Redox chemistry in thin layers of organometallic complexes prepared using ion soft landing†
Wen-Ping
Peng
a,
Grant E.
Johnson
b,
Ivy C.
Fortmeyer‡
b,
Peng
Wang
b,
Omar
Hadjar
b,
R. Graham
Cooks
*c and
Julia
Laskin
*b
aDepartment of Physics, National Dong Hwa University, Hualien, 974, Taiwan, Republic of China
bChemical and Materials Sciences Division, Pacific Northwest National Laboratory, Richland, Washington 99352, USA. E-mail: Julia.Laskin@pnl.gov
cDepartment of Chemistry and Center for Analytical Instrumentation Development, Purdue University, 560 Oval Drive, West Lafayette, Indiana 47907, USA. E-mail: cooks@purdue.edu
First published on 12th November 2010
Abstract
Soft landing (SL) of mass-selected ions is used to transfer catalytically-active metal complexes complete with organic ligands from the gas phase onto an inert surface. This is part of an effort to prepare materials with defined active sites and thus achieve molecular design of surfaces in a highly controlled way. Solution-phase electrochemical studies have shown that VIVO(salen) reacts in the presence of acid to form VVO(salen)+ and the deoxygenated VIII(salen)+ complex—a key intermediate in the four electron reduction of O2 by vanadium–salen. In this work, the VVO(salen)+ and [NiII(salen) + H]+ complexes were generated by electrospray ionization and mass-selected before being deposited onto an inert fluorinated self-assembled monolayer (FSAM) surface on gold. A time dependence study after ion deposition showed loss of O from VVO(salen)+ forming VIII(salen)+ over a four-day period, indicating a slow interfacial reduction process. Similar results were obtained when other protonated molecules were co-deposited with VVO(salen)+ on the FSAM surface. In all these experiments oxidation of the VIII(salen)+ product occurred upon exposure to oxygen or to air. The cyclic regeneration of VVO(salen)+ upon exposure to molecular oxygen and its subsequent reduction to VIII(salen)+ in vacuum completes the catalytic cycle of O2reduction by the immobilized vanadium–salen species. Moreover, our results represent the first evidence of formation of reactive organometallic complexes on substrates in the absence of solvent. Remarkably, deoxygenation of the oxo-vanadium complex, previously observed only in highly acidic non-aqueous solvents, occurs on the surface in the UHV environment using an acid which is deposited into the inert monolayer. This acid can be a protonated metal complex, e.g. [NiII(salen) + H]+, or an organic acid such as protonated diaminododecane.
Introduction
Thin films of organometallic complexes on substrates play important roles in a variety of applications in heterogeneous catalysis and materials science.1–5 Preparation of catalytic substrates by immobilization of organometallic complexes on surfaces is advantageous because it combines the high selectivity of solution-phase catalysts with the ease of separation of products from catalyst that characterizes heterogeneous catalysts. Furthermore, enhanced catalytic activity observed for a number of organometallic complexes immobilized on surfaces indicates a clear pathway for controlled preparation of highly selective catalysts by proper tuning of molecule–substrate interactions.2 Other applications involving immobilization of organometallic complexes include synthesis of hybrid organic–inorganic polymers using molecular layer deposition6 and interface synthesis of layers for light-emitting diodes or coatings for biocompatible materials.3Preparation of organometallic films typically involves chemical vapor deposition or self-assembly from solution.7 Here we investigate the use of soft landing (SL) of mass-selected ions, i.e. ions of well-controlled mass, and hence chemical composition to create organometallic films. Ion SL instrumentation and various applications have been reviewed.8,9 The SL methodology is used to prepare high purity materials with well-defined structures and active sites and thus to achieve molecular design of surfaces in a highly controlled way. The ion deposition approach has advantages in relation to solution-phase techniques, avoiding clumping of materials seen as solvent evaporates and allowing deposition of uniform films while preserving the structure of deposited molecules.10–15
Our experiments present a first step towards the development of catalytically active organometallic materials using SL and also extend the work of others16–21 on ion deposition as an approach to catalyst preparation. Metal–salen complexes [salen = N,N′-ethylenebis(salicylideneaminato) ligand] were chosen as model systems in this study because they are effective homogeneous catalysts for the epoxidation of a variety of non-functionalized olefins with high levels of enantioselectivity,22 for ring-opening polymerization of six-membered cyclic carbonates,23 for the oxygenation of heteroatom containing organic compounds24 and for the hydroxylation of organic substrates.25 In a previous study, we showed that metal–salen complexes display redox activity in the gas phase.26 The protonated mixed complex [NiIIVIVO(salen) + H]+, generated by atmospheric pressure thermal desorption ionization, was found to undergo formal oxidation (hydrogen atom loss) to produce Ni(salen) monomer as the molecular radical cation upon collision-induced dissociation (CID). In this paper, we study redox chemistry in thin layers of vanadium–salen complexes initiated by the presence of a proton donor deposited from the gas-phase.
First studies of the reactivity of vanadium–salen complexes in strongly acidic solution were focused on understanding of vanadium bioaccumulation in blood cells of ascidians.27 Bonadies et al. demonstrated that in an acidic solution VIVO(salen) undergoes a disproportionation reaction shown in eqn (1):28
| 2VIVO(salen) + 2H+ → VIII(salen)+ + VVO(salen)+ + H2O | (1) |
| 2VIII(salen)+ + O2 → 2VVO(salen)+ + 4e− | (2) |
In this study we examine the immobilization and reactivity of VVO(salen)+ deposited on an inert fluorinated self-assembled monolayer surface (FSAM) on gold using SL of mass-selected ions. [We use the term immobilization to indicate that the landed species are bound to the surface strongly enough that desorption from the substrate is very slow.] Ions were generated by electrospray ionization (ESI) and guided to a mass analyzer where species with m/z values corresponding to the desired chemical entities were selected and gently deposited onto an FSAM surface. SAM surfaces are convenient substrates, the chemical properties of which can be easily tailored.33 It has been demonstrated that FSAM surfaces prevent neutralization of immobilized ions34 including metal–salen complexes that undergo very slow charge reduction through electron transfer to the surface.35 SL also allows convenient co-deposition of protonated ions which serve as proton donors on the surface enabling investigation of acid-mediated reactivity. Secondary ion mass spectrometry (SIMS) was chosen for the analysis of the SL prepared surfaces both before and after exposure to gaseous reagents. We demonstrate facile proton-initiated reduction of VVO(salen)+ to VIII(salen)+ in the absence of solvent and regeneration of VVO(salen)+ by exposure to O2 (Scheme 1). The role of a proton donor is examined by depositing protonated [NiII(salen) + H]+ complexes or protonated diaminododecane molecules into the SAM containing the VVO(salen)+ species.
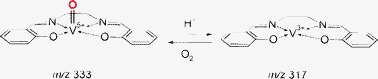 | ||
| Scheme 1 | ||
Experimental
The kinetics of transformation and reactivity of metal–salen complexes immobilized by soft-landing were examined using FT-ICR SIMS and TOF-SIMS as described below. The differences in the methods employed for SIMS analysis, viz. FT-ICR and TOF-SIMS discussed in detail in ref. 36, have no effect on the conclusions of this study.36FT-ICR SIMS experiments
The kinetics of transformation of soft-landed VVO(salen)+ in the presence of the [NiII(salen) + H]+ was examined using 8 keV Cs+ SIMS experiments in an FT-ICR mass spectrometer specially designed for studying ion–surface interactions.37,38 In these experiments [NiII(salen) + H]+ and VVO(salen)+ were simultaneously deposited from a mixed ion beam on the FSAM surface positioned at the rear trapping plate of the ICR cell. The experimental approach for SL studies has been described in detail elsewhere.36–39 Briefly, ions were produced in a high-transmission electrospray source, efficiently thermalized in the collision quadrupole and mass-selected prior to acceleration and collision with the surface. Ion kinetic energy was controlled by varying the voltage difference between the collisional quadrupole of the ion source and the surface and was maintained at 20 eV in these experiments. During ion SL the surface positioned at the rear trapping plate of the ICR cell was exposed to a continuous beam of mass-selected ions. A relatively wide mass window of ca. 15 amu was used for co-deposition of [NiII(salen) + H]+ and VVO(salen)+. The solution composition was optimized to generate a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 mixture of metal–salen complexes in the gas phase. Typical ion current of 7 pA was delivered onto a ca. 3.5 mm diameter spot on the target. The deposition time was 325 min. The maximum coverage of soft-landed ions obtained in these experiments does not exceed 15% of a monolayer, suggesting that the total ion dose is well below the SIMS saturation threshold.37
∶1 mixture of metal–salen complexes in the gas phase. Typical ion current of 7 pA was delivered onto a ca. 3.5 mm diameter spot on the target. The deposition time was 325 min. The maximum coverage of soft-landed ions obtained in these experiments does not exceed 15% of a monolayer, suggesting that the total ion dose is well below the SIMS saturation threshold.37
In situ analysis of surfaces following SL was performed by combining 8 keV Cs+ secondary ion mass spectrometry with FT-ICR detection of the sputtered ions (FT-ICR-SIMS) as described elsewhere.37–39 Static SIMS conditions with a total ion flux of about 2 × 1010 ions per cm2 (current 4 nA, duration 80 μs, spot diameter 4.6 mm, 15 shots per spectrum, 100–200 data points) were used in these experiments. Data acquisition was accomplished using a MIDAS data station.40
TOF-SIMS experiments
Ion deposition combined with TOF-SIMS analysis was performed using two different instruments. Initial soft-landing experiments followed by ex situTOF-SIMS characterization were performed using the apparatus described elsewhere41 while subsequent in situTOF-SIMS experiments were conducted using the ion deposition chamber coupled to a commercial TOF-SIMS instrument (PHI THIFT II, Physical Electronics, Eden Prairie, MN) that enables characterization of surfaces modified by soft-landing without breaking vacuum.36 In both instruments ions were generated and deposited on surfaces as described earlier.For ex situTOF-SIMS experiments mass-selected ion currents of 300 pA and 200 pA were obtained for VVO(salen)+ and [NiII(salen) + H]+, respectively. During the deposition (deposition times of 20 min and 30 min for VVO(salen)+ and [NiII(salen) + H]+, respectively) 2 × 1012 ions of each species were delivered onto a 5 mm diameter spot on the target. In situTOF-SIMS experiments utilized a different apparatus, in which 1012 ions of VVO(salen)+ and [NiII(salen) + H]+ were deposited onto a 3 mm spot. Ion currents of 50 pA and 20 pA were obtained and deposition times of 60 min and 120 min were used for VVO(salen)+ and [NiII(salen) + H]+, respectively.
In these experiments sequential deposition of metal–salen complexes on the FSAM surface was followed by characterization using 15 keV Ga+ SIMS and subsequent reactivity studies. Ex situTOF-SIMS analysis was performed after a brief exposure of surfaces to laboratory air because it took approximately 10 min to remove the sample from the soft-landing apparatus and introduce it into the TOF-SIMS.41In situTOF-SIMS experiments were performed ca. 5 min after soft-landing—the time required to transfer the surface from the ion deposition apparatus into the TOF-SIMS and position it on the stage. ESI conditions for ion soft-landing experiments were as follows: spray voltage, 3 kV; flow rate, 30 μL h−1; capillary temperature, 160°.
Self-assembled monolayer (SAM) surface
FSAM surfaces were prepared on gold coated silicon wafers. FT-ICR-SIMS experiments utilized a gold coated silicon wafer (5 nm chromium adhesion layer and 100 nm of polycrystalline vapor-deposited gold) from SPI Supplies (Westchester, PA) that was custom laser cut into 4.8 mm diameter substrates by Delaware Diamond Knives (Wilmington, DE). For TOF-SIMS experiments 10 × 10 mm gold substrates (525 μm thick Si, 50 Å Ti adhesion layer, 1000 Å Au layer) were purchased from Platypus Technologies (Madison, WI). 1H, 1H, 2H, 2H-perfluorodecanethiol was purchased from Sigma-Aldrich (St. Louis, MO) and used to form the FSAM surface by exposure of the gold surface to a 1 mM ethanol solution of the thiol for at least 12 h. The surface was removed from the thiol solution, ultrasonically washed in ethanol for 5 min to remove extra layers of the reagent, and dried under nitrogen gas before being introduced into the instrument.Chemicals
Ni(salen) and diaminododecane were purchased from Sigma-Aldrich (St. Louis, MO); RGDGG was purchased from Peptron. VVO(salen) was prepared according to the method reported by Choudhary et al.42 The metal–salen complexes were dissolved in a MeOH/H2O spray solvent to a concentration of 10−4 M. The solutions were analyzed using an HCT-Ultra ion trap (Bruker Daltonics, Billerica). Typical ESI mass spectra are shown in Fig. S1 (ESI†).Results and discussion
Evidence for VVO(salen)+ reduction to VIII(salen)+
Ex situ TOF-SIMS characterization was performed to examine the reactivity of VVO(salen)+ and [NiII(salen) + H]+ soft landed onto FSAM surfaces. Fig. 1 compares TOF-SIMS spectra obtained in two separate experiments, in which similar amounts of VVO(salen)+ and [NiII(salen) + H]+ were deposited onto different FSAM surfaces. In this figure the TOF-SIMS spectrum of the FSAM surface prior to ion deposition is shown as a black trace. The TOF-SIMS spectrum obtained following SL of VVO(salen)+ contains an abundant peak of VVO(salen)+ at m/z 333, a peak at m/z 666 corresponding to the [VIVOVV(salen)2]+ dimer ion, and a minor peak at m/z 317 assigned as VIII(salen)+. In this spectrum the small abundance of VIII(salen)+ is most likely produced as a result of gas-phase fragmentation of vibrationally excited VVO(salen)+ species in the SIMS plume. The TOF-SIMS spectrum obtained following SL of [NiII(salen) + H]+ contains the [NiII(salen) + H]+ ion at m/z 325, an abundant NiIII(salen)+ ion, the protonated [NiII2(salen)2 + H]+ dimer at m/z 649, and a number of minor features in the trimer region.![Comparison of ex situTOF-SIMS spectra obtained after the deposition of 2.2 × 1012 ions of VO(salen)+ (blue) and 1.6 × 1012 ions of [Ni(salen) + H]+ (red) on the FSAM surface in two separate experiments; black trace corresponds to the FSAM background.](/image/article/2011/CP/c0cp01457e/c0cp01457e-f1.gif) | ||
| Fig. 1 Comparison of ex situTOF-SIMS spectra obtained after the deposition of 2.2 × 1012 ions of VO(salen)+ (blue) and 1.6 × 1012 ions of [Ni(salen) + H]+ (red) on the FSAM surface in two separate experiments; black trace corresponds to the FSAM background. | ||
Sequential deposition of VVO(salen)+ and [NiII(salen) + H]+ onto the same FSAM surface yields a TOF-SIMS spectrum shown as the red trace in Fig. 2 that closely resembles the superposition of individual VVO(salen)+ and [NiII(salen) + H]+TOF-SIMS spectra shown in Fig. 1. However, the TOF-SIMS spectrum of the same surface re-examined after 4 days in UHV (blue trace in Fig. 2) shows reactivity of the deposited molecules, while the spectra of the individually deposited VVO(salen)+ and [NiII(salen) + H]+ do not change a great deal with time (data not shown). This reactivity is manifested by substantial increases in the abundances of VIII(salen)+ (m/z 317), [VIVOVIII(salen)2]+ (m/z 650), [NiIIVIII(salen)2]+ (m/z 641), [Ni2V(salen)3 + 3H]+ (m/z 968), and [Ni2VO(salen)3 + 2H]+ (m/z 983) ions in the TOF-SIMS spectrum recorded four days after ion deposition. In this paper we will limit the discussion to the monomeric species observed in SIMS spectra. Deposition and analysis of dimer ions will be discussed in a separate publication.
![Ex
situ
TOF-SIMS characterization of the FSAM surface before the deposition (black), 10 min after the sequential deposition of 2.5 × 1012 ions of VO(salen)+ and 2.5 × 1012 ions of [Ni(salen) + H]+ on the FSAM surface (red), and 4 days later (blue).](/image/article/2011/CP/c0cp01457e/c0cp01457e-f2.gif) | ||
| Fig. 2 Ex situ TOF-SIMS characterization of the FSAM surface before the deposition (black), 10 min after the sequential deposition of 2.5 × 1012 ions of VO(salen)+ and 2.5 × 1012 ions of [Ni(salen) + H]+ on the FSAM surface (red), and 4 days later (blue). | ||
The observed increase in the abundance of VIII(salen)+ with time and the concomitant decrease in the abundance of VVO(salen)+ could be attributed to a slow interfacial reduction of the VVO(salen)+ on the FSAM surface in UHV. It is remarkable that the large increase in the VIII(salen)+ peak is seen only when [NiII(salen) + H]+ is present on the surface, but such an effect is not observed in its absence.
Kinetics of VO(salen)+ reduction to V(salen)+
Further evidence for the interfacial reduction reaction was obtained from the kinetics experiment conducted using FT-ICR SIMS.37,39 In this experiment simultaneous deposition of VVO(salen)+ and [NiII(salen) + H]+ on FSAM in an ultrahigh vacuum system (<2 × 10−9 Torr) was examined using 8 keV Cs+ SIMS. Fig. 3a shows the composition of the ion beam used for SL experiments. The primary ion beam is dominated by VVO(salen)+ and [NiII(salen) + H]+ ions with a small amount of NiIII(salen)+ at m/z 324. SIMS spectra shown in Fig. 3(b–d) were recorded in situ during and after ion deposition. The SIMS spectra contain peaks corresponding to the deposited VVO(salen)+ and [NiII(salen) + H]+ ions at m/z 333 and m/z 325, respectively. In addition, the spectrum contains an abundant VIII(salen)+ peak at m/z 317 and the NiIII(salen)+ peak at m/z 324 that is significantly more abundant than the NiIII(salen)+ in the precursor ion beam.![(a) FT-ICR spectrum of the primary beam of a mixture of VVO(salen)+ and [NiII(salen) + H]+; FT-ICR-SIMS spectra of the FSAM surface after (b) 170 min and (c) 320 min of soft-landing (at the end of the deposition), (d) 87 h after the end of soft-landing, and (e) after exposure of the sample to laboratory air for 1 h.](/image/article/2011/CP/c0cp01457e/c0cp01457e-f3.gif) | ||
| Fig. 3 (a) FT-ICR spectrum of the primary beam of a mixture of VVO(salen)+ and [NiII(salen) + H]+; FT-ICR-SIMS spectra of the FSAM surface after (b) 170 min and (c) 320 min of soft-landing (at the end of the deposition), (d) 87 h after the end of soft-landing, and (e) after exposure of the sample to laboratory air for 1 h. | ||
The intensity ratio of VIII(salen)+ and VVO(salen)+, I(317)/I(333), increases from the value of 0.85 ± 0.15 at the beginning of the deposition to 2.0 ± 0.2 at the end of the deposition and continues to increase after the deposition is finished reaching the value of 3.8 ± 0.2 after 100 h (data not shown). A dramatic decrease in the I(317)/I(333) ratio is observed after exposure of the surface to laboratory air (Fig. 3e); the I(317)/I(333) ratio drops to a value of ca. 0.2.
VIII(salen)+ ions observed in the SIMS spectrum may be produced through several processes. First, a small fraction of VIII(salen)+ ions is produced as a result of dissociation of VVO(salen)+ in the SIMS analysis. The percentage of VIII(salen)+ fragments is expected to remain constant at low surface coverage used in our experiments. The upper limit for the amount of fragmentation in the FT-ICR-SIMS plume of 20% was estimated from the I(317)/I(333) ratio obtained after exposure of the FSAM surface to air. This value is higher than 3.5% fragmentation efficiency observed in the TOF-SIMS experiment (Fig. 1). Lower fragmentation efficiency in TOF-SIMS spectra was previously attributed to the shorter residence time (few microseconds) for ions in the TOF-SIMS as compared to hundreds of milliseconds residence time in FT-ICR SIMS experiments.43
The second process involves dissociation (crash landing)39,44,45 of VVO(salen)+ during deposition followed by trapping of the VIII(salen)+ fragment in the monolayer. Similar to dissociation in SIMS, the fraction of VIII(salen)+ ions resulting from crash landing should remain constant during ion deposition and cannot contribute to the observed increase in the yield of VIII(salen)+ after the deposition is complete. Finally, VIII(salen)+ can be produced through reactions between ions and neutral molecules trapped on the surface. Surface reactivity is the only process that can rationalize the increase in the I(317)/I(333) ratio after the end of ion deposition. From the above discussion it follows that a significant fraction of the VIII(salen)+ (m/z 317) is produced as a result of the loss of O from the VVO(salen)+ (m/z 333) deposited on the surface and the I(317)/I(333) ratio is a measure of the extent of the reduction of VVO(salen)+ to VIII(salen)+. A significant drop in the I(317)/I(333) ratio upon exposure of the surface to laboratory air (Fig. 3e) indicates a facile oxidation of VIII(salen)+ to VVO(salen)+ upon exposure to O2.
The abundance ratio of NiIII(salen)+ (m/z 324) and [NiII(salen) + H]+ (m/z 325) also increases with time. It is reasonable to assume that the NiIII(salen)+ ion is produced in SIMS through ionization of the neutral NiII(salen). Our previous studies demonstrated that FSAM surfaces are characterized by the highest efficiency for charge retention when protonated molecules are deposited on the substrate.43 We found that both instantaneous proton loss during ion deposition and a slow charge reduction process that continues for many hours after SL are responsible for neutralization of the deposited protonated species.38 It is reasonable to assume that [NiII(salen) + H]+ undergoes a similar slow charge reduction on the FSAM surface. The resulting neutral NiII(salen) complex is subsequently re-ionized in SIMS and observed as NiIII(salen)+ species.
Fig. 4a shows the time dependence of the I(317)/I(333) ratio. In the first 165 min of the kinetics experiment the ratio remains almost constant while an almost linear increase is observed after the deposition of ca. 7% of a monolayer of metal–salen species on the surface (4.5 × 1011 ions). This trend continues after the deposition is finished. Our results can be rationalized assuming that VVO(salen)+ deposited onto the FSAM surface is slowly converted into VIII(salen)+. The observed threshold behavior shown in Fig. 4a indicates that slow diffusion-limited interfacial reduction occurs on the inert FSAM surface and confirms that the formation of the reaction product, VIII(salen)+, is not associated with the SIMS analysis step.
![Time dependent intensities of (a) VIII(salen)+ (m/z 317), (b) [NiII(salen) + H]+ (m/z 325), and (c) NiIII(salen)+ (m/z 324) in FT-ICR SIMS spectra normalized to the intensity of the VVO(salen)+ (m/z 333). The dashed line shows the end of ion deposition.](/image/article/2011/CP/c0cp01457e/c0cp01457e-f4.gif) | ||
| Fig. 4 Time dependent intensities of (a) VIII(salen)+ (m/z 317), (b) [NiII(salen) + H]+ (m/z 325), and (c) NiIII(salen)+ (m/z 324) in FT-ICR SIMS spectra normalized to the intensity of the VVO(salen)+ (m/z 333). The dashed line shows the end of ion deposition. | ||
Role of proton donor
By analogy with the acid-mediated solution-phase reactivity of VIVO(salen) we hypothesized that the observed reduction of VVO(salen)+ is facilitated by the presence of a proton donor. To examine whether or not the loss of oxygen from VVO(salen)+ is a proton-mediated process, we conducted several experiments, in which VVO(salen)+ was deposited in sequence with different proton donors on the FSAM surface. Both the time and the order of deposition were varied in these experiments. The results indicated that the initial I(317)/I(333) ratio varies significantly as a function of the deposition time, the type of the proton donor, and the presence of [VIVO(salen) + H]+ that can be produced in the ESI source and co-deposited with VVO(salen)+.Table 1 lists the I(317)/I(333) ratios observed in TOF-SIMS spectra obtained in situ immediately after sequential deposition of 1012 ions of VVO(salen)+ and 1012 ions of a proton donor. The lowest initial yield of VIII (salen)+ was obtained when [NiII(salen) + H]+ was used as a proton donor. The I(317)/I(333) ratio of 0.5–0.7 was obtained in a number of repeated experiments. This ratio increases to 1.0 when VVO(salen)+ is co-deposited with [VIVO(salen) + H]+ which may also serve as a proton donor. Interestingly, the I(317)/I(333) ratio of 2.5–3.2 was obtained when soft-landing of VVO(salen)+/[VIVO(salen) + H]+ was followed by a fairly long deposition (2–3 h) of [NiII(salen) + H]+. However, a much lower ratio of 1.2 was obtained when the deposition time of [NiII(salen) + H]+ was reduced to 1.5 hours consistent with the presence of an initial induction period for the reduction reaction.
| Deposition 1 (ion) | Time/h | Deposition 2 (ion) | Time/h | I(317)/I(333) |
|---|---|---|---|---|
| a In these experiments both VVO(salen)+ and [VIVO(salen)+H] were produced in the ESI source and were not separated prior to deposition. | ||||
| VVO(salen)+ | 0.75 | [NiII (salen) + H]+ | 2 | 0.5 |
| VVO(salen)+ | 0.75 | [NiII (salen) + H]+ | 3 | 0.7 |
| VVO(salen)+ | 0.75 | [Diaminododecane + H]+ | 7 | 3.1 |
| [Diaminododecane + H]+ | 12 | VVO(salen)+ | 0.75 | 1.6 |
| VVO(salen)+ | 1 | [RGDGG + H]+ | 2 | 3.7 |
| [RGDGG + H]+ | 3 | VVO(salen)+ | 0.75 | 1.3 |
| VVO(salen)+/[VIVO(salen) + H]+a | 0.75 | — | — | 1.0 |
| VVO(salen)+/[VIVO(salen) + H]+a | 1 | [NiII (salen) + H]+ | 2 | 3.2 |
| VVO(salen)+/[VIVO(salen) + H]+a | 2 | [NiII (salen) + H]+ | 3 | 2.5 |
| VVO(salen)+/[VIVO(salen) + H]+a | 3 | [NiII (salen) + H]+ | 3 | 2.9 |
| VVO(salen)+/[VIVO(salen) + H]+a | 3 | [NiII (salen) + H]+ | 1.5 | 1.2 |
Deposition of VVO(salen)+ with protonated diaminododecane resulted in a fairly high initial ratio of 3.1. However, a much lower ratio of 1.6 was obtained when the order of the deposition was reversed and diaminododecane was deposited prior to VVO(salen)+. Similar results were obtained when a small protonated pentapeptide, RGDGG, was used as a proton donor. We attribute the observed dependence on the order of deposition to the shorter residence time of VVO(salen)+ on the surface when it is deposited after the proton donor.
Oxidation of VIII(salen)+ and regeneration of VVO(salen)+
The decrease in the I(317)/I(333) ratio observed following exposure of the FSAM surface to laboratory air (Fig. 3e) could be attributed to the oxidation reaction of VIII(salen)+ that regenerates VVO(salen)+ species. This process was further investigated using in situTOF-SIMS analysis of surfaces modified by SL in a recently constructed ion deposition apparatus coupled to a TOF-SIMS instrument. Fig. 5 shows TOF-SIMS spectra obtained after co-deposition of VVO(salen)+ and [VIVO(salen) + H]+ that could not be separated in our quadrupole mass filter without a significant loss of the ion current. Therefore, the only protons present on the monolayer are derived from [VIVO(salen) + H]+ as [NiII(salen) + H]+ was not deposited. As discussed earlier the initial I(317)/I(333) ratio of 0.88 indicates facile formation of VIII(salen)+ on the surface during SL. The ratio remains unchanged after four hours (Fig. 5b) in UHV (2 × 10−9 Torr). Exposure of the surface to 10−4 Torr of molecular oxygen for 20 min results in a significant depletion of the VIII(salen)+ peak and an increase in the VVO(salen)+ peak. The I(317)/I(333) ratio then remains constant during 65 h residence time in UHV. These results indicate that the reduction reaction resulting in formation of VIII(salen)+ is complete and all the [VIVO(salen) + H]+ ions that may serve as proton donors are consumed by the end of the ion deposition step and that exposure to oxygen converts a significant fraction of VIII(salen)+ into VO(salen)+. The fact that no further reduction of VVO(salen)+ occurs on the surface confirms that this process requires the presence of an additional proton donor deposited from the gas phase. | ||
| Fig. 5 In situ TOF-SIMS characterization of the FSAM surface (a) after the deposition of 1 × 1012 ions of VO(salen)+, (b) after 4 h in vacuum, (c) after exposure to 10−4 Torr of O2 for 20 min, (d) after 65 h in vacuum. | ||
Fig. 6a shows the I(317)/I(333) ratio derived from TOF-SIMS spectra obtained following deposition of VVO(salen)+ and [VIVO(salen) + H]+. As discussed earlier, the ratio changes only upon initial exposure to oxygen and remains constant when the system is kept in vacuum. In contrast, both the reduction of VVO(salen)+ to VIII(salen)+ and the oxidation of VIII(salen)+ to VVO(salen)+ by exposure to O2 were observed when VVO(salen)+ was co-deposited with [NiII(salen) + H]+. The cyclic regeneration of VVO(salen)+ upon repeated exposures to O2 is evident in the oscillating abundance ratios shown in Fig. 6b. Similar oxidation/reduction cycles were observed when protonated diaminododecane was used as a proton donor. The I(317)/I(333) ratio obtained in this experiment is shown in Fig. 6c. We note that the initial yield of VIII(salen)+ obtained after SL was quite high. Exposure to oxygen resulted in a significant decrease in the I(317)/I(333) ratio. Subsequent increase in the I(317)/I(333) ratio indicates that the reduction of VVO(salen)+ to VIII(salen)+ on the surface continues even after exposure of the surface to oxygen. This behavior is reproducible and continues for at least three cycles when [NiII(salen) + H]+ is used as a proton donor. However, the reactivity is clearly suppressed upon multiple exposures of the surface to oxygen. This is likely due to depletion of the population of [NiII(salen) + H]+ on the surface which results in a smaller number of proton donors, and consequently, a lower overall acidity in the monolayer.
![The intensity ratio of peaks at m/z 317 and m/z 333 observed in situTOF-SIMS spectra showing oxidation and reduction cycles on the FSAM surface for (a) VO(salen)+, (b) sequential deposition of VO(salen)+ and [Ni(salen) + H]+, (c) sequential deposition of the singly protonated diaminododecane and VO(salen)+. Individual points were obtained immediately after SL (labeled as End of SL), after one or multiple O2 exposures (labeled as O2), and at various times after each processing step, SL or O2 deposition (labels indicate the time between the corresponding processing step and the analysis). Lines are shown to guide the eye.](/image/article/2011/CP/c0cp01457e/c0cp01457e-f6.gif) | ||
| Fig. 6 The intensity ratio of peaks at m/z 317 and m/z 333 observed in situTOF-SIMS spectra showing oxidation and reduction cycles on the FSAM surface for (a) VO(salen)+, (b) sequential deposition of VO(salen)+ and [Ni(salen) + H]+, (c) sequential deposition of the singly protonated diaminododecane and VO(salen)+. Individual points were obtained immediately after SL (labeled as End of SL), after one or multiple O2 exposures (labeled as O2), and at various times after each processing step, SL or O2 deposition (labels indicate the time between the corresponding processing step and the analysis). Lines are shown to guide the eye. | ||
Comparison with solution-phase reactivity
It is remarkable that deoxygenation of the oxo-vanadium complex previously observed only in highly acidic non-aqueous solvents occurs on the surface in the UHV (0.5–2 × 10−9 Torr) environment.31,32 Bonadies et al. proposed that in a strongly acidic solution VIVO(salen) undergoes a disproportionation reaction (eqn (1)) forming VIII(salen)+ and VVO(salen)+.28 Tsuchida et al. demonstrated that VIII(salen)+ is the key intermediate responsible for the electroreduction of O2 in acidic nonaqueous solvents.46 In this process the vanadium–salen complex cycles are between the VIII(salen)+ and VVO(salen)+ states. However, the mechanism of the O2 electroreduction proposed in that study did not involve the disproportionation reaction but rather assumed the formation of doubly and triply charged monomers and dimers of VO(salen) and the reduced species. Because of the high Coulomb barrier for close approach of positively charged ions in the absence of solvent it is unlikely that such multiply charged intermediates are formed on the surface in our experiments.Liu and Anson re-examined the mechanism of the acid-mediated reactivity of VO(salen) in solution.47 They provided further support for the disproportionation reaction and showed that VIII(salen)+ in acetonitrile reacts with O2via the reaction shown in eqn (2). The reaction is fast and the conversion of VIII(salen)+ into VVO(salen)+ is quantitative. They also showed that the disproportionation reaction is a multistep process as shown in eqn (3) and (4).
| VIVO(salen) + 2H+ → VIV(salen)2+ + H2O | (3) |
| VIVO(salen) + VIV(salen)2+ → VIII(salen)+ + VVO(salen)+ | (4) |
From the above discussion it follows that the slow interfacial reactivity observed in this study could be attributed to reactions between the ionic and neutral species trapped on or in the FSAM. The reaction rate is determined either by the rate of migration of vanadium–salen complexes on the surface or by the rate of the proton loss by the protonated nickel–salen complex. Previous studies demonstrated that both small ions and organometallic clusters can be trapped between adjacent SAM chains.21,34 Penetration of the soft-landed species into the layer may significantly reduce the rate of migration and slow down the reaction. Alternatively, the reaction rate may be determined by the availability of protons in the layer. Previous work showed that the proton loss from protonated molecules soft-landed onto FSAM surfaces is rather slow.38 Similarly, the decay of the [Ni(salen) + H]+ signal observed in FT-ICR SIMS experiments occurs on a timescale of several hours.
Conclusions
This study presents a first step towards immobilization of catalytically active organometallic complexes onto suitable supports using soft-landing of mass-selected ions. The results presented here indicate that VVO(salen)+ ions soft-landed onto inert FSAM surfaces undergo slow proton-mediated reduction forming VIII(salen)+ species. The data show that reactivity continues for several days after the deposition, indicating that reaction is interfacial rather than beam-related. In situSIMS data suggest that the reduction reaction proceeds within the deposited monolayer and is characterized by an induction period, during which the ratio of VIII(salen)+ to VVO(salen)+ remains constant. The presence of the induction period indicates that the reduction reaction is diffusion-limited. The dependence of the initial yield of VIII(salen)+ observed after the deposition on the properties of the proton donor and its residence time on the surface indicates that SL of protonated molecules on inert FSAM surfaces can be used as a tool for controlled preparation of monolayers of varying acidity.The reverse reaction in which VIII(salen)+ is oxidized upon exposure to molecular oxygen regenerating the original VVO(salen)+ complex is fairly efficient and continues for several cycles. This cyclic regeneration of VVO(salen)+ was evident in the oscillating abundance ratios of VIII(salen)+ and VVO(salen)+. The yield of the reduction reaction is limited by the amount of protonated molecules co-deposited with VVO(salen)+ and the rate of the proton loss by the proton donor. Our results indicate that in the presence of proton donors soft-landed VVO(salen)+ initiates reduction of molecular oxygen that is similar to the catalytic electrochemical reduction of O2 observed in strongly acidic solutions. Although this study focused on a specific reduction reaction we believe that the SL methodology can be utilized for preparation of model catalysts of controlled chemical composition and hence, in conjunction with SIMS analysis and other surface characterization techniques, it should enable detailed fundamental studies of the surface reactivity of metal complexes on solid supports and perhaps allow screening and increased understanding of a wider variety of catalysts.48
Acknowledgements
This work was supported by grants from the Chemical Sciences Division, Office of Basic Energy Sciences (BES) of the U.S. Department of Energy, DE-FG02-06ER15807 (RGC), National Science Council NSC 96-2112-M-259-010-MY3 (WPP), and the grant from the Chemical Sciences Division of BES (JL). Ivy Fortmeyer acknowledges support from the DOE Science Undergraduate Laboratory Internship (SULI) program at Pacific Northwest National Laboratory (PNNL). The work was performed at the W. R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the U.S. DOE of Biological and Environmental Research and located at PNNL. PNNL is operated by Battelle for the U.S. DOE.References
- A. K. Kakkar, Chem. Rev., 2002, 102, 3579–3588 CrossRef CAS.
- J. M. Notestein and A. Katz, Chem.–Eur. J., 2006, 12, 3954–3965 CrossRef CAS.
- J. Schwartz and S. L. Bernasek, Catal. Today, 2001, 66, 3–13 CrossRef CAS.
- C. Coperet, M. Chabanas, R. P. Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef CAS.
- R. T. Baker, S. Kobayashi and W. Leitner, Adv. Synth. Catal., 2006, 348, 1337–1340 CrossRef CAS.
- S. M. George, B. Yoon and A. A. Dameron, Acc. Chem. Res., 2009, 42, 498–508 CrossRef CAS.
- M. Chabanas, A. Baudouin, C. Coperet and J. M. Basset, J. Am. Chem. Soc., 2001, 123, 2062–2063 CrossRef CAS.
- B. Gologan, J. M. Wiseman and R. G. Cooks, in Principles of Mass Spectrometry Applied to Biomolecules, ed. J. Laskin and C. Lifshitz, John Wiley and Sons, Hoboken, NJ, 2006 Search PubMed.
- V. Grill, J. Shen, C. Evans and R. G. Cooks, Rev. Sci. Instrum., 2001, 72, 3149–3179 CrossRef CAS.
- P. Wang, O. Hadjar, P. L. Gassman and J. Laskin, Phys. Chem. Chem. Phys., 2008, 10, 1512–1522 RSC.
- G. R. Blacken, M. Volny, T. Vaisar, M. Sadílek and F. Turecek, Anal. Chem., 2007, 79, 5449–5456 CrossRef CAS.
- S. Lee, C. Fan, T. Wu and S. L. Anderson, J. Am. Chem. Soc., 2004, 126, 5682–5683 CrossRef CAS.
- X. Tong, L. Benz, P. Kemper, H. Metiu, M. T. Bowers and S. K. Buratto, J. Am. Chem. Soc., 2005, 127, 13516–13518 CrossRef CAS.
- S. Rauschenbach, F. L. Stadler, E. Lunedei, N. Malinowski, S. Koltsov, G. Costantini and K. Kern, Small, 2006, 2, 540–547 CrossRef CAS.
- J. L. P. Benesch, B. T. Ruotolo, D. A. Simmons, N. P. Barrera, N. Morgner, L. Wang, H. R. Saibil and C. V. Robinson, J. Struct. Biol., 2010, 172, 161–168 CrossRef CAS.
- F. Zaera, J. Phys. Chem. Lett., 2010, 1, 621–627 Search PubMed.
- R. E. Winans, S. Vajda, G. E. Ballentine, J. W. Elam, B. Lee, M. J. Pellin, S. Seifert, G. Y. Tikhonov and N. A. Tomczyk, Top. Catal., 2006, 39, 145–149 CrossRef CAS.
- Z. Wang, S. Shingubara, H. Sakaue and T. Takahagi, J. Electrochem. Soc., 2005, 152, C684–C687 CrossRef CAS.
- W. E. Kaden, T. Wu, W. A. Kunkel and S. L. Anderson, Science, 2009, 326, 826–829 CrossRef CAS.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS.
- M. Mitsui, S. Nagaoka, T. Matsumoto and A. Nakajima, J. Phys. Chem. B, 2006, 110, 2968–2971 CrossRef CAS.
- J. El-Bahraoui, J. O. Wiest, D. Feichtinger and D. A. Plattner, Angew. Chem., Int. Ed., 2001, 40, 2073–2076 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS.
- N. S. Venkataramanan, G. Kuppuraj and S. Rajagopal, Coord. Chem. Rev., 2005, 249, 1249–1268 CrossRef CAS.
- J. T. Groves, W. J. Kruper, T. E. Nemo and R. S. Myers, J. Mol. Catal., 1980, 7, 169–177 CrossRef CAS.
- W.-P. Peng, M. Goodwin, H. Chen, R. G. Cooks and J. Wilker, Rapid Commun. Mass Spectrom., 2008, 22, 3540–3548 CrossRef CAS.
- M. J. Smith, D. Kim, B. Horenstein, K. Nakanishi and K. Kustin, Acc. Chem. Res., 1991, 24, 117–124 CrossRef CAS.
- J. A. Bonadies, W. M. Butler, V. L. Pecoraro and C. J. Carrano, Inorg. Chem., 1987, 26, 1218–1222 CrossRef CAS.
- Z. Liu and F. C. Anson, Inorg. Chem., 2000, 39, 274–280 CrossRef CAS.
- E. Tsuchida and K. Oyaizu, Coord. Chem. Rev., 2003, 237, 213–228 CrossRef CAS.
- E. Tsuchida, K. Yamamoto, K. Oyaizu, N. Iwasaki and F. C. Anson, Inorg. Chem., 1994, 33, 1056–1063 CrossRef CAS.
- K. Yamamato, K. Oyaizu and E. Tsuchida, J. Am. Chem. Soc., 1996, 118, 12665–12672 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS.
- S. A. Miller, H. Luo, S. J. Pachuta and R. G. Cooks, Science, 1997, 275, 1447–1450 CrossRef CAS.
- J. Laskin, P. Wang and O. Hadjar, J. Phys. Chem. C, 2010, 114, 5305–5311 CrossRef CAS.
- G. Johnson, M. Lysonski and J. Laskin, Anal. Chem., 2010, 82, 5718–5727 CrossRef CAS.
- J. Alvarez, R. G. Cooks, S. E. Barlow, D. J. Gaspar, J. H. Futrell and J. Laskin, Anal. Chem., 2005, 77, 3452–3460 CrossRef CAS.
- O. Hadjar, J. H. Futrell and J. Laskin, J. Phys. Chem. C, 2007, 111, 18220–18225 CrossRef CAS.
- J. Alvarez, J. H. Futrell and J. Laskin, J. Phys. Chem. A, 2006, 110, 1678–1687 CrossRef CAS.
- M. W. Senko, J. D. Canterbury, S. Guan and A. G. Marshall, Rapid Commun. Mass Spectrom., 1996, 10, 1839–1844 CrossRef CAS.
- O. Hadjar, P. Wang, J. H. Futrell, Y. Dessiaterik, Z. H. Zhu, J. P. Cowin, M. J. Iedema and J. Laskin, Anal. Chem., 2007, 79, 6566–6574 CrossRef CAS.
- N. Choudhary, D. L. Hughes, U. Kleinkes, L. F. Larkworthy, G. J. Leigh, M. Maiwald, C. J. Marmion, J. R. Sander, G. W. Smith and C. Sudbrake, Polyhedron, 1997, 16, 1517–1528 CrossRef.
- J. Laskin, P. Wang, O. Hadjar, J. H. Futrell, J. Alvarez and Z. G. Cooks, Int. J. Mass Spectrom., 2007, 265, 237–243 Search PubMed.
- Z. Ouyang, Z. Takats, T. A. Blake, B. Gologan, A. J. Guymon, J. M. Wiseman, J. C. Oliver, J. Davisson and R. G. Cooks, Science, 2003, 301, 1351–1354 CrossRef CAS.
- J. Shen, Y. H. Yim, B. Feng, V. Grill, C. Evans and R. G. Cooks, Int. J. Mass Spectrom., 1999, 182/183, 423–435 Search PubMed.
- E. Tsuchida, K. Oyaizu, E. L. Dewi, T. Imai and F. C. Anson, Inorg. Chem., 1999, 38, 3704–3708 CrossRef CAS.
- Z. Liu and F. C. Anson, Inorg. Chem., 2000, 39, 274–280 CrossRef CAS.
- X. Wang, N. Na, S. Zhang, Y. Wu and X. Zhang, J. Am. Chem. Soc., 2007, 129, 6062–6065 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Electrospray ionization mass spectra of solutions used in soft-landing experiments. See DOI: 10.1039/c0cp01457e |
| ‡ Undergraduate student from Columbia University |
| This journal is © the Owner Societies 2011 |