Solvation structure of coronene–transition metal complex: a RISM-SCF study
Hirofumi
Sato
*,
Chisa
Kikumori
and
Shigeyoshi
Sakaki
Department of Molecular Engineering, Kyoto University, 615-8510 Japan. E-mail: hirofumi@moleng.kyoto-u.ac.jp
First published on 19th November 2010
Abstract
Coronene (C24H12) is a flat polyaromatic hydrocarbon consisting of seven peri-fused benzene rings and attracts lots of attention as a fragment of graphene. Using a hybrid method of quantum chemistry and statistical mechanics called RISM-SCF, which is an alternative to QM/MM, the electronic structure and solvation structure of a coronene–transition metal complex were computed in a self-consistent manner. The binding of a ruthenium complex ([C5H5Ru]+) was extensively studied, especially the changing of the solvation structure.
1. Introduction
Coronene consists of seven peri-fused benzene rings (Scheme 1), regarded as a small fragment of graphene, which has been attracting increased interest due to its huge potential in electronic applications.1 A class of carbon-based materials per se is promising, but numerous synthetic studies were also carried out on a novel class of compounds of polycyclic aromatic hydrocarbons (PAHs) with transition metal complexes because the functionalisation by the transition metal further expands the capability of the material.![(a) Structure of coronene, its unique sites and their numbering. (b) η6out complex and (c) η6in complex of [RuCp]+ and coronene.](/image/article/2011/CP/c0cp01464h/c0cp01464h-s1.gif) | ||
| Scheme 1 (a) Structure of coronene, its unique sites and their numbering. (b) η6out complex and (c) η6in complex of [RuCp]+ and coronene. | ||
Several interesting characteristics of the interaction between a carbon atoms and a metal have become clearer. For example, it is known that the metal is always bonded with two carbon atoms (η2) shared between two six membered rings in fullerene (C60) but η6-type coordination, in which the metal is located above the centre of the benzene ring, has never been observed yet. Recently, Alvarez et al. reported2 that η6–Cp*Ir complex of 1,2,5,6-tetramethylcorannulene exhibits migration of the Cp*Ir2+ unit on the surface of corannulene (C20H10), which is regarded as a curved-surface fragment of C60. On the other hand, several η6-type coordination complexes have been reported for flat polyaromatic hydrocarbons such as coronene.3–5 The bonding nature is basically governed by quantum chemistry, but solvation often plays essential role in reality. When a molecule is dissolved into solvent, the electronic structure is affected by the surrounding solvent molecules. It means the electronic structure (which is described by quantum chemistry) and the solvation structure (which is governed by statistical mechanics) are coupled with each other.
We have been developing RISM-SCF,6–9 which combines two ab initio methods in theoretical chemistry: one is the reference interaction site model (RISM),10,11 and the other is ab initio molecular orbital (MO) theory. The method determines the electronic structure and solvent distribution around a solute molecule in a self-consistent manner. It is regarded as an alternative to QM/MM method because of its capability to provide information on the electronic structure and microscopic solvation structure. However, one of the remarkable advantages of RISM-SCF is that it enables the use of highly sophisticated quantum chemical methods such as CCSD(T) due to its analytical treatment in statistical mechanics.12,13 RISM-SCF has been successfully applied to numerous molecular phenomena including chemical reactions, chemical equilibria, charge electron processes and so on.14 In this article, the electronic structure of coronene and its transition-metal complex is considered together with the solvation effect. We would like to emphasise that many of the experimental studies have been performed in the solution phase but the effect from solvent, especially on the transition metal, is not sufficiently understood. This is because a hybrid type computation such as QM/MM is generally too time-consuming to treat transition metal complexes at a reasonably accurate level of theory. Thanks to the analytical nature of the integral equation theory for a molecular liquid, RISM, the electronic structure described in high-level quantum chemical method is obtained together with information about solvation at the molecular level. The polarizable continuum model (PCM)15 is the popular method for an electronic structure study of solvation. Since we are focusing also on the changing of the solvation structure, RISM-SCF is preferable in the present study.
2. Methods
2.1 RISM-SCF theory
The RISM-SCF method compiles ab initio electronic structure theory and the statistical mechanical theory of molecular liquids. The total energy of the solvation system is defined as the sum of the quantum chemical energy of the solute (Esolute) and the solvation free energy (Δμ).7![[scr A, script letter A]](https://www.rsc.org/images/entities/char_e520.gif) = Esolute + Δμ = 〈Ψ|H0|Ψ〉 + Δμ, = Esolute + Δμ = 〈Ψ|H0|Ψ〉 + Δμ, | (1) |
| Ereorg = Esolute − Eisolated = 〈Ψ|H|Ψ〉 − 〈Ψ0|H|Ψ0〉, | (2) |
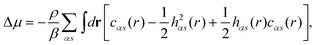 | (3) |
Applying the variational principle to eqn (1), the Fock operator of the RISM-SCF theory (Fsolv) including a solute–solvent interaction, V, is naturally derived.7
| Fsolv = Fgas + V. | (4) |
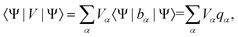 | (5) |
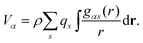 | (6) |
Recently, we developed a new-generation method of RISM-SCF called RISM-SCF-SEDD.9 In this method, auxiliary basis sets on each atom are prepared to divide electron density into the components assigned on each atom. The great advantage of the procedure is to directly treat spatial electron density distribution (SEDD); it does not require the set of grid points that was necessary to fit to electrostatic potential (ESP) and it is free from these artificial parameters. Furthermore the RISM-SCF-SEDD is quite robust in the computational procedure. The numerical stability significantly expands the versatility of the RISM-SCF family, for example, in treating molecules with buried atoms16 and transition metals.17
2.2 Computational details
The complex formation of [CpRu]+ (Cp = C5H5) to coronene in dichloromethane solvent (CH2Cl2) was considered in the present study. All the computations were performed with density functional theory using GAMESS program package (US version),18 which was modified by us to enable the RISM-SCF-SEDD method. B3PW91 functional was chosen as the exchange–correlation term. 6-31G(d) basis set (6d10f) was employed for C and H atoms, and ECP with the corresponding basis set parameters was used for the metal.19 The gas-phase geometries were adopted to highlight the solvation effect throughout the study.The RISM integral equation was solved with hyper-netted chain (HNC) closure with the Lennard-Jones parameters listed in Table 1. These for dichloromethane and for the solute C and H are standard ones taken from the literature.20 The Ru one was the same as the previous study.21 The density of dichloromethane was assumed to be 0.009339 molecules per Å. Temperature was taken to be 298.15 K.
3. Results
3.1 Energy change on the complex formation
On the complexation of [CpRu]2+ and coronene, two binding sites were found. One is η6out, in which the metal unit binds to a benzene ring of the edge site, and the other is η6in, in which the unit is located at the centre of the coronene. In both cases, the Cp-ring is coplanar with the π-surface of coronene with about 2.2 Å of Ru–C distance. Their formation energies are summarised in Table 2. The formation energy in solvent is calculated as the difference between the complex and infinitely separated coronene and [CpRu]2+.| Δ = ΔEsolute + ΔEreorg + ΔΔμ, | (7) |
values are −85.95 kcal mol−1 (η6out) and −75.11 kcal mol−1 (η6in), indicating that the binding is strengthened by the solvation. Unfortunately, it is difficult to judge which estimation is closer to the values in the real system, but GF is known to often show better agreement with observations.
| Gas phase | Dichloromethane | |||
|---|---|---|---|---|
| ΔEsolute | ΔEreorog | ΔΔμ | Total (Δ) |
|
| a All the values are given in kcal mol−1. | ||||
| η 6out | −81.92 | −0.39 | 17.49 | −64.82 |
| η 6in | −68.40 | −0.36 | 19.82 | −48.94 |
The MP2 computations in the gas phase were also performed to estimate the binding energy, showing considerable enhancements: −115.39 kcal mol−1 (η6in) and −125.13 kcal mol−1 (η6out). The estimated energy differences between the two isomers are 13.5 kcal mol−1 (DFT in gas phase), 15.9 kcal mol−1 (DFT in solution) and 9.7 kcal mol−1 (MP2 in gas phase). Unfortunately, the MP2 computation in solvent is infeasible but, based on these results, it is very likely that the energy difference is similar to the gas phase one. Anyway, the salient point is that [CpRu]2+ is η6-bonded to coronene in the edge position (η6out), consistent with experimental reports.3
The edge-preference may be simply understood in terms of charge distribution in π-system of coronene. Indeed, the edge-carbon atom (Cedge) is negatively charged compared to the central ones (Table 3). The edge-hydrogen atoms also show noticeable change on the formation. Interestingly the changes in Cvoid are considerable compared to the directly bonding Ccentre on the complexation of η6in. On the other hand, the change in η6out formation does not look so significant partly due to a simple averaging over all the atoms in different situations. The solvation effect in electronic structure is moderate except for the isolated [CpRu]+, in which all the atoms are exposed to solvent. Presumably, the changing in electronic structure on the formation is neutralised by the π-system. This is a reason why the energy difference between η6out and η6in in dichloromethane is essentially unchanged from the gas phase value (Table 2).
| [CpRu]+, C24H12a | η 6in | η 6out | ||||
|---|---|---|---|---|---|---|
| q gas | Δq | q gas | Δq | q gas | Δq | |
| a The first two rows are for the [CpRu]+ moiety and the remaining rows for the coronene moiety. qgas: Mulliken charge in gas phase. Δq: the difference of Mulliken charge in dichloromethane from the gas phase value. The averaged values of the equivalent atoms are listed for the C24H12 moiety. | ||||||
| Ru | 0.521 | 0.036 | 0.013 | −0.006 | −0.044 | −0.008 |
| Cp | 0.479 | −0.036 | 0.335 | 0.018 | 0.320 | 0.018 |
| Ccentre | 0.007 | −0.003 | −0.001 | −0.003 | 0.009 | −0.002 |
| Cvoid | 0.123 | −0.006 | 0.155 | −0.004 | 0.127 | −0.004 |
| Cedge | −0.230 | −0.006 | −0.226 | −0.003 | −0.214 | −0.004 |
| H | 0.165 | 0.011 | 0.204 | 0.006 | 0.206 | 0.006 |
The scheme of orbital interaction is another important viewpoint. As is well known, both donation (charge transfer from HOMO of PAH to the metal d-orbital) and back-donation (from the metal to LUMO of PAH) could occur between the metal and π-conjugated system, and the former is dominant in the present complex. The amplitude of coronene HOMO is localised at its edge-side, whose phase is matching with the metal's d-orbital (LUMO).
3.2 Migration of the Ru moiety on the coronene surface
It would be interesting to compare the planarity with the curved surface by seeing whether or not the migration occurs on the planar surface of coronene. Scheme 2 indicates the positions of the [CpRu]+ moiety on the surface corresponding to three local minima and two saddle points computed with DFT. All these points were checked through vibrational frequency calculations and two transition states (TSs) were confirmed, namely TS1 and TS2. The former is related to an isomerisation (a) from η6in to η6out, and the latter is located on the migration (displacement) path (b) between two different η6out. An intermediate (IM) was found on the path. Table 4 lists the computed energy of these structures together with MP2 computations.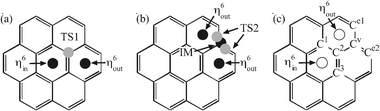 | ||
| Scheme 2 (a) Isomerisation from η6in to η6out; (b) displacement between η6out values. The gray circles correspond to transition state (TS1 and TS2), while the black ones are stable structures including intermediate (IM). (c) The labeling to specify the geometry. See Table 5. | ||
| η 6in | TS1 | IM | TS2 | |
|---|---|---|---|---|
| a Relative energies with respect to η6out given in kcal mol−1. Total energies of η6out are −1208.80070 Eh (DFT) and −1205.38496 Eh (MP2). b Imaginary frequencies obtained by the DFT computations. | ||||
| DFT (gas) | 13.52 | 40.21 | 33.48 | 33.65 |
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) im
b/cm−1
im
b/cm−1 |
— | (90.6i) | — | (67.8i) |
| DFT (sol) | 15.88 | 41.45 | 31.19 | 31.23 |
| MP2 (gas) | 9.74 | 49.83 | 51.22 | 51.01 |
The barrier height of the isomerisation path from η6in to η6out through TS1 is computed as 26.7 kcal mol−1 (DFT) and 40.1 kcal mol−1 (MP2) in the gas phase. In dichloromethane solvent, the barrier height is slightly decreased to 25.6 kcal mol−1 (DFT). Because of the great binding energy (Table 2), the value of TS1 energy is still lower than the reactant, namely separated coronene and [CpRu]+. At all events, η6in is considered to gradually isomerise to η6out. Note that the [CpRu]+ moiety does not cross over the C–C bond via η2-coordination, Instead, the η1-bond with Ccentre is the structure of TS1.
On the displacement path, an intermediate was found, in which the Ru moiety is located just above one of Cedge in the vicinity of a TS (TS2). The computed activation barriers are higher than those in the curved surface, corannulene complex. The barrier obtained by DFT is more than 30 kcal mol−1, and even higher by MP2 estimation. As seen in Table 5, the computed energies of IM and TS2 are very close to each other, though the MP2 energy of IM is slightly higher than that of TS2. We cannot rule out the possibility that IM is an artifact in the DFT computation, which might be replaced with TS. It should be stressed that both TS and IM are too high in energy to migrate through these structures in reality. The population analysis shows that the donation to the metal is relatively weak near the TSs compared to the corannulene case, which may be the reason for the high barrier in the present coronene system.
A set of selected bond distances between Ru and carbon atoms at the critical geometries are listed in Table 5. Though the symmetry is slightly broken because of the Cp ring, the shortest bond listed in the table clearly characterises the structure of each state. The Ru moiety in η6out and η6in are positioned at the centre of the benzene ring; all the Ccentre–Ru bonds are equivalent in η6in while the moiety is slightly shifted from the centre outward in η6out (Cedge–Ru is the shorter than Ccentre–Ru). As mentioned above, IM and TS2 are very close to each other in energy but their geometries are evidently different. The energy surface in this region is considered to be flat.
3.3 Solvation structure
Solvation structure is described based on the probability of finding a solvent molecule in the vicinity of a solute molecule. In the RISM-SCF, radial distribution function (RDF) represents a microscopic character of solvation.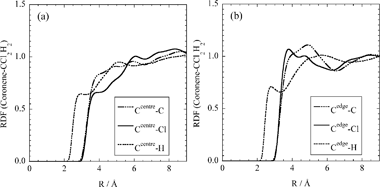 | ||
| Fig. 1 RDFs between coronene and dichloromethane solvent. (a) the central carbon atom (Ccentre) and (b) the edge carbon (Cedge). | ||
Fig. 2 is the RDF around Ru of [RuCp]+. Because of its positive charge, [RuCp]+ attracts solvent molecule, especially Cl atom, and a distinct peak appears around 4.0 Å. The peak corresponds to a direct contact of Ru and Cl according to their radius, (4.68 + 3.47)/2 = 4.08 Å (see Table 1). Due to the delocalisation of positive charge over the complex, Ru and Cp-ring share evenly the excess positive charge. As mentioned above, solvation slightly enhances the positive charge on Ru, probably caused by the specific solvation of Cl site. It should be noted, however, that the peak height is rather low, less than 2. The number of solvating Cl atom is estimated to be 3.3 by integration up to the minimum beyond the first peak (4.7 Å). Unfortunately, the enormously broad and obscure first peak of Ru–C RDF makes it difficult to directly estimate the coordination number of the solvent molecule.
![RDFs between Ru moiety and dichloromethane solvent. (a) the isolated [RuCp]+ and (b) the moiety in the complex of η6out and η6in.](/image/article/2011/CP/c0cp01464h/c0cp01464h-f2.gif) | ||
| Fig. 2 RDFs between Ru moiety and dichloromethane solvent. (a) the isolated [RuCp]+ and (b) the moiety in the complex of η6out and η6in. | ||
Fig. 2b illustrates the RDFs of Ru moiety in η6out and η6in complex. The rising of the RDF becomes moderated and the peak-top is distinctly shifted backward. This is caused by the exclusion of directly attaching solvent molecules on the complexation. As a result, the Ru atom is sandwiched by Cp and coronene, hence the solvent is allowed access to the Ru only through the interspace between the π-surfaces. Compared to η6in, the Ru moiety in η6out is exposed to the solvent, and more accessible. Hence the RDF of η6out is higher than that of η6in.
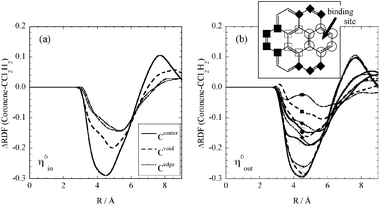 | ||
| Fig. 3 Change of RDF with carbon (dichloromethane) on the complexation of (a) η6in and (b) η6out; Ccentre (solid), Cvoid (dashed-dotted) and Cedge (dotted). The positions in η6out (right-hand side) are indicated with marks; for example, corresponding to the binding site. | ||
According to the RDFs shown above, the interaction between the solute (coronene, [RuCp]+ and their complex) and solvent is essentially governed by a simple excluded volume effect, and the solvation is readily replaced with the Ru moiety on the binding. Additionally, the effect from the solvent without a specific interaction such as hydrogen bonding does not strongly change the electronic structure of the solute.
4. Conclusions
In this contribution, the solvation structure and electronic structure of a coronene–transition metal complex were reported using a hybrid method of quantum chemistry and statistical mechanics for molecular liquid called RISM-SCF-SEDD. The solvation effect modifies the electronic structure of the solute molecule, and, at the same time, this modification affects the structure of the solvent molecules surrounding the solute. The present method enables us to evaluate the relative stability of isomers and the possibility of the migration among them.Because of the relatively mild solvation effect of dichloromethane, the relative energy among the isomers is almost unchanged upon solvation. In reality, it is likely that both of η6in and η6out are formed very quickly, but the latter is very stable. η6in is then gradually isomerised into η6outvia the barrier with about 27 kcal mol−1.
Acknowledgements
This work has been financially supported by Grant-in Aids (19350010, 20550013, 452-20031014 and 461), all supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) Japan. We acknowledge Marvin Jose Fernandez for his careful reading of the manuscript.References
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS.
- C. M. Alvarez, R. J. Angelici, A. Sygula, R. Sygula and P. W. Rabideau, Organometallics, 2003, 22, 624 CrossRef CAS.
- T. Jon Seiders, K. K. Baldridge, J. M. O'Conor and J. S. Siegel, J. Am. Chem. Soc., 1997, 119, 4781 CrossRef CAS.
- M. W. Stoddart, J. H. Brownie, M. C. Baird and H. L. Schmider, J. Organomet. Chem., 2005, 690, 3440 CrossRef.
- I. Chávez, A. Cisternas, M. Otero and E. Román, Z. Naturforsch., 1990, 45b, 658 Search PubMed.
- S. Ten-no, F. Hirata and S. Kato, Chem. Phys. Lett., 1994, 225, 202 CrossRef.
- H. Sato, F. Hirata and S. Kato, J. Chem. Phys., 1996, 105, 1546 CrossRef CAS.
- H. Sato, A. Kovalenko and F. Hirata, J. Chem. Phys., 2000, 112, 9463 CrossRef CAS.
- D. Yokogawa, H. Sato and S. Sakaki, J. Chem. Phys., 2007, 126, 244504 CrossRef.
- D. Chandler and H. C. Andersen, J. Chem. Phys., 1972, 57, 1930 CrossRef CAS.
- F. Hirata and P. J. Rossky, Chem. Phys. Lett., 1981, 83, 329 CrossRef CAS.
- K. Iida, D. Yokogawa, A. Ikeda, H. Sato and S. Sakaki, Phys. Chem. Chem. Phys., 2009, 11, 8556 RSC.
- S. Hayaki, K. Kido, H. Sato and S. Sakaki, Phys. Chem. Chem. Phys., 2010, 12, 1822 RSC.
- Molecular Theory of Solvation, ed. F. Hirata, Kluwer Academic Publishers, Dordrecht Hardbound, 2003 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS.
- K. Iida, D. Yokogawa, H. Sato and S. Sakaki, Chem. Phys. Lett., 2007, 443, 264 CrossRef CAS.
- S. Hayaki, D. Yokogawa, H. Sato and S. Sakaki, Chem. Phys. Lett., 2008, 458, 329–332 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111 CrossRef CAS; M. Couty and M. B. Hall, J. Comput. Chem., 1996, 17, 1359 CrossRef CAS.
- T. Fox and P. A. Kollman, J. Phys. Chem. B, 1998, 102, 8070 CrossRef CAS; W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M. Fergunson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179 CrossRef CAS.
- H. Sato and F. Hirata, J. Phys. Chem. A, 2002, 106, 2300 CrossRef CAS; H. Sato, I. Kawamoto, D. Yokogawa and S. Sakaki, J. Mol. Liq., 2007, 136, 194 CrossRef CAS.
| This journal is © the Owner Societies 2011 |