Enhanced reactivity of Pt nanoparticles supported on ceria thin films during ethylene dehydrogenation
Yaroslava
Lykhach
*a,
Thorsten
Staudt
a,
Nataliya
Tsud
b,
Tomáš
Skála
c,
Kevin Charles
Prince
c,
Vladimír
Matolín
b and
Jörg
Libuda
ad
aLehrstuhl für Physikalische Chemie II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: yaroslava.lykhach@chemie.uni-erlangen.de; Fax: +49 9131 8528867; Tel: +49 9131 8520944
bCharles University, Faculty of Mathematics and Physics, Department of Plasma and Surface Science, V Holešovičkách 2, 18000 Prague 8, Czech Republic
cSincrotrone Trieste, Strada Statale 14, km 163.5, 34149 Basovizza-Trieste, Italy
dErlangen Catalysis Resource Center, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany
First published on 10th November 2010
Abstract
The adsorption and reaction of ethylene on Pt/CeO2−x/Cu(111) model catalysts were studied by means of high resolution photoelectron spectroscopy (HR-PES) in conjunction with resonant photoemission spectroscopy (RPES). The dehydrogenation mechanism is compared to the HR-PES data obtained on a Pt(111) single crystal under identical conditions. It was found that the Pt nanoparticle system shows a substantially enhanced reactivity and several additional reaction pathways. In sharp contrast to Pt(111), partial dehydrogenation of ethylene on the supported Pt nanoparticles already starts at temperatures as low as 100 K. Similar to the single crystal surface, dehydrogenation occurs via the isomer ethylidene (CHCH3) and then mainly viaethylidyne (CCH3). In the temperature region between 100 and 250 K there is strong evidence for spillover of hydrocarbon fragments to the ceria support. In addition, splitting of ethylene to C1 fragments is more facile than on Pt(111), giving rise to the formation of CH species and CO in the temperature region between 250 and 400 K. Upon further annealing, carbonaceous deposits are formed at 450 K. By heating to 700 K, these carbon deposits are completely removed from the surface by reaction with oxygen, provided by reverse spillover of oxygen from the ceria support.
1. Introduction
The use of ceria in many catalytic applications involving dynamic oxygen uptake and release is well established.1–3 This includes for example its application as an oxygen storage compound (OSC) in automotive exhaust catalysis or its application as a water gas shift catalyst (WGS) to remove CO during the generation of high-grade hydrogen streams. Potentially, the reducible CeO2 may endow a supported catalyst with additional self-cleaning functionality. The gradual accumulation of inactive surface carbon during conversion of hydrocarbons is a common problem for many noble metal catalysts. The reducible ceria directly participates in surface chemical reactions by the release of active oxygen species.4,5 As a result, carbonaceous species may be removed oxidatively, before the aggregation and formation of graphitic carbon occurs.6–8In spite of the importance of ceria supported metal catalysts, little is known on the underlying reaction mechanisms at the microscopic scale. It is anticipated that boundary sites or spillover/reverse spillover generally play an important role on ceria-supported catalysts. Clear evidence for reverse spillover of oxygen from the ceria substrate to the supported Rh metal particles was, for example, provided by Zafiris and Gorte.9 The phenomenon was later also observed by other groups on ceria–zirconia supported Pd catalyst.10
In contrast to work on powder materials, studies on single crystal based model catalysts hold the potential of providing insights into the elementary processes on these systems at a more detailed level. The reason is that the complexity of systems can be kept minimal and undesired contamination is largely avoided, because most experimental methods of surface science can be applied to these model systems in a straightforward fashion. For ceria surfaces a broad spectrum of model surfaces has been investigated, including both single crystals11–14 and ordered thin films on metal single crystal supports.15–27 Mullins and coworkers have performed pioneering work, studying the reactivity of a broad spectrum of small molecules, both on pure CeO2 films and metal nanoparticles on ceria.28–32
In our recent work, we have used Pt nanoparticles on ordered CeO2 films grown on Cu(111) to study the elementary steps of methane activation and dehydrogenation.33,34 It turned out that there are actually two critical issues related to the understanding of surface chemistry of these oxide-supported nanoparticle systems: the first is the intrinsically modified chemistry on nanoparticles. For the example of CH4, we have recently shown that the energy profile along the dehydrogenation pathway is indeed strongly modified on small particles, giving rise to a substantially enhanced activity.34 The second point is the involvement of the support, which may come into play for example via spillover and reverse spillover processes. For the present model system, for example, both spillover of hydrogen and reverse spillover of oxygen could be observed.33 Coupling of both phenomena will add further complexity and may open new reaction pathways, which are not observed on simple metal surfaces.
In order to obtain a more detailed insight into these processes, we have chosen the dehydrogenation of ethylene as a simple model reaction. Ethylene adsorption and decomposition on Pt(111) has been systematically investigated in both experimental35–38 and theoretical studies.39 At temperatures below 90 K ethylene molecules adsorb with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond parallel to the surface through a weak π–donor interaction. Above 90 K, however, ethylene chemisorption occurs via the formation of di-σ bound species with sp3 hybridization. Density Functional Theory (DFT) calculations identified adsorption sites of the di-σ ethylene as bridge positions.39 Saturation coverage of these species at 95 K is 0.25 ML.40 With increasing surface temperature, ethylene partly desorbs and partly dehydrogenates to ethylidyne (CCH3) populating fcc hollow sites. Intermediates of dehydrogenation have been observed and it has been argued that these species could be vinyl (CHCH2)41 or ethylidene (CHCH3).42–46 DFT calculations, however, favor the formation of ethylidene.39
C bond parallel to the surface through a weak π–donor interaction. Above 90 K, however, ethylene chemisorption occurs via the formation of di-σ bound species with sp3 hybridization. Density Functional Theory (DFT) calculations identified adsorption sites of the di-σ ethylene as bridge positions.39 Saturation coverage of these species at 95 K is 0.25 ML.40 With increasing surface temperature, ethylene partly desorbs and partly dehydrogenates to ethylidyne (CCH3) populating fcc hollow sites. Intermediates of dehydrogenation have been observed and it has been argued that these species could be vinyl (CHCH2)41 or ethylidene (CHCH3).42–46 DFT calculations, however, favor the formation of ethylidene.39
To facilitate a direct comparison between single crystal and supported particles, we have repeated experiments on Pt(111) and compared them to data on Pt/CeO2−x/Cu(111) model catalysts obtained under identical conditions. It is shown that the nanoparticles on the one hand show a strongly enhanced dehydrogenation activity. On the other hand completely new reaction channels are opened as a result of spillover and reverse spillover processes.
2. Experimental section
High resolution X-ray synchrotron radiation photoelectron spectroscopy studies on ethylene decomposition over Pt/CeO2−x/Cu(111) model catalysts were performed at the Materials Science Beamline at the Elettra synchrotron facility in Trieste, Italy. The source was a bending magnet that produces linearly polarized light in the energy range of 21–1000 eV. The ultrahigh vacuum (UHV) end-station (base pressure 1 × 10−10 mbar) was equipped with a multichannel electron energy analyzer (Specs Phoibos 150), a rear view Low Energy Electron Diffraction (LEED) optics, a quadrupole mass spectrometer, an argon sputter gun, and a gas inlet system. The basic setup of the chamber includes a dual Mg/Al X-ray source used for the calibration of the energy of the synchrotron light and off-line work. Additionally, two electron-beam evaporators for metal deposition were installed on the chamber.A single crystal Pt(111) disc (MaTecK) was cleaned by Ar+ sputtering (at 300 K for 20 min) and annealing at 973 K in an oxygen atmosphere for 1 min and in UHV for 2 min. Still, a trace of carbon was found on the surface of the sample (less than 3%). The carbon contribution was numerically subtracted from the C 1s spectra of Pt(111) samples discussed below.
A single crystal Cu(111) disc (MaTecK) was used as a substrate for the Pt/CeO2 samples. First, the Cu(111) was cleaned by several cycles of Ar+ sputtering (at 300 K for 60 min) and annealing (723 K for 5 min) until no traces of carbon or any other contaminant were found in the photoelectron spectra. Epitaxial layers of CeO2 were grown on clean Cu(111) by physical vapor deposition of Ce metal (Goodfellow, 99.99%) in an oxygen atmosphere (pO2 = 5 × 10−7 mbar, Linde, 99.995%) at 523 K, followed by annealing of the films at 523 K in an oxygen atmosphere of the same pressure for 10 min. This preparation procedure follows the recipe previously described by Matolín et al.15,16 The preparation method yields a continuous CeO2(111) film with a thickness of approximately 1.5 nm as determined from the attenuation of the Cu 2p3/2 intensity. LEED observations on the prepared films confirm the epitaxial growth of CeO2(111) with the characteristic (1.5 × 1.5) superstructure in relation to the Cu(111) substrate.
Pt (Goodfellow, 99.99%) was deposited onto the CeO2/Cu(111) at 300 K. The nominal thickness of the deposited Pt layer was 0.4 nm as determined from the attenuation of the Cu 2p3/2 intensity. The deposited samples were further treated to yield different stoichiometries of cerium oxide. Thus, Pt/CeO1.95/Cu(111) was prepared by annealing of the freshly deposited Pt/CeO2/Cu(111) sample to 700 K in vacuum. The corresponding treatment procedure results in the mild reduction of the cerium oxide due to the activation of reverse oxygen spillover from the substrate onto the Pt nanoparticles.33 The samples of Pt/CeO1.90/Cu(111) were obtained by annealing of the fresh Pt/CeO2/Cu(111) to 700 K after it was exposed to a saturation dose of ethylene. This procedure results in a significant reduction of ceria as a result of interaction of the reverse spilt over oxygen with the products of the hydrocarbon dehydrogenation. After following the preparation procedures both samples contained active oxygen on the surface of the Pt particles.33 No traces of carbon were found on the surfaces of the prepared samples.
Pt 4f and C 1s core level spectra were acquired with photon energies of 180 and 410 eV. Al Kα radiation (1486.6 eV) was used to measure the core levels of Pt 4f, Ce 3d and Cu 2p3/2. Additionally, valence band spectra were recorded on the Pt/CeO2−x/Cu(111) samples at three different photon energies, 115.0, 121.4, and 124.8 eV. All spectra were acquired at a constant pass energy at emission angles for the photoelectrons of 20° and 0° with respect to the sample normal, while using the X-ray source or synchrotron radiation as appropriate. The total spectral resolutions achieved with Al Kα (1486.6 eV) and synchrotron radiation were 1 eV and 150–200 meV, respectively. The core level spectra were fitted with Voigt (Ce 3d) and Doniach–Sunjic convoluted with a Gaussian (Pt 4f, C 1s) line profiles after subtraction of a Shirley background. During the experiment the sample temperature was controlled by a DC power supply passing a current through the Ta wires holding the sample. The actual temperature was measured by K-type thermocouple attached to the rear surface of the sample. Stable temperature and fast cooling after the annealing steps were achieved by simultaneous resistive heating and cooling of the manipulator with liquid nitrogen. The investigated samples were exposed to a total dose of ethylene (Linde, 99.8%) of 50 L (1 L = 1.33 × 10−6 mbar × s) at 100 K. Ethylene was dosed by backfilling the UHV chamber to a pressure of 1.33 × 10−7 mbar.
The stoichiometry of Pt/CeO2−x/Cu(111) was quantified by the fraction of the reduced Ce2O3 oxide estimated following the fitting procedure of the Ce 3d spectra similar to that applied by Skála et al.47
3. Results and discussion
3.1 Ethylene adsorption and decomposition over Pt(111)
As a reference for the experiments on ceria supported Pt nanoparticles under identical conditions, core level spectra for ethylene adsorption and reaction on Pt(111) were recorded. In Fig. 1a and b the C 1s and Pt 4f regions after exposure to a saturation dose of ethylene (50 L) at 100 K and subsequent annealing steps are shown. The spectra are in good agreement with the data published previously by Fuhrmann et al.36 The C 1s spectrum (upper trace) of the chemisorbed ethylene molecules reveals fine structure due to excitation of the C–H stretching mode in the photoemission process. The spectrum contains three peaks with the major adiabatic component located at binding energy (BE) 283.3 eV.36 The separation of the adiabatic and the first and second vibrationally excited peaks is 0.37 eV and 0.73 eV, with intensity ratios of 0.37 and 0.08, respectively. The shape of the C 1s line remained unchanged upon annealing to 200 K. At higher temperatures a novel feature in the C 1s spectra develops at 284.1 eV, resulting from dehydrogenation of ethylene to ethylidyne (CCH3). Here, the dominant adiabatic component is accompanied by a small contribution from the first vibrationally excited component. The peaks are separated by 0.40 eV and the intensity ratio is 0.17. An intermediate species, usually identified as ethylidene (CHCH3), appears in the C 1s spectra at 250 K. We fit this peak with a single component (green line) located at 283.8 eV. It is the ethylidyne species, however, which dominates the C 1s spectra in the temperature range between 300 K and 400 K. Upon annealing above 400 K, further dehydrogenation and simultaneous C–C bond cleavage result in the formation of CH and finally of residual surface carbon. | ||
| Fig. 1 Development of C 1s (a) and Pt 4f7/2 (b) core level spectra observed upon annealing of Pt(111) exposed to 50 L of C2H4 at 100 K. | ||
The development of the Pt 4f7/2spectra is shown in Fig. 1b. The Pt 4f signal obtained from clean Pt(111) exhibits two components due to a surface (Pts) and a bulk (Ptb) contribution located at 70.75 eV and 71.10 eV, respectively (spectrum is not shown, see, for example ref. 48). The adsorption of ethylene attenuates the surface component and causes a small BE shift of the bulk component towards higher values. An additional small component (gray line) added to the spectra accounts for the presence of carbon contamination on the surface. Formation of ethylidyne on Pt(111) is associated with the appearance of the new component in the Pt 4f region located at 71.50 eV. The corresponding peak intensity increases with an increasing amount of ethylidyne on the surface and disappears after ethylidyne decomposition above 400 K. It is known that the complete dehydrogenation of ethylene on Pt(111) results in the formation of amorphous carbon particles.49 During successive annealing of these deposits to 700 K, the corresponding C 1s peak becomes narrower, but retains its BE in the range of 284.0–284.1 eV (see Fig. 1a). The carbonaceous species that were formed during the decomposition of ethylidyne on Pt between 450 K and 700 K are mainly carbidic carbon.50–52 It was reported that a rearrangement of the carbonaceous species to a graphitic layer occurs above 700 K.49,53,54
3.2 Ethylene adsorption and decomposition over Pt/CeO2−x/Cu(111)
In a second step, we examine the adsorption of ethylene on two Pt/CeO2−x/Cu(111) samples. Both samples correspond to the same nominal Pt coverage (nominal thickness 0.4 nm). The thickness of the epitaxial CeO2 films on Cu(111) was about 1.5 nm. Concerning the growth and the morphology of the Pt/CeO2/Cu(111) we refer to a recent publication.33 As a result of different sample pretreatments (see Experimental Section for details) the average Ce oxidation state of both samples was different, however. The stoichiometries of the Pt/CeO2−x samples were estimated to be Pt/CeO1.95 and Pt/CeO1.90, where x was determined as the half of the ratio of the Ce3+ contribution to the total intensity of the Ce 3d spectra (see e.g.ref. 47 for fitting details). It should be pointed out that both samples were subjected to pre-annealing steps to 700 K. As has been shown in a previous study, pre-annealing leads to the reverse spillover of oxygen from the ceria film to the Pt nanoparticles.33 Thus the presence of a small amount of co-adsorbed oxygen has to be taken into consideration, when discussing the interaction with C2H4.The C 1s spectra of ethylene adsorbed at 100 K on the two Pt/CeO2−x/Cu(111) samples are displayed in Fig. 2. At first glance it is apparent that the structure of the corresponding spectra is more complicated than the reference spectra for clean Pt(111) (Fig. 1). Immediately after adsorption at 100 K, we resolve at least four distinct components for both the Pt/CeO1.95 and the Pt/CeO1.90 sample. The dominant contribution is located at 283.40 eV (Pt/CeO1.95; 283.45 eV for Pt/CeO1.90). It is straightforwardly attributed to molecularly chemisorbed di-σ ethylene. From the experiments on Pt(111) we know that the spectral signature of this species contains three peaks due to vibrational excitations. We use the BE shifts and relative intensity ratios from the single crystal experiment to fit the corresponding spectra for Pt/CeO2−x/Cu(111) (see Fig. 2, black lines). An additional well-resolved component emerges for Pt/CeO2−x/Cu(111) at 283.80 eV (Pt/CeO1.95; 283.90 eV for Pt/CeO1.90). It is ascribed to the intermediate ethylidene species (green line), which is also found in the spectra on Pt(111) but only after heating to 250 K (compare Fig. 1a). The next component appears at 284.20 eV (both for Pt/CeO1.95 and Pt/CeO1.90, red line in Fig. 2). According to the single crystal data this feature is attributed to ethylidyne. It should be noted at this point that an alternative interpretation may be taken into consideration: the oxygen traces on the surface, generated by reverse spillover, may induce the adsorption state of ethylene corresponding to the π configuration.55 The latter species may give rise to a C 1s signal in the observed region. For example, a C 1s BE of 283.9 eV was observed during adsorption of ethylene on sulfated Pt(111)37 or Pt2Sn.38,56 However, a careful analysis of the corresponding Pt 4f signals (see Fig. 3) disproves this possibility. The Pt 4f spectra obtained after the exposure to ethylene at 100 K contain two components. The main peak is slightly shifted by 0.10 eV towards higher BE when compared to the Pt 4f signal before exposure (71.10 eV on Pt/CeO1.95 and 71.15 eV on Pt/CeO1.90, upper traces in Fig. 3). This shift is consistent with molecular adsorption of di-σ ethylene (compare also the behavior on Pt(111), see Fig. 1). The second component in the Pt spectra emerges at 71.6 eV. This BE shift is characteristic for the strongly bound ethylidyne species (compare Pt(111), Fig. 1) and would not be expected for weakly adsorbed π-ethylene. At this point we can conclude that the Pt nanoparticles indeed show a strongly enhanced reactivity, if compared to the Pt(111) single crystal surface. Partial decomposition of C2H4 is observed already at temperatures as low as 100 K, whereas temperatures exceeding 200 K are required to initiate this reaction on the Pt(111) surface. Recently, we observed a similar effect for the dehydrogenation of CH4, with specific dehydrogenation steps showing strongly reduced activation barriers on Pt nanoparticles.34 Based on DFT calculations, the reduced barriers were attributed to specific sites such as particle edges. These sites show strongly modified BEs and, as a result, lead to reduced activation barriers for specific elementary reaction steps.
 | ||
| Fig. 2 Development of C 1s core level spectra observed upon annealing of two samples, (a) Pt/CeO1.95/Cu(111) and(b) Pt/CeO1.90/Cu(111), exposed to 50 L of C2H4 at 100 K. | ||
 | ||
| Fig. 3 Development of Pt 4f7/2 core level spectra observed upon annealing of two samples, (a) Pt/CeO1.95/Cu(111) and (b) Pt/CeO1.90/Cu(111), exposed to 50 L of C2H4 at 100 K. | ||
Note that a close inspection of the C 1s spectra obtained for C2H4 adsorption on Pt/CeO2−x/Cu(111) finally reveals that there is a fourth C 1s component, which appears as a broad feature at 285.40 eV. Since this peak has no corresponding feature in the Pt single crystal spectra, we tentatively assign it to both molecularly adsorbed C2H4 and the CxHy fragments situated on CeO2−x. This assignment may be rationalized as follows: the C 1s spectrum observed during adsorption of C2H4 on pure CeO2 shows a single peak at a similar BE (285.30 eV) (data not shown). However, when no metal is present, the C2H4 desorbs at temperatures as low as 150 K,57 whereas the peak at 285.40 persists at 200 K. Therefore, it appears likely that it is at least in part due to the hydrocarbon fragments, which may originate from spillover from the Pt particles to the ceria support. Mullins and coworkers have studied a variety of carbon containing species on ceria films.32 They found BEs in the range between 287 and 290 eV for C atoms bound to oxygen, whereas C atoms without direct bonds to oxygen typically appeared at BEs between 285 and 286 eV.58 Thus, the observed BE would be consistent with a CxHy fragment spilt over to the support bound to a Ce3+ site. This hypothesis is corroborated by the results of the RPES experiments (see Section 3.3) that show a strong involvement of the ceria support in the reaction, exactly in the temperature region where these species appear.
Next we consider the thermal behavior of Pt/CeO2−x/Cu(111) after C2H4 adsorption. During annealing of the sample to 200 K, the intensity of the peak at 284.20 eV—associated with ethylidyne—slightly decreases, while the intensity of the peak at 283.40 eV (Pt/CeO1.95; 283.45 eV for Pt/CeO1.90) grows. Upon further heating to 250 K the components associated with ethylidyne (284.20 eV) and the ethylidene intermediate (283.80 eV for Pt/CeO1.95; 283.90 eV for Pt/CeO1.90) decrease substantially in intensity. Surprisingly, the peaks at 283.40 eV (Pt/CeO1.95) and 283.45 eV (Pt/CeO1.90) retain their high intensity up to temperatures of 300 K. For Pt(111) this peak was exclusively ascribed to molecularly adsorbed ethylene, but molecular ethylene should desorb or decompose at temperatures above 250 K (see Fig. 1). In view of the enhanced reactivity of the Pt nanoparticles it therefore appears likely that this peak includes not only the signature of molecular ethylene, but also contributions from more strongly bound decomposition products of ethylene. One of the possible species could be vinylidene (CCH2). Vinylidene is found upon ethylene decomposition on (100) surfaces of Pt,59Pd,60 and Rh,61 and can possibly rearrange to quad sigma acetylene (CH–CH) upon annealing to about 400 K.35,59 A simpler alternative, however, would imply scission of the C–C bond and the formation of C1 fragments. CH species, for instance, are found at 283.6 eV on Pt(111),36 which is close to the BEs observed on the present sample. It should be noted that also the Pt 4f spectra (see Fig. 3) support the above suggestion that strongly bound fragments like CH and C are dominant at temperatures of 250 K and above. The associated peak at 71.60 eV reaches the highest intensity at 300 K. At this temperature, the features at 284.20 and 283.60 eV, associated with C and CH, dominate the C 1s spectrum.
Further indications supporting the formation of C1 species can be derived from two new features appearing in the C 1s spectra upon heating to 300 K. One of those appears at 286.50 eV (Pt/CeO1.95; ∼286.80 eV for Pt/CeO1.90, see Fig. 2). Based on its BE, this may be associated with CO on Pt.62–64 The second additional feature around BEs of 285 eV (see Fig. 2) could be associated with an oxygen containing hydrocarbon fragments adsorbed on Pt. For example, species such as CHO have been theoretically predicted65 and experimentally observed as intermediates of CH oxidation to CO and H2 on different noble metal surfaces.66,67
Turning our focus now to the behavior at higher temperatures, we observe that at 400 K, a single peak at 284.20 eV dominates the C 1s spectrum, together with a weaker shoulder at 283.40 eV (Pt/CeO1.95; 283.45 eV for Pt/CeO1.90). According to the above discussion, the shoulder is attributed to a CH fragment on Pt, whereas the main peak (which starts to grow already at 300 K) is associated with carbidic carbon.
3.3 On the involvement of the ceria support in C2H4 dehydrogenation and oxidation on Pt/CeO2−x/Cu(111)
Finally, we investigate the role of the ceria support in C2H4 dehydrogenation and oxidation in more detail. In a recent publication we have investigated the activation and dehydrogenation of methane on Pt/CeO2−x/Cu(111).33 Here, we have shown that both spillover of hydrogen from the Pt particles to the ceria support (at temperatures below 300 K) and reverse spillover of oxygen from ceria to the Pt particles (mainly activated at temperatures above 400 K) play an important role in the surface chemistry of these systems. However, all information on the ceria oxidation state was derived from the analysis of laboratory X-ray spectra of the Ce 3d region. The sensitivity of this method is low, however, in particular at low concentrations of Ce3+. Here, we use RPES, to directly monitor changes in the Ce oxidation state at the highest sensitivity (see e.g.ref. 15 and 68 for a more detailed discussion). Essentially, the method is based on measuring the valence bands at photon energies corresponding to the 4d → 4f resonance either in the Ce3+ or the Ce4+ ions (see also ref. 69 and 70 for more details). The Ce3+ resonance at a photon energy of 121.4 eV is caused by an Auger decay involving electron emission from Ce 4f states located about 1.4 eV below the Fermi edge. The Ce4+ resonance at a photon energy of 124.80 eV involves emission of O 2p electrons (hybridized with Ce states) from valence band states around 4.0 eV BE. Additionally, we measure the valence band spectrum at a photon energy of 115 eV, corresponding to an ‘off-resonance’ condition. Typical examples of valence band spectra acquired at Ce3+ resonance, Ce4+ resonance and off-resonance are shown in Fig. 4a.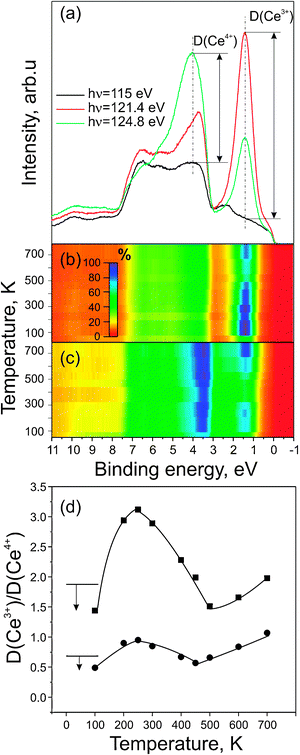 | ||
| Fig. 4 (a) Typical valence band spectra obtained on partially reduced Pt/CeO2−x/Cu(111) in off-resonance mode (115 eV, black), at Ce3+ resonance (121.4 eV, red), and at Ce4+ resonance (124.8 eV, green). Resonant enhancement ratios, D(Ce3+) and D(Ce4+), are determined as intensity differences between the valence bands measured in- and off-resonance (see text); color intensity map of the valence band spectra at Ce3+ resonance on (b) Pt/CeO1.95/Cu(111) and (c) Pt/CeO1.90/Cu(111) during thermal treatment after exposure to 50 L of C2H4 at 100 K (d) Evolution of the resonant enhancement ratio D(Ce3+)/D(Ce4+) as a function of annealing temperature on Pt/CeO1.95/Cu(111) (circles) and Pt/CeO1.90/Cu(111) (squares). The arrows indicate the initial drop of the enhancement ratio upon exposure of the samples to 50 L of C2H4 at 100 K. | ||
In order to follow the reaction steps involving the ceria support, RPE spectra were measured as a function of temperature during decomposition of C2H4 on the Pt/CeO2−x/Cu(111) samples. The intensity of the valence band spectra obtained on both samples in Ce3+ resonance are shown in Fig. 4b and c, represented as a color intensity map. It is immediately apparent that the ceria support undergoes strong changes in oxidation state as a function of the annealing temperature. To further analyze these changes, the so-called resonant enhancement ratio (RER) was calculated. Towards this aim, the resonant enhancements for Ce3+ (denoted as D(Ce3+)) and for Ce4+ (D(Ce4+)) are individually quantified by calculating the difference of the corresponding spectra in- and off-resonance. The resonant enhancement ratio (RER), calculated as (D(Ce3+))/(D(Ce4+), directly reflects the variations of the ceria oxidation state as monitored by RPES. The corresponding data as a function of temperature are displayed in Fig. 4d.
The first and somewhat surprising finding is that the RER decreases at 100 K upon initial adsorption of C2H4. Formally, this behavior would correspond to a decrease in Ce3+ concentration, i.e. a partial oxidation of the ceria. It is hard to rationalize such an effect, however, as a result of hydrocarbon decomposition, which should rather lead to reduction of the support than to oxidation. We therefore suggest that the apparent decrease in the RER is rather due to a damping effect, leading to preferential attenuation of the Ce3+ signal upon C2H4 adsorption. The hydrocarbon spillover from Pt to ceria, which was presented as a hypothesis in Section 3.2, could explain this observation: hydrocarbon fragments formed on the Pt particles could migrate onto the ceria surface and preferentially adsorb on Ce3+ sites, thus selectively attenuating the signal from these sites. The suggestion is consistent with the assumption that the majority of Ce3+ is situated at the perimeter of the deposited metal particles.68
Upon annealing to 200 K, we observe a very strong increase in the RER. This effect is direct evidence for the involvement of the support in hydrocarbon dehydrogenation on Pt/CeO2−x/Cu(111). In our previous work, we have attributed this effect mainly to hydrogen spillover from the Pt particle to ceria sites.33 It should be noted that hydrogen spillover is a well-documented phenomenon on supported metal catalysts.71 On ceria, hydrogen spillover leads to the formation of OH groups and, simultaneously, to the reduction of adjacent Ce4+ centers to Ce3+. It should also be pointed out that spillover of hydrocarbon fragments from Pt to ceria and decomposition of hydrocarbon fragments on ceria (leading to the release of hydrogen) could also contribute to reduction.
The behavior of the RER in the temperature window from 200 K to 400 K is somewhat surprising. Here we observe a drastic decrease of the RER, indicating a partial re-oxidation of ceria. A similar tendency for the reduction/re-oxidation of ceria during adsorption and decomposition of methanol was observed by Matolín et al.72 The authors associated the increase of RER at 200 K to the desorption of water and its following decrease at 300 K to the restoration of the topmost ceria layers by the oxygen provided from the bulk. A more straightforward explanation could be obtained assuming that hydrogen spillover on Pt/CeO2−x/Cu(111) is a reversible process, i.e.hydrogen could migrate in both directions via the Pt–ceria boundary. After a reverse spillover of hydrogen from the support to the Pt nanoparticles, molecular desorption of hydrogen would occur at a sufficiently high temperature. After the release of the molecular hydrogen, the ceria temporarily involved in the reaction would be left in its initial state, i.e. as Ce4+. It should be pointed out that a similar behavior has previously been observed, for example on ceria supported Rh catalysts.73 According to the C 1s spectra presented in Fig. 2, Pt nanoparticles are covered by amorphous carbon at a temperature of 450 K. In the temperature window from 450 K to 700 K we observe a monotonic increase in the RER, indicating an increasing degree of reduction of the ceria support. Simultaneously, the C 1s associated with the amorphous carbon decreases, until it completely vanishes at 700 K.
We attribute the increasing Ce3+ concentration to the reverse spillover of oxygen. The spilt over oxygen recombines with the surface carbon species. CO immediately desorbs upon formation. We have earlier shown that oxygen reverse spillover occurs on Pt/CeO2−x/Cu(111), even in the absence of surface carbon.33 It should be pointed out that oxygen release from the ceria and reverse spillover leads to complete removal of all surface carbon from the catalyst surface. This self-cleaning effect is one of the unique properties of ceria-based noble metal catalysts.
4. Conclusions
We have investigated the adsorption, dehydrogenation and oxidation of ethylene on a Pt/CeO2−x/Cu(111) model catalyst by means of HR-PES and RPES. The dehydrogenation mechanism of ethylene observed on supported nanoparticles is discussed with regard to the behavior on a Pt(111) single crystal. The comparison reveals characteristic differences in activity and novel reaction pathways for the supported particle system:(1) In contrast to pure molecular adsorption on Pt(111) at 100 K, ethylene partially decomposes on supported Pt nanoparticles on CeO2−x/Cu(111), forming a series of different reaction products. HR-PES supports the initial formation of ethylidene, ethylidyne on the Pt particles and hydrocarbon fragments on the ceria support, the latter formed via spillover.
(2) At higher temperatures, new decomposition pathways are observed in addition to the decomposition pathway viaethylidyne found for Pt(111). It is shown that on the particles a higher tendency for C–C scission is observed. In addition, we find oxygen containing intermediates, as a result of the reaction with oxygen provided by reverse spillover. The new pathways lead to larger amounts of CO and CH.
(3) At temperatures exceeding 250 K, CH decomposes and forms carbidic carbon. These carbon species readily react with the oxygen provided by reverse spillover at temperatures exceeding 400 K. At 700 K carbon is completely removed, leaving a partially reduced cerium oxide surface.
(4) RPES allows monitoring of the changes of the ceria oxidation state with the highest sensitivity. The phenomena of reduction and re-oxidation occur mainly as a result of spillover and reverse spillover. We show that in the low temperature regime (100–200 K) spillover of hydrogen occurs. In addition there are experimental indications for the spillover of hydrocarbon fragments. In the temperature range between 300 K and 400 K hydrogen reverse spillover occurs, followed by molecular desorption. This process leads to partial re-oxidation of the ceria support. At temperatures of 450 K and above, reverse spillover of oxygen leads to further reduction of the ceria support. The spilt-over oxygen is irreversibly consumed by recombination with carbon on the Pt particles.
Acknowledgements
The authors gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG) within the ERACHEM program (“NanoFunC” project), as well as additional support from the DFG within the Excellence Cluster “Engineering of Advanced Materials” in the framework of the excellence initiative. Furthermore, we acknowledge support by the Fonds der Chemischen Industrie, the DAAD (PPP, Acciones Integradas Hispano-Alemanas), the European Union (COST D-41) and the Ministry of Education of the Czech Republic (LA08022).References
- X. Wang and R. J. Gorte, Catal. Lett., 2001, 73, 15–19 CrossRef CAS.
- X. Wang and R. J. Gorte, Appl. Catal., A, 2002, 224, 209–218 CrossRef CAS.
- R. Craciun, B. Shereck and R. J. Gorte, Catal. Lett., 1998, 51, 149–153 CrossRef CAS.
- J. W. Gosselink and W. H. J. Stork, Ind. Eng. Chem. Res., 1997, 36, 3354–3359 CrossRef CAS.
- J. R. Rostrup-Nielsen, Catal. Today, 1997, 37, 225–232 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, Catal. Rev. Sci. Eng., 1999, 41, 1–42 CrossRef CAS.
- Y. H. Hu and E. Ruckenstein, Adv. Catal., 2004, 48, 297–345 CAS.
- A. Trovarelli, Catalysis by Ceria and Related Metals, Imperial College Press, London, 2002 Search PubMed.
- G. S. Zafiris and R. J. Gorte, J. Catal., 1993, 139, 561–567 CrossRef CAS.
- M. Y. Smirnov and G. W. Graham, Catal. Lett., 2001, 72, 39–44 CrossRef CAS.
- H. Nörenberg and G. A. D. Briggs, Phys. Rev. Lett., 1997, 79, 4222–4225 CrossRef.
- S. Gritschneder, Y. Namai, Y. Iwasawa and M. Reichling, Nanotechnology, 2005, 16, S41–S48 CrossRef CAS.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS.
- S. Torbrugge, M. Cranney and M. Reichling, Appl. Phys. Lett., 2008, 93, 0561011–0561014.
- F. Šutara, M. Cabala, L. Sedláček, T. Skála, M. Škoda, V. Matolín, K. C. Prince and V. Cháb, Thin Solid Films, 2008, 516, 6120–6124 CrossRef CAS.
- V. Matolín, J. Libra, I. Matolínová, V. Nehasil, L. Sedláček and F. Šutara, Appl. Surf. Sci., 2007, 254, 153–155 CrossRef CAS.
- U. Berner and K. Schierbaum, Thin Solid Films, 2001, 400, 46–49 CrossRef CAS.
- E. L. Wilson, R. Grau-Crespo, C. L. Pang, G. Cabailh, Q. Chen, J. A. Purton, C. R. A. Catlow, W. A. Brown, N. H. de Leeuw and G. Thornton, J. Phys. Chem. C, 2008, 112, 10918–10922 CrossRef CAS.
- M. Alexandrou and R. M. Nix, Surf. Sci., 1994, 321, 47–57 CrossRef CAS.
- S. Eck, C. Castellarin-Cudia, S. Surnev, M. G. Ramsey and F. P. Netzer, Surf. Sci., 2002, 520, 173–185 CrossRef CAS.
- J.-L. Lu, H.-J. Gao, S. Shaikhutdinov and H.-J. Freund, Surf. Sci., 2006, 600, 5004–5010 CrossRef CAS.
- C. J. Weststrate, R. Westerstrom, E. Lundgren, A. Mikkelsen and J. N. Andersen, J. Phys. Chem. C, 2009, 113, 724–728 CrossRef CAS.
- D. R. Mullins, P. V. Radulovic and S. H. Overbury, Surf. Sci., 1999, 429, 186–198 CrossRef CAS.
- J. Zhou, A. P. Baddorf, D. R. Mullins and S. H. Overbury, J. Phys. Chem. C, 2008, 112, 9336–9345 CrossRef CAS.
- W. Xiao, Q. Guo and E. G. Wang, Chem. Phys. Lett., 2003, 368, 527–531 CrossRef CAS.
- A. Siokou and R. M. Nix, J. Phys. Chem. B, 1999, 103, 6984–6997 CrossRef CAS.
- R. Wrobel, Y. Suchorski, S. Becker and H. Weiss, Surf. Sci., 2008, 602, 436–442 CrossRef CAS.
- D. R. Mullins and S. H. Overbury, J. Catal., 1999, 188, 340–345 CrossRef CAS.
- D. R. Mullins and K. Z. Zhang, Surf. Sci., 2002, 513, 163–173 CrossRef CAS.
- S. D. Senanayake, J. Zhou, A. P. Baddorf and D. R. Mullins, Surf. Sci., 2007, 601, 3215–3223 CrossRef CAS.
- D. R. Mullins, M. D. Robbins and J. Zhou, Surf. Sci., 2006, 600, 1547–1558 CrossRef CAS.
- S. D. Senanayake and D. R. Mullins, J. Phys. Chem. C, 2008, 112, 9744–9752 CrossRef CAS.
- Y. Lykhach, T. Staudt, M. P. A. Lorenz, R. Streber, A. Bayer, H.-P. Steinrück and J. Libuda, ChemPhysChem, 2010, 11, 1496–1504 CrossRef CAS.
- F. Viñes, Y. Lykhach, T. Staudt, M. P. A. Lorenz, C. Papp, H.-P. Steinrück, J. Libuda, K. M. Neyman and A. Görling, Chem.–Eur. J., 2010, 16, 6530–6539 CAS.
- R. I. Masel, Principles of Adsorption and Reaction on Solid Surfaces, John Wiley & Sons, Inc., New York, 1996 Search PubMed.
- T. Fuhrmann, M. Kinne, B. Tränkenschuh, C. Papp, J. F. Zhu, R. Denecke and H.-P. Steinrück, New J. Phys., 2005, 7, 1–19 CrossRef.
- A. F. Lee and K. Wilson, J. Vac. Sci. Technol., A, 2003, 21, 563–568 CrossRef CAS.
- J. M. Essen, J. Haubrich, C. Becker and K. Wandelt, Surf. Sci., 2007, 601, 3472–3480 CrossRef CAS.
- Q. Ge and D. A. King, J. Chem. Phys., 1999, 110, 4699–4702 CrossRef CAS.
- K. Griffiths, W. N. Lennard, I. V. Mitchell, P. R. Norton, G. Pirug and H. P. Bonzel, Surf. Sci. Lett., 1993, 284, L389–L393 Search PubMed.
- F. Zaera, J. Am. Chem. Soc., 1989, 111, 4240–4244 CrossRef CAS.
- H. Steininger, H. Ibach and S. Lehwald, Surf. Sci., 1982, 117, 685–698 CrossRef CAS.
- F. Zaera, T. V. W. Janssens and H. Ofner, Surf. Sci., 1996, 368, 371–376 CrossRef CAS.
- M. Salmeron and G. A. Somorjai, J. Phys. Chem., 1982, 86, 341–350 CrossRef CAS.
- J. Stöhr, F. Sette and A. L. Johnson, Phys. Rev. Lett., 1984, 53, 1684–1687 CrossRef CAS.
- P. Cremer, C. Stanners, J. W. Niemantsverdriet, Y. R. Shen and G. Somorjai, Surf. Sci., 1995, 328, 111–118 CrossRef CAS.
- T. Skála, F. Šutara, M. Škoda, K. C. Prince and V. Matolín, J. Phys.: Condens. Matter, 2009, 21, 0550051–0550059.
- E. Janin, M. Bjorkqvist, T. M. Grehk, M. Gothelid, C. M. Pradier, U. O. Karlsson and A. Rosengren, Appl. Surf. Sci., 1996, 99, 371–378 CrossRef CAS.
- T. A. Land, T. Michely, R. J. Behm, J. C. Hemminger and G. Comsa, J. Chem. Phys., 1992, 97, 6774–6783 CrossRef.
- X.-L. Zhou, X.-Y. Zhu and J. M. White, Surf. Sci., 1988, 193, 387–416 CrossRef CAS.
- B. Vermang, M. Juel and S. Raaen, J. Vac. Sci. Technol., A, 2007, 25, 1512–1518 CrossRef CAS.
- F. Vines, K. M. Neyman and A. Gorling, J. Phys. Chem. A, 2009, 113, 11963–11973 CrossRef CAS.
- R. I. Kvon, S. V. Koscheev and A. I. Boronin, J. Mol. Catal. A: Chem., 2000, 158, 297–300 CrossRef CAS.
- V. Johánek, A. B. De la Ree and J. C. Hemminger, J. Phys. Chem. C, 2009, 113, 4441–4444 CrossRef CAS.
- W. H. Hung and S. L. Bernasek, Surf. Sci., 1995, 339, 272–290 CrossRef CAS.
- Y. L. Tsai, C. Xu and B. E. Koel, Surf. Sci., 1997, 385, 37–59 CrossRef CAS.
- D. R. Mullins and K. Zhang, J. Phys. Chem. B, 2001, 105, 1374–1380 CrossRef CAS.
- S. D. Senanayake, W. O. Gordon, S. H. Overbury and D. R. Mullins, J. Phys. Chem. C, 2009, 113, 6208–6214 CrossRef CAS.
- G. H. Hatzikos and R. I. Masel, Surf. Sci., 1987, 185, 479–494 CAS.
- E. M. Stuve and R. J. Madix, J. Phys. Chem., 1985, 89, 105–112 CrossRef CAS.
- D. L. S. Nieskens, A. P. van Bavel, D. C. Ferre and J. W. Niemantsverdriet, J. Phys. Chem. B, 2004, 108, 14541–14548 CrossRef CAS.
- J. G. Wang, W. X. Li, M. Borg, J. Gustafson, A. Mikkelsen, T. M. Pedersen, E. Lundgren, J. Weissenrieder, J. Klikovits, M. Schmid, B. Hammer and J. N. Andersen, Phys. Rev. Lett., 2005, 95, 2561021–2561024.
- J. Zarraga-Colina and R. M. Nix, Surf. Sci., 2006, 600, 3058–3071 CrossRef CAS.
- A. Ramstad, F. Strisland, S. Raaen, A. Borg and C. Berg, Surf. Sci., 1999, 440, 290–300 CrossRef CAS.
- T. Bhattacharjee, O. R. Inderwildi, S. J. Jenkins, U. Riedel and J. Warnatz, J. Phys. Chem. C, 2008, 112, 8751–8753 CrossRef CAS.
- M. Borasio, O. R. de la Fuente, G. Rupprechter and H. J. Freund, J. Phys. Chem. B, 2005, 109, 17791–17794 CrossRef CAS.
- D. T. P. Watson, J. J. W. Harris and D. A. King, Surf. Sci., 2002, 505, 58–70 CrossRef CAS.
- M. Škoda, M. Cabala, I. Matolínová, K. C. Prince, T. Skála, F. Šutara, K. Veltruská and V. Matolín, J. Chem. Phys., 2009, 130, 0347031–0347037.
- V. Matolín, M. Cabala, V. Cháb, I. Matolínová, K. C. Prince, M. Škoda, F. Šutara, T. Skála and K. Veltruská, Surf. Interface Anal., 2008, 40, 225–230 CrossRef CAS.
- V. Matolín, I. Matolínová, L. Sedláček, K. C. Prince and T. Skála, Nanotechnology, 2009, 20, 2157061–2157067.
- J. Libuda and H. J. Freund, Surf. Sci. Rep., 2005, 57, 157–298 CrossRef CAS.
- V. Matolín, J. Libra, M. Škoda, N. Tsud, K. C. Prince and T. Skála, Surf. Sci., 2009, 603, 1087–1092 CrossRef CAS.
- S. Bernal, J. J. Calvino, G. A. Gifredo, J. M. Rodriguez-Izquiero, V. Perrichon and A. Laachir, J. Catal., 1992, 137, 1–11 CrossRef CAS.
| This journal is © the Owner Societies 2011 |