Crystal structure of anhydrous 5-aminotetrazole and its high-pressure behavior†
Hiroshi
Fujihisa
*a,
Kazumasa
Honda
a,
Shigeaki
Obata
a,
Hiroshi
Yamawaki
a,
Satoshi
Takeya
a,
Yoshito
Gotoh
a and
Takehiro
Matsunaga
b
aResearch Institute of Instrumentation Frontier (RIIF)National Institute of Advanced Industrial Science and Technology (AIST), AIST Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: hiroshi.fujihisa@aist.go.jp
bResearch Institute of Science for Safety and Sustainability (RISS)National Institute of Advanced Industrial Science and Technology (AIST), AIST Tsukuba Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
First published on 27th September 2010
Abstract
Anhydrous 5-aminotetrazole (5-amino-1H-tetrazole, 5-ATZ) is an energetic material that produces a large amount of nitrogen gas by thermal decomposition and is often used as a gas generator agent. Although it is used widely as an air bag inflator, its crystal structure has not been determined yet. Thus, we performed a powder X-ray diffraction experiment, a Rietveld analysis, and the density functional theory calculation to investigate its structure and determined it to be an orthorhombic P212121 with a 1H-form molecule. We also investigated the high-pressure behavior and found that the orthorhombic phase is stable up to at least 11.6 GPa. Furthermore, the quantum molecular dynamics calculations at high-temperature and high-pressure of 5-ATZ were carried out and predicted the phase change including molecular decomposition.
Introduction
5-Aminotetrazole (5-amino-1H-tetrazole, 5-ATZ, CH3N5) is an energetic material containing 80 weight% nitrogen that is used as a gas generator agent for air bag inflators. Its molecular structure is shown in Fig. 1(a). It melts at 480 K, then the five-membered rings begin to break shortly thereafter and is followed by the production of nitrogen gas.1 It does not show any explosive feature within itself. 5-ATZ monohydrate has been known to form a monoclinic P21/c crystal by single crystal experiments.2,3 However, the crystal structure of anhydrous 5-ATZ has not been determined yet despite its wide use in air bags, probably because the anhydrous form can only be obtained in powder form.![Molecular structures of (a) 5-amino-1H-tetrazole [5-ATZ 1H-form], (b) 5-amino-2H-tetrazole [5-ATZ 2H-form], and (c) 5-imino-1H,4H-tetrazole [5-ITZ, imino-form].](/image/article/2011/CE/c0ce00278j/c0ce00278j-f1.gif) | ||
| Fig. 1 Molecular structures of (a) 5-amino-1H-tetrazole [5-ATZ 1H-form], (b) 5-amino-2H-tetrazole [5-ATZ 2H-form], and (c) 5-imino-1H,4H-tetrazole [5-ITZ, imino-form]. | ||
There are some experimental1,4–8 and theoretical9–11 studies for the decomposition of 5-ATZ at high temperature. The decomposition mechanisms were considered with a monomolecule in these studies. Recently, Kiselev and Gritsan12 calculated the decomposition path with a bimolecular reaction and concluded that the 2H-form shown in Fig. 1(b) is more preferable at a high temperature than the imino-form shown in Fig. 1(c), contrary to Lesnikovich et al.'s report.7 Thus, the molecular form and the decomposition process at a high temperature are still unclear. Our group recently performed differential calorimetry and powder X-ray diffraction experiments to investigate the phase behavior from room temperature to 195 °C13 and found the presence of anhydrous crystalline phases I, II, and III. Phase I is identical to the one in the present report. Unfortunately, the crystal structures for phases II and III have not been determined yet at higher temperatures. We believe that the thermal decomposition mechanism would become much clearer if they can be found.
The purpose of this study is to determine the crystal and molecular structures of anhydrous 5-ATZ when single crystals are not available. Then, we investigate the pressure dependence of the crystal structure and the different behavior that can be expected up to high temperatures. To achieve this, we employ the density functional theory (DFT) calculation to optimize the atomic coordinates including hydrogen and evaluate the small change in the intra-molecular bond lengths. The quantum molecular dynamics (MD) calculation is performed on the obtained structure to simulate the high-temperature high-pressure behavior of the 5-ATZ crystal to investigate the phase transition including molecular decomposition toward nitrogen gas.
Crystal structure at ambient temperature and pressure
The anhydrous 5-amino-1H-tetrazole powder specimen was donated by Chugoku-Kayaku Co. The powder was ground finely in mortar and placed into a hole with a 0.8 mm diameter and a 0.5 mm thickness. The X-ray was generated by a MoKα rotating anode (Rigaku, Co. Ltd., normal focus, 50 kV/200 mA, λ = 0.71070 Å), monochromatized by pyrolytic graphite (002) reflection, and collimated by a 200 µm pinhole. The powder X-ray diffraction image was detected by an imaging plate detector with a typical exposure time of 1 hour. Two-dimensional Debye–Scherrer rings were obtained and converted to one-dimensional 2θ-intensity patterns by the software PIP.14A good lattice candidate could not be obtained from the initial diffraction pattern. However, another diffraction pattern taken several hours later changed to the one for 5-ATZ monohydrate.2,3 Thus, the anhydrous 5-ATZ was found to absorb water gradually in the atmosphere, and the initial pattern contained both peaks of the anhydrous 5-ATZ and the monohydrate.
To prevent the absorption of water, we ground the sample in a nitrogen gas atmosphere and mixed some paraffin oil with the sample powder and were able to obtain the diffraction of pure anhydrous 5-ATZ successfully as shown by the dots in Fig. 2. The pattern was indexed with 28 peaks by the software X-Cell15 of Accelrys, Inc. The most probable candidate for the unit cell was found to be an orthorhombic lattice of a = 5.088 Å, b = 3.664 Å, and c = 18.040 Å with a cell volume of V = 336.3 Å3.
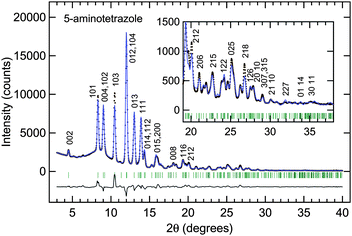 | ||
| Fig. 2 Diffraction pattern and Rietveld analysis of the anhydrous crystal of 5-aminotetrazole at ambient pressure and temperature. The closed circles show the experimental values. The blue solid lines show the calculated diffraction pattern of the crystal structure model with the orthorhombic space groupP212121. The numerical values on the peaks of the calculated diffraction pattern indicate the Miller indices. Rwp, Rwp (without background), and Rp are 7.4, 13.0, and 5.4%, respectively. | ||
The initial model was constructed by the software FOX.16 The molecular shape without hydrogen was referred from the one in the 5-ATZ monohydrate by Bray and White.3 The space group numbers 16–19, 25, 26, 28, 31, 47, 51, and 59 were allowed from the extinction rule. Only P212121 (no. 19) was able to fit the diffraction intensity. After the nitrogen and carbon positions were obtained, three hydrogen atoms (H7, H8, and H9) were attached manually to complete the 5-ATZ 1H-form. Thus, the constructed initial model was refined by a Rietveld analysis with the software Materials Studio Reflex of Accelrys, Inc. The molecule with hydrogen was treated as a rigid body with no internal degrees of freedom. Three translational and three rotational degrees of freedom were optimized for the molecule in the unit cell. A good Rietveld fit was finally obtained as shown in Fig. 2.
The atomic positions were optimized by the DFT calculation software CASTEP17 to reduce the error of the atomic positions that originated from the difference in the molecular shape between the monohydrate and the anhydrate and the error in the powder diffraction intensities. This allows us to evaluate the hydrogen bond distances and the small pressure change in the covalent bonds precisely. The lattice parameters were set to the experimental values, and the atomic positions were optimized to minimize the total energy. We employed the Wu-Cohen GGA18 and used an ultrasoft pseudopotential.19 The energy cutoff for the plane wave basis set was 350 eV. The 10 × 15 × 3 Monkhorst-Pack grid was chosen for the k-point set that produced k-separations of approximately 0.02 Å−1. Furthermore, the hydrogen positions were examined to find out whether or not hydrogen moved to a neighboring molecule when forming a solid. When hydrogen was added to produce the 2H-form as shown in Fig. 1(b), the DFT optimized atomic position of the 5-membered ring moved 0.39 Å from that of the 1H-form. The enthalpy of the optimized structure with the 2H-form was 0.64 eV higher than that of the 1H-form. The difference between the diffraction pattern and the experimental pattern of the 2H-form resulted in Rwp = 12.7%. Since the 1H-form has a smaller more suitable Rwp of 7.4%, we rejected the 2H-form as a candidate for the model. When the 5-ITZ imino-form was used as shown in Fig. 1(c), the hydrogen automatically moved to a neighboring molecule, and eventually back to the 1H-form. Thus, the original 1H-form was confirmed to be maintained in the solid. The final crystal structure determined by the Rietveld refinement and the DFT calculation is shown in Fig. 3. Fig. 3(a) and (b) show the two-dimensional hydrogen bond network in the ac-plane and the stacking of the planar molecules along the b-axis, respectively. Their structure data are provided in the CIF file in the ESI†.
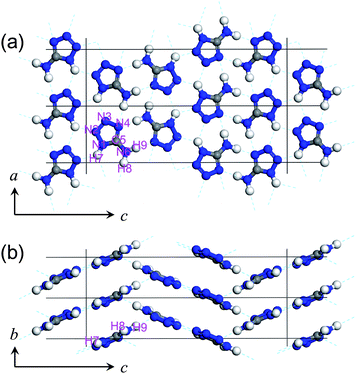 | ||
| Fig. 3 Structure of 5-aminotetrazole at ambient pressure and temperature in (a) ac- and (b) bc-planes. The lattice constants and the cell volume were refined to be a = 5.090 ± 0.001 Å, b = 3.666 ± 0.001 Å, c = 18.073 ± 0.002 Å and V = 337.2 ± 0.2 Å3. The atomic coordinates were optimized by the DFT calculation. | ||
Crystal structure at high pressure
The 5-ATZ powder with paraffin oil that was prepared in the same way as that in the previous section was sealed in the diamond anvil high-pressure cell. The culet of the anvils was 400 µm in diameter. The sample chamber was 120 µm in diameter and 50 µm in thickness. The pressures were determined by the ruby fluorescence method.20 The X-ray beams from the MoKα target were collimated to 80 µm, and the typical exposure time was 48 to 96 hours.The diffraction peak intensities gradually weakened, till only six strong peaks were observed at the highest pressure of 11.6 GPa in this experiment; however, the crystal still maintained the P212121 structure. The pressure induced decomposition could not have occurred since the peak intensities recovered after attaining ambient pressure. In the Rietveld analysis under pressure, the molecular size was fixed to the one at ambient pressure, and treated as a rigid body. However, the intra-molecular bond lengths and angles could be changed with pressure. Furthermore, the reliability of the diffraction intensities would worsen due to the uniaxial component of the stress. Therefore, the atomic positions after the Rietveld analysis at each pressure were optimized by the DFT calculation.17 The lattice parameters were set to the experimental values, and the calculations were made under the same conditions as that at ambient pressure.
Fig. 4(a) shows the pressure dependence of the lattice parameters and the cell volume normalized by the values at ambient pressure. Their numerical values are shown in Table S1 in the ESI†. Representative structure data at 4.8 GPa and 11.6 GPa are provided in the CIF file in the ESI†. The a- and c-axes shrunk at the same rate, whereas the b-axis shrunk rapidly. The ac-plane has many hydrogen bonds as in Fig. 3(a); however, only the van der Waals contact seems to exist along the b-axis as in Fig. 3(b). Therefore, the large anisotropy found in Fig. 4(a) and the crystal structure are consistent. The cell volume decreased to 77% from ambient pressure to 11.6 GPa. The inter-atomic distances are shown in Fig. 4(b–d). The 5-ATZ molecule has three intra-molecular N–H distances which all elongated with pressure. This means that the hydrogen bonds became stronger, i.e., the hydrogen atoms are pulled by the neighboring molecule. The distance N1–H7 increased the most rapidly. On the other hand, all the intra-molecular N–N, C–N distances decreased monotonically, as shown in Fig. 4(c). Three inter-molecular hydrogen bond distances decreased and only one distance H7⋯N2 remained constant as shown in Fig. 4(d). We do not have any good explanation for these behaviors at the moment.
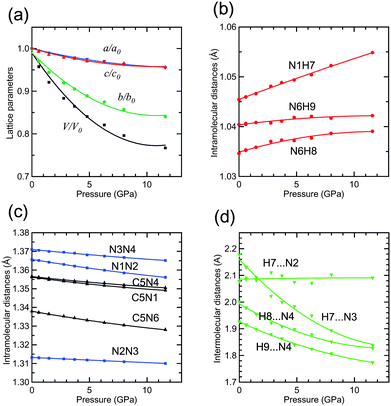 | ||
| Fig. 4 Pressure dependence of (a) lattice parameters and volume, (b) and (c) intramolecular distances, and (d) interatomic hydrogen bond distances of 5-aminotetrazole. | ||
Molecular dynamics simulation
Based on the structural model of 5-ATZ determined in this study, we performed a quantum MD calculation by the CASTEP code17 up to 3000 K and 200 GPa. The 2 × 2 × 1 super-cell containing 16 molecules was simulated with the isothermal–isobaric constant–NPT ensemble at a given temperature and pressure with 0.5 fs time-step up to 2 ps.At a pressure below 10 GPa and temperatures from 1000 to 1500 K, the hydrogen bonds became disconnected and the molecules rotated and eventually became a liquid state as shown in Fig. 5(a). Since the number of atoms and the simulation time were quite limited in the quantum calculation, physical phenomena are expected to occur at a much lower temperature and pressure in the experiment. For example 5-ATZ crystal melts at 480 K in the experiment; however, temperatures above 1000 K were required to recognize clear molecular rotations within 2 ps in simulations.
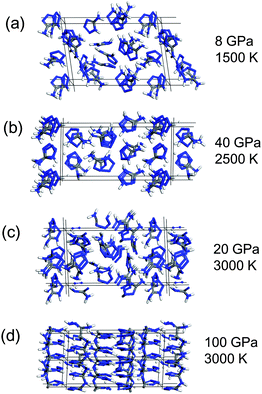 | ||
| Fig. 5 Representative state of 5-aminotetrazole at high-temperature and high-pressure predicted by the quantum molecular dynamics simulation after 2 ps simulation time: (a) liquid state, (b) proton migration, (c) molecular decomposition, and (d) polymerization. | ||
From 1500 K to 2500 K, the hydrogen atom H7 detached from N1 and started to migrate as shown in Fig. 5(b). The hydrogen atoms H8 and H9 rarely disconnected from N6. As a result, the 2H-form often appeared instead of the imino-form. This is very similar to the calculation result by Kiselev and Gritsan12 and also agrees with the expansion of distance N1H7 with pressure in Fig. 4(b), showing that the covalent bond N1H7 is weaker than that of N6H8 and N6H9.
Above 2500 K, the 5-membered rings opened and gradually fell apart as shown in Fig. 5(c). The fragments which were predicted by Zhang et al.9 and Paul et al.11 including the N2 molecule appeared. The simulation found that 2500 K was required for the decomposition, and this temperature did not depend on pressure.
Above 100 GPa at room temperature, the sp2 to sp3 conversion of carbon atom and polymerization were found to occur as shown in Fig. 5(d). The polymerization required a high pressure above 100 GPa probably because there was not enough space for the atomic motion that remained in the cell.
Conclusion
We determined the crystal structure for anhydrous 5-ATZ to be an orthorhombic P212121. This could be achieved by shutting out the moisture by paraffin oil. The pressure dependence of the crystal structure was determined up to 11.6 GPa. The 5-ATZ 1H-form was maintained in the phase. The anisotropic pressure behavior of the lattice parameters was consistent with the crystal structure of the planar molecule and the stacking along the b-axis. The pressure dependence of the intra-molecular covalent and the inter-molecular hydrogen bonds was analyzed precisely by the combination of the Rietveld analysis and the DFT calculation. We performed a MD calculation of 5-ATZ at high temperature and high pressure and observed a variety of changes in the bonding state. The structure analyses for the other two anhydrous phases II and III found at high temperature13 are still in progress.References
- S. V. Levchik, O. A. Ivashkevich, A. I. Balabanovich, A. I. Lesnikovich, P. N. Gaponik and L. Costa, Thermochim. Acta, 1992, 207, 115 CrossRef CAS.
- K. Britts and I. L. Karle, Acta Crystallogr., 1967, 22, 308 CrossRef CAS.
- D. D. Bray and J. G. White, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 3089 CrossRef.
- A. Gao, Y. Oyumi and T. B. Brill, Combust. Flame, 1991, 83, 345 CrossRef CAS.
- S. V. Levchik, A. I. Balabanovich, O. A. Ivashkevich, A. I. Lesnikovich, P. N. Gaponik and L. Costa, Thermochim. Acta, 1993, 225, 53 CrossRef CAS.
- T. B. Brill and H. Ramanathan, Combust. Flame, 2000, 122, 165 CrossRef CAS.
- A. I. Lesnikovich, O. A. Ivashkevich, S. V. Levchik, A. I. Balabanovich, P. N. Gaponik and A. A. Kulak, Thermochim. Acta, 2002, 388, 233 CrossRef CAS.
- A. A. Paletsky, N. V. Budachev and O. P. Korobeinichev, Kinet. Catal., 2009, 50, 627 CrossRef CAS.
- J.-G. Zhang, L.-N. Feng, S.-W. Zhang, T.-L. Zhang and H.-H. Zheng, J. Mol. Model., 2008, 14, 403 CrossRef CAS.
- J.-G. Zhang, L.-N. Feng, Y.-J. Shu, S.-W. Zhang, T.-L. Zhang, L. Yang and M. Wu, J. Mol. Model., 2009, 15, 67 CrossRef CAS.
- K. W. Paul, M. M. Hurley and K. K. Irikura, J. Phys. Chem. A, 2009, 113, 2483 CrossRef CAS.
- V. G. Kiselev and N. P. Gritsan, J. Phys. Chem. A, 2009, 113, 3677 CrossRef CAS.
- S. Obata, S. Takeya, H. Fujihisa, K. Honda and Y. Gotoh, J. Phys. Chem. B, 2010 DOI:10.1021/jp1035376.
- H. Fujihisa, Rev. High Press. Sci. Technol., 1999, 9, 65 Search PubMed.
- M. A. Neumann, J. Appl. Crystallogr., 2003, 36, 356 CrossRef CAS.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CrossRef CAS.
- Z. Wu and R. E. Cohen, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 235116 CrossRef.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter, 1990, 41, 7892 CrossRef.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., 1986, 91, 4673 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structure data at ambient, 4.8 GPa, and 11.6 GPa (CIF) and numerical data for pressure evolution of lattice parameters and interatomic distances (PDF). CCDC reference numbers 780133, 790420 and 790421. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ce00278j |
| This journal is © The Royal Society of Chemistry 2011 |