DOI:
10.1039/C0CE00530D
(Paper)
CrystEngComm, 2011,
13, 103-108
A time-resolved diffraction study of a window of stability in the synthesis of a copper carboxylate metal–organic framework†
Received
12th August 2010
, Accepted 8th September 2010
First published on 1st October 2010
Abstract
An in situ, time-resolved energy dispersive powder X-ray diffraction study of the solvothermal crystallisation of the copper(II) 4,4′,4″-benzene-1,3,5-triyl-tris(benzoate) metal–organic framework MOF-14 shows how reaction conditions must be carefully chosen to allow successful preparation of the material, since on prolonged heating at ≥120 °C the material irreversibly collapses into Cu2O under solvothermal conditions in less than 2 hours. This situation is in contrast to the related Cu(II)-containing metal–organic framework HKUST-1, which shows solvothermal stability over similar temperatures and reaction times. The kinetics of crystallisation of both MOFs are examined using a mathematical model proposed by Gualtieri for zeolite crystallisation: this allows separation of the nucleation and growth regimes to yield two rate constants. Arrhenius analysis gives activation energies that reveal in both cases the crystallisations are nucleation controlled. For MOF-14 we can additionally simulate its decomposition as dissolution of the first-formed interpenetrating structure: this produces a complete picture of the solvothermal stability of MOF-14 as nucleation-growth crystallisation, with some evidence of secondary nucleation, followed by dissolution.
Introduction
Metal–organic framework (MOF) materials are currently attracting great attention.1 These hybrid solids are constructed from metals or metal–oxy clusters linked by polydentate organic ligands (often carboxylates) to give a huge variety of open framework structures that may possess porosity ranging from the nanoporous to the mesoporous.2 Particular focus is placed on applications of these materials in gas storage and separation (such as hydrogen, methane or carbon dioxide),3 but now functionalities of MOFs include those long associated with conventional inorganic solid materials4 such as electronic,5 magnetic,6 multiferroic7 and catalytic properties.8 The attraction of MOFs lies in the scope for the ‘design’ of new materials by combination of a metal centre that has a particular coordination preference with an organic ligand of specific geometry and binding configuration to yield a network with some degree of predictability of both structural and chemical properties. This has proved possible to some degree where ‘isoreticular’ structures based on the principle of sequential modifications of ligands can be used to modify rationally pore dimensions, for example.9–12
There are as yet few detailed experimental studies of the crystallisation mechanism of MOFs: these are required if the true potential for the design of new materials is to be realised.13 Techniques such as EXAFS14 and light scattering15,16 have been used to study the local structure of species found in reactive solutions and AFM has been applied to examine growing crystals faces of a copper MOF.17 These are important studies since they examined in situ the evolution of reactive solutions and their interaction with surfaces in the case of AFM. The use of time-resolved powder X-ray diffraction is an additional powerful method for examining crystal growth under solvothermal conditions, and has been widely demonstrated for the study of the crystal growth of silicate zeolites,18–20 inorganic zeotypes,21–24 metal oxides,25,26 complex chalcogenides27,28 and hydroxides.29 We recently showed that in the solvothermal crystallisation of MOF materials, time-resolved diffraction could also reveal new information not possible from other methods, such as the observation of crystalline intermediate materials that form in situ transiently.30
In this paper we now show how the stability of MOFs can depend critically on the reaction conditions (time and temperature, for example) used: this is an important consideration in the future discovery of new materials which may be kinetically stable. In this work, we have used time-resolved diffraction experiments using the energy-dispersive X-ray diffraction (EDXRD) technique. This is a low d-spacing resolution method whose advantage lies in the combination of intense synchrotron-generated white X-rays that can penetrate large-scale reaction vessels and a fixed angle solid-state detector that can accumulate data rapidly (minutes to seconds).18,31 We have studied the solvothermal formation of MOF-14, a copper(II) material that contains dimeric paddle-wheel units, connected by the tridentate linker 4,4′,4″-benzene-1,3,5-triyl-tris(benzoate) to yield a complex structure that contains a pair of interwoven nets,32Fig. 1. MOF-14 was originally shown to possess high porosity and able to uptake a variety of small gases32 and has been studied because of potential for the separation of hydrogen from methane.33 Our interest in the material lay in studying the crystallisation of a complex, interpenetrating structure, to compare with the work so far done on simple MOF networks, with the aim of identifying any differences in crystallisation kinetics.
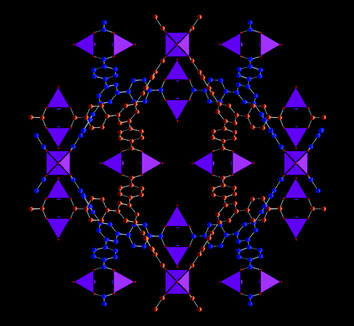 |
| | Fig. 1 A view of the structure of MOF-14 showing the two interpenetrating network of tridentate links in blue and orange. The purple polyhedra represent five-coordinate Cu(II)-centres as found in the fully solvated material. Drawn using crystal data from ref. 32. | |
Results and discussion
Fig. 2 shows contour maps of EDXRD data recorded in 2 minute intervals during the formation of MOF-14 at two temperatures. At the lowest temperature studied, 110 °C or less, the characteristic product Bragg peaks reach their maximum intensities after around three hours and this allows for the successful isolation of phase-pure polycrystalline samples of MOF-14 to be identified. At higher temperatures, although crystallisation does occur, complete disappearance of the solid product is observed on prolonged heating: for example at 130 °C the product vanishes after around 100 minutes, Fig. 2b. It is well known from zeolite chemistry that open-framework materials can be metastable and collapse upon prolonged heating to give condensed, crystalline silicates: for example zeolite A will transform into the dense phase hydroxysodalite under hydrothermal conditions with time or at high pH.19 In the case of MOF-14, the metal–organic framework is completely destroyed on prolonged heating to yield Cu2O as the only crystalline product identified by ex situpowder X-ray diffraction (ESI†). Cu2O has previously been reported as a by-product in the synthesis of other Cu(II) containing MOFs if reaction temperatures are high.34,35
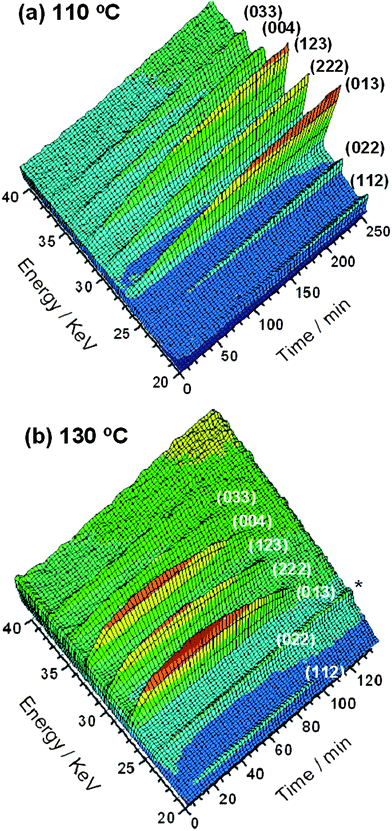 |
| | Fig. 2 Three-dimensional views of MOF-14 crystallisation seen using EDXRD at (a) 110 °C and (b) 130 °C with Miller indices of Bragg peaks labelled. * denotes a feature due to the reaction cell that is present throughout the crystallisation. | |
Fig. 3a shows integrated peak areas of the strongest (013) Bragg reflection for each temperature studied. Integration of all observed Bragg peaks was performed and the normalised curves suggest that crystal growth is isotropic, see Fig. 3b for the case of 110 °C for four separate Bragg peaks. Fig. 3a indicates the striking dependence on the stability of MOF-14 with reaction conditions and shows clearly a ‘window of stability’ that needs to be considered for the preparation of a phase-pure sample of this particular MOF. The in situ diffraction measurement uniquely allows this information to be determined efficiently without making any assumptions about the material being unaltered in numerous quenching experiments.
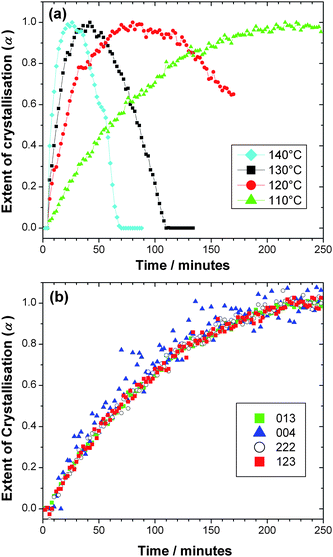 |
| | Fig. 3 (a) Crystallisation curves for MOF-14 at four temperatures obtained by integrating the strongest (013) Bragg reflections and (b) Extent of crystallisation determined for four Bragg reflections at 110 °C indicating the isotropic crystal growth. | |
Kinetic analysis of crystallisation curves of materials under solvothermal conditions has so far largely been performed using empirical mathematical expressions. The most popular of these is the Avrami–Erofe'ev equation, eqn (1).36–39
| | | α = 1 − exp{−[k(t − t0)]n} | (1) |
This expression is versatile enough to describe the form of many extent of crystallisation curves and allows calculation of a rate constant of crystallisation:40 indeed this model has been used to study the solvothermal formation of a number of materials including zeolites, zeotype phosphates and transition-metal oxides.19–25 This model and other Avrami-type expressions, however, have severe limitations in these situations: in particular that they were originally developed for the specific case of solid–solid reactions in powdered compacts so the fitted parameters have little physical significance in the situation of heterogeneous crystallisation of a solid from a liquid, and furthermore they do not separate the processes of nucleation and crystal growth, instead simulate ‘extent of crystallisation’ by a single set of parameters. In order to improve understanding of MOF crystallisation, we have therefore sought a more suitable mathematical crystallisation model. Gualtieri studied the crystallisation of three zeolites from amorphous clay and silica mixtures in aqueous NaOH solution at 90–150 °C using time-resolved XRD and developed a crystallisation model that was able to simulate solution-mediated crystal growth of zeolites, separating nucleation and growth as two separate processes with different activation energies.41 He considered nucleation and growth separately to develop a model for extent of crystallisation (α) vs. time (t).
| | 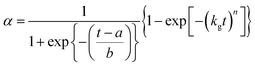 | (2) |
In order to apply this model it is assumed that zeolite crystal growth takes place in 3 dimensions (n = 3; note this is not the same exponent as the Avrami exponent). This may be tested by monitoring the emergence of several different hkl Bragg peaks and also by examining the crystal form using microscopy (see below). kg is the rate constant of crystal growth, while a and b are constants related to nucleation. Importantly, if these values are known then a probability function for nucleation, PNvs. time can be calculated.
| | 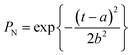 | (3) |
This approach to modeling the sigmoidal curves associated with crystal growth uniquely separates nucleation and crystal growth to give two rate constants, since:
The relative magnitudes of kg and kn can then indicate the rate determining process in crystallisation. In addition the value of b can be used to infer information about the nature of nucleation: if b ≈ 15 nucleation is heterogeneous, if b ≈ 20 nucleation is homogeneous, and if b > 20 then nucleation is autocatalytic.41
Gualtieri studied systems where one of three different zeolites crystallised and although these were not necessarily stable, two going on to dissolve at long times, he did not consider the case of dissolution. Dissolution kinetics have been studied in a variety of situations and the simplest (empirical) model is to assume a power law with a rate constant of dissolution, kd.42
Thus although the earlier stages of reaction is likely to be dominated by crystal growth, we can simulate the two competing processes of crystallisation and dissolution as time proceeds by using two terms in our fitted expression for extent of crystallisation:
| | 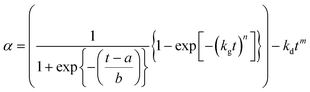 | (6) |
Fig. 4 shows the fits obtained by using this kinetic model (eqn (6)) for MOF-14 crystallisation. Using the temperature dependence of the rate constants for both nucleation and growth we have calculated activation energies for each process: the corresponding Arrhenius plots are shown in Fig. 5. Table 1 contains all of the calculated parameters from this analysis. At the highest temperature studied the kinetic model breaks down, indicated by physically unreasonable parameters being calculated (see ESI†), suggesting that the initial growth of MOF-14 is soon followed by the decay of the first formed material, and growth and dissolution/collapse are intimately linked. Indeed, MOF-14 is observed for little more than 1 hour at 140 °C. At other temperatures excellent fits are achieved.
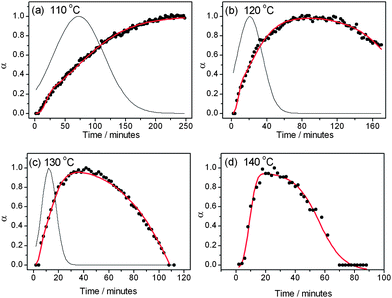 |
| | Fig. 4 Fits of the growth curves (points) of MOF-14 at four temperatures. The grey line is the simulated nucleation and the red line extent of crystal growth. At 120, 130 and 140 °C the Gualtieri equation combined with a dissolution term is used. At 110 °C the Gualtieri equation alone has been used. Note that in (d) the simulated nucleation has not been plotted due to physically unreasonable fitted parameters (see text). | |
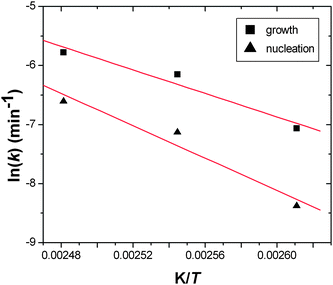 |
| | Fig. 5 Arrenhius plots for MOF-14 crystallisation for nucleation and for crystal growth. The red lines are those determined by linear regression to calculate activation energies. | |
Table 1 Fitted kinetic parameters for the crystallisation of the copper(II) carboxylates MOF-14 and HKUST-1 along with activation energies determined from Arrhenius plots
|
T/°C |
a/min |
b/min |
k
g/min−1 |
k
n/min−1 |
E
a(growth)/kJ mol−1 |
E
a(nucleation)/kJ mol−1 |
|
HKUST-1
|
| 85 |
104.1(6) |
66.6(6) |
0.0274(5) |
0.0096(1) |
|
|
| 95 |
52.8(3) |
28.9(3) |
0.068(2) |
0.0189(1) |
|
|
| 105 |
26.5(2) |
15.8(2) |
0.118(4) |
0.0378(3) |
|
|
| 115 |
14.8(2) |
8.8(2) |
0.164(7) |
0.067(1) |
|
|
| 125 |
9.2(1) |
4.1(1) |
0.26(2) |
0.108(1) |
|
|
| |
|
|
|
|
63.8 |
71.6 |
|
MOF-14
|
| 110 |
72.5(5) |
42.0(5) |
0.051(2) |
0.0138(1) |
|
|
| 120 |
20.8(5) |
14.2(5) |
0.128(8) |
0.048(1) |
|
|
| 130 |
12.35(3) |
5.7(3) |
0.19(2) |
0.0810(2) |
|
|
| |
|
|
|
|
82.8 |
113.9 |
For comparison, we have re-analysed extent of crystallisation curves we recently reported30 for a related copper(II) material, HKUST-1, that contains the same paddle-wheel dimers but linked by 1,3,5-benzene-tri-carboxylate. Fig. 6 shows the fits to the extent of crystallisation data along with the simulated nucleation curve, Fig. 7 shows the Arrhenius plots used to determine activation energy and Table 1 contains the calculated parameters from the kinetic analysis. It is noteworthy that the values of activation energies for HKUST-1 are rather similar to the value we recently calculated using the Avrami–Erofe'ev model using the same extent of crystallisation data (73.3 kJ mol−1).30 The values of activation energies (both nucleation and growth) we have now determined for both HKUST-1 and MOF-14 are comparable although larger than those reported previously for the hydrothermal preparation of materials that have been proved to form under nucleation control such as barium titanate (55 kJ mol−1)43 and magnesium-aluminium double hydroxide (41 kJ mol−1).29 The value of activation energy of nucleation for MOF-14 is noticeably larger than for HKUST-1 and this may reflect the more complex, interpenetrating structure.
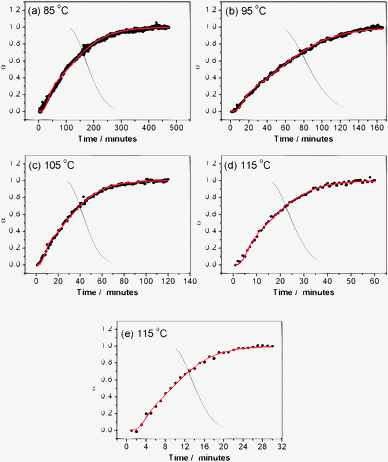 |
| | Fig. 6 Fits of the extent of crystallisation curves (points) of HKUST-1 using the Gualtieri equation at five temperatures. The grey curve is the simulated nucleation and the red line extent of crystal growth. | |
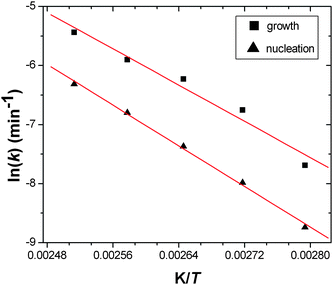 |
| | Fig. 7 Arrenhius plots for HKUST-1 crystallisation for nucleation and for crystal growth. The red lines are those determined by linear regression to calculate activation energies. | |
It is noteworthy that for both materials we have studied here the nucleation extends well into the crystal growth (see the simulated nucleation curves on Fig. 4 and 6) and this is consistent with light scattering studies of HKUST-1 crystallisation performed independently by Zacher et al. who also reported that the continued formation of nucleation sites continued late into the period of emergence of crystalline material.16Table 1 importantly shows that the rate constants for nucleation are always lower than those of crystal growth: this shows how nucleation is rate determining for these reactions. Gualtieri further proposed that the constant b reflects the nature of nucleation with b > 20 indicating autocatalytic nucleation (the increase of nucleation rate by the release of further nucleation sites upon dissolution of a gel, for example) while b ≤ 15 suggests that heterogeneous nucleation predominates.25 Thus for MOF-14 with increasing temperature, heterogeneous nucleation (suggesting nucleation at the surfaces of already formed crystallites, for example) becomes important. This situation was also seen for the silicate zeolites studied by Gualtieri.25 In order to provide further evidence for heterogeneous nucleation we used SEM to examine specimens of MOF-14 prepared using the same conditions as in the in situ studies: Fig. 8 shows a SEM image from a specimen prepared reaction at 110 °C for 3 hours that indeed reveals the presence of small particles on the surface or larger well-formed crystallites: this suggests that at later stages of crystallisation some heterogeneous nucleation may occur at the surface of already formed crystallites. The micrograph also shows a cube-like crystal form that validates the assumption of isotropic crystal growth in the kinetic fitting (see above).
 |
| | Fig. 8
SEM image of MOF-14 crystals prepared at 110 °C for 3 hours. | |
Experimental
In situ EDXRD spectra were recorded using Beamline F3 of the HASYLAB facility, Germany. This beamline receives white-beam radiation with energy 13.5–65 keV and the incident X-ray beam is collimated to 20 × 20 µm2. Reactions were performed with 12 mm diameter DURAN® tubes fitted with PBT screw caps and PTFE-coated gaskets. Copper(II) nitrate hydrate Cu(NO3)2·H2O (0.5245 g) and 4,4′,4″-benzene-1,3,5-triyl-tris(benzoic acid) (0.6570 g) in a mixture of DMF (10 mL), dioxane (1 mL) and water (1 mL) were stirred a few minutes before introducing 2 mL of the resulting suspension into the tube which was placed in a circulating oil heater equipped with a magnetic stirring device. EDXRD patterns were recorded in 120 s intervals using a fixed angle solid-state Ge detector whose angle (2θ) was calibrated using the characteristic Bragg peaks of a pre-made solid sample of MOF-14. In the EDXRD experiment, a Bragg peak is characterised by a characteristic energy, E/keV, related to its d-spacing, d/Å, according to:| | 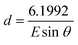 | (7) |
Data were normalised to the incident beam intensity by using the logged synchrotron beam current. The program PowDLL44 was used to process the data into a format suitable for peak fitting using the program XFIT45 using Pseudo-Voigt functions. The identification of the final solid product was also confirmed using ex situ high-resolution powder XRD (see ESI†). Kinetic analysis was performed using least-squares refinement to the expressions outlined above.
Conclusions
In summary, we have demonstrated using in situ X-ray diffraction how reaction conditions may have to be very carefully chosen to maximise the yield of MOF materials under solvothermal conditions, and that materials with interesting properties may be kinetic products. This has implications for the discovery of new MOF materials since a wide exploration of reaction conditions must be undertaken in order to isolate kinetic phases that may have practical applications. The kinetic analysis approach we have adopted, using a model previously developed for zeolite formation, provides more physical insight than other simple empirical mathematical models that have been used to interpret solvothermal crystallisation data since it separates nucleation and growth into two separate terms: we suggest that it could be more widely applicable for the interpretation of crystallisation data from a variety of solvothermal reactions.
Acknowledgements
We thank DESY for award of beamtime at HASYLAB, Mark Feyand of the Christian-Albrechts-Universität zu Kiel for his assistance with use of Beamline F3, and Cristelle Thouvenot (Versailles) for her help with SEM experiments.
References
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213 RSC.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- C. N. R. Rao, A. K. Cheetham and A. Thirumurugan, J. Phys.: Condens. Matter, 2008, 20, 083202 CrossRef.
- C. G. Silva, A. Corma and H. Garcia, J. Mater. Chem., 2010, 20, 3141 RSC.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC.
- P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS.
- S. Surblé, C. Serre, C. Mellot-Draznieks, F. Millange and G. Férey, Chem. Commun., 2006, 284 RSC.
- X. Lin, J. H. Jia, X. B. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schroder, Angew. Chem., Int. Ed., 2006, 45, 7358 CrossRef.
- R. E. Morris, ChemPhysChem, 2009, 10, 327 CrossRef CAS.
- S. Surblé, F. Millange, C. Serre, G. Férey and R. I. Walton, Chem. Commun., 2006, 1518 RSC.
- S. Hermes, T. Witte, T. Hikov, D. Zacher, S. Bahnmuller, G. Langstein, K. Huber and R. A. Fischer, J. Am. Chem. Soc., 2007, 129, 5324 CrossRef CAS.
- D. Zacher, J. N. Liu, K. Huber and R. A. Fischer, Chem. Commun., 2009, 1031 RSC.
- M. Shoaee, M. W. Anderson and M. R. Attfield, Angew. Chem., Int. Ed., 2008, 47, 8525 CrossRef CAS.
- J. Munn, P. Barnes, D. Hausermann, S. A. Axon and J. Klinowski, Phase Transitions, 1992, 39, 129 CrossRef CAS.
- R. I. Walton, F. Millange, D. O'Hare, A. T. Davies, G. Sankar and C. R. A. Catlow, J. Phys. Chem. B, 2001, 101, 83 CrossRef.
- R. I. Walton, R. I. Smith and D. O'Hare, Microporous Mesoporous Mater., 2001, 48, 79 CrossRef CAS.
- R. J. Francis, S. J. Price, J. S. O. Evans, S. O'Brien, D. O' Hare and S. M. Clark, Chem. Mater., 1996, 8, 2102 CrossRef CAS.
- R. I. Walton, T. Loiseau, D. O'Hare and G. Férey, Chem. Mater., 1999, 11, 3201 CrossRef CAS.
- G. Sankar, T. Okubo, W. Fan and F. Meneau, Faraday Discuss., 2007, 136, 157 RSC.
- M. G. O'Brien, M. Sanchez-Sanchez, A. M. Beale, D. W. Lewis, G. Sankar and C. R. A. Catlow, J. Phys. Chem. C, 2007, 111, 16951 CrossRef CAS.
- R. Kiebach, N. Pienack, M. E. Ordolff, F. Studt and W. Bensch, Chem. Mater., 2006, 18, 1196 CrossRef CAS.
- D. R. Modeshia, R. J. Darton, S. E. Ashbrook and R. I. Walton, Chem. Commun., 2009, 68 RSC.
- L. Engelke, M. Schaefer, F. Porsch and W. Bensch, Eur. J. Inorg. Chem., 2003, 506 CrossRef CAS.
- L. Engelke, M. Schaefer, M. Schur and W. Bensch, Chem. Mater., 2001, 13, 1383 CrossRef CAS.
- F. Millange, R. I. Walton and D. O'Hare, J. Mater. Chem., 2000, 10, 1713 RSC.
- F. Millange, M. Medina, N. Guillou, G. Férey, K. M. Golden and R. I. Walton, Angew. Chem., Int. Ed., 2010, 49, 763 CAS.
- R. I. Walton and D. O'Hare, Chem. Commun., 2000, 2283 RSC.
- B. L. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021 CrossRef CAS.
- M. Gallo and D. Glossman-Mitnik, J. Phys. Chem. C, 2009, 113, 6634 CrossRef CAS.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81 CrossRef CAS.
- E. Biemmi, S. Christian, N. Stock and T. Bein, Microporous Mesoporous Mater., 2009, 117, 111 CrossRef CAS.
- M. J. Avrami, Chem. Phys. Lett., 1939, 7, 1103 CAS.
- M. J. Avrami, Chem. Phys. Lett., 1940, 8, 212 CAS.
- M. J. Avrami, Chem. Phys. Lett., 1941, 9, 177 CAS.
- B. V. Eroféev, C. R. (Dokl.) Acad. Sci. URSS, 1946, 52, 511 Search PubMed.
- E. E. Finney and R. G. Finke, Chem. Mater., 2009, 21, 4692 CrossRef CAS.
- A. F. Gualtieri, Phys. Chem. Miner., 2001, 28, 719 CrossRef CAS.
- P. J. Skrdla, J. Pharm. Biomed. Anal., 2007, 25, 251 CrossRef.
- R. I. Walton, F. Millange, R. I. Smith, T. C. Hansen and D. O'Hare, J. Am. Chem. Soc., 2001, 123, 12547 CrossRef CAS.
-
N. Kourkoumelis, PowDLL: A program for the interconversion of powder diffraction data files, Version 2.19, 2009, http://users.uoi.gr/nkourkou/powdll.htm Search PubMed.
-
R. W. Cheary and A. A. Coelho, Program XFIT, deposited in CCP14 Powder Diffraction Library, Engineering and Physical Sciences Research Council, Daresbury Laboratory, Warrington, England, 1996, http://www.ccp14.ac.uk/ Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterisation data for MOF-14. See DOI: 10.1039/c0ce00530d |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.