nanci, D. F. Perepichka and F. Rosei, J. Am. Chem. Soc., 2006, 128, 4212. | ||
5. Examples of applications
5.1 Anchoring, ordering, immobilization
The specificity of the H-bonding interaction of carboxylic groups makes them useful for anchoring molecules of interest to the surface or nanoobjects. For example, it was employed in design of photoresponsive surfaces based on a light-switchable rotaxane linked to a self-assembled monolayer (SAM) of 11-mercaptoundecanoic acid on Au(111) (Fig. 39).56 The carboxylic groups of the SAM molecules form H-bonded hetero-dimers (D) with pyridine moieties resulting in a monolayer of randomly oriented rotaxane molecules. The UV irradiation switches the conformation of the rotaxane molecule altering macroscopic properties of the surface. The associated changes in the surface wettability allow transporting droplets of CH2I2 over the surface along the light exposure direction (Fig. 39, right).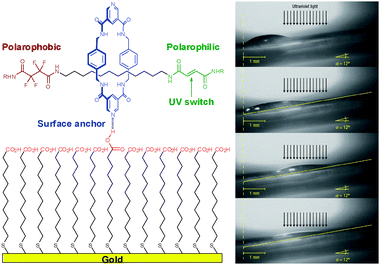 | ||
| Fig. 39 Droplet-transporting phenomenon enabled by photoswitchable rotaxane molecules immobilized by H-bonding on COOH-functionalized surface. Adopted from ref. 56 with permission. | ||
If the molecule of interest does not have strong H-bond donor or acceptor functionality it still might be possible to control its assembly using host–guest interactions within supramolecular H-bonding structures. For example, we have used the pores of TMA chicken wire network to order functional (semiconducting) molecules, octathio[8]circulene and tetrathiotetraseleno[8]circulene on a surface of graphite (Fig. 40).57 A very tight fit between the host and the guest freezes the orientation of the later and allows achieving an excellent sub-molecular resolution in room-temperature STM studies at solid–liquid interface.
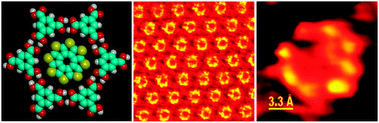 | ||
| Fig. 40 Immobilization of the heterocirculene semiconducting molecules within the H-bonded matrix of TMA (STM images at solid–liquid interface). Adopted with permission from ref. 57. | ||
Highly directional nature of the R22(8) synthons can be instrumental to design ordered assemblies of not only small molecules, but also –COOH functionalized nanostructures. This allowed for the first single crystal X-ray analysis of monodisperse gold nanoparticles (Au102). As depicted in Fig. 41, both the position and the orientation of the nanoparticles is fixed by mutliple R22(8)H-bonding of p-mercaptobenzoic acid ligands.58
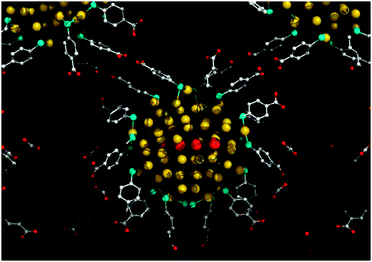 | ||
| Fig. 41 Single crystal structure of Au102(p-MBA)44 showing interparticle interactions mediated through hydrogen bonding between p-mercaptobenzoic acid (p-MBA) residues. Reproduced with permission from ref. 58. | ||
Simplicity and versatility of H-bonding based supramolecular synthesis found numerous applications in the design of various soft materials, including polymers59 and liquid crystals.60 Recently H-bonding of COOH- and pyridine-functionalized mesogens has been used to design supramolecular liquid crystals exhibiting a very wide temperature range of a so-called “blue” phase (Fig. 42).61
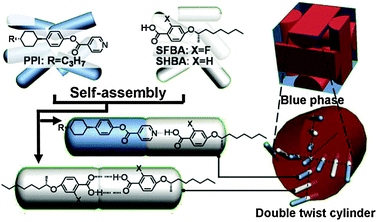 | ||
| Fig. 42 Schematic illustration of the formation of blue phase in the H-bonded LCs. Adopted from ref. 61 with permission. | ||
Carboxylic acids with a long hydrophobic tail also represent the simplest and the most renowned class of amphiphilic molecules (surfactants). Aggregation behavior of such amphiphiles is strongly pH-sensitive due to possibility of deprotonation of the carboxylic group. At high pH, when all carboxylic groups are ionized, aggregation is minimal due to significant electrostatic repulsion between COO−groups. At lower pH partial/full protonation of carboxylic anions decreases the electrostatic repulsion and restores the H-bonding interactions between the surfactant molecules leading to aggregation. Such pH-dependent behavior of the COOH group incorporated in peptidic amphiphiles can be utilized to control formation of various nanostructures, as shown by Stupp et al.62
5.2 Solid-state reactivity
Supramolecular interactions and H-bonding specifically have been extensively exploited to construct reactive co-crystals in which a topological arrangement of reactants allows to perform unusual and difficult chemical transformations.63,64The first topotactic single-crystal to single crystal (SCSC) polymerization of diacetylene-containing carboxylic acids, that leads to π-conjugated polymers, was pioneered by Wegner in 1960s.63 More recently, MacGillivray et al. used 1,8-naphthalenedicarboxylic acid to template assembly of trans-1-(3-pyridyl)-2-(4-pyridyl)ethylene in a co-facial orientation, via O–Hio⋯N H-bonding.64 UV-irradiation of thus preorganized molecules led to quantitative regio- and stereocontrolled [2+2] photodimerization yielding exclusively the head-to-head photoproduct (Fig. 43). Accommodating flexibility of a non-covalently binding template is important and even critical in achieving smooth single-crystal to single-crystal (SCSC) transformations.
![Template-controlled [2+2] photodimerization of trans-1-(3-pyridyl)-2-(4-pyridyl)ethylene: (a) ACOGEE, (b) ACOGOO in single crystals. Reproduced with permission from T. Friščič and L. R. MacGillivray, Making Crystals by Reactions in Crystals. Supramolecular Approaches to Crystal-to-Crystal Transformations within Molecular Co-Crystals, in Making Crystals by Design, ed. D. Braga and F. Grepioni, Wiley-VCH, Weinheim, 2007, pp. 176–192.](/image/article/2011/CS/c0cs00022a/c0cs00022a-f43.gif) | ||
| Fig. 43 Template-controlled [2+2] photodimerization of trans-1-(3-pyridyl)-2-(4-pyridyl)ethylene: (a) ACOGEE†, (b) ACOGOO† in single crystals. Reproduced with permission from T. Friščič and L. R. MacGillivray, Making Crystals by Reactions in Crystals. Supramolecular Approaches to Crystal-to-Crystal Transformations within Molecular Co-Crystals, in Making Crystals by Design, ed. D. Braga and F. Grepioni, Wiley-VCH, Weinheim, 2007, pp. 176–192. | ||
Not only topotactic reactions but solid state reactivity in general is controlled by mutual orientation of the molecules.65 It is thus not surprising that different polymorphs and even different faces of the same crystal can have different reactivity. For example, crystals of 4-aminobenzoic acid do not diazotize on the (001) face with NOCl since infinite hydrogen-bonded strings hinder migration pathways. Conversely, on (100) surface of the same crystals, where amino groups are freely available, the reaction occurs quite easily.66
5.3 Recognition
The design of synthetic molecules capable of enzyme-like recognition and binding to small substrates has been intensively studied since the beginning of supramolecular chemistry pioneered by Cram, Pedersen and Lehn (Nobel Prize 1987).67 In this respect, the carboxylic group can be thought of as one of the simplest recognition sites, as it usually requires simultaneous interaction with an H-bond donor and an H-bond acceptor centers in a specific geometry, for the most efficient binding. Two examples of such recognition enabled by COOH group are shown in Fig. 44.68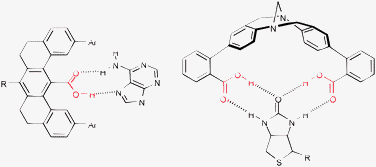 | ||
| Fig. 44 Examples of supramolecular receptors based on carboxylic acids. | ||
In turn, receptors containing H-bond donors and acceptors could be used to recognize the COOH groups and even specific carboxylic acids. This was shown by Moore et al. by examples of aminopyridine and aminopyrimidine derivatives which selectively and strongly (Ka > 106 M−1 in CDCl3) bind isophthalic acid (17) through hetero-R22(8) synthons (Fig. 45).69
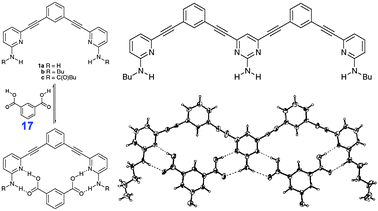 | ||
| Fig. 45 Receptors for recognition of isophthalic acids (17). Adopted from ref. 69 with permission. | ||
Many effects encountered in complex biological systems can be modeled using artificial receptors. An interesting positive allosteric effect was observed in bis(porphyrinate) double-decker 55 and used to detect chirality of dicarboxylic acids (Fig. 46).70 Binding a chiral diacid to this receptor dissymmetrizes its structure and increases the association constant for the diacids of the same charility. Collectively, it leads to an asymmetric induction that is manifested in an amplified circular dichroism (CD) signal in the spectral region of the porphirin chromophore.
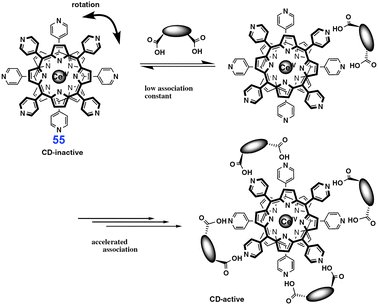 | ||
| Fig. 46 Chiral sensing of diacids by cerium(IV) bis(porphyrinate) double-decker 55. Reproduced from ref. 70 with permission. | ||
Unfortunately the above approach lacks generality in the sense that it requires a custom design, often involving multistep synthesis, for every substrate. Much more versatile approach is the so-called molecular imprinting which has been successfully used in the design of various chemical and biosensors.59,71 In a non-covalent version of this method, a functional monomer displaying specific non-covalent interactions with the target molecule, is polymerized in the presence of a template (the target molecule). During the polymerization process, the structure of the template is “memorized” (“imprinted”) in the resultant polymer providing the desired receptor site upon removal of the template (Fig. 47).
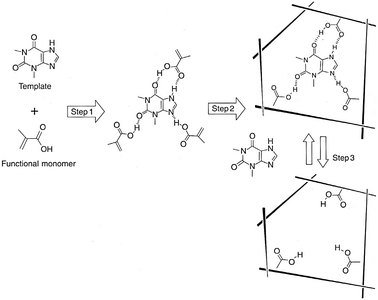 | ||
| Fig. 47 Noncovalent imprinting using theophylline drug molecule as a template. Step1: Preorganization of functional monomers through noncovalent interactions. Step 2: Polymerization of preorganized functional monomers. Step 3: Removal of the template. Reproduced with permission from M. Komiyama, in Molecular Imprinting: in Supramolecular Polymers, ed. A. Ciferri, CRC Press, Boca Raton, FL, 2nd edn, 2005, pp. 711–723. | ||
5.4 Catalysis
Catalysis is perhaps one of the most important aspects of chemical industry. Supramolecular chemistry offers interesting alternatives to the conventional design of catalytic systems, mimicking highly efficient mechanisms found in biological systems. The role of the COOH group self-assembly can vary from design of chiral auxiliaries,53 to the dynamic H-bonding that preferentially stabilizes a desired stereoisomer or an intermediate and is responsible for the catalytic activity, as realized in several organic catalysts, e.g.proline 56 (Fig. 48).72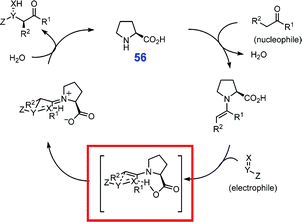 | ||
| Fig. 48 General mechanism of asymmetric addition reactions catalyzed by proline (56). H-bonded transition state responsible for the observed stereospecificity is shown in the red box. Reproduced from ref. 72 with permission. | ||
Enzymes use molecular recognition to orient a substrate in the active site, leading to full regio- and stereocontrol of the subsequent reaction. Borrowing this concept from the nature, Crabtree et al. have used R22(8)H-bonding of carboxylic groups to position remote C–H bonds of ibuprofen (a popular anti-inflammatory drug molecule) next to the oxidation site, thus facilitating the reaction and providing high regioselectivity (Fig. 49).73
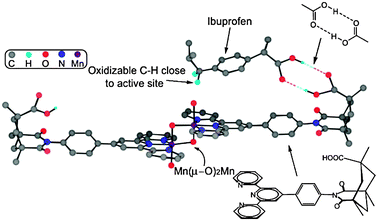 | ||
| Fig. 49 The structure of the Mn-terpyridine molecular recognition catalyst with ibuprofen substrate docked at the recognition site. Reproduced from ref. 73 with permission. | ||
Combination of such biomimetic strategy with H-bonding, controlled supramolecular assembly of reactants and industrially appealing molecular imprinting technology (mentioned above) resulted in development of ‘synthetic catalytic antibodies’ which selectively recognize the transition state of a reaction, providing high catalytic efficiency.71 Even a higher degree of functional complexity in COOH groups containing H-bonded supramolecular systems was realized in design of artificial self-replicating systems.74
6. Outlook
Molecular self-assembly as an enabling tool of Supramolecular Chemistry continues to gain importance as a key fabrication method (‘bottom-up’ approach) for nano-scale structures ranging from molecular electronics to regenerative medicine. In this review we took an example of one of the simplest and most accessible hydrogen bonding functionality, a carboxylic group, to expose the diversity of supramolecular architectures enabled by self-assembly. Tuning the (i) valency, (ii) symmetry and (iii) the size of the carboxylic acid building block, as well as (iv) secondary interactions (tailored by substituents), we showed control of the dimensionality and the exact topology of self-assembly. Supramolecular cycles, cages, linear and helical chains, ladders, sheets, diamond-like networks and other structures of 0D, 1D, 2D and 3D architectures can be created in this way. Co-assembly of carboxylic acids with other building blocks viahydrogen bonding, electrostatic or van-der-Waals interactions, provides another degree of freedom to design a targeted structure.Granted, the robustness of such control is not perfect and it is not always possible to select the desired architecture versus its many close-energy alternatives. The acidity of carboxylic acid may pose further limitations in certain systems. Nevertheless, we believe that the molecular structure—self-assembly motif relationships discussed in this paper can be applied to design new functional materials, as well as to predict supramolecular chemistry of other hydrogen bonding functionalities. Indeed, interchangeability between different synthons and building blocks is one of the great powers of supramolecular design. The principles discussed here for self-assemblyviaR22(8) homo-dimerization of carboxylic groups are also valid for self-assemblyvia other linear supramolecular synthons and even for some covalent systems (e.g, covalent organic frameworks).
Acknowledgements
We thank all our collaborators who have contributed to our own research in this area and acknowledge continuous support from the NSERC of Canada and FQRNT.References
- G. R. Desiraju, Crystal Engineering. The Design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed.
- V. R. Thalladi, B. S. Goud, V. J. Hoy, F. H. Allen, J. A. K. Howard and G. R. Desiraju, Chem. Commun., 1996, 401 RSC , and references therein.
- Cambridge Structural Database (CSD) from Cambridge Crystallographic Data Centre (http://www.ccdc.cam.ac.uk): F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 Search PubMed.
- L. Leiserowitz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 775 CrossRef.
- M. Lackinger and W. M. Heckl, Langmuir, 2009, 25, 11307 CrossRef CAS.
- Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. Mac-Nicol, F. Vogtle and J.-M. Lehn, Elsevier, Oxford, 1996, vol. 1–11, p. 8500 Search PubMed.
- C. V. K. Sharma, K. Panneerselvam, T. Pilati and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1993, 2209 RSC; C. B. Aakeröy, J. Desper and B. A. Helfrich, CrystEngComm, 2004, 6, 19 RSC , and references therein.
- M. Gdaniec, W. Jankowski, M. J. Milewska and T. Połoñski, Angew. Chem., Int. Ed., 2003, 42, 3903 CrossRef CAS.
- M. Polito, E. D'Oria, L. Maini, P. G. Karamertzanis, F. Grepioni, D. Braga and S. L. Price, CrystEngComm, 2008, 10, 1848 RSC.
- N. Yamamoto, T. Taga and K. Machida, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45, 162 CrossRef.
- A. T. ten Cate, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2004, 126, 3801 CrossRef , and references therein.
- Y. Miyake, A. Hosoda, M. Takagaki, E. Nomura and H. Taniguchi, Chem. Commun., 2002, 132 RSC.
- T. Yokoyama, T. Kamikado, S. Yokoyama and S. Mashiko, J. Chem. Phys., 2004, 121, 11993 CrossRef CAS.
- S. Valiyaveettil and K. Müllen, New J. Chem., 1998, 22, 89 RSC.
- S. Lei, M. Surin, K. Tahara, J. Adisoejoso, R. Lazzaroni, Y. Tobe and S. De Feyter, Nano Lett., 2008, 8, 2541 CrossRef CAS.
- J. Adisoejoso, K. Tahara, S. Okuhata, S. Lei, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7353 CrossRef CAS.
- M. Tichý, T. Kraus, J. Závada, I. Císařová and J. Podlaha, Tetrahedron: Asymmetry, 1999, 10, 3277 CrossRef CAS.
- J. E. Field, M. Y. Combariza, R. W. Vachet and D. Venkataraman, Chem. Commun., 2002, 2260 RSC.
- S. C. Zimmerman, F. Zeng, D. E. C. Reichert and S. V. Kolotuchin, Science, 1996, 271, 1095 CrossRef CAS.
- B. T. Ibragimov, K. M. Beketov and E. Weberc, J. Supramol. Chem., 2002, 2, 353 CrossRef CAS.
- S. L. Childs and K. S. Hagen, CrystEngComm, 2002, 4, 265 RSC.
- J. E. Field and D. Venkataraman, Chem. Commun., 2002, 306 RSC.
- E. Tamanini, G. D. Pantoş and J. K. M. Sanders, Chem.–Eur. J., 2010, 16, 81 CrossRef CAS , and refs therein.
- P. R. Ashton, M. C. T. Fyfe, S. K. Hickingbottom, S. Menzer, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.–Eur. J., 1998, 4, 577 CrossRef CAS.
- G. Mezei, J. W. Kampf and V. L. Pecoraro, New J. Chem., 2007, 31, 439 RSC.
- A. A. Salaudeen, C. A. Kilner and M. A. Halcrow, Chem. Commun., 2008, 5200 RSC.
- J. Lu, S. Lei, Q. Zeng, S. Kang, C. Wang, L. Wan and C. Bai, J. Phys. Chem. B, 2004, 108, 5161 CrossRef CAS.
- I. Goldberg and J. Bernstein, Chem. Commun., 2007, 132 RSC.
- A. I. Kitaigorodsky, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed; J. Perlstein, J. Am. Chem. Soc., 1994, 116, 11420 Search PubMed , and references therein.
- A. D. Burrows, Crystal Engineering Using Multiple Hydrogen Bonds in Structure and Bonding, Springer, New York, 2004, vol. 108, p. 55 Search PubMed.
- W. Xiao, X. Feng, P. Ruffieux, O. Gröning, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2008, 130, 8910 CrossRef CAS.
- J. M. MacLeod, O. Ivasenko, C. Fu, T. Taerum, F. Rosei and D. F. Perepichka, J. Am. Chem. Soc., 2009, 131, 16844 CrossRef CAS.
- S. V. Kolotuchin, P. A. Thiessen, E. E. Fenlon, S. R. Wilson, C. J. Loweth and S. C. Zimmerman, Chem.–Eur. J., 1999, 5, 2537 CrossRef CAS.
- F. Guo and K. D. M. Harris, J. Am. Chem. Soc., 2005, 127, 7314 CrossRef CAS , and references therein.
- S. Yoshimoto, N. Yokoo, T. Fukuda, N. Kobayashi and K. Itaya, Chem. Commun., 2006, 500 RSC.
- H. Zhou, H. Dang, J.-H. Yi, A. Nanci, A. Rochefort and J. D. Wuest, J. Am. Chem. Soc., 2007, 129, 13774 CrossRef CAS.
- M. O. Blunt, J. C. Russell, M. del Carmen Giménez-López, J. P. Garrahan, X. Lin, M. Schröder, N. R. Champness and P. H. Beton, Science, 2008, 322, 1077 CrossRef CAS.
- M. Blunt, X. Lin, M. del Carmen Gimenez-Lopez, M. Schröder, N. R. Champness and P. H. Beton, Chem. Commun., 2008, 2304 RSC.
- P. Holý, P. Sehnal, M. Tichý, J. Závada and I. Císařová, Tetrahedron: Asymmetry, 2003, 14, 245 CrossRef CAS.
- K. Kobayashi, T. Shirasaka, E. Horn and N. Furukawa, Tetrahedron Lett., 2000, 41, 89 CrossRef CAS.
- O. Ermer and A. Eling, Angew. Chem., Int. Ed. Engl., 1988, 27, 829 CrossRef , and references therein.
- A. Lazar, O. Danylyuk, K. Suwinska, F. Perret and A. W. Coleman, Chem. Commun., 2006, 903 RSC.
- M. Tsutsui, N. Kakimoto, D. D. Axtell, H. Oikawa and K. Asai, J. Am. Chem. Soc., 1976, 98, 8287 CrossRef CAS.
- Themed issue: Metal–organic frameworks, Chem. Soc. Rev., 2009, 38, 1201–1508.
- W. D. Xiao, Y. H. Jiang, K. Aït-Mansour, P. Ruffieux, H.-J. Gao and R. Fasel, J. Phys. Chem. C, 2010, 114, 6646 CrossRef CAS.
- M. Lackinger, S. Griessl, W. M. Heckl, M. Hietschold and G. W. Flynn, Langmuir, 2005, 21, 4984 CrossRef CAS.
- J. MacLeod, O. Ivasenko, D. F. Perepichka and F. Rosei, Nanotechnology, 2007, 18, 424031 CrossRef.
- Y. Ye, W. Sun, Y. Wang, X. Shao, X. Xu, F. Cheng, J. Li and K. Wu, J. Phys. Chem. C, 2007, 111, 10138 CrossRef CAS.
- M. Gantenberg, M. Halupka and W. Sander, Chem.–Eur. J., 2000, 6, 1865 CrossRef CAS.
- J. N. Moorthy and P. Natarajan, Cryst. Growth Des., 2008, 8, 3360 CrossRef CAS.
- J. N. Moorthy, R. Natarajan, P. Mal and P. Venugopalan, J. Am. Chem. Soc., 2002, 124, 6530 CrossRef CAS.
- M. D. Ward, Struct. Bonding, 2009, 132, 1 CAS.
- T. Hasegawa, Y. Furusho, H. Katagiri and E. Yashima, Angew. Chem., Int. Ed., 2007, 46, 5885 CrossRef CAS; H. Katagiri, Y. Tanaka, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2007, 46, 2435 CrossRef CAS.
- C. B. Aakeröy, A. M. Beatty and B. A. Helfrich, Angew. Chem., Int. Ed., 2001, 40, 3240 CrossRef CAS; A. Aakeröy and N. Schultheissin Assembly of Molecular Solids via Non-covalent Interactions: in Making Crystals by Design, ed. D. Braga and F. Grepioni, Wiley-VCH, Weinheim, 2007, pp. 209–240 Search PubMed.
- K. G. Nath, O. Ivasenko, J. M. MacLeod, J. A. Miwa, J. D. Wuest, A. Nanci, D. F. Perepichka and F. Rosei, J. Phys. Chem. C, 2007, 111, 16996 CrossRef CAS.
- J. Berná, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Pérez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704 CrossRef CAS.
- O. Ivasenko, J. M. MacLeod, K. Yu. Chernichenko, E. S. Balenkova, R. V. Shpanchenko, V. G. Nenajdenko, F. Rosei and D. F. Perepichka, Chem. Commun., 2009, 1192 RSC.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430 CrossRef CAS.
- W. H. Binder and R. Zirbs, Adv. Polym. Sci., 2007, 207, 1 CAS.
- T. Kato, N. Mizoshita and K. Kanie, Macromol. Rapid Commun., 2001, 22, 797 CrossRef CAS; C. M. Paleos and D. Tsiourvas, Liq. Cryst., 2001, 28, 1127 CrossRef CAS.
- W. He, G. Pan, Z. Yang, D. Zhao, G. Niu, W. Huang, X. Yuan, J. Guo, H. Cao and H. Yang, Adv. Mater., 2009, 21, 2050 CrossRef CAS.
- H. G. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1 CrossRef CAS.
- G. Wegner, Z. Naturforsch., 1969, 24b, 824 Search PubMed; J. W. Lauher, F. W. Fowler and N. S. Goroff, Acc. Chem. Res., 2008, 41, 1215 CrossRef CAS.
- L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D.-K. Bučar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280 CrossRef CAS.
- G. Kaupp, J. Phys. Org. Chem., 2008, 21, 630 CrossRef CAS.
- G. Kaupp, A. Herrmann and J. Schmeyers, Chem.–Eur. J., 2002, 8, 1395 CrossRef CAS.
- J.-M. Lehn, Science, 1993, 260, 1762 CrossRef CAS , and references therein.
- E. Fan and A. D. Hamilton, Molecular recognition: Synthetic Approaches to Artificial Receptors: in Crystal Engineering: The Design and Application of Functional Solids, ed. K. R. Seddon and M. Zaworotko, Kluwer Academic Publishers, Netherlands, 1999, pp. 357–369 Search PubMed.
- C. Bielawski, Y.-S. Chen, P. Zhang, P.-J. Prest and J. S. Moore, Chem. Commun., 1998, 1313 RSC.
- M. Takeuchi, T. Imada and S. Shinkai, Angew. Chem., Int. Ed., 1998, 37, 2096 CrossRef CAS.
- M. Patek and M. Drew, Curr. Opin. Chem. Biol., 2008, 12, 332 CrossRef CAS , and references therein.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS , and references therein.
- S. Das, G. W. Brudvig and R. H. Crabtree, Chem. Commun., 2008, 413 RSC.
- A. Vidonne and D. Philp, Eur. J. Org. Chem., 2009, 593 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full references to discussed crystal structures corresponding to the given CSD structure codes.3 See DOI: 10.1039/c0cs00022a |
| This journal is © The Royal Society of Chemistry 2011 |