A crossed molecular beam study on the reaction of methylidyne radicals [CH(X2Π)] with acetylene [C2H2(X1Σg+)]—competing C3H2 + H and C3H + H2 channels
Pavlo
Maksyutenko
,
Fangtong
Zhang
,
Xibin
Gu
and
Ralf I.
Kaiser
*
Department of Chemistry, University of Hawaii at Manoa, Honolulu, HI 96822, USA. E-mail: ralfk@hawaii.edu
First published on 16th November 2010
Abstract
We carried out the crossed molecular beam reaction of ground state methylidyne radicals, CH(X2Π), with acetylene, C2H2(X1Σg+), at a nominal collision energy of 16.8 kJ mol−1. Under single collision conditions, we identified both the atomic and molecular hydrogen loss pathways forming C3H2 and C3H isomers, respectively. A detailed analysis of the experimental data suggested the formation of c-C3H2 (31.5 ± 5.0%), HCCCH/H2CCC (59.5 ± 5.0%), and l-HCCC (9.0 ± 2.0%). The reaction proceeded indirectly via complex formation and involved the unimolecular decomposition of long-lived propargyl radicals to form l-HCCC plus molecular hydrogen and HCCCH/H2CCC plus atomic hydrogen. The formation of c-C3H2 was suggested to be produced via unimolecular decomposition of the cyclopropenyl radical, which in turn could be accessed via addition of the methylidyne radical to both carbon atoms of the acetylene molecule or after an initial addition to only one acetylenic carbon atom via ring closure. This investigation brings us closer to unraveling of the reaction of important combustion radicals—methylidyne—and the connected unimolecular decomposition of chemically activated propargyl radicals. This also links to the formation of C3H and C3H2 in combustion flames and in the interstellar medium.
1. Introduction
The energetics and dynamics of reactions of resonantly stabilized free radicals (RSFRs) are of paramount importance in untangling the formation of soot particles,1–3 polycyclic aromatic hydrocarbons (PAHs) and their hydrogen deficient precursors from the ‘bottom up’ in combustion processes.3–15 In RSFRs, such as propargyl (C3H3; X2B1), allyl (C3H5; X2A2), 1-buten-3-yn-1-yl (C4H3; X2A′), and the 2,4-pentadiynyl-1 radical (C5H3; X2B1) (Fig. 1), the unpaired electron is delocalized and spread out over two or more sites in the molecule. This results in a number of resonant electronic structures of comparable importance. Owing to the delocalization, resonantly stabilized free hydrocarbon radicals are more stable than ordinary radicals16–18 and can reach high concentrations in flames. These high concentrations make them important reactants to be involved in the formation of soot and PAHs. Combustion models suggest that the synthesis of small carbon-bearing molecules and radicals is linked to the formation of PAHs and to the production of soot in hydrocarbon flames.19–24 Reaction mechanisms currently in favor are thought to involve RSFRs like propargyl.9,12,25–39 The self-reaction of propargyl is considered to be one of the most significant cyclization steps in flames of aliphatic fuels.40–46 Electronic structure calculations imply that the acyclic collision complex(es) initially formed in the recombination of two propargyl radicals may isomerize ultimately forming benzene which decomposes to the phenyl radical plus a hydrogen atom.47,48 The models suggest further that consecutive reactions of the phenyl radical with unsaturated hydrocarbons can form more complex structures, possibly bicyclic aromatic hydrocarbon molecules like indene (C9H8) and naphthalene (C10H8).49–52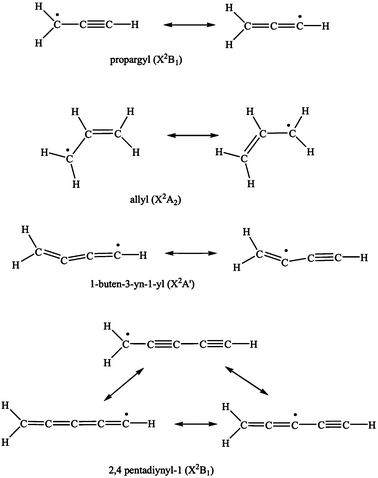 | ||
| Fig. 1 Resonant structures of propargyl, allyl, 1-buten-3-yn-1-yl, and 2,4-pentadiynyl-1. | ||
Although the propargyl radical represents the most studied RSFR due to its potential role in the formation of the first aromatic ring in hydrocarbon flames, its stability and hence unimolecular decomposition has been poorly understood so far.42,44,48,53–65 Here, the unimolecular decomposition of propargyl66 has been suggested to form predominantly C3H2 isomers cyclopropenylidene (c-C3H2; X1A1), propargylene (HCCCH; X3B), and vinylidene carbene (H2CCC; X1A1) (Fig. 2).33,43,67–77 These isomers present also important building blocks in the formation of PAHs and related molecules due to the equilibrium reaction with hydrogen atoms, which access the C3H3 surface via the generic process C3H2 + H ↔ C3H3.78 In this context it is very important to highlight that Hansen et al. assigned propargylene (HCCCH) and cyclopropenylidene (c-C3H2) in cyclopentene flames via photoionization mass spectrometry.79 A re-analysis of previous flame studies80–83 suggested that the nature of the C3H2 isomers formed critically depended on the hydrocarbon fuel.
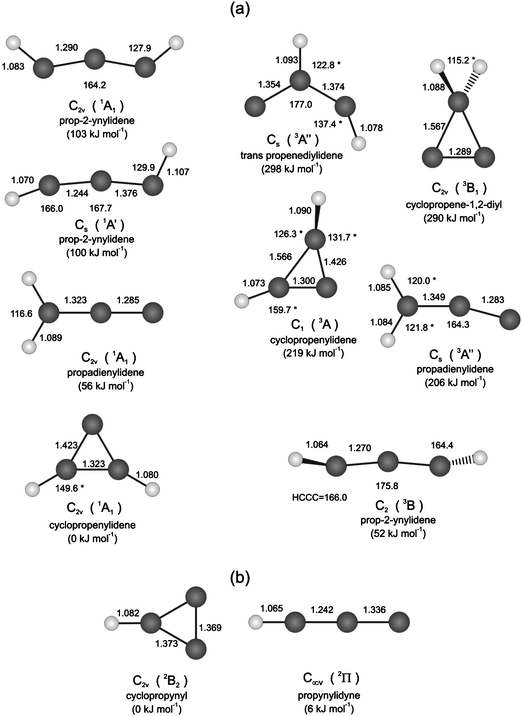 | ||
| Fig. 2 Geometries of C3H2 (a) and C3H (b) isomers; singlet and triplet spin states of the C3H2 isomers are grouped in the left and right column, respectively. The enthalpies of formation of the C3H2 and C3H isomers are indicated relative to the lowest energy isomers (cyclopropenylidene and cyclopropynyl). Bond lengths are in angstroms and bond angles in degrees. Geometries and energies are compiled from Mebel et al.89 Dimensions with asterisk (*) are taken from Peeters et al.86 | ||
Besides flame studies, the formation of C3H2 isomers was also studied experimentally and theoretically by investigating the unimolecular decomposition of chemically activated C3H3 molecules formed in the reaction of methylidyne radicals, CH(X2Π), with acetylene, C2H2 (X1Σg+), and of ground state carbon atoms, C(3Pj), with the vinyl radical, C2H3 (X2A′). Most of the attention has been focused on the reaction of the methylidyne radical; this species presents one of the most reactive carbon-bearing radicals with unsaturated hydrocarbons holding rate constants of a few 10−10 cm3 s−1.84,85 It possesses a Π symmetric vacant, non-bonding molecular orbital which is localized at the carbon atom directed perpendicular to the C–H bond. This empty orbital suggests that the methylidyne radical acts as a Lewis acid in reactions with unsaturated hydrocarbons such as acetylene. Nevertheless, no conclusion has been reached on the reaction mechanism of the methylidyne radical with acetylene to form distinct C3H3 isomers. Three entrance channels have been considered (Fig. 3): (i) an insertion into a carbon–hydrogen bond leading to the propargyl radical (3), (ii) addition to a single carbon atom forming 1-propene-1-yl-3-ylidene (1), and (iii) cyclo addition to the π-electron system leading to cycloprop-2-enyl (4). The initial collision complexes can undergo multiple isomerization and fragment via atomic and molecular hydrogen loss forming C3H2 and C3H isomers, respectively. From the theoretical viewpoint, Peeters et al.86 expanded a theoretical study by Walch et al. on the C3H3 PES87 thus providing a detailed potential energy surface (PES) for the reaction at the B3LYP-DFT/6-31G** level of theory (Table 1). Accompanied by RRKM calculations,88 the authors predicted that at pressures of up to several atmospheres, the initial insertion and cyclo addition had comparable probabilities, while the terminal addition pathway was insignificant (<10%). Over a temperature range from 300 K to 2000 K, the triplet propargylene radical (HCCCH) was determined to be the dominant product (90–82%) followed by the thermodynamically more stable singlet cyclopropenylidene (c-C3H2) isomer (7–11%). The formation of singlet vinylidene carbene (H2CCC) was predicted to be only a minor channel (∼1%) similar to the molecular hydrogen loss forming the linear C3H molecule (propenylidyne; ∼2%). The latter pathway was found to have an exit barrier; the calculations give low yields of this channel despite the greater exoergicity by about 46 kJ mol−1 to form propargylene (HCCCH) plus atomic hydrogen. Vereecken and Peeters suggested a tight transition state for the molecular hydrogen loss, which has a comparable energy to HCCCH plus atomic hydrogen, in contrast to a very loose variational transition state for HCCCH plus atomic hydrogen pathway. An earlier study by Walch et al.87 yielded a similar hydrogen atom yield of 100%, but predicted exclusively singlet propargylene (H2CCC) production; however, multiple reaction pathways were not considered in this study. A third theoretical study by Mebel et al.89 predicted a similar trend as Peeters et al.; branching ratio of HCCCH + H (84–87%), c-C3H2 + H (10–13%), and H2CCC + H (∼1%) were predicted with the molecular hydrogen elimination to linear tricarbon hydride isomer of minor importance (∼2%). This theoretical study located a transition state from the cyclic C3H3 isomer to form c-C3H2 + H, which could not be found by Peeters et al.86 The most recent high-level electronic structure and RRKM calculations predicted higher contributions of the c-C3H2 isomer (27.0%) versus triplet propargylene (HCCCH; 63.5%) with minor contributions from vinylidenecarbene (H2CCC) plus atomic hydrogen and linear C3H (HCCC) plus molecular hydrogen (less than 9.5% combined).90
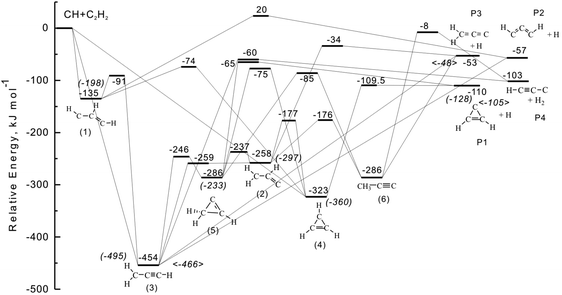 | ||
| Fig. 3 Potential energy diagram of the reaction of methylidyne radicals with acetylene compiled from Mebel et al.,89 Goulay et al.90 (values in round brackets), and Vazquez et al.132 (values in angle brackets). | ||
| Reference | Method | Temperature, pressure | Branching ratio |
|---|---|---|---|
| a branching ratios corresponding to our experimental collision energy are only shown | |||
| Walch et al. (1998)87 | RRKM on internally confined CI PES | 1000 K, 0 Torr | 100% CCCH2 + H |
| Vereecken and Peeters (1999)88 | RRKM on B3LYP-DFT PES | 1400 K*, 0 Torr | 86% HCCCH + H |
| 9% c-C3H2 + H | |||
| 3.3% HCCC + H2 | |||
| 1.7% CCCH2 + H | |||
| Mebel et al. (2001)89 | RRKM on B3LYP and RCCSD(T) PES | 0 K, 0 Torr | 84.5–87% HCCCH + H |
| 10.2–12.8% c-C3H2 + H | |||
| 1.8–1.9% HCCC + H2 | |||
| 0.9% CCCH2 + H | |||
| Goulay et al. (2009)90 | RRKM on CBS-APNO PES | 298 K, 0 Torr | 27% c-C3H2 + H |
| 63.5% HCCCH + H | |||
| <9.5% CCCH2 + H and C3H + H2 | |||
| Neumark et al. (2008)93 | Photofragment translational spectroscopy of propargyl (248 nm) | 0 K, 0 Torr | 97.6 ± 1.2% H loss |
| 2.4 ± 1.2% H2 loss | |||
| Mebel et al. (2001)89 | Theoretical study of photodissociation of propargyl (193 nm) | 0 K, 0 Torr | 86.5% HCCCH + H |
| 3.6% c-C3H2 + H | |||
| 5.5% HCCC + H2 | |||
| 3.5% CCCH2 + H | |||
| 0.9% C2H2 + CH | |||
| Theoretical study of photodissociation of propargyl (242 nm) | 0 K, 0 Torr | 90.2% HCCCH + H | |
| 5.1% c-C3H2 + H | |||
| 3% HCCC + H2 | |||
| 1.6% CCCH2 + H | |||
| 0.1% C2H2 + CH | |||
| Boullart et al. (1996)96 | Low pressure acetylene/atomic oxygen flame (600 K), threshold ionization MS | 600 K, 2 Torr | 85+15−9 H loss |
| 15+9−15 H2 loss | |||
| McKee et al. (2003)97 | Helium reaction flow, LIF of H atoms | 295 K, 25 Torr | 105 ± 9% H loss |
| Loison and Bergeat (2009)85 | Helium reaction flow, LIF of H atoms | 295 K, 2 Torr | 90 ± 9% H loss |
| Goulay et al. (2009)90 | Helium gas flow, tunable VUV (synchrotron) conformer-specific ionization of products; time-resoled MS | 298 K, 4 Torr | >90% c-C3H2 + H |
| <10% HCCCH + H |
Experimentally, the unimolecular decomposition of C3H3 radicals can also be studied under molecular beam conditions via photodissociation of the propargyl radical. Photodissociation of the propargyl radical in the range of 242 to 248 nm generates the propargyl radical with a similar energy to that obtained via chemical activation in the reaction of methylidyne plus acetylene. Deyerl et al.91 used 242 nm to photodissociate internally cold propargyl radicals; the hydrogen atom products were ionized by Lyman-α radiation at 121 nm; Doppler profiles were analyzed to obtain the center-of-mass translational energy distribution. The peak close to zero translational energy suggests that electronic excitation is followed by internal conversion to the ground state surface followed by a statistical decay to the products. From the energetics, the authors concluded that the most likely product was cyclopropenylidene (c-C3H2). Photodissociation studies of isotopically substituted propargyl (D2CCCH) radicals showed complete isotopic scrambling. The authors suggested a unimolecular decomposition of a C3H2D intermediate formed via an internal1,2hydrogen shift followed by cyclization. In a more recent study, Butler et al.92 examined the unimolecular dissociation of propargyl (C3H3) over a wide range of internal energies. The authors proposed that H2CCC is preferentially contributing on the fast side of the time-of-flight (TOF) distribution; c-C3H2 is likely the dominant isomer formed in the dissociation of propargyl radicals with energies near the dissociation threshold. A photodissociation study of propargyl at 248 nm by Neumark et al.93 suggested that the propargyl radical fragments via atomic and molecular hydrogen loss with branching ratios of about 97.6 to 2.4. Under these conditions, theory predicts that only 5% of the atomic hydrogen loss produces cyclo propenylidene (c-C3H2) with propargylene (HCCCH) being dominant.93 The P(ET) distribution of the molecular hydrogen loss leading to the C3H product was distinct from the atomic hydrogen loss; it peaked away from zero translational energy at about 76 kJ mol−1, with a high energy tail extending to about 112 kJ mol−1 as characteristic for the formation of C3H products. This distribution is indicative of a unimolecular decomposition of a C3H3 intermediate via a tight exit transition state. Unfortunately, the center-of-mass translational energy distribution (P(ET)) of the C3H2 and C3H products at m/z = 38 and 37 could not distinguish well between the isomers, except for the small amount of signal at m/z = 38 of 4 ± 2% with translational energy above the maximum for the propargylene (HCCCH) plus atomic hydrogen products; these were suggested to originate from the c-C3H2 isomer. A theoretical study of propargyl photodissociation at 193 and 242 nm by Mebel et al.89 supports the overall branching ratio of the atomic versus molecular hydrogen loss (97%/3%) measured by Neumark et al. at 242 nm and predicts a ∼5% contribution of c-C3H2 and 1.6% of H2CCC.
Alternatively, the reaction of methylidyne radicals with acetylene was also investigated in kinetics studies indicating a barrier-less addition of methylidyne to the acetylene molecule.84,94,95 Canosa et al.84 investigated the temperature dependence of the global rate constant down to 23 K; their data supported the absence of any entrance barrier; however, this work could not determine any reaction product. Kinetic studies in gas flow tubes presented ambiguous results. For an elevated temperature of 600 K and 2 Torr, Boullart et al.96 suggested atomic and molecular hydrogen pathways leading to C3H2 and C3H isomers of about 85% and 15%, respectively, as derived from isothermal discharge flow reactor studies. However, McKee et al. employing the detection of hydrogen atoms via laser induced fluorescence (LIF) indicated an almost exclusive formation of C3H2 molecules of unknown structure.97 On the other hand, Loison and Bergeat reported in a low-pressure fast flow reactor study that the hydrogen atom loss channel contributes to only 90%.85 The most recent kinetic study by Goulay et al.90 was conducted in a slow flow reactor at 4 Torr at 293 K coupled with tunable vacuum ultraviolet (VUV) photoionization and time resolved mass spectrometry to monitor the products. An analysis of the photoionization efficiency curve permitted an isomer-specific detection of the reaction products and allowed an estimation of the reaction product branching ratios. The raw data suggested a predominant fraction of the cyclic C3H2 isomer (90%) and about 10% propargylene (HCCCH). Electronic structure calculations on the C3H3 and C3H2D surfaces revealed the presence of facile hydrogen-atom assisted isomerization processes of the nascent C3H2 products, in particular propargylene (HCCCH), to the cyclic C3H2 isomer; note that under these experimental conditions, single collision conditions do not exist.
Summarized, the previous theoretical and experimental (kinetics, photodissociation) studies suggest that no complete picture has emerged on the unimolecular decomposition of propargyl radicals. Consequently, a high-level experimental investigation on the unimolecular decomposition of the propargyl radical is warranted to shed light on this issue. These are experiments under single collision conditions, in which particles of one supersonic beam are made to ‘collide’ only with particles of a second beam. The crossed molecular beam technique represents the most versatile approach in the elucidation of the energetics, dynamics, and potential energy surfaces (PES) of elementary reactions under single collision conditions without successive reactions of the nascent reaction products.98–101 Here, we present crossed molecular beam data on the reaction of ground state methylidyne radicals with acetylene (CH/C2H2) at a collision energy of 16.8 kJ mol−1. These data are compared then with previous experimental and theoretical studies of this system in an attempt to gain a more coherent picture of the reaction of methylidyne radicals with acetylene. Our results can be placed in the context of hydrocarbon combustion,102,103,104–107 interstellar chemistry108–114 and the chemistry of hydrocarbon rich atmospheres of planets (Uranus, Neptune)115 and moons (Titan, Triton)116,117 where methylidyne reactions with acetylene are important.
2. Experimental
The crossed beam reactions of methylidyne, CH(X2Π), with acetylene, C2H2(X1Σg+), were conducted in a universal crossed molecular beams machine under single collision conditions.118–121 We generated a pulsed supersonic beam of ground state methylidyne radicals, CH(X2Π), via photolysis of helium-seeded bromoform (CHBr3). Helium gas (99.9999%; Gaspro) at a pressure of 2.2 atm was bubbled through a stainless steel container, which acted as a reservoir for the bromoform held at a temperature of 283 K. This resulted in seeding fractions of 0.12% bromoform in helium. This mixture was introduced into a pulsed piezoelectric valve operated at a repetition rate of 60 Hz, pulse widths of 80 μs, and a peak voltage of −400 to −450 V. The output of an excimer laser (KrF, 248 nm, 60 mJ per pulse) was focused with a 1 meter lens downstream of the nozzle to an area of about 4 mm by 0.7 mm. Based on calibration with noble gases, we estimated that a few 1012 radicals cm−3 can be formed in the interaction region of the scattering chamber. Metastable species formed from the tight focus conditions during the photolysis with the 248 nm laser beam are one potential source of background interference. These neutral electronically highly excited species can travel within the beam and pass the QMS filter; they get field ionized by the high voltage doorknob target held at −22.5 kV in the detector. In order to eliminate them, we introduced an electrostatic plate in the region between the pulsed valve nozzle and the skimmer operated at −2000 V. The pulsed beam of the methylidyne radicals pass through a skimmer; a four-slit chopper wheel selects a part of this beam with a well-defined velocity. In the interaction region, this section of the pulse intercepts the most intense section of a pulsed acetylene beam perpendicularly. The peak velocities (vp) and speed ratios (S) of the segments of the interacting beams together with corresponding collision energies and center-of-mass angles are 1747 ± 17 ms−1 and 17 ± 3 for methylidyne and 902 ± 20 ms−1 and 16 ± 1 for acetylene, respectively. This results in a nominal collision energy of 16.8 ± 0.4 kJ mol−1 and a center-of-mass angle of 45.9 ± 0.9°. The time sequence of the experiment is shown in Fig. 4. Note that we determined the velocity and the speed ratio of the methylidyne radical beam on-axis in the TOF mode. Since signal at m/z = 13 (CH+) also originates from dissociative ionization of non-photolyzed bromoform in the ionizer, even a laser on minus laser off subtraction cannot eliminate this contribution. Therefore, we utilized the electron impact ionizer in the soft ionization mode at an electron energy which still allowed sufficient signal from ionization of the methylidyne radical, but a significantly reduced signal from dissociative ionization of the bromoform precursor. The TOF spectra of the methylidyne beam were obtained at electron energy of 34 eV. Note that the photodissociation of bromoform is a multiphoton process initiated by the cleavage of the carbon–bromine bond to yield CHBr2 + Br122,123 (σ(248 nm) = 1.9 × 10−18 cm2).124 Utilizing photoionization photofragment translational spectroscopy, North et al. observed also CHBr, CBr, HBr, and Br2 fragments which were attributed to higher-order photodissociation processes of CHBr2 and CHBr. Mebel computed the photodissociation cross sections of CHBr2 and CHBr at 248 nm to form the methylidyne radical plus molecular and atomic bromine to be 1.6 ± 0.4 × 10−18 and 2.0 ± 0.3 × 10−18 cm2, respectively.125 Therefore, the photodissociation of bromoform produces apart from methylidyne radical possibly the reactive species CHBr2, CHBr, and CBr. When crossing with the acetylene beam, these systems have considerably lower center-of-mass angles due to the heavy bromine atom. For example, the CHBr fragment travelling with the same velocity as the methylidyne radical has a center-of-mass angle of only 8° compared to 45.9° in the methylidyne–acetylene system. Hence, the dynamics of bromine-containing radicals can be distinguished from those of the methylidyne reactions based on the distinct scattering angular ranges of the products and also by the different masses of the products.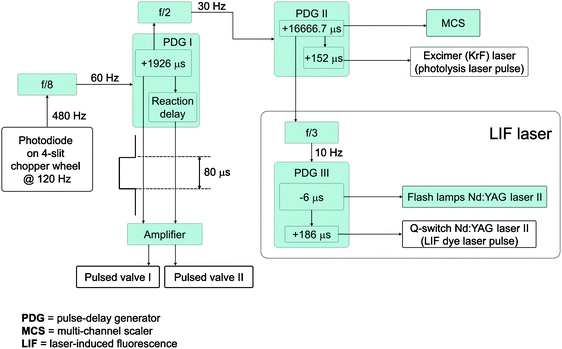 | ||
| Fig. 4 Time sequence of the crossed beam experiments and LIF detection. The delay generator PDG I defines the time delay between the photodiode (time zero) and the pulsed valves; this delay generator triggers PDG II at a rate of 30 Hz. PDG II sets the delay for the excimer laser and—after division by three—triggers PDG III at 10 Hz; the latter triggers the Q-switch of the Nd:YAG laser utilized to pump the dye laser of the LIF laser system. | ||
The reactively scattered species were mass-filtered using a quadrupole mass spectrometric detector in the time-of-flight (TOF) mode after electron-impact ionization of the neutral molecules at 80 eV electron energy. The detector can be rotated within the plane defined by the primary and the secondary reactant beams to allow taking angular resolved TOF spectra. At each angle, up to 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 TOF spectra were accumulated to obtain good signal-to-noise ratios. The recorded TOF spectra were then integrated and normalized to extract the product angular distribution in the laboratory frame (LAB). In order to acquire information on the scattering dynamics, the laboratory data (TOF, LAB) were transformed into the center-of-mass reference frame utilizing a forward-convolution routine.126,127 This iterative method employs a parametrized or point-form angular flux distribution, T(θ), and translational energy flux distribution, P(ET), in the center-of-mass system (CM). Laboratory TOF spectra and the laboratory angular distributions (LAB) are calculated from the T(θ) and P(ET) functions and are averaged over a grid of Newton diagrams accounting for the apparatus functions, beam divergences, and velocity spreads.
000 TOF spectra were accumulated to obtain good signal-to-noise ratios. The recorded TOF spectra were then integrated and normalized to extract the product angular distribution in the laboratory frame (LAB). In order to acquire information on the scattering dynamics, the laboratory data (TOF, LAB) were transformed into the center-of-mass reference frame utilizing a forward-convolution routine.126,127 This iterative method employs a parametrized or point-form angular flux distribution, T(θ), and translational energy flux distribution, P(ET), in the center-of-mass system (CM). Laboratory TOF spectra and the laboratory angular distributions (LAB) are calculated from the T(θ) and P(ET) functions and are averaged over a grid of Newton diagrams accounting for the apparatus functions, beam divergences, and velocity spreads.
Only methylidyne radicals in the 2Π ground electronic state participate in the reaction. The A and B states that might be populated in photolysis process have lifetimes of 440 ± 20 ns and 470 ± 20 ns128 and hence relax to the ground state before they reach the collision center. We characterized the rotational and vibrational modes of the methylidyne radical CH(X2Π) in the interaction region of the scattering chamber utilizing laser induced fluorescence (LIF). Methylidyne radicals are detected using A2Δ–X2Π transitions: (0,0) vibrational band for excitation near 431 nm and (0,1) band for detection near 490 nm. Laser light was produced by Lambda Physics Scanmate dye laser using Stilbene 420 dye pumped by the third harmonic (355 nm) of an integrated neodymium-doped yttrium aluminium garnet (Nd:YAG) laser. The output was attenuated to ∼10 μJ to avoid saturation of the electronic transition. The interference filter (Andover corp., centered at 490 nm, 10 nm bandwidth) in front of the photomultiplier tube (PMT, Hamamatsu R955) discriminated against scattered laser light. The incoming detection laser beam is mainly absorbed by a piece of polished black glass (ThorLabs, neutral density filter, 40–20 surface quality), and the reflected part travels back into the baffles tube. The fluorescence spot in the interaction region is projected by a 35 mm focus lens onto the center of the iris in front of the PMT which is mounted on top and centered at the axis of QMS detector rotation. This vertical orientation of light detector minimizes the collection of Rayleigh scattered light of vertically polarized laser on the atoms and molecules in the beam. Another piece of polished black glass is placed under the interaction region onto the mount for the cold shield to minimize the propagation of scattered laser light in the light collection cone. The fluorescence signal detected by the PMT was then amplified by a built-in preamplifier of the Hamamatsu C7247 PMT socket assembly prior to feeding into a digital oscilloscope. Typically, 16 laser shots were averaged for each data point and sent to a computer viaGPIB interface. An actual LIF spectrum of supersonic jet cooled methylidyne radicals is shown in Fig. 5. The spectrum was analyzed utilizing a LIFBASE database and spectral simulation for diatomic molecules by Jorge Luque.129 Best fits were achieved with a rotational temperature of 14 K; the relative populations of the first vibrationally excited level (ν = 1) was estimated to be less than 6% based on the (1,1), R1(1) peak. Note that we cannot distinguish between different spin–orbit states of methylidyne radical (Ω = 1/2 vs. Ω = 3/2) because for the observed transitions, the largest spectroscopic splitting (0.11 cm−1 for R2(1) transition) would be still smaller than the line width of the detection laser of 0.15 cm−1.
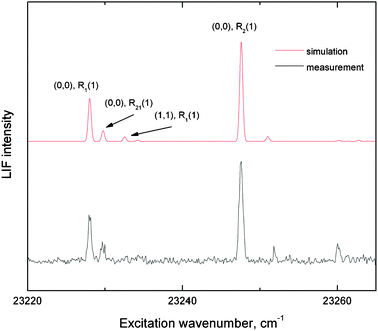 | ||
| Fig. 5 LIF spectrum of methylidyne radicals in a supersonic helium beam (bottom) and the best-fit simulation (top). The simulation suggests a rotational temperature of 14 K; the relative populations of the first vibrationally excited level is less than 6%. | ||
3. Results
Scattering signal for the reaction of methylidyne with acetylene was monitored at mass-to-charge ratios (m/z) of 38 (C3H2+), 37 (C3H+), and 36 (C3+); the time-of-flight (TOF) spectra and the corresponding laboratory angular distributions are shown in Fig. 6 and 7, respectively. Signal at m/z = 38 (C3H2+) originates from the atomic hydrogen loss channel forming C3H2 isomers. The TOF and laboratory angular distribution for this channel could not be fit with a single channel, parametrized center-of-mass translational energy distribution (P(ET)). However, a one channel fit could be achieved by successfully utilizing a P(ET) in point form and a parametrized center-of-mass angular distribution (T(θ)). The TOF spectra at m/z = 37 (C3H+) are very interesting and after scaling—do not overlap with those obtained at m/z = 38. This finding alone suggests that signal at m/z = 37 does not only originate from dissociative electron impact ionization of the C3H2 parent molecules. In order to fit the laboratory data at m/z = 37 (C3H+), it was necessary to include two additional channels with the mass combination 37 amu (C3H) plus 2 amu (H2) from the methylidyne plus acetylene reactants and a second pathway with the mass combination 37 amu (C3H) plus 1 amu (H). These findings deserve some comments. First, the need to incorporate the channel with the mass combination 37 amu plus 2 amu indicates that besides the methylidyneversus atomic hydrogen exchange pathway leading to molecules of the generic formula C3H2, a second reaction channel to form C3H isomer(s) via molecular hydrogen loss is also open. Secondly, it was imperative to include a reaction channel with the mass combination 37 amu plus 1 amu originating from reaction of ground state carbon atoms with acetylene to fit the data. Since the reaction dynamics of this system were studied in our group over a broad range of collision energies from 8 to 31 kJ mol−1,57 the incorporation of this reaction channel does not present a complication. Note that due to the required tight laser focus necessary to generate methylidyne radicals in the primary source via multi photon dissociation of bromoform in the primary source chamber, this tight laser focus likely induced a photolysis of a fraction of the methylidyne radicals to generate ground state carbon atoms plus atomic hydrogen as well. Finally, let us have a look at the TOF data and laboratory angular distributions of m/z = 36 (C3+). It is obvious that neither the TOF nor the LAB distribution of m/z = 36 overlaps with those obtained at m/z = 37 or 38. Therefore, we have to conclude that the signal at m/z = 36 does not originate solely from dissociative ionization of the C3H2 and C3H parent molecules. A closer look at the TOF spectra depicts pronounced shoulders from about 200 to 250 μs; these shoulders are also reflected in enhanced signal at the laboratory angular distribution of m/z = 36. This fast component could be fit with a product mass combination of 36 amu (C3) plus 2 amu (H2) originating from the reaction of ground state carbon atoms with acetylene. Recall the molecular hydrogen plus tricarbon channel was also observed previously in our group during the dynamics studies of the reaction of ground state carbon atoms plus acetylene. Therefore, the center-of-mass functions of this pathway were extracted from ref. 57 to adequately fit the laboratory data.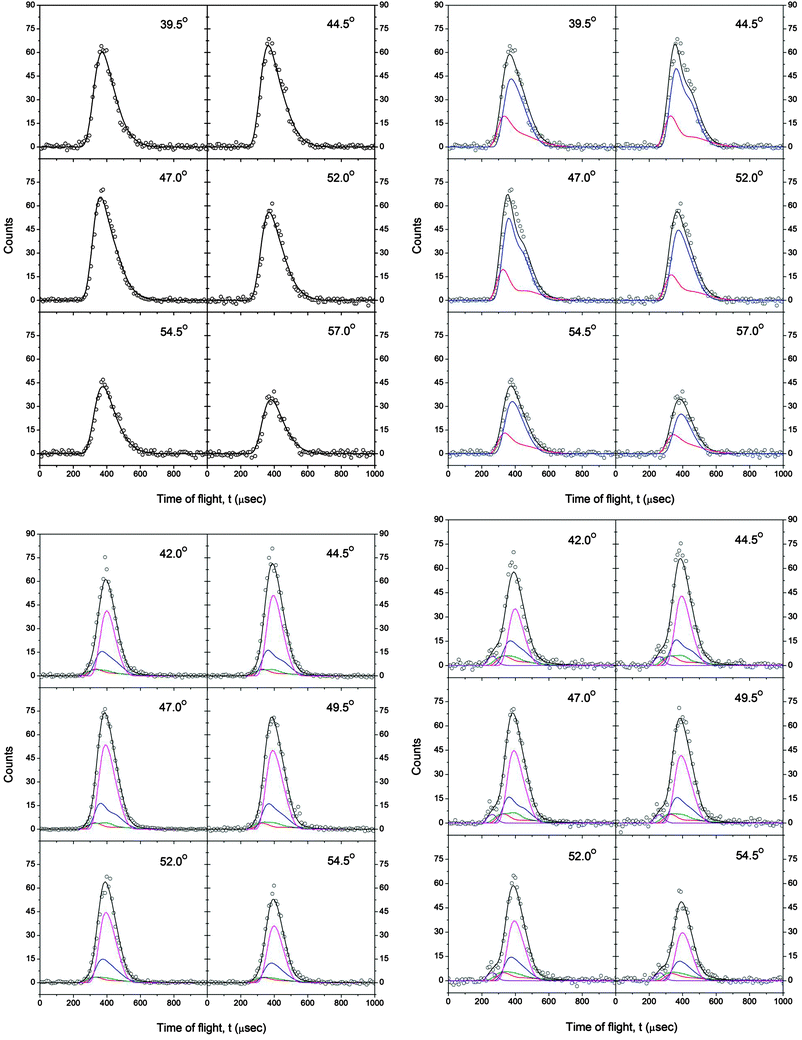 | ||
| Fig. 6 TOF spectra recorded during the reaction of methylidyne radicals with acetylene. Top left m/z = 38 (C3H2+) (single channel fit), top right: m/z = 38 (C3H2+) (two channels fit; red: c-C3H2; blue: HCCCH/H2CCC; black: sum), bottom left: m/z = 37 (C3H+; red: fragmentation from c-C3H2; blue: fragmentation from HCCCH/H2CCC; olive: C3H + H2; magenta: C3H + H; black: sum), bottom right: m/z = 36 (C3+; violet: C3 + H2). The open circles are the experimental data, the solid lines the fits. | ||
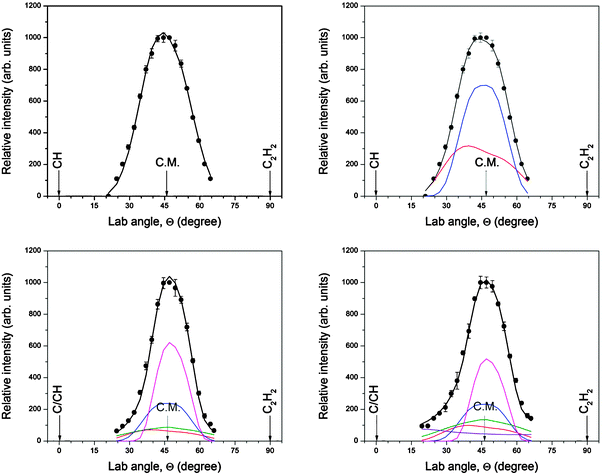 | ||
| Fig. 7 Laboratory angular distributions of scattering signal recorded during the reaction of methylidyne radicals with acetylene. Top left m/z = 38 (C3H2+) (single channel fit), top right: m/z = 38 (C3H2+) (two channels fit), bottom left: m/z = 37 (C3H+), bottom right: m/z = 36 (C3+). C.M. indicates the center-of-mass angle of the reaction of methylidyne with acetylene. The circles are the experimental data, the solid lines the fits. The color code is identical to Fig. 6. | ||
We would like to point out that although the laboratory data at m/z = 38 (C3H2+) were nicely fit with a single channel utilizing center-of-mass translational energy and angular distributions in point and parameter form, respectively, we also investigated if a two-channel fit using all center-of-mass functions in parameter form could lead to an acceptable fit. This approach was guided by the results of the electronic structure calculations as compiled in Fig. 2 and the computed reaction energies to form the c-C3H2 isomer (exoergicity: 105–128 kJ mol−1) and the HCCCH/H2CCC isomers (exoergicity: 48–57 kJ mol−1). The results of the fits are overlaid in Fig. 6 and 7. The data at m/z = 38 could be nicely fit with a two-channel system accounting for the formation of c-C3H2 and HCCCH/H2CCC; the corresponding center-of-mass functions are discussed below.
To summarize these findings, the laboratory data suggest that the reaction of methylidyne radicals with acetylene involves atomic and molecular hydrogen loss pathways leading to C3H2 and C3H isomers as detected via m/z = 38 and 37. The scattering signal at m/z = 38 could be also fit with two channels representing the formation of two classes of isomers: c-C3H2 and HCCCH/H2CCC. Following the procedure as outlined in ref. 57 we extracted the branching ratios of the products formed under single collision conditions in the reaction of methylidyne radicals plus acetylene to be as follows: c-C3H2: 31.5 ± 5.0%, HCCCH/H2CCC: 59.5 ± 5.0%, and HCCC: 9.0 ± 2.0%.
4. Discussion
To provide information on the reaction dynamics, we have to analyze the derived center-of-mass functions as compiled in Fig. 8. Please note that we discuss only those functions relevant to the reaction of methylidyne radicals with acetylene; the atomic carbon–acetylene system, whose center-of-mass functions were necessary to fit data at m/z = 37 and 36, was disseminated previously and the reader is referred to the original literature.57 Let us investigate the center-of-mass translational energy distributions first. The P(ET) of the one-channel depicts a high energy cutoff at 118 ± 8 kJ mol−1. For those molecules born with no internal excitation, the maximum translational energy allowed presents the arithmetic sum of the collision energy and the absolute of the reaction exoergicity. Accordingly, the reaction exoergicity can be determined by subtracting the collision energy from the maximum translational energy observed to be 101 ± 8 kJ mol−1. A comparison of this value with those computed for distinct C3H2 isomers suggests the formation of at least the thermodynamically most stable c-C3H2 molecule. Further, the P(ET) peaks away from zero translational energy; no acceptable fits were achieved with monotonically decreasing functions peaking at zero translational energy. This pattern suggests the existence of a tight exit transition state upon formation of c-C3H2 plus atomic hydrogen or alternatively dynamical effects as the consequence of the gas phase bimolecular collision. Recall that we were able to split up the one-channel fit into two channels (Fig. 8, center column). As evident from the fits of the laboratory data, both the one and the two channel fits nicely replicate the experimental data. The corresponding two channel fit was achieved with P(ET)s extending to maxima of 73 ± 7 kJ mol−1 and 120 ± 10 kJ mol−1. A subtraction of the collision energy leads to reaction exoergicities of 56 ± 7 kJ mol−1 and 103 ± 10 kJ mol−1, respectively. These values are in good agreement with those attributable to the formation of the HCCCH/H2CCC (channel 1) and the c-C3H2 isomers (channel 2) by comparison with the computed exoergicities of 48–57 kJ mol−1 and 105–128 kJ mol−1, respectively. Note that within the error limits, we cannot discriminate between the HCCCH and H2CCC isomers, with the latter energetically slightly unfavorable. It is also important to indicate that the P(ET) related to the formation of the HCCCH/H2CCC isomer(s) peaks closer to zero at 10–20 kJ mol−1 than the function accounting for the c-C3H2 molecule which shows a distribution maximum at about 35–50 kJ mol−1. Therefore, either the transition state leading to c-C3H2 is tighter compared to those leading to HCCCH/H2CCC or dynamical effects are more pronounced in the formation of the c-C3H2 isomer. Having accounted for the atomic hydrogen channel, we are turning our attention now to the molecular hydrogen loss pathway. Here, the P(ET) extends to translational energies of 115 ± 8 kJ mol−1; a subtraction of the collision energy results in an exoergicity of 98 ± 8 kJ mol−1, to form the linear C3H isomer, which is in close agreement with the theoretically predicted value of 103 kJ mol−1 (Fig. 3). Likewise, this distribution peaks well away from zero translational energy at about 25–30 kJ mol−1 indicating a tight exit transition state involved in the formation of l-C3H plus molecular hydrogen. Note that the calculations suggest that the cyclic C3H isomer cannot be formed in the reaction of methylidene with acetylene. The translational energy distributions also allow for the determination of the percentage of available energy partitioned into the translational degrees of freedom. These values were determined to be 39 ± 5% for c-C3H2, 30 ± 3% for HCCCH/H2CCC, and 35 ± 3% for the linear C3H isomer—fractions which indicate indirect scattering dynamics involving collision complexes.130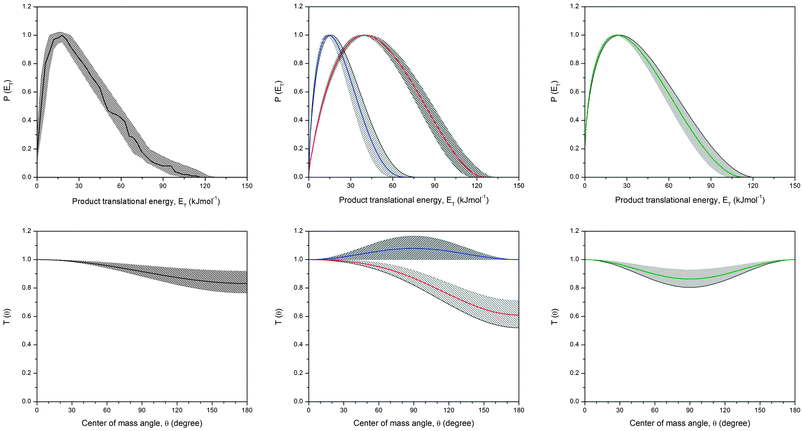 | ||
| Fig. 8 Center-of-mass translational energy (top row) and angular distributions (bottom row) derived for the reaction channels in the methylidyne–acetylene system. Left column: one-channel fit of the C3H2 (38 amu) plus atomic hydrogen (1 amu) channel; center column: two channel fits of the C3H2 (38 amu) plus atomic hydrogen (1 amu) channel, blue: HCCCH/H2CCC isomer, red: c-C3H2 isomer, right column: molecular hydrogen elimination channel leading to C3H isomer(s) (37 amu). The hatched areas define the acceptable fits within the experimental error limits. | ||
The center-of-mass angular distributions reveal important additional information on the reaction dynamics. Upon first inspection, we notice that for all channels, intensity of the center-mass angular distributions is always greater than zero for all angles. This finding implies rather indirect scattering dynamics involving C3H3 intermediates. Secondly, the angular distributions leading to HCCCH/H2CCC and l-C3H are forward–backward symmetric about 90°. The symmetry suggests that the intermediate has a lifetime longer than the rotational period of the decomposing complex.130 Most importantly, data for the molecular elimination channel leading to l-C3H could be only fit with a T(θ) with a minimum at 90°. This indicates geometrical constraint upon complex decomposition with the molecular hydrogen being emitted preferentially within the rotation plane of the decomposing C3H3 intermediate almost perpendicularly to the total angular momentum vector.130 The channel leading to HCCCH/H2CCC involved isotropic or slightly peaking center-of-mass angular distributions. However, the T(θ) leading to c-C3H2 plus atomic hydrogen is distinct from the previously discussed functions as it shows a slight forward peaking. Ratios of the flux intensities at the respective maxima and minima, I(0°)/I(180°), were found to be 1.3 ± 0.2 indicating the possible existence of an osculating complex which decomposes to c-C3H2 plus atomic hydrogen. Further, the incorporated methylidyne radical and the leaving hydrogen atom have to be located on opposite sides of the rotational axis to account for this forward scattering.
Having analyzed the derived center-of-mass functions, we are proposing now the underlying chemical dynamics leading to the formation of c-C3H2, HCCCH/H2CCC, and l-C3H by combining our results with those obtained from recent electronic structure calculations as compiled in Fig. 3. For this, we are correlating the structures of the reaction products with the intermediates involved and also with the experimentally found dynamics. The results suggest that the propargyl radical intermediate (3) presents the central decomposing intermediate forming HCCCH/H2CCC plus atomic and l-C3H plus molecular hydrogenvia a complex-forming reaction mechanism (indirect scattering dynamics as predicted by the center-of-mass angular distributions). Here, propargyl can undergo unimolecular decompositionvia molecular hydrogen loss leading to the l-C3H isomer plus molecular hydrogen through a tight exit transition state located about 43 kJ mol−1 above the energy of the separated reactants. The computed geometry of the exit transition state86 suggests that molecular hydrogen is ejected almost in the plane of the decomposing intermediate. Here, one hydrogen atom of the leaving molecular hydrogen was calculated to depart at an angle of about 1.1° below and the second hydrogen atom 7.4° above the molecular plane. This theoretical prediction is fully supported by our experimental finding of a T(θ) holding a minimum at 90°, i.e. a preferential emission of molecular hydrogen within the plane of the decomposing complex. Note that based on the PES, an alternative pathway connects intermediate CH3CC (6) to l-C3H plus molecular hydrogen. This mechanism would also involve a tight exit transition state, which is located 95 kJ mol−1 above the separated reactants. However, the computed geometry of the transition state connecting intermediate (6) and l-C3H indicates that molecular hydrogen is lost perpendicularly to the rotational plane at an angle of about 86.7°. This clearly contradicts our experimental finding, and we can dismiss that intermediate (6) plays a role in the chemical dynamics of this channel. Therefore, we can conclude that the l-C3H plus molecular hydrogen are formed via unimolecular decomposition of the propargyl radical, but not from intermediate (6). Recall further that the T(θ) of this channel is indicative of a long-lived complex; this can be rationalized by the deep potential energy well of 454 to 495 kJ mol−1, in which the propargyl radical intermediate resides. To summarize, the existence of the propargyl radical (3), decomposing to l-C3H plus molecular hydrogen, can account for the experimental findings of a forward–backward symmetric center-of-mass angular distribution, an experimentally predicted tight exit transition state (recall that the P(ET) of this channel was found to peak away from zero translational energy in the range of 25–30 kJ mol−1), and the in-plane emission of molecular hydrogen.
A closer look at the pertinent PES indicates that the propargyl radical (6) can—besides molecular hydrogen emission to form l-C3H—also fragment via atomic hydrogen elimination to H2CCC and/or HCCCHvia loose transition states. Since the transition state connecting (6) with l-C3H plus molecular hydrogen is almost isoenergetic with the energies of H2CCC and/or HCCCH, but of tight nature compared to the loose transition states from propargyl via atomic hydrogen loss, we expect that the atomic hydrogen loss to H2CCC and/or HCCCH dominates over the molecular hydrogen loss channel. This is confirmed by our experimental findings suggesting fractions of 59.5 ± 5.0% for HCCCH/H2CCC, but only 9.0 ± 2.0% for l-HCCC. Further, the inherent center-of-mass angular distribution for the hydrogen atom loss channel leading to HCCCH/H2CCC suggested the existence of a long-lived reaction intermediate. Consequently, we can conclude that a long-lived propargyl radical presents a common reaction intermediate; its unimolecular decay leads predominantly to HCCCH/H2CCCvia atomic and to a lesser amount to l-HCCCvia molecular hydrogen loss pathways. However, we should point out that the P(ET) of the atomic hydrogen loss channel was found to peak at 10–20 kJ mol−1. A long-lived complex behavior should be reflected in a P(ET) peaking at zero translational energy. Could other reaction intermediates also contribute to the atomic hydrogen loss and inherent formation of HCCCH/H2CCC? Based on the PES, intermediate (1) could in principle decay to HCCCH plus atomic hydrogen. However, the inherent barrier of 155 kJ mol−1 cannot be verified experimentally. Likewise, if (1) is formed, it rather isomerizes to the propargyl intermediate (3) via a barrier of only 44 kJ mol−1. Hence, we can exclude intermediate (1) as a source of HCCCH plus atomic hydrogen. On the other hand, intermediate (2), which could be formed viaisomerization of (3) and/or (3) → (5) → (2) can connect to H2CCC plus atomic hydrogenvia a barrier located 9 to 14 kJ mol−1 above the energy of the separated products. Therefore, at the present stage, we cannot disregard that the decomposition of intermediate (2), which is formed from the already identified propargyl radical intermediate (3) viaisomerization leads to H2CCC plus atomic hydrogen.
Finally, we are addressing the possible dynamics to form the c-C3H2 molecule. The PES offers two possible routes. The first pathway proceeds via addition of the methylidyne radical to the carbon–carbon triple bond of the acetylene molecule forming a cyclic intermediate (4) which decomposes through a loose exit transition state. The second mechanism would involve a decomposing intermediate (5), which is formed viaisomerization of the propargyl radical. Recall that the propargyl radical was found to be long-lived with respect to its rotational period. On the other hand, the T(θ) is slightly forward-scattered and indicative of an osculating complex. Therefore, if c-C3H2 is formed from (5), and the latter from (3), we expect a forward–backward symmetric center-of-mass angular distribution. Therefore, we can conclude that intermediate (4) decomposed preferentially to c-C3H2. However, does the experimentally found off-zero-peaking of the corresponding P(ET) contradict the loose transition state predicted from calculations? It should be recalled that dynamical factor can lead to center-of-mass translational energy distributions peaking well away from zero translational energy. This was observed previously in the forward scattered channels involved in the formation of c-C3H and the propargyl isomer (C3H3) plus a light hydrogen atom co-fragment as observed in the crossed beam reaction of ground state carbon atoms with acetylene57 and ethylene131 studied earlier. The off-zero peaking micro channels were also correlated with slightly forward-peaking center-of-mass angular distributions. Therefore, a comparison of the presently observed dynamics with those extracted for the c-C3H + H and C3H3 + H channels in the C(3P)/C2H2 and C(3P)/C2H4 reactions indicates that in the CH/C2H2 system, the c-C3H2 is likely formed via a cyclic intermediate (4), which in turn is accessed via addition of the methylidyne radical to the carbon–carbon triple bond of the acetylene reactant. An analysis of the possible rotational axes of (4) suggests that a rotation around A/B axes can account for the forward scattering since in this case, the incorporated methylidyne reactant and the leaving hydrogen atom are located on opposite sides of the rotational axis.
How do our results of c-C3H2 (31.5 ± 5.0%), HCCCH/H2CCC (59.5 ± 5.0%), and HCCC (9.0 ± 2.0%) compare with previous experiments and calculations as compiled in Table 1? It is important to note that none of the prior experiments was conducted under single collision conditions. Nevertheless, the preceding experiments by Boullart et al., McKee et al., Loison et al., and Goulay et al. agree with our finding that the atomic hydrogen loss channel (105 ± 9%–85 ± 15%) dominates over the molecular hydrogen loss. Previous works suggested upper limits of about 10% by predominantly monitoring the hydrogen atom yield quantitatively with respect to a well-known reference system. Our crossed beam experiments, on the other hand, were able to explicitly verify the molecular hydrogen loss pathway directly by monitoring TOF spectra at m/z = 37 and by a comparison of their patterns and fits with those of m/z = 38 and 36. Based on the two channel fit, we were also able to estimate the branching ratios of the cyclic versus non-cyclic C3H2 isomers to be about 2∶3. It should be recalled that based on our experimental data, we cannot discriminate between the HCCCH and H2CCC isomers.
5. Summary and conclusion
We conducted the crossed molecular beam reaction of ground state methylidyne radicals, CH(X2Π), with acetylene, C2H2(X1Σg+), at a collision energy of 16.8 kJ mol−1. Under single collision conditions, we identified both the atomic and molecular hydrogen loss pathways leading to C3H2 and C3H isomers, respectively. A detailed analysis of the experimental data suggested the formation of c-C3H2 (31.5 ± 5.0%), HCCCH/H2CCC (59.5 ± 5.0%), and l-HCCC (9.0 ± 2.0%) and identified for the first time the formation of C3H2and C3H isomers under single collision conditions. The reaction proceeds indirectly via complex formation and proceeds via unimolecular decomposition of long-lived propargyl radicals (3) to form l-HCCC plus molecular hydrogen and HCCCH/H2CCC plus atomic hydrogen. The involvement of two higher energy, C3H3 intermediates (5) and (2) formed viaisomerization of propargyl (3) cannot be discounted at the present stage. On the other hand, the c-C3H2 was suggested to be produced via unimolecular decomposition of the cyclopropenyl radical (4), which in turn can be accessed via addition of the methylidyne radical to both carbon atoms of the acetylene molecule or after an initial addition to only one acetylenic carbon atom via ring closure. Future studies will investigate the CH/C2D2 and CD/C2H2 systems to answer the remaining questions, such as the determination of an explicit branching ratio of HCCCHversusH2CCC and the role of higher energy C3H3 isomers (5) and (2). Nevertheless, our present studies bring us closer to the understanding of the reaction of important combustion radicals—methylidyne—and the connected unimolecular decomposition of chemically activated propargyl radicals. This also links to the formation of C3H and C3H2 in combustion flames and in the interstellar medium.Acknowledgements
This project was supported by the US Department of Energy Office of Science (DE-FG02-03-ER15411).References
- C. C. Austin, D. Wang, D. J. Ecobichon and G. Dussault, J. Toxicol. Environ. Health, Part A, 2001, 63, 437 Search PubMed.
- A. Ergut, Y. A. Levendis, H. Richter, J. Carlson, Fourth Jt. Meet. U.S. Sect. Combust. Inst.: West. States, Cent. States, East. States, Philadelphia, PA, United States, Mar. 20–23, 2005, A05/1.
- A. Kazakov and M. Frenklach, Combust. Flame, 1998, 112, 270 CrossRef CAS.
- R. I. Kaiser, T. N. Le, T. L. Nguyen, A. M. Mebel, N. Balucani, Y. T. Lee, F. Stahl, P. v. R. Schleyer and H. F. Schaefer III, Faraday Discuss., 2002, 119, 51 RSC.
- J. Appel, H. Bockhorn and M. Frenklach, Combust. Flame, 2000, 121, 122 CrossRef CAS.
- C. E. Baukal, Oxygen Enhanced Combustion, 1998 Search PubMed.
- M. F. Budyka, T. S. Zyubina, A. G. Ryabenko, V. E. Muradyan, S. E. Esipov and N. I. Cherepanova, Chem. Phys. Lett., 2002, 354, 93 CrossRef CAS.
- F. Cataldo, Editor Polyynes: Synthesis, Properties, and Applications, Interdisciplinary Meeting on Polyynes and Carbyne held 30–31 October 2003 in Naples, Italy, 2006.
- M. Frenklach, Phys. Chem. Chem. Phys., 2002, 4, 2028 RSC.
- A. Kazakov and M. Frenklach, Combust. Flame, 1998, 114, 484 CrossRef CAS.
- Y.-T. Lin, R. K. Mishra and S.-L. Lee, J. Phys. Chem. B, 1999, 103, 3151 CrossRef CAS.
- A. Necula and L. T. Scott, J. Am. Chem. Soc., 2000, 122, 1548 CrossRef CAS.
- A. M. Nienow and J. T. Roberts, Annu. Rev. Phys. Chem., 2006, 57, 105 CrossRef CAS.
- H. Richter and J. B. Howard, Prog. Energy Combust. Sci., 2000, 26, 565 CrossRef CAS.
- L. Vereecken, H. F. Bettinger and J. Peeters, Phys. Chem. Chem. Phys., 2002, 4, 2019 RSC.
- D. B. Atkinson and J. W. Hudgens, J. Phys. Chem. A, 1999, 103, 4242 CrossRef CAS.
- D. K. Hahn, S. J. Klippenstein and J. A. Miller, Faraday Discuss., 2001, 119, 79 Search PubMed.
- D. J. Mann and W. L. Hase, J. Am. Chem. Soc., 2002, 124, 3208 CrossRef CAS.
- A. B. Fialkov, J. Dennebaum and K. H. Homann, Combust. Flame, 2001, 125, 763 CrossRef CAS.
- K. J. Higgins, H. Jung, D. B. Kittelson, J. T. Roberts and M. R. Zachariah, J. Phys. Chem. A, 2002, 106, 96 CrossRef CAS.
- V. V. Kislov, A. M. Mebel and S. H. Lin, J. Phys. Chem. A, 2002, 106, 6171 CrossRef CAS.
- A. Kitajima, T. Hatanaka, M. Takeuchi, H. Torikai and T. Miyadera, Combust. Flame, 2005, 142, 72 CrossRef CAS.
- J. Kou, T. Mori, Y. Kubozono and K. Mitsuke, Phys. Chem. Chem. Phys., 2005, 7, 119 RSC.
- P. Mitchell and M. Frenklach, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 061407 CrossRef.
- B. Apicella, R. Barbella, A. Ciajolo and A. Tregrossi, Combust. Sci. Technol., 2002, 174, 309 CrossRef CAS.
- V. I. Babushok and A. W. Miziolek, Combust. Flame, 2004, 136, 141 CrossRef CAS.
- F. Battin-Leclerc, Phys. Chem. Chem. Phys., 2002, 4, 2072 RSC.
- G. A. Dolgonos and G. H. Peslherbe, Int. J. Mass Spectrom., 2005, 241, 261 Search PubMed.
- N. S. Goroff, Acc. Chem. Res., 1996, 29, 77 CrossRef CAS.
- K.-H. Homann, Angew. Chem., Int. Ed., 1998, 37, 2435 CrossRef.
- M. M. Maricq, Combust. Flame, 2004, 137, 340 CrossRef CAS.
- B. Oektem, M. P. Tolocka, B. Zhao, H. Wang and M. V. Johnston, Combust. Flame, 2005, 142, 364 CrossRef.
- H. Richter and J. B. Howard, Phys. Chem. Chem. Phys., 2002, 4, 2038 RSC.
- L. Shebaro, S. R. Bhalotra and D. Herschbach, J. Phys. Chem. A, 1997, 101, 6775 CrossRef CAS.
- K. Siegmann and K. Sattler, J. Chem. Phys., 2000, 112, 698 CrossRef CAS.
- R. Taylor, G. J. Langley, H. W. Kroto and D. R. M. Walton, Nature, 1993, 366, 728 CAS.
- B. V. Unterreiner, M. Sierka and R. Ahlrichs, Phys. Chem. Chem. Phys., 2004, 6, 4377 RSC.
- C. L. Morter, S. K. Farhat, J. D. Adamson, G. P. Glass and R. F. Curl, J. Phys. Chem., 1994, 98, 7029 CrossRef CAS.
- R. D. Kern, H. Chen, J. H. Kiefer and P. S. Mudipalli, Combust. Flame, 1995, 100, 177 CrossRef CAS.
- Y. Georgievskii, J. A. Miller and S. J. Klippenstein, Phys. Chem. Chem. Phys., 2007, 9, 4259 RSC.
- L. B. Harding, S. J. Klippenstein and J. A. Miller, J. Phys. Chem. A, 2008, 112, 522 CrossRef CAS.
- R. D. Kern and K. Xie, Prog. Energy Combust. Sci., 1991, 17, 191 CrossRef CAS.
- J. A. Miller and C. F. Melius, Combust. Flame, 1992, 91, 21 CrossRef CAS.
- J. A. Miller, M. J. Pilling and J. Troe, Proc. Combust. Inst., 2005, 30, 43 CrossRef.
- J. A. Miller, J. P. Senosiain, S. J. Klippenstein and Y. Georgievskii, J. Phys. Chem. A, 2008, 112, 9429 CrossRef CAS.
- J. P. Senosiain, S. J. Klippenstein and J. A. Miller, Proc. Combust. Inst., 2007, 31, 185 CrossRef.
- C. F. Melius, J. A. Miller and E. M. Evleth, Symp. (Int.) Combust., [Proc.], 1992, 24, 621 Search PubMed.
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2003, 107, 7783 CrossRef CAS.
- Here, PAHs may also form without involving six-membered aromatic precursors via reactions of polyacetylenes with C4Hx species (x = 1–5) and through reactions of two C5H5 radicals followed by rearrangements and hydrogen emission(s) to form naphthalene and/or the azulene isomer.
- C. A. Arrington, C. Ramos, A. D. Robinson and T. S. Zwier, J. Phys. Chem. A, 1998, 102, 3315 CrossRef CAS.
- C. Cavallotti, S. Fascella, R. Rota and S. Carra, Combust. Sci. Technol., 2004, 176, 705 CrossRef CAS.
- C. S. McEnally, L. D. Pfefferle, A. G. Robinson and T. S. Zwier, Combust. Flame, 2000, 123, 344 CrossRef CAS.
- N. Balucani, G. Capozza, F. Leonori, E. Segoloni and P. Casavecchia, Int. Rev. Phys. Chem., 2006, 25, 109 CrossRef CAS.
- E. Buonomo and D. C. Clary, J. Phys. Chem. A, 2001, 105, 2694 CrossRef CAS.
- D. C. Clary, E. Buonomo, I. R. Sims, I. W. M. Smith, W. D. Geppert, C. Naulin, M. Costes, L. Cartechini and P. Casavecchia, J. Phys. Chem. A, 2002, 106, 5541 CrossRef CAS.
- M. Frenklach and E. D. Feigelson, Astrophys. J., 1989, 341, 372 CrossRef CAS.
- X. Gu, Y. Guo, F. Zhang and R. I. Kaiser, J. Phys. Chem. A, 2007, 111, 2980 CrossRef.
- E. Ionescu and S. A. Reid, THEOCHEM, 2005, 725, 45 CrossRef CAS.
- W. M. Jackson, D. S. Anex, R. E. Continetti, B. A. Balko and Y. T. Lee, J. Chem. Phys., 1991, 95, 7327 CrossRef CAS.
- R. I. Kaiser, C. Oschsenfeld, M. Head-Gordon, Y. T. Lee and A. G. Suits, Science, 1996, 274, 1508 CrossRef CAS.
- A. M. Mebel, Rev. Mod. Quantum Chem., 2002, 1, 340 Search PubMed.
- A. M. Mebel, W. M. Jackson, A. H. H. Chang and S. H. Lin, J. Am. Chem. Soc., 1998, 120, 5751 CrossRef CAS.
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2001, 105, 7254 CrossRef CAS.
- C. Ochsenfeld, R. I. Kaiser, Y. T. Lee, A. G. Suits and M. Head-Gordon, J. Chem. Phys., 1997, 106, 4141 CrossRef CAS.
- W. K. Park, J. Park, S. C. Park, B. J. Braams, C. Chen and J. M. Bowman, J. Chem. Phys., 2006, 125, 081101 CrossRef.
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2003, 107, 2680 CrossRef CAS.
- J. Aguilera-Iparraguirre, A. D. Boese, W. Klopper and B. Ruscic, Chem. Phys., 2008, 346, 56 CrossRef CAS.
- A. Bergeat, T. Calvo, F. Caralp, J.-H. Fillion, G. Dorthe and J.-C. Loison, Faraday Discuss., 2001, 119, 67 Search PubMed.
- I. Fernandez and G. Frenking, Faraday Discuss., 2007, 135, 403 RSC.
- M. Hausmann and K. H. Homann, Ber. Bunsen–Ges., 1997, 101, 651 CAS.
- G. Maier, H. P. Reisenauer, W. Schwab, P. Carsky, B. A. Hess, Jr. and L. J. Schaad, J. Am. Chem. Soc., 1987, 109, 5183 CrossRef CAS.
- J. A. Miller, J. V. Volponi and J.-F. Pauwels, Combust. Flame, 1996, 105, 451 CrossRef CAS.
- A. Mohajeri and M. J. Jenabi, THEOCHEM, 2007, 820, 65 CrossRef CAS.
- H. P. Reisenauer, G. Maier, A. Riemann and R. W. Hoffman, Angew. Chem., 1984, 96, 596 CrossRef CAS.
- R. A. Seburg, J. T. DePinto, E. V. Patterson and R. J. McMahon, J. Am. Chem. Soc., 1995, 117, 835 CrossRef CAS.
- R. A. Seburg, E. V. Patterson and R. J. McMahon, J. Am. Chem. Soc., 2009, 131, 9442 CrossRef CAS.
- R. A. Seburg, E. V. Patterson, J. F. Stanton and R. J. McMahon, J. Am. Chem. Soc., 1997, 119, 5847 CrossRef CAS.
- L. B. Harding, S. J. Klippenstein and Y. Georgievskii, J. Phys. Chem. A, 2007, 111, 3789 CrossRef CAS.
- C. A. Taatjes, S. J. Klippenstein, N. Hansen, J. A. Miller, T. A. Cool, J. Wang, M. E. Law and P. R. Westmoreland, Phys. Chem. Chem. Phys., 2005, 7, 806 RSC.
- A. Bhargava and P. R. Westmoreland, Combust. Flame, 1998, 113, 333 CrossRef CAS.
- J. D. Bittner, PhD Thesis, MIT, 1981 Search PubMed.
- G. E. Oulundsen, PhD Thesis, Univ. Massachussets, 1999 Search PubMed.
- P. R. Westmoreland, J. B. Howard, J. P. Longwell and A. M. Dean, AIChE J., 1986, 32, 1971 CAS.
- A. Canosa, I. R. Sims, D. Travers, I. W. M. Smith and B. R. Rowe, Astron. Astrophys., 1997, 323, 644 CAS.
- J.-C. Loison and A. Bergeat, Phys. Chem. Chem. Phys., 2009, 11, 655 RSC.
- L. Vereecken, K. Pierloot and J. Peeters, J. Chem. Phys., 1998, 108, 1068 CrossRef CAS.
- R. Guadagnini, G. C. Schatz and S. P. Walch, J. Phys. Chem. A, 1998, 102, 5857 CrossRef CAS.
- L. Vereecken and J. Peeters, J. Phys. Chem. A, 1999, 103, 5523 CrossRef CAS.
- T. L. Nguyen, A. M. Mebel, S. H. Lin and R. I. Kaiser, J. Phys. Chem. A, 2001, 105, 11549 CrossRef CAS.
- F. Goulay, A. J. Trevitt, G. Meloni, T. M. Selby, D. L. Osborn, C. A. Taatjes, L. Vereecken and S. R. Leone, J. Am. Chem. Soc., 2009, 131, 993 CrossRef CAS.
- H.-J. Deyerl, I. Fischer and P. Chen, J. Chem. Phys., 1999, 111, 3441 CrossRef CAS.
- L. R. McCunn, B. L. FitzPatrick, M. J. Krisch, L. J. Butler, C.-W. Liang and J. J. Lin, J. Chem. Phys., 2006, 125, 133306 CrossRef.
- S. J. Goncher, D. T. Moore, N. E. Sveum and D. M. Neumark, J. Chem. Phys., 2008, 128, 114303 CrossRef.
- A compilation of rate constants is given in http://kinetics.nist.gov/kinetics/index.jsp.
- H. Thiesemann, J. MacNamara and C. A. Taatjes, J. Phys. Chem. A, 1997, 101, 1881 CrossRef CAS.
- W. Boullart, K. Devriendt, R. Borms and J. Peeters, J. Phys. Chem., 1996, 100, 998 CrossRef CAS.
- K. McKee, M. A. Blitz, K. J. Hughes, M. J. Pilling, H.-B. Qian, A. Taylor and P. W. Seakins, J. Phys. Chem. A, 2003, 107, 5710 CrossRef CAS.
- Atomic and Molecular Beam Methods, ed. G. Scoles,Oxford University Press, Oxford, 1988 Search PubMed.
- P. Casavecchia, G. Capozza and E. Segoloni, Adv. Ser. Phys. Chem., 2004, 14, 329 Search PubMed.
- R. I. Kaiser, Chem. Rev., 2002, 102, 1309 CrossRef CAS.
- K. Liu, J. Chem. Phys., 2006, 125, 132307 CrossRef.
- J. A. Miller and C. T. Bowman, Prog. Energy Combust. Sci., 1989, 15, 287 CrossRef CAS.
- J. A. Miller, R. J. Kee and C. K. Westbrook, Annu. Rev. Phys. Chem., 1990, 41, 345 CrossRef CAS.
- C. H. Wu and R. D. Kern, J. Phys. Chem., 1987, 91, 6291 CrossRef CAS.
- R. D. Kern, C. H. Wu, J. N. Yong, K. M. Pamidimukkala and H. J. Singh, Energy Fuels, 1988, 2, 454 CrossRef CAS.
- J. Peeters, W. Boullart and K. Devriendt, J. Phys. Chem., 1995, 99, 3583 CrossRef CAS.
- J. Peeters, Bull. Soc. Chim. Belg., 1997, 106, 337 CAS.
- R. A. Abramovich, Editor Reactive Intermediates, 1980, vol. 1 Search PubMed.
- H. E. Matthews and W. M. Irvine, Astrophys. J., 1985, 298, L61 CrossRef.
- P. Thaddeus, J. M. Vrtilek and C. A. Gottlieb, Astrophys. J., 1985, 299, L63 CrossRef CAS.
- J. Cernicharo, C. A. Gottlieb, M. Guelin, T. C. Killian, G. Paubert, P. Thaddeus and J. M. Vrtilek, Astrophys. J., 1991, 368, L39 CrossRef CAS.
- R. I. Kaiser, C. Ochsenfeld, D. Stranges, M. Head-Gordon and b. p. Y. T. Lee, Faraday Discuss., 1998, 109, 183 RSC.
- E. Herbst, H.-H. Lee, D. A. Howe and T. J. Millar, Mon. Not. R. Astron. Soc., 1994, 268, 335 CAS.
- T. B. H. Kuiper, J. B. Whiteoak, R. S. Peng, W. L. Peters, III and J. E. Reynolds, Astrophys. J., 1993, 416, L33 CrossRef CAS.
- R. V. Yelle, F. Herbert, B. R. Sandel, R. J. Vervack, Jr. and T. M. Wentzel, Icarus, 1993, 104, 38 CrossRef CAS.
- V. A. Krasnopolsky and D. P. Cruikshank, J. Geophys. Res., [Planets], 1995, 100, 21 CAS.
- P. P. Lavvas, A. Coustenis and I. M. Vardavas, Planet. Space Sci., 2008, 56, 67 CrossRef CAS.
- X. B. Gu, Y. Guo, H. Chan, E. Kawamura and R. I. Kaiser, Rev. Sci. Instrum., 2005, 76, 116103 CrossRef.
- X. B. Gu, Y. Guo, E. Kawamura and R. I. Kaiser, Rev. Sci. Instrum., 2005, 76, 083115 CrossRef.
- Y. Guo, X. Gu, E. Kawamura and R. I. Kaiser, Rev. Sci. Instrum., 2006, 77, 034701 CrossRef.
- Y. Guo, X. Gu and R. I. Kaiser, Int. J. Mass Spectrom., 2006, 249/250, 420 Search PubMed.
- W. S. McGivern, O. Sorkhabi, A. G. Suits, A. Derecskei-Kovacs and S. W. North, J. Phys. Chem. A, 2000, 104, 10085 CrossRef CAS.
- P. Zou, J. Shu, T. J. Sears, G. E. Hall and S. W. North, J. Phys. Chem. A, 2004, 108, 1482 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, Jr., J. A. Kerr and J. Troe, J. Phys. Chem. Ref. Data, 1992, 21, 1548.
- A. M. Mebel, Florida International University, unpublished results, 2010 Search PubMed.
- M. Vernon, PhD Thesis, University of California, 1981 Search PubMed.
- M. S. Weiss, PhD Thesis, University of California, 1986 Search PubMed.
- M. Danielsson, P. Erman, A. Hishikawa, M. Larsson and E. Rachlew-Kaellne, J. Chem. Phys., 1993, 98, 9405 CrossRef CAS.
- http://www.sri.com/psd/lifbase/ .
- R. D. Levine , Molecular Reaction Dynamics, Cambridge University Press , Cambridge, UK , 2005, p. 568 Search PubMed.
- R. I. Kaiser, Y. T. Lee and A. G. Suits, J. Chem. Phys., 1996, 105, 8705 CrossRef CAS.
- J. Vazquez, M. E. Harding, J. Gauss and J. F. Stanton, J. Phys. Chem. A, 2009, 113, 12447 CrossRef CAS.
| This journal is © the Owner Societies 2011 |