Empty level structure of boryl-substituted pentacyclic heteroaromatics
Alberto
Modelli
*ab and
Derek
Jones
c
aUniversità di Bologna, Dipartimento di Chimica “G. Ciamician”, via Selmi 2, 40126 Bologna, Italy. E-mail: alberto.modelli@unibo.it; Fax: +39 051 2099456; Tel: +39 051 2099522
bCentro Interdipartimentale di Ricerca in Scienze Ambientali (CIRSA), Università di Bologna, via S. Alberto 163, 48123 Ravenna, Italy
cISOF, Istituto per la Sintesi Organica e la Fotoreattività, C.N.R., via Gobetti 101, 40129, Bologna, Italy. E-mail: d.jones@isof.cnr.it
First published on 18th November 2010
Abstract
Extended π-conjugation in molecular systems is being extensively exploited in a rapidly expanding range of electronic and photonic applications. The modification of such extended systems through the use of heteroatoms allows their tailoring to specific requirements. In particular, the use of the vacant boron pπ orbital can increase the electron affinity of the extended π system in an analogous fashion to the p-doping of crystalline silicon used in more classical microelectronic devices. Experimental data on such modifications of the empty level structures of boron-containing π-electron materials leading to an increase in their electron affinity are not available in the literature. Using Electron Transmission Spectroscopy, the energies of the vertical anion formation of 5-methyl-2-furanboronic acid pinacol ester (1) and 4-methyl-3-thiopheneboronic acid pinacol ester (2) are measured and compared with those of the reference unsubstituted heteroaromatics furan and thiophene. The results are interpreted with the support of density functional theory (DFT) calculations with the B3LYP functional. DFT calculations are also used to predict the effects of boryl substituents on the electronic and geometrical structures of trans and cisthienyl thiazole.
Introduction
Polymeric organic compounds have been attracting a great deal of interest for several decades due to their remarkable electronic and optical properties. The potential uses of π-conjugated systems range from light emitting diodes, energy storage systems (batteries), nonlinear optical materials, electroluminescent devices, organic field-effect transistors, electrochromic windows.1,2Polymers deriving from aromatic and heteroaromatic compounds such as benzene, thiophene, and pyrrole increase their electrical conductivity by several orders of magnitude when properly p- or n-doped.3–7 More recently, the interest in organic semiconductors received a further boost with the use of metal-free dyes for photovoltaic devices, where the semiconducting materials between the electrodes are polymers or oligomers with reduced band gaps,8–10 or large π-systems combining conjugated donor and acceptor blocks.11–14The insertion of substituents can improve the π-system properties (solubility in common solvents, electronic structure, electrical conductivity) and provides an opportunity for modulating these properties by modifying the stereoelectronic effects of substituents. Among main-group heteroatom derivatives, three-coordinate organoboron compounds have recently emerged as a very promising class of materials extensively exploited in device and sensor applications. The incorporation of electron-deficient boryl groups into conjugated polymeric structures results in attractive features,15 such as unusual photoluminescent16 and electron-conducting properties.17–19 Organic compounds with fluorene as a π bridge and the dimesitylboryl group as an electron acceptor show strong two-photon excited blue fluorescence.20 Three-coordinate boron derivatives of conjugated organic molecules possess interesting linear and nonlinear optical properties, exhibit fluoride ion sensing properties,21 and can serve as emissive and/or electron-transport layers in organic light-emitting diodes and thin-film transistors.22–26
The peculiar properties of three-coordinate organoboron compounds can be traced back to the empty and relatively low-lying 2pπ orbital on the boron center. Its interaction with the empty π* orbitals of the organic system stabilizes the lowest unoccupied molecular orbital (LUMO), thus enhancing the electron affinity (EA). Because of the intrinsic donor properties (low ionization energy of the highest occupied molecular orbital, HOMO) of large conjugated π-systems, their boron derivatives can thus easily undergo polarized electronic transitions.
According to Wakamiya et al.,15,23 incorporation of boryl groups at the appropriate positions of a nitrogen-containing π-conjugated framework such as thienylthiazole, in addition to direct electronic effects, would allow B–N intramolecular coordination constraining the two pentacyclic rings into a coplanar cis conformation, thus causing a further EA increase.
In spite of the fact that the above-mentioned properties of organoboron compounds are associated with the electron-acceptor properties due to the electron-deficient boron atom, experimental data on the empty level structure of similar compounds and the EA increase produced by boron substitution on adjacent π-systems are not available in the literature. Only for borazine, to our knowledge, have such data been reported. An electron transmission and X-ray absorption spectroscopy study27 showed that the vertical EA (EAv) of B3N3H6 is almost 0.5 eV smaller than that of its carbon analogue benzene.
The electron transmission spectroscopy (ETS) technique devised by Sanche and Schulz28 is still one of the most suitable means for observing the formation of temporary anions in the gas phase and measuring negative EAvs. The ETS technique takes advantage of the sharp variations in the total electron-molecule scattering cross-section caused by resonance processes, namely, temporary capture of electrons with appropriate energy and angular momentum into empty MOs.29Electron attachment is rapid with respect to nuclear motion, so that temporary anions are formed in the equilibrium geometry of the neutral molecule. The measured vertical attachment energies (VAEs) are the negative of the EAvs.
In this ETS study the energies of vertical anion formation of 5-methyl-2-furanboronic acid pinacol ester (1) and 4-methyl-3-thiopheneboronic acid pinacol ester (2) are measured and compared with those of the reference unsubstituted heteroaromatics furan and thiophene. The results are interpreted with the support of density functional theory (DFT) calculations with the B3LYP functional. DFT calculations are also used to predict the effects of boryl substituents on the electronic and geometrical structures of trans and cisthienyl thiazole.
Experimental section
Our electron transmission apparatus is in the format devised by Sanche and Schulz28 and has been previously described.30 An electron beam energy selected with a trochoidal monochromator31 is directed through a gas cell containing a sufficient density of the target compound to partially attenuate the beam. The scattered electrons are rejected, while the transmitted current is collected. To enhance the visibility of the sharp resonance structures, corresponding to a dip in the current transmitted, the impact energy of the electron beam is modulated with a small ac voltage, and the derivative of the electron current transmitted through the gas sample is measured directly by a synchronous lock-in amplifier. Each resonance is characterized by a minimum and a maximum in the derivative signal. The energy of the midpoint between these features is assigned as the VAE. The spectra were obtained using the apparatus in the “high-rejection” mode32 and are, therefore, related to the nearly total scattering cross-section. The electron beam resolution was about 50 meV (fwhm). The energy scale was calibrated with reference to the (1s12s2)2S anion state of He. The estimated accuracy is ±0.05 or ±0.1 eV, depending on the number of decimal digits reported. The samples (commercially available) were heated to about 50 °C to obtain a suitable vapour pressure.Ab initio HF and B3LYP calculations for geometry optimization and evaluation of the orbital energies and total energies were performed using the Gaussian 03W Rev.D.0133 program package. The EAv was calculated as the difference of the total energy of the neutral and the lowest anion state, both in the optimized geometry of the neutral state, using the B3LYP hybrid functional34 with the standard 6-31G(d) and 6-31+G(d) basis sets. The adiabatic EA (EAa) was obtained as the energy difference between the neutral and the lowest anion state, each in its optimized geometry.
Results and discussion
Empty level structure: ET spectra, calculated VAEs and EAs
Compounds 1 and 2, as well as their parent compounds furan and thiophene, possess two empty antibonding π* MOs. Fig. 1 displays a representation of the two lowest unoccupied π* MOs of thiophene (of b1 and a2 symmetry, respectively, in the C2v point group notation), as supplied by B3LYP/6-31G(d) calculations. Essentially equivalent results are obtained with Hartree–Fock (HF) calculations. Similar localization properties are displayed by the π* MOs of furan. In addition, because of the presence of a third-row element (sulfur atom), thiophene also possesses a low-energy σ* MO, mainly localized on the sulfur atom and the two adjacent carbon atoms. In the ET spectrum of thiophene the resonance associated with electron capture into this σ* MO occurs around 2 eV,35 but is obscured by the low-energy side of the more intense π*2 resonance, while a distinct σ* resonance is observed in its aza derivatives isothiazole and thiazole.35 The B3LYP/6-31G(d) virtual orbital energies (VOEs) correctly predict the energy of the lowest σ* MO of thiophene to be intermediate between those of the two π* MOs (see Table 1). By contrast, in furan the first σ* MO is predicted to lie well above the second π* MO and to possess different localization properties (mainly CH character, with small contributions from the heteroatom).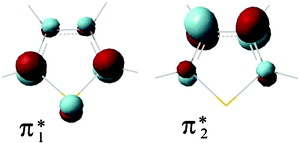 | ||
| Fig. 1 Representation of the two lowest unoccupied π* MOs of thiophene, as supplied by B3LYP/6-31G(d) calculations. | ||
| MO | VOE | Scaled VOE | Expt. VAE | |
|---|---|---|---|---|
| a VAEs from ref. 35. | ||||
| Furan a | σ*CH | 3.197 | ||
| π*2 | 2.139 | 2.93 | 3.15 | |
| π*1 | 0.537 | 1.64 | 1.73 | |
| 2-Methylfuran | σ*CH | 3.027 | ||
| π*2 | 2.286 | 3.05 | ||
| π*1 | 0.634 | 1.72 | ||
| 5-Methyl-2-furan boronic acid pinacol ester (1) | σ*CH | 2.030 | ||
| π*2 | 1.868 | 2.72 | 2.87 | |
| π*1 | −0.187 | 1.07 | 1.05 | |
| Thiophene a | π*2 | 1.945 | 2.78 | 2.63 |
| σ*CS | 0.966 | |||
| π*1 | −0.207 | 1.04 | 1.15 | |
| 3-Methylthiophene | π*2 | 1.964 | 2.79 | |
| σ*CS | 0.999 | |||
| π*1 | −0.171 | 1.07 | ||
| 4-Methyl-3-thiopheneboronic acid pinacol ester (2) | σ*CS | 1.122 | ||
| π*2 | 0.975 | 2.00 | 1.92 | |
| π*1 | −0.354 | 0.93 | 0.82 |
Fig. 2 reports the ET spectra of the boryl derivatives 1 and 2. The measured VAEs are given in Table 1 and in the diagram of Fig. 3, together with those of furan and thiophene for comparison. The ET spectra of 1 and 2 display two intense resonances associated with electron capture into the empty π*1 and π*2 MOs, as in their corresponding unsubstituted pentacyclic parents furan and thiophene.
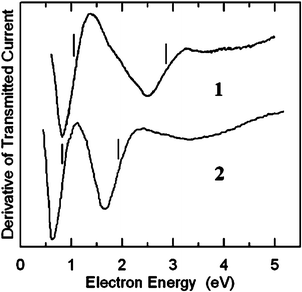 | ||
| Fig. 2 Derivative of transmitted current, as a function of electron energy, in gas-phase 5-methyl-2-furanboronic acid pinacol ester (1) and 4-methyl-3-thiopheneboronic acid pinacol ester (2). Vertical lines locate the VAEs. | ||
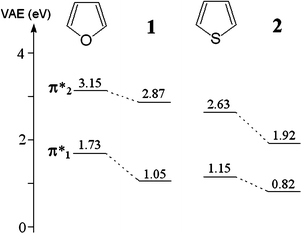 | ||
| Fig. 3 Correlation diagram of the VAEs (eV) measured in the boryl derivatives 1 and 2 and the reference molecules furan and thiophene. | ||
An adequate approach for describing unstable anion states involves difficulties not encountered for neutral or cation states.36–39 The most correct method is, in principle, the calculation of the total scattering cross-section with the use of continuum functions, but complications arise from the lack of an accurate description of the electron–molecule interactions.40EAs can be obtained as the difference between the energy of the lowest anion state and that of the neutral ground state, both at the optimized geometry of the neutral species (EAv) or each with its optimized geometry (EAa). A proper description of the spatially diffuse electron distributions of anions normally requires a basis set with diffuse functions.41,42 However, calculated anion state energies decrease as the basis set is expanded, so that the choice of a basis set which gives a satisfactory description of the energy and nature of the anion is a priori not obvious.43 In general, the more unstable the anion state, the greater the need to augment the basis set with diffuse functions, increasing the risk that the singly occupied MO (SOMO) of the anion is described as a diffuse function with no physical significance with respect to anion formation.36,37,43,44
It has, however, been demonstrated36,38 that good linear correlations can be obtained between the π*C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C VAEs measured in unsaturated hydrocarbons and the corresponding virtual orbital energies (VOEs) of the neutral molecules obtained with simple HF calculations, using basis sets which do not include diffuse functions. More recently, it has been shown43 that the π* VOEs obtained with DFT B3LYP/6-31G(d) calculations also supply a good linear correlation with the corresponding VAEs measured over a variety of unsaturated compounds, including hetero-substituted hydrocarbons. A more accurate correlation is expected if the scaling equation is calibrated with “training” compounds structurally close to the subject molecule. For this reason, the use of the B3LYP scaling43 (VAE = 0.8054 × VOE + 1.2110) to predict the π* VAEs of 1 and 2 seems to be more appropriate.
C VAEs measured in unsaturated hydrocarbons and the corresponding virtual orbital energies (VOEs) of the neutral molecules obtained with simple HF calculations, using basis sets which do not include diffuse functions. More recently, it has been shown43 that the π* VOEs obtained with DFT B3LYP/6-31G(d) calculations also supply a good linear correlation with the corresponding VAEs measured over a variety of unsaturated compounds, including hetero-substituted hydrocarbons. A more accurate correlation is expected if the scaling equation is calibrated with “training” compounds structurally close to the subject molecule. For this reason, the use of the B3LYP scaling43 (VAE = 0.8054 × VOE + 1.2110) to predict the π* VAEs of 1 and 2 seems to be more appropriate.
Table 1 compares the scaled π* VOEs with the corresponding measured VAEs of furan, thiophene and their boryl derivatives 1 and 2. There is a nice agreement (generally within ±0.1 eV) between the calculated and experimental VAEs. The scaled VOEs of 2-methylfuran and 3-methylthiophene (see Table 1) show that the stabilization of the π* anion states on going from furan and thiophene to 1 and 2, respectively, is completely due to the boryl substituents, the methyl substituents causing a very small effect in the opposite direction. A representation of the LUMO localization properties of 1 is given in Fig. 4.
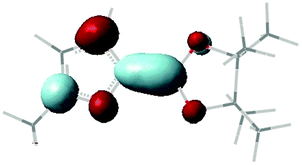 | ||
| Fig. 4 Representation of the LUMO localization properties of 1, as supplied by B3LYP/6-31G(d) calculations. | ||
Table 2 reports the calculated energies of the first vertical and adiabatic anion states of 1 and 2 relative to the corresponding neutral ground states. As expected, the minimal basis set (6-31G(d)) overestimates the anion energies. In contrast, the 6-31+G(d) basis set, which includes the smallest addition of diffuse functions, overestimates the EAv of both compounds by about 0.2 eV. Moreover, the solution associated with the SOMO of the vertical anion state of 1 (lying 0.780 eV above the neutral ground state) is described as a diffuse function. The value (0.866 eV) reported in Table 2 was obtained by adding the energy (0.086 eV) of the first excited state of the vertical anion, as obtained with TD-B3LYP calculations,45 where the extra electron resides in the lowest valence π* MO.
According to the B3LYP optimized geometries of 1 and 2 (with both the 6-31G(d) and 6-31+G(d) basis sets) the B–O bonds and the pentacyclic ring are nearly coplanar, so that the ring π-system mixes with the pπ orbital of the boron atom. Fig. 3 clearly shows that the stabilizing effects caused on the π*1 and π*2 MOs by the boryl substituent are different. The LUMO of furan is stabilized by 0.7 eV, while the π*2 MO only by 0.3 eV. The π* MOs of thiophene are also stabilized by 0.7 and 0.3 eV, but in this case the larger effect is exerted on the π*2 MO. The different stabilizing effects can be easily explained on the basis of the wavefunction coefficient of each MO on the site of substitution (see Fig. 1). On this point, it is to be noted that in the dimers (and oligomers) position (2), where the LUMO has the largest coefficient, is normally used for ring coupling. However, the calculated localization properties show that on going from the monomers to the dimers the relative magnitude of the LUMO wavefunction coefficient in position (3) (available for a substituent group) increases.
As a final comment, according to the DFT calculations, not only does the boryl substituent cause a large stabilization of the b1 (π*) LUMO of furan and thiophene but also acts in the opposite direction on the a2 (π) HOMO, destabilized by 0.36 eV in furan and 0.28 eV in thiophene (at the B3LYP/6-31G(d) level). Therefore, boryl substitution causes an energy decrease (1.19 eV from furan to 1 and 0.73 eV from thiophene to 2) of the first π → π* singlet excited state even larger than the EA increase.
Yamaguchi et al.15,23 have recently proposed (3-boryl-2-thienyl)-2-thiazole (3, see Chart 1) as a π-conjugated building unit for electron-transporting materials. Aza substitution is expected to increase the EA (with respect to the dithiophene analogue), and in the cis isomer the same effect would be produced by the Lewis base/Lewis acid (N → B) intramolecular interaction, both by constraining the π-conjugated framework into a planar configuration and affecting the electronic structure by lowering the LUMO level.15 Consistently, cyclic voltammetry showed15 that the reduction potential of (3-BR2-2-thienyl)-2-thiazole, with R = mesityl, is about 0.5 eV lower than that of 2-thienyl-2-thiazole.
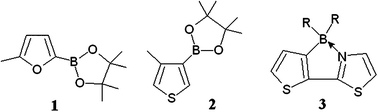 | ||
| Chart 1 | ||
Table 3 reports the B3LYP/6-31G(d) energies of the first three empty MOs and the corresponding scaled π* VOEs of trans-2,2′-dithiophene, trans and cis 2-thienyl-2-thiazole and their 3-BR2 derivatives, where R is a methyl (Me) or a phenyl (Ph) group. According to the calculations the trans conformers of the thiophene dimer and 2-thienyl-2-thiazole are slightly more stable (0.025 and 0.055 eV, respectively) than the corresponding cis conformers, and in both the trans and cis conformers of 2-thienyl-2-thiazole the thiophene and thiazole rings are coplanar.
| MO | VOE | Scaled VOE | |
|---|---|---|---|
| trans 2,2′-Thiophene dimer | σ*CS | 0.678 | |
| π*2 | 0.494 | 1.61 | |
| π*1 | −1.245 | 0.21 | |
| trans-2-Thienyl-2-thiazole | σ*CS | 0.380 | |
| π*2 | 0.319 | 1.47 | |
| π*1 | −1.573 | −0.06 | |
| cis-2-Thienyl-2-thiazole | π*2 | 0.421 | 1.55 |
| σ*CS | −0.122 | ||
| π*1 | −1.572 | −0.06 | |
| trans-(3-B(Me)2-2-thienyl)-2-Thiazole | π*3 | 0.260 | 1.42 |
| π*2 | −0.539 | 0.78 | |
| π*1 | −1.782 | −0.22 | |
| cis-(3-B(Me)2-2-thienyl)-2-Thiazole | π*2 | 0.149 | 1.33 |
| σ*CS | −0.258 | ||
| π*1 | −1.791 | −0.23 | |
| trans-(3-B(Ph)2-2-thienyl)-2-Thiazole | π*ben | −0.146 | 1.09 |
| π*tien | −1.212 | 0.23 | |
| π*B/tien/ben | −2.012 | −0.41 | |
| cis-(3-B(Ph)2-2-thienyl)-2-Thiazole | π*tien | −0.036 | 1.18 |
| σ*CS | −0.418 | ||
| π*tien | −2.009 | −0.41 |
The scaled energy (0.21 eV) of the π* LUMO of dithiophene is in excellent agreement with the measured6 first VAE (0.2 eV), those (−0.06 eV) of trans and cis 2-thienyl-2-thiazole are equal to each other and indicate that the first vertical anion state is slightly stable (slightly positive EAv). The calculated EAv increase (≤0.3 eV) caused by 3-aza substitution on dithiophene is in line with the VAEs measured in the monomers thiophene and thiazole.35
According to the scaled VOEs (see Table 3), insertion of a 3-BR2 substituent in the thiophene ring of 2-thienyl-2-thiazole increases the EAv by about 0.2 eV with R = Me, and 0.4 eV with R = Ph, in line with the above-mentioned EA increase of 0.5 eV found15 in solution with R = mesityl (i.e., 2,4,6-trimethylphenyl). It is to be noted that, according to the scaled VOEs, in the boryl derivatives the trans and cis conformers are predicted to possess the same EAv. However, at variance with unsubstituted thienylthiazole, where both conformers are calculated to be coplanar, the B3LYP optimized geometries of the trans and cis forms of the 3-boryl derivatives present pronounced differences. In both cases the cis conformer is predicted to be significantly more stable than the trans isomer (0.519 eV with R = Me, 0.332 eV with R = Ph; similar values are obtained with the 6-31+G(d) basis set).
In the cis conformers of both the BMe2 and BPh2 derivatives the thienylthiazole π-framework is calculated to be coplanar (see Table 4). This geometrical factor maximizes the conjugation between the two π rings and thus, other conditions being the same, the EA. The importance of the N lone pair (σ) → B intramolecular interaction is revealed by the B–N distance (1.65 Å, see Table 4), which is only slightly larger than the (B–Cring) bond length between the boron atom and the thiophene carbon atom. The B–N distance calculated in the gas phase is very close to that (1.67 Å) experimentally (X-ray) determined in the solid state.23 The B–C bonds of the boryl substituent (with both R = Me and Ph) are predicted to be nearly perpendicular to the thienylthiazole plane. It is to be noted that with this geometry the empty p orbital of the boron atom mixes with the σ orbital framework. The optimized geometry of the cisBPh2 derivative and the calculated localization properties of its LUMO are reported in Fig. 5. The LUMO is essentially localized on the thienylthiazole moiety, the contributions of the boron atom and the phenyl rings being very small.
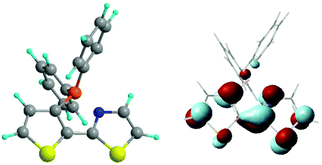 | ||
| Fig. 5 Optimized geometry of the cis isomer of (3-BPh2-2-thienyl)-2-thiazole and localization properties of its LUMO, as supplied by B3LYP/6-31G(d) calculations. | ||
| trans | cis | |||
|---|---|---|---|---|
| R = Me | R = Ph | R = Me | R = Ph | |
| dih. SCCN | 27.1 (32.1) | 24.3 (28.5) | 180.0 (180.0) | 178.2 (178.2) |
| dih. CCBC | 44.5 (43.2) | 47.5 (46.5) | 113.1 (113.1) | 112.7 (112.8) |
| d(B–Cring) | 1.568 (1.569) | 1.573 (1.574) | 1.625 (1.624) | 1.629 (1.629) |
| d(B–N) | 1.654 (1.654) | 1.653 (1.652) | ||
In contrast, in the trans isomers of both the Me and Ph derivatives the two pentacyclic rings of the thienylthiazole moiety are not perfectly coplanar, forming a dihedral angle of about 30° (see Table 4). However, the B–C bonds of the boryl substituent form a dihedral angle of about 45° with the plane of the thiophene ring, thus allowing mixing between the boron p orbital and the thienylthiazole π* LUMO. Moreover, the B–Cring bond length is significantly shorter than that of the cis isomer. The optimized geometry of the transBPh2 derivative and the calculated localization properties of its LUMO are reported in Fig. 6. At variance with the cis isomer, the LUMO is largely localized on the boron atom and the phenyl substituents.
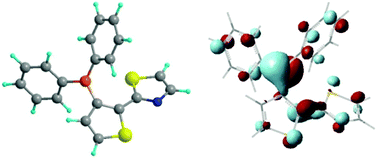 | ||
| Fig. 6 Optimized geometry of the trans isomer of (3-BPh2-2-thienyl)-2-thiazole and localization properties of its LUMO, as supplied by B3LYP/6-31G(d) calculations. | ||
Conclusions
The electron transmission spectra of π-systems substituted by boryl groups are presented for the first time and they demonstrate the strong stabilising effect of pπ/π* mixing on the π* anion states of furan and thiophene. The measured vertical electron affinities (EAv) are quantitatively reproduced by the virtual orbital energies calculated for the neutral molecules and scaled with empirical equations.This approach can then be utilized for predicting the electronic structures of more extended π-systems interacting with boryl groups such as the thienylthiazole derivative 3, recently synthesized and used as a monomer for producing even larger systems.15 The results for 3 do, in fact, show an EAv increase caused by aza substitution, in line with the changes in the measured EAv of thiophene and thiazole, as well as a strong N → B interaction leading to significant energy differences between the cis and trans conformers. The results show that for the more stable cis conformers the LUMO is localised on the thienylthiazole moiety, whereas for the trans isomer the LUMO is largely localized on the boron atom and the phenyl substituents.
This approach will undoubtedly be useful in the design and choice of better candidates for specific uses in the continually expanding range of applications in the field of molecular electronics.
Acknowledgements
Thanks are due to the Italian Ministero dell'Istruzione, dell'Università e della Ricerca for financial support.References
- D. Fichou, Handbook of Oligo- and Polythiophenes, Wiley-VCH, Weinheim, Germany, 1999 Search PubMed.
- G. Hadziioannou and P. F. van Hutten, Semiconducting Polymers, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- Conjugated Polymeric Materials: Opportunities in Electronics, Optoelectronics, and Molecular Electronics, ed. J.-L. Bredas and R. R. Chance, NATO ASI Series E, Kluver, Dordrecht, The Netherlands, vol. 182, 1990 Search PubMed.
- Quantum Chemistry Aided Design of Organic Polymers, ed. J. M. André, J. Delhalle and J.-L. Bredas, World Scientific, Singapore, 1991 Search PubMed.
- J. Roncali, Chem. Rev., 1992, 92, 711 CrossRef CAS.
- D. Jones, M. Guerra, L. Favaretto, A. Modelli, M. Fabrizio and G. Distefano, J. Phys. Chem., 1990, 94, 5761 CrossRef CAS.
- M. Dal Colle, C. Cova, G. Distefano, D. Jones, A. Modelli and N. Comisso, J. Phys. Chem. A, 1999, 103, 2828 CrossRef CAS.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338 CrossRef CAS.
- T. W. Hamann, R. A. Jensen, A. B. F. Martinson, H. van Ryswyk and J. T. Hupp, Energy Environ. Sci., 2008, 1, 66 RSC.
- J.-L. Brédas, J. E. Norton, J. Cornil and V. Cropceanu, Acc. Chem. Res., 2009, 42, 1691 CrossRef CAS.
- J. L. Segura, N. Martín and D. M. Guldi, Chem. Soc. Rev., 2005, 34, 31 RSC.
- S. Roquet, A. Cravino, P. Leriche, O. Aléveque, P. Frère and J. Roncali, J. Am. Chem. Soc., 2006, 128, 3459 CrossRef CAS.
- M. Liang, W. Xu, F. Cai, P. Chen, B. Peng, J. Chen and Z. Li, J. Phys. Chem. C, 2007, 111, 4465 CrossRef CAS.
- W. Xu, B. Peng, J. Chen, M. Liang and F. Cai, J. Phys. Chem. C, 2008, 112, 874 CrossRef CAS.
- S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413 CrossRef CAS.
- W.-L. Jia, D.-R. Bai, T. McCormick, Q.-D. Liu, M. Motala, R.-Y. Wang, C. Seward, Y. Tao and S. Wang, Chem.–Eur. J., 2004, 10, 994 CrossRef CAS.
- H. Kobayashi, N. Sato, Y. Ichikawa, M. Miyata, Y. Chujo and T. Matsuyama, Synth. Met., 2003, 135–136, 393 CAS.
- A. Sundararaman, M. Victor, R. Varughese and F. Jäkle, J. Am. Chem. Soc., 2005, 127, 13748 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927 CrossRef CAS.
- D. X. Cao, Z. Q. Liu, Q. Fang, G. B. Xu, G. Xue, G. Q. Liu and W. T. Yu, J. Organomet. Chem., 2004, 689, 2201 CrossRef CAS.
- S. Yamaguchi, T. Shirasak, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2002, 124, 8816 CrossRef CAS.
- G. Hughes and M. R. Bryce, J. Mater. Chem., 2005, 15, 94 RSC.
- A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., Int. Ed., 2006, 45, 3170 CrossRef CAS.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574 CrossRef CAS.
- M. Mazzeo, V. Vitale, F. Della Sala, M. Anni, G. Barbarella, L. Favaretto, G. Sotgiu, R. Cingolani and G. Gigli, Adv. Mater., 2005, 17, 34 CrossRef CAS.
- H. Doi, M. Kinoshita, K. Okumoto and Y. Shirota, Chem. Mater., 2003, 15, 1080 CrossRef CAS.
- J. P. Doering, A. Gedanken, A. P. Hitchcock, P. Fisher, J. H. Moore, J. K. Olthoff, J. A. Tossell, K. Raghavachari and M. B. Robin, J. Am. Chem. Soc., 1986, 108, 3602 CrossRef CAS.
- L. Sanche and G. J. Schulz, Phys. Rev. A: At., Mol., Opt. Phys., 1972, 5, 1672 CrossRef CAS.
- G. J. Schulz, Rev. Mod. Phys., 1973, 45, 378–423 CrossRef CAS.
- A. Modelli, D. Jones and G. Distefano, Chem. Phys. Lett., 1982, 86, 434 CrossRef CAS.
- A. Stamatovic and G. J. Schulz, Rev. Sci. Instrum., 1970, 41, 423 CrossRef.
- A. R. Johnston and P. D. Burrow, J. Electron Spectrosc. Relat. Phenom., 1982, 25, 119 CrossRef CAS.
- M. J. Frisch, et al., GAUSSIAN 03, revision D.01, Gaussian Inc., Wallingford, CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. Modelli and P. D. Burrow, J. Phys. Chem. A, 2004, 108, 5721 CrossRef CAS.
- S. W. Staley and J. T. Strnad, J. Phys. Chem., 1994, 98, 161.
- M. Guerra, Chem. Phys. Lett., 1990, 167, 315 CrossRef CAS.
- D. A. Chen and G. A. Gallup, J. Chem. Phys., 1990, 93, 8893 CrossRef CAS.
- J. Simons and K. D. Jordan, Chem. Rev., 1987, 87, 535 CrossRef CAS.
- N. F. Lane, Rev. Mod. Phys., 1980, 52, 29 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, 1986, Wiley, New York, Search PubMed.
- T. H. Dunning, Jr, K. A. Peterson and D. E. Woon, Basis Sets: Correlation Consistent Sets in The Encyclopedia of Computational Chemistry, ed. P. v. R. Schleyer, John Wiley, Chichester, 1998 Search PubMed.
- A. Modelli, Phys. Chem. Chem. Phys., 2003, 5, 2923 RSC.
- A. Modelli, B. Hajgató, J. F. Nixon and L. Nyulászi, J. Phys. Chem. A, 2004, 108, 7440 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS.
| This journal is © the Owner Societies 2011 |