Coordination polymers of flexible tetracarboxylic acids with metal ions. I. Synthesis of CH2- and (CH2)2-spaced bis(oxy)isophthalic acid ligands, and structural characterization of their polymeric adducts with lanthanoid ions†
Anirban
Karmakar
and
Israel
Goldberg
*
School of Chemistry, Sackler Faculty of Exact Sciences, Tel-Aviv University, Ramat-Aviv, 69978, Tel-Aviv, Israel. E-mail: anirban@iitg.ernet.in; goldberg@post.tau.ac.il
First published on 8th September 2010
Abstract
We report herein on the synthesis and structure of new tetracarboxylic ligands, 5,5′-methylene-bis(oxy)diisophthalic acid and 5,5′-(ethane-1,2-diyl)-bis(oxy)diisophthalic acid, bearing flexible spacers between the two isophthalic acid fragments, as well as of the coordination polymers of these two ligands with various lanthanoid metal ions. The formed compounds were characterized by X-ray crystallography, IR spectroscopy and thermal analysis. The nine hybrid organic–inorganic supramolecular assemblies reveal polymeric architectures of either two-dimensional or three-dimensional connectivity.
Introduction
The design and synthesis of Metal–Organic Frameworks (MOFs) have attracted great interest due their potential use as gas storage, magnetism, catalysis activity, ion exchange, molecular recognition, and optical properties.1 In this respect, various theoretical predictions and network-based approaches to control the network topology and geometries have been made to produce useful functional materials.2 A remarkable progress has been made in recent years in the development of porous metal–organic frameworks by using multidentate aromatic carboxylic acid ligands as building blocks, due to their robustness and thermal stability.3 Moreover, these ligands can be readily deprotonated to balance the charge of the metal ions they interact with, without the need to include in the resulting framework lattice additional uncoordinated counter ions which could occupy and block the channel voids. These features of the metal–carboxylate lattices could be quite useful in various applications such as size- and shape-selective separations and catalysis.4 Until now, MOFs utilizing common transition metal ions as inter-ligand connectors have been studied in more depth, most probably due to their well defined crystal field effects and preferred coordination geometries. Analogous chemistry of the lanthanoid ions is still lacking in scope.5 On the other hand, the high and variable coordination numbers, flexible coordination geometries, oxophilic and hard nature, and the presence of multi-single electrons of the lanthanoids provide exceptional opportunities for the discovery of unusual networking features and unique structural properties.6,7 Indeed, the reported MOFs based on lanthanoid ion connectors reveal interesting topological structures, such as 1D chains,8 2D grids,9 3D porous frameworks, and interpenetrating networks.10 Some of these lanthanoid materials exhibit also unique photophysical properties, which can be attributed to f–f transitions with an extremely narrow bandwidth.11 The interplay between rigidity and flexibility of the organic component may have various implications on the formed lattice. Rigidity is beneficial for the generation of porosity, because it cannot permit structural distortion of the framework.12 On the other hand the use of flexible multicarboxylate ligands as building blocks in the assembly of coordination frameworks is attractive because some conformational freedom of the ligand may offer various possibilities for release of the steric strain imposed by the metal–ligand association and relaxation of the network architecture.13In the latter context we describe the coordination polymerization features of two newly designed flexible tetracarboxylic acid ligands. To this end we used methane or ethane spacer units for interconnection between two oxyisophthalic acid moieties through their ether linkage and generation of the tetracarboxylic acid precursors: 5,5′-methylene-bis(oxy)diisophthalic acid (H4L′) and 5,5′-(ethane-1,2-diyl)-bis(oxy)diisophthalic acid (H4L″) (Schemes 1 and 2). This molecular design of the organic component was based on the following considerations: (a) the flexible and multifunctional coordination sites may generate multidimensional structures; (b) the ligand can be singly, doubly, triply or quadruply deprotonated to the corresponding carboxylate species (H3L−, H2L2−, H1L3−, and L4−) to allow its diverse coordination modes to the inorganic connectors. We report herein on the synthesis and structure of the two ligands, H4L′ and H4L″, and on the versatile architectures of the coordination polymers obtained by reacting these ligands with a series of lanthanoid(III) nitrates under hydrothermal conditions. Prior to the polymerization reactions with the lanthanoid ions, H4L′ could be crystallized as a co-crystal of this ligand with 1,2-bi(4-pyridyl)ethane (bpe) and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:3), while H4L″ could be crystallized as a monohydrate. The empirical formulae of the resulting analyzed compounds include:
:3:3), while H4L″ could be crystallized as a monohydrate. The empirical formulae of the resulting analyzed compounds include:
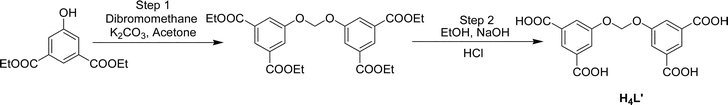 | ||
| Scheme 1 Synthesis of 5,5′-methylene-bis(oxy)diisophthalic acid (H4L′). | ||
(1) H4L′·3(bpe)·3H2O
(2) H4L″·H2O
(3) [La3+(H2O)·(HL′)3−]
(4) [Nd3+(H2O)·(HL′)3−]
(5) [Sm3+(H2O)2·(HL′)3−]·5H2O
(6) [Dy3+(H2O)2·(HL′)3−]·3H2O
(7) [Eu3+(H2O)2·(HL′)3−]·3H2O
(8) [La3+(H2O)3·(HL″)3−]·H2O
(9) [Nd3+(H2O)2·(HL″)3−]·3½H2O
(10) [Sm3+(H2O)2·(HL″)3−]·3½H2O
(11) [Ce3+(H2O)2·(HL″)3−]·xH2O
Experimental
All reagents and solvents were purchased from commercial sources and used without further purification. FT-IR spectra were recorded in the range of 400–4000 cm−1 from KBr pellets (Aldrich 99+%, FT-IR grade) using a Bruker PS15 spectrophotometer.Synthesis of H4L′ and H4L″ ligands
The H4L′ compound was synthesized by a two step procedure (Scheme 1).In the first step diethyl ester of 5-hydroxyisophthalic acid (2.38 g, 10 mmol), dibromomethane (0.86 g, 5 mmol) and anhydrous K2CO3 (1.65 g, 12 mmol) were placed in a round bottom flask and then added dry acetone (20 mL). The reaction mixture was stirred for 8 h at 60 °C (the reaction progress being monitored at regular intervals by TLC). After completion of the reaction, it was filtered off (to remove unreacted K2CO3). The solvent was removed under reduced pressure and a white solid was obtained. The isolated product was washed with NaOH (5%) solution in water and then extracted with dichloromethane. The organic extracts were collected over anhydrous sodium sulfate; subsequent removal of the solvent gave the ethyl ester of H4L′. Yield: 73%. In the second step the isolated ester (2.44 g, 5 mmol) and NaOH (0.6 g, 15 mmol) were dissolved in 20 mL of EtOH:water (4:1). The reaction mixture was refluxed for 4 h at 80 °C, after which the solvent was removed under reduced pressure, 10 mL of water was added into it and the solution was acidified (pH ≈ 2) with dilute HCl solution. The obtained white solid product H4L′ was filtered and washed with water until free from acid. Yield: 62%. FT-IR (KBr, cm−1): 3375 (bs, νO–H), 2987 (mb, νC–H), 1700 (s, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric), 1598 (s, νCC), 1456 (m, νCC), 1402 (m, νCO symmetric), 1269 (bs, νC–O), 1128 (w), 1103 (w), 1024 (s), 907 (w), 759 (s), 727 (w), 687 (m), 666 (m), 609 (w), 545 (w), 449 (w); 1H-NMR (DMSO-d6): 8.58 (2H, s), 8.26 (4H, s), 6.51 (2H, s); 13C-NMR (DMSO-d6): 166.58, 156.59, 133.47, 124.39, 121.06, 90.69.
O asymmetric), 1598 (s, νCC), 1456 (m, νCC), 1402 (m, νCO symmetric), 1269 (bs, νC–O), 1128 (w), 1103 (w), 1024 (s), 907 (w), 759 (s), 727 (w), 687 (m), 666 (m), 609 (w), 545 (w), 449 (w); 1H-NMR (DMSO-d6): 8.58 (2H, s), 8.26 (4H, s), 6.51 (2H, s); 13C-NMR (DMSO-d6): 166.58, 156.59, 133.47, 124.39, 121.06, 90.69.
The ligand H4L″ was synthesized by a similar pathway (Scheme 2). In the first stage diethyl ester of 5-hydroxyisophthalic acid (2.38 g, 10 mmol), dibromoethane (0.93 g, 5 mmol) and anhydrous K2CO3 (1.65 g, 12 mmol) were placed in a round bottom flask and then added dry acetone (20 mL). The reaction mixture was stirred for 24 h at 60 °C, the reaction progress being monitored at regular intervals by TLC. Then, after removing unreacted K2CO3 by filtration, and removing the solvent under reduced pressure, a white solid was isolated. It was washed with NaOH(aq.) (5%), extracted with dichloromethane, yielding the ethyl ester of H4L″ in 64% yield. At the second stage 2.44 g (5 mmol) of the latter and 0.6 g (15 mmol) of NaOH were dissolved in 20 mL of EtOH:water (4:1). As in the previous procedure, after reflux of the reaction mixture for 4 h at 80 °C, followed by removal of the solvent at reduced pressure, acidification with dilute HCl, washing with water and filtering of the product, white solid of H4L″ was obtained in 81% yield. FT-IR (KBr, cm−1): 3355 (bs, νO–H), 2970 (mb, νC–H), 1701 (s, νCO asymmetric), 1596 (s, νCC), 1421 (s, νCO symmetric), 1280 (s, νC–O), 1184 (m), 1134 (m), 1106 (w), 1057 (s), 887 (m), 760 (s), 738 (m), 689 (m), 666 (m), 578 (w), 444 (w); 1H-NMR (DMSO-d6): 8.09 (2H, s), 7.70 (4H, s), 4.48 (2H, s); 13C-NMR (DMSO-d6): 166.34, 158.46, 132.66, 122.51, 119.22, 67.02.
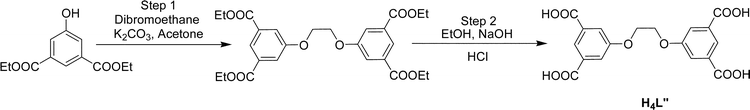 | ||
| Scheme 2 Synthesis of 5,5′-(ethane-1,2-diyl)-bis(oxy)diisophthalic acid (H4L″). | ||
Crystallization experiments of the two ligands proceeded as follows.
C), 1709 (s, νCO asymmetric), 1602 (s, νCC), 1450 (m, νCC), 1420 (m, νCO symmetric), 1273 (bs, νC–O), 1228 (s), 1028 (s), 893 (w), 832 (m), 759 (m), 690 (w), 547 (s), 490 (w).
Synthesis of the coordination polymers
All the metal–ligand coordination reactions were carried out in aqueous environments.O), 1613 (m, νCO asymmetric), 1536 (s), 1462 (m, νCC), 1380 (s, νCO symmetric), 1320 (m), 1243 (s, νC–O), 1198 (s), 1144 (m), 1062 (m), 1025 (s), 933 (w), 778 (s), 722 (m), 683 (s), 501 (w), 442 (w).
O), 1615 (m, νCC), 1571 (s, νCO asymmetric), 1545 (s), 1466 (m, νCC), 1386 (s, νCO symmetric), 1234 (w, νC–O), 1145 (w), 1026 (s), 932 (m), 773 (s), 681 (s).
O), 1620 (s, νCC), 1573 (s, νCO asymmetric), 1537 (s), 1469 (s, νCC), 1387 (s, νCO symmetric), 1225 (m, νC–O), 1146 (w), 1027 (s), 932 (m), 888 (w), 773 (s), 716 (m), 682 (s).
O), 1626 (s, νCO asymmetric), 1532 (s), 1457 (m, νCC), 1396 (s, νCO symmetric), 1283 (w), 1245 (m, νC–O), 1150 (w), 1027 (s), 925 (w), 774 (s), 692 (m), 570 (w).
O), 1598 (s, νCO asymmetric), 1540 (s), 1457 (m, νCC), 1396 (s, νCO symmetric), 1309 (w), 1240 (m, νC–O), 1142 (w), 1024 (s), 921 (w), 769 (s), 700 (m), 541 (w).
O), 1537 (s, νCO asymmetric), 1448 (s, νCC), 1386 (s, νCO symmetric), 1280 (m, νC–O), 1229 (w), 1076 (w), 1036 (m), 985 (m), 784 (m), 722 (m), 682 (w), 521 (w).
O), 1608 (m) 1545 (s, νCO asymmetric), 1454 (s, νCC), 1390 (s, νCO symmetric), 1332 (w), 1265 (m, νC–O), 1132 (m), 1063 (s), 930 (w), 772 (s), 733 (w), 684 (m). For (10), FT-IR (KBr, cm−1): 3421 (bs, νO–H coordinated water), 2945 (w, νC–H), 1667 (s, νCO), 1607 (m) 1554 (s, νCO asymmetric), 1455 (s, νCC), 1391 (s, νCO symmetric), 1267 (m, νC–O), 1132 (m), 1065 (s), 1005 (w), 772 (s), 728 (w). For (11), FT-IR (KBr, cm−1): 3428 (bs, νO–H coordinated water), 3172 (bs, νC–H), 1714 (s, νCO), 1608 (m) 1551 (s, νCO asymmetric), 1387 (s, νCO symmetric), 1330 (w), 1273 (m, νC–O), 1203 (s), 1133 (s), 1065 (m), 1002 (m), 894 (w), 820 (w), 769 (m), 728 (s).
Crystal structure determinations
The X-ray measurements (Nonius KappaCCD diffractometer, MoKα radiation) were carried out at approximately 110 K on crystals coated with a thin layer of amorphous oil to minimize crystal deterioration, possible structural disorder and related thermal motion effects, and to optimise the precision of the structural results. These structures were solved by direct methods (SIR-97) and refined by full-matrix least-squares (SHELXL-97).15 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms attached to carbon were located in idealized/calculated positions and were refined using a riding model, with Uiso = 1.2Ueq of the parent atom. Those attached to O and N atoms, which are involved in hydrogen bonding (except for the disordered species) were located in difference-Fourier maps; then their O–H and N–H distances were restrained to common values. Crystallographic refinements of all the structures converged to acceptable R-values, representing precisely determined structural models of compounds 1–11 and allowing reliable characterizations of the molecular structures and supramolecular binding motifs. Crystals of the two ligands and most of the polymeric assemblies contain non-coordinated molecules of water as crystallization solvent, which engage in extensive supramolecular hydrogen bonding schemes. In 1 (co-crystal of H4L′ and bpe), the bipyridyl moieties (the central bond in one of them being orientationally disordered) are also involved in extensive H-bonding with the tetracarboxylic ligand. In 11, most of the water solvent was found severely disordered, and could not be modelled by discrete atoms. Correspondingly, the contribution of the disordered solvent moieties was subtracted from the diffraction pattern by the SQUEEZE procedure and PLATON software.16 The crystallographic and experimental data for 1–11 are given in Table 1. The uniform identity of the formed crystal lattices (1–11) in a given reaction was confirmed in each case by repeated measurements of the unit-cell dimensions from different single crystallites.| Compound | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 a |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Excluding the non-coordinated water species diffused between the polymeric assemblies (see Experimental). b Data in parentheses refer to refinement based on the uncorrected diffraction data with severely disordered and poorly modelled water solvent. | |||||||||||
| Formula | C53H54N6O13 | C18H16O11 | C17H11LaO11 | C17H11NdO11 | C17H23O17Sm | C17H19DyO15 | C17H19EuO15 | C18H19LaO14 | C18H19NdO15.5 | C18H19O15.5Sm | C18H17CeO13 |
| F w | 983.02 | 408.31 | 530.17 | 535.50 | 649.70 | 625.82 | 615.28 | 598.24 | 627.56 | 633.68 | 581.44 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Triclinic | Triclinic | Monoclinic | Monoclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
P21/c | P21/c |
P |
P |
P |
P |
| a/Å | 19.3767(3) | 7.9466(2) | 8.8934(2) | 8.8278(1) | 9.9205(2) | 14.8023(6) | 14.8422(3) | 8.5147(2) | 8.5679(3) | 8.5589(2) | 8.0901(3) |
| b/Å | 23.1849(7) | 8.6064(2) | 8.9328(2) | 8.8751(2) | 10.6776(3) | 20.1819(6) | 20.2605(3) | 10.2584(2) | 9.9939(3) | 9.9686(3) | 10.1509(4) |
| c/Å | 13.7787(3) | 14.9515(4) | 12.6735(3) | 12.6582(3) | 11.7567(4) | 7.1981(2) | 7.2273(1) | 12.4540(5) | 12.5472(5) | 12.5240(4) | 14.6134(7) |
| α/° | 90.00 | 79.112(1) | 81.260(1) | 81.004(1) | 96.664(1) | 90.00 | 90.00 | 68.415(1) | 91.596(2) | 91.399(1) | 82.840(2) |
| β/° | 126.742(1) | 76.353(1) | 69.658(1) | 69.825(1) | 108.338(1) | 103.297(1) | 103.630(1) | 89.338(1) | 94.883(1) | 94.958(1) | 88.141(2) |
| γ/° | 90.00 | 62.502(1) | 60.961(1) | 61.238(2) | 106.837(2) | 90.00 | 90.00 | 72.780(2) | 93.405(2) | 93.485(1) | 66.521(3) |
| V/Å3 | 4960.3(2) | 877.63(4) | 825.19(3) | 815.98(2) | 1101.63(5) | 2092.7(2) | 2112.12(6) | 960.37(5) | 1068.00(7) | 1062.11(5) | 1091.94(8) |
| Z | 4 | 2 | 2 | 2 | 2 | 4 | 4 | 2 | 2 | 2 | 2 |
| ρ calcd/Mg m−3 | 1.316 | 1.545 | 2.134 | 2.180 | 1.959 | 1.986 | 1.935 | 2.069 | 1.952 | 1.981 | 1.768 |
| µ/mm−1 | 0.095 | 0.131 | 2.656 | 3.250 | 2.753 | 3.650 | 3.048 | 2.306 | 2.514 | 2.848 | 2.150 |
| T/K | 110(2) | 110(2) | 110(2) | 110(2) | 110(2) | 110(2) | 110(2) | 110(2) | 110(2) | 110(2) | 110(2) |
| F(000) | 2072 | 424 | 516 | 552 | 646 | 1228 | 1216 | 592 | 622 | 626 | 574 |
| θ max/° | 26.00 | 27.91 | 26.39 | 27.91 | 26.35 | 27.86 | 27.87 | 27.99 | 27.90 | 27.83 | 26.50 |
| Refl. collected | 18609 |
9750 | 8617 | 9148 | 13401 |
17040 |
18767 |
16654 |
10107 |
10883 |
13557 |
| Refl. unique | 4872 | 4145 | 3362 | 3846 | 4445 | 4962 | 4992 | 4565 | 4997 | 4966 | 4472 |
| R int | 0.064 | 0.038 | 0.043 | 0.037 | 0.038 | 0.064 | 0.041 | 0.060 | 0.047 | 0.043 | 0.079 |
| Completeness (%) | 100 | 99 | 100 | 98 | 99 | 99 | 99 | 99 | 98 | 98 | 99 |
| Refl. with I > 2σ(I) | 3000 | 2855 | 2976 | 3435 | 4174 | 4170 | 4076 | 3924 | 4113 | 4221 | 3728 |
| Refined param. | 337 | 263 | 262 | 262 | 294 | 299 | 309 | 317 | 315 | 316 | 289 |
| R 1 [I > 2σ(I)] | 0.074 | 0.052 | 0.036 | 0.035 | 0.028 | 0.052 | 0.033 | 0.038 | 0.048 | 0.040 | 0.043 (0.053)b |
| wR2 [I > 2σ(I)] | 0.140 | 0.125 | 0.084 | 0.082 | 0.064 | 0.096 | 0.086 | 0.084 | 0.122 | 0.102 | 0.089 (0.134)b |
| R 1 [all data] | 0.129 | 0.084 | 0.043 | 0.042 | 0.032 | 0.068 | 0.046 | 0.050 | 0.064 | 0.053 | 0.058 (0.071)b |
| wR2 [all data] | 0.160 | 0.142 | 0.088 | 0.085 | 0.066 | 0.101 | 0.095 | 0.090 | 0.129 | 0.110 | 0.093 (0.144)b |
| g.o.f. | 1.036 | 0.994 | 1.059 | 1.064 | 1.037 | 1.157 | 0.913 | 0.928 | 1.016 | 0.904 | 1.013 |
| ±Δρmax/e Å−3 | +0.34, −0.24 | +0.30, −0.35 | +1.57, −1.26 | +2.10, −1.44 | +1.17, −1.05 | +1.46, −1.29 | +1.45, −1.45 | +1.93, −1.78 | +2.07, −2.02 | +1.82, −1.56 | +1.24, −1.07 |
Results and discussion
General features of the FT-IR spectra for compounds 1–11 are almost identical through the region from 400 to 4000 cm−1.14 The characteristic strong bands of coordinated carboxylate groups are shown in the range of 1537–1626 cm−1 for asymmetric stretching and 1386–1421 cm−1 for symmetric stretching. C–O stretching of coordinated carboxylate groups comes into view in between 1225 and 1280 cm−1. A strong absorption band between 1660 and 1714 cm−1 appeared due to the CO stretching frequency of uncoordinated carboxylate groups. The O–H stretching frequency of carboxylic acid and water molecules are visible in the region of 3209–3464 cm−1. The νC–H vibration modes of –CH2– groups of methane or ethane spacers unit appear as a weak band in between 2925 and 3172 cm−1. The bands in the region 1607–1620 cm−1 and 1448–1469 cm−1 are attributed to the CC stretching frequency of aromatic rings. In compound 1, weak absorption was observed at 3058 cm−1 due to νC–H of the free 1,2-bis(4-pyridyl)ethane.
The molecular structures of the two ligands in compounds 1 and 2 are shown in Fig. 1, revealing their conformations. H4L′ is characterized by a V-shape geometry and has a C2 symmetry, with the peripheral carboxylic functions at the top of the molecular framework pointing sideways in opposite directions. In 1, they are engaged in extensive COOH⋯N and COOH⋯O hydrogen bonding interactions with the surrounding bpe and water moieties (Fig. 2a), forming a 3D hydrogen-bonded motif throughout the crystal. The overall conformation of H4L′ can be best characterized by two parameters: the dihedral angle between the two phenyl rings of δ′ = 61.4(1)°, and the Cphenyl–O⋯O–Cphenyl torsion angle in the central part of the molecule of τ′ = 124.7(3)°. It should be pointed out, however, that the actual conformation of the free ligand when not ion-paired with the bpe Lewis base could be somewhat different. The observed structure of H4L″ in 2 is characterized by gauche conformation around the central CH2–CH2 bond with the O–C–C–O torsion angle of τ″ = 69.2(2)°, and dihedral angle δ″ between the two C6-phenyl rings of 59.97(4)°. As a result, the four carboxylic acid molecular recognition groups point roughly at tetrahedral directions with respect to the central C–C bond. The intermolecular organization in 2 is directed by numerous ligand–ligand as well as ligand–water O–H⋯O hydrogen bonds (Fig. 2b), which represents a continuous supramolecular assembly of the component species. The isophthalic acid paddle-like residues in 1 and 2 are approximately planar, with only a small twist of the carboxylic groups with respect to the plane of their proxime phenyl rings.
 | ||
| Fig. 1 Ball-and-stick illustrations of the molecular structures of (a) H4L′ in 1 and (b) H4L″ in 2. | ||
 | ||
| Fig. 2 Hydrogen bonding interactions (dashed lines) of the H4L′ and H4L″ ligands with the surrounding moieties. Wireframe representation, except for the water species, (spheres) and the central H4L″ molecule in (b) (ball-and-stick). (a) In 1, H4L′ forms four O–H⋯N and two O⋯H–O (water) hydrogen bonds at 2.579(3)–2.586(3) Å and 2.808(4) Å, respectively. (b) In 2, each H4L″ molecule is directly involved in six O–H⋯O ligand–ligand hydrogen bonds at 2.588(2)–2.678(2) Å, and additional O–H⋯O (water) = 2.593(2) Å, O–H (water)⋯O (carboxy) = 2.732(2) Å, and O–H (water)⋯O (ether) = 3.019(2) Å interactions. | ||
The extensive hydrogen bonding interactions in all the analyzed compounds 1–11 are detailed in the Crystallographic Information Files (CIFs)† of the corresponding structures.
All the lanthanoid metal ions that reacted with H4L′ and H4L″ appear in their most common oxidation state of 3+. A 1:1 reaction between the organic and the inorganic components is associated, due to charge balance requirements, with triple deprotonation of the organic species to a HL′3− or HL″3− state. The hydrogen bonding capacity of the remaining carboxylic acid function is then normally utilized in hydrogen bonding to neighboring ligand moieties or to the water solvent. Compounds 3 and 4 are isomorphous (Table 1), representing 1:1 two-dimensional coordination polymerization of H4L′ with La and Nd ions, respectively (Fig. 3, hydrothermal reactions between Ce(NO3)3·6H2O or PrCl3 with H4L′ also led to isomorphous materials, but their crystals were of lower quality). The metal ions are nine-coordinate and arrange in pairs across centers of inversion. Eight carboxylate groups are coordinated to the two inversion-related metal centers. The latter constitute a di-nuclear core acting as a secondary building block unit in the construction of the two-dimensional polymeric assembly. Every metal ion coordinates through one monodentate, one chelating, one µ2–η1–η1-bridging and finally one µ2–η2–η1-bridging carboxylate groups (see below). In addition its ninth coordination site is occupied by a water molecule.
 | ||
| Fig. 3 The two-dimensional bilayered coordination polymerization in 3 (4 is isomorphous). The metal ions and their water ligands are denoted by small spheres. | ||
The Ln–O bond lengths are within 2.431(3)–2.671(3) Å in 3 and 2.374(3)–2.620(3) Å in 4. The La⋯La and Nd⋯Nd distances between the inversion-related ions are 4.1850(5) and 4.0958(4) Å, respectively. The polymeric array adopts a bilayered nature with respect to the metal-connected ligand components. The latter are characterized by a flattened geometry to conform to the 2D coordination scheme, as it is reflected in the corresponding conformation indicators δ′ = 30.2(2)° and 29.5(5)°, and τ′ = 142.3(3)° and 142.5(3)° in 3 and 4, respectively. The upper and lower parts of the bilayered ensemble are related to one another by crystallographic inversion, as are the individual metal linkers within the dinuclear lanthanoid cluster. The water ligands are oriented perpendicular to the polymeric bilayers as flag poles on both sides of the bilayer. They provide additional molecular recognition sites for further intermolecular association, and impart roughness to layer surface. In fact, neighboring bilayers interdigitate one into the other with their water “poles”. Water molecules of one layer penetrate between those of an adjacent layer and hydrogen bond to the carboxylate groups that line the concave zones of the latter.
A bilayered two-dimensional polymerization motif, which extends parallel to the (ab) plane of the crystal, characterizes also compound 5, wherein dinuclear clusters of inversion-related Sm ions interconnect by coordination between six H4L′ ligand units (Fig. 4a). The metal ions are nine-coordinate, with four ligand species bridging between them. The coordination sphere of every metal ion is complemented by an additional carboxylate group and two water ligands. The Sm–O coordination bonds are within 2.356(2)–2.631(2) Å, and the Sm⋯Sm distance across the inversion is 4.0205(3) Å.
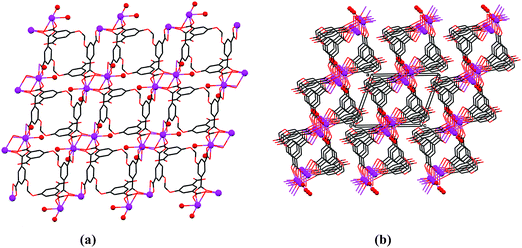 | ||
| Fig. 4 Compound 5. (a) Face-on view of the two-dimensional polymeric assembly. (b) Edge-on view (down the b-axis) of three adjacent polymers, illustrating the tight packing of the corrugated layered ensembles along the c-axis (horizontal). The Sm ions and the water ligands bound to them are denoted by small spheres. The non-coordinated water species residing in the interligand void grids are omitted. | ||
However, in 5 the observed metal–ligand connectivity scheme is somewhat different than in 3 and 4. The ligand adopts a considerably more bent V-geometry with δ′ = 85.9(1)° and τ′ = 103.3(3)°, and utilizes only three of its carboxylate groups for coordination to the lanthanoid linkers. The fourth carboxylic group as well as the methylene center of every ligand are turned away from the metal center, lining the surface of the polymeric layers on both sides. The top and bottom layers of the polymeric assembly are related to each other by inversion, the organic ligands converging on the dinuclear metal–ion clusters in the center of the bilayer. Edge-on view of the formed layers (Fig. 4b) shows a grid-like structure of the polymeric array associated with the sharp bending of the ligands (with a nearly perpendicular alignment of the two phenyl residues). It consists of a square-type organization with the organic ligands and inorganic connectors located in opposite corners of a given square. The metal-coordinated water ligands point inward into the interligand voids, which are also accommodated by molecules of non-coordinated water moieties. All the water entities are involved in an extensive array of hydrogen bonds. The corrugated polymeric layers pack tightly along the c-axis, with the convex zones of one layer fitting tightly into the concave surfaces of adjacent layers from above and below.
The isomorphous structures 6 and 7 with heavier lanthanoid ions represent a different polymeric aggregation than in compounds 3–5. They exhibit continuous coordination polymerization in three (rather than two) dimensions, forming genuine framework solids (Fig. 5).
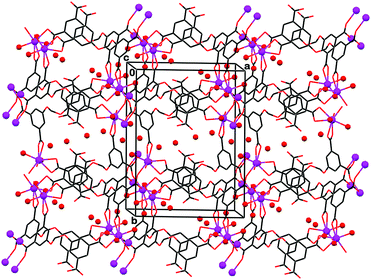 | ||
| Fig. 5 Crystal packing in framework-3D coordination polymer 7 (6 is isomorphous), viewed approximately down the c-axis. The metal ions and the water molecules (metal-ligated as well as uncoordinated solvent) are denoted by small spheres. | ||
Molecules of the non-coordinated water solvent occupy intra-lattice channels that propagate through the polymeric assembly parallel to the c-axis. These two structures are characterized by mononuclear inorganic connectors (Dy or Eu, the distance between closest ions in the polymeric lattice are longer than 5.0 Å), and a V-shape conformation of the H4L′ ligand is very similar to that observed in 1. It is characterized by the C–O⋯O–C torsion angle in the central part of the ligand, and the dihedral angle between the two phenyl rings, of τ′ = 125.5(5)° (in 6) and 123.3(4)° (in 7), and δ′ = 58.2(2)° (in 6) and 59.3(1)° (in 7), correspondingly. The metal ions have a coordination number of 8, doubly coordinating to two carboxylate groups of different ligands, and singly coordinating to the carboxylates of two other ligand species and to two water molecules at Dy/Eu–O distances within 2.234(14)–2.2445(4) Å in 6 and 2.282(3)–2.479(3) Å in 7. The three carboxylate groups of H4L′ are coordinated to four metal ions. One of the carboxylates on each phenyl ring is doubly coordinated to Dy/Eu, while the third one is bound to, and bridges between, two ions. The carboxylic acid function of H4L′ is oriented into the periodically spaced water-filled channels and hydrogen bonds to them.
Ligand H4L″ is considerably more flexible than H4L′ due to the extended aliphatic bridge that connects between the two phenyl fragments. Correspondingly, the relative disposition of the carboxylic molecular recognition functions can vary from coplanar to tetrahedral. The latter is favorable for the induction of three-dimensional coordination patterns, particularly when combined with metal ion connectors of complimentary tetrahedral binding geometry. With the lanthanoid metals that lack crystal field effects, the situation is less predictable. A single example of a 3D-framework polymerization of H4L″ with La ions is provided by compound 8 (Fig. 6). In this compound the La ion is nine-coordinate and binds to eight ligating molecules (three water molecules, one doubly coordinated H4L″, and four singly coordinated H4L″), with La–O distances ranging from 2.457(3) Å to 2.622(3) Å. The relatively large size of the metal ion allows it to coordinate simultaneously to five different H4L″ ligands, giving rise to the expansion of the coordination polymerization in three dimensions. Moreover, every ligand species coordinates to five different La ions, one of the carboxylate groups bridging between two of the ions 5.813(1) Å apart from each other. The 3D architecture formed in 8 can be attributed mainly to the high coordination capacity of the La ion, as the H4L″ organic ligand in it is characterized by a relatively flat shape. The conformation of H4L″ in 8 is characterized by a τ″ = 58.4(4)° torsion angle about the central bond and a dihedral angle between the phenyl rings of only δ″ = 23.2(2)°. As in the previous example, molecules of non-coordinated water are trapped in the interstitial voids of, and hydrogen bond to, the polymeric assembly.
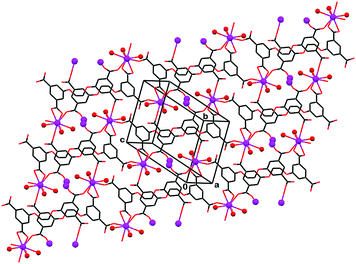 | ||
| Fig. 6 Crystal structure of the framework coordination polymer 8. The metal ions and the coordinated water molecules are denoted by small spheres. Molecules of the non-coordinated water solvent are omitted for clarity. Note that flat shape of the organic ligand which extends with its four carboxylic/carboxylate groups along the horizontal direction, while the metal–ligand binding the other two directions is facilitated by the spatially extended coordination valency of the La ions. | ||
Only 2D coordination polymerization takes place, however, in the reactions of H4L″ with Ce, Nd and Sm ions (9–11). It is associated with an almost entirely flat conformation of the organic ligand in the three compounds. The latter is characterized primarily by anti torsion angles about the central fragment and a nearly coplanar orientation of the two phenyl rings of the ligand: τ″(O–C–C–O) = 178.7(4)° in 9, 179.4(4)° in 10, and 177.8(4)° in 11; δ″ (phenyl–phenyl) = 13.4(3)° in 9, 13.7(3)° in 10, and 16.6(2)° in 11. The coordination pattern is also similar in the three compounds. The metal ion connectors are nine-coordinate. They coordinate doubly to two carboxylate groups of two different ligands, and singly to the carboxylate groups of three other H4L″ molecules as well as to two water ligands. The corresponding Ln–O bond distances are within 2.359(4)–2.770(4) Å in 9, 2.325(3)–2.780(3) Å in 10, and 2.414(3)–2.651(3) Å in 11. Then, in each of these three compounds the organic ligands connect to five different metal ions, two of the carboxylates bind doubly to two of the ions, the third to another metal ion, and the fourth carboxylate function links to, and bridges between, two additional metal ions. These metals do not seem to interact strongly with one another, the distances between the carboxylate-bridged ions being Nd⋯Nd = 5.293(1) Å in 9, Sm⋯Sm = 5.303(1) Å in 10, and Ce⋯Ce = 6.043(1) Å in 11. As observed in compounds 3–5, the coordination polymers have a double-layer nature (in terms of the organic ligand arrangement in them), with the metal-bound water ligands directed outward on both sides (Fig. 7). Compounds 9 and 10 are isomorphous and isometric. Compound 11 crystallizes with a unit-cell of somewhat different dimensions and reveals slightly different ligand conformation and metal–ligand coordination distances. However, these variations have little effect on the overall coordination motif. All three compounds contain non-coordinated and partly disordered water solvent species (hydration layer) in the interface between the layered polymeric assemblies.
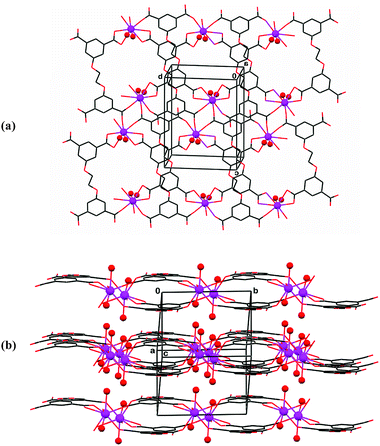 | ||
| Fig. 7 Compound 9. (a) Face-on view of the two-dimensional polymeric assembly. (b) Edge-on view (approximately down the a–c axis) of three adjacent polymers, illustrating the outward orientation of the metal-bound water ligands into the interface between the bilayered ensembles. The Sm ions and the water ligands bound to them are denoted by small spheres. The non-coordinated water species residing in the interligand void grids as well as at the interface between adjacent polymeric arrays are omitted. Similar two-dimensional coordination polymerization characterizes compounds 10 and 11. | ||
In order to understand the structures of metal–organic frameworks, it would be valuable to explore the binding mode variety between the metal centers and the carboxylate ligands.17 In this study we observe that the two ligands H4L′ and H4L″ are linked to the lanthanoid centers in four different ways (Fig. 8): (i) in a monodentate coordination mode the carboxylic acid coordinates to one metal center via the carbonyl O-atom without deprotonation; (ii) in a chelating bidentate mode, two oxygen atoms of a carboxylate group are chelating the same metal center; (iii) in the µ2–η1–η1-bridging coordination mode, two oxygen atoms of the carboxylate group connect with, and bridge between, two different lanthanoid ions; and (iv) in the µ2–η2–η1-bridging coordination mode one O-atom of the carboxylate group forms a bifurcated bond with two different lanthanoid ions, the other O-atom connects to only a single metal ion. In this case, then, the carboxylate group binds simultaneously to two metals.
 | ||
| Fig. 8 Coordination modes of H4L′ and H4L″ with lanthanoid ions in compounds 3–11. | ||
Similar type of torsionally flexible tetracarboxylic acids, with central butyl and butenyl spacer units are reported in literature.18 The ligand with butyl spacer exhibited a planar conformation upon coordination with metal ions, while the acid with butenyl spacer adopted either a planar or a gauche arrangement depending on the particular coordination mode. Tetracarboxylic acid with a flexible aryloxy group as spacer has been also reported in the literature.19 Two selected literature examples are of a particular relevance to our present observations. One relates to a 3D lanthanide–organic framework involving Eu3+ ions and the 2,5-pyridinedicarboxylic and 1,4-phenylenediacetic acids as organic ligands,20 as their polymeric assembly is tessellated by di-nuclear cores of similar type to that found in compound 3. The Eu ions in this compound are nine-coordinate and are paired across centers of crystallographic inversion. The di-nuclear Eu⋯Eu node coordinates through one monodentate, one chelating (involving also the N-atom), and one µ2–η1–η1-bridging pyridinedicarboxylic acids. The remaining coordination sites are occupied by one µ2–η2–η1-bridging phenylenediacetic acid and one water molecule. Another example involves a 3D coordination polymer of Yb3+ with 1,2,4,5-benzenetetracarboxylic acid, which has a structural resemblance with compounds 6 and 7, revealing a rather similar binding pattern around the metal ion.5a Two types of coordination modes of benzenetetracarboxylic acid ligands are present in that structure: (i) a bidentate chelating mode and (ii) a µ2–η2–η1-bridging coordination mode. Each Yb-atom is coordinated by eight oxygens, four from two chelating carboxylate groups, two from bridging carboxylate groups of different benzenetetracarboxylate units, and two coordinated water molecules.5a
In addition to lanthanoid polymeric compounds with benzene–polycarboxylates,5c,5d,22–25 a few examples of polymeric arrays constructed with lanthanide centers and multiphenyl carboxylates ligands have also been reported in literature.26 Among them difunctional organic linkers as 4,4′-biphenyldicarboxylate27 and naphthalene dicarboxylate28 have been widely investigated. Other interesting coordination networks of lanthanoid ions with 6,6′-dichloro-2,2′-diethoxy-1,1′-binaphthalene-4,4′-dicarboxylic acid,29S,S-dioxodibenzothio-phen-3,7-dicarboxylic acid,30 1,3,5-benzenetrisbenzoate,31 and 3,3′,4,4′-biphenyltetracarboxylate17b have also been reported. These compounds exhibit a wide diversity of lanthanoid–carboxylate coordination patterns, the correlation of which is beyond the scope of the present discussion. The focus in this article is on the design and application of new functional building blocks for crystal engineering of related hybrid organic–inorganic coordination networks, while utilizing similar coordination synthons, and to demonstrate if and how they materialize.
The topology of the coordination networks described above has been analyzed with the TOPOS (v. 4.0) software.21 The relatively high complexity of these arrays evolves from the multiple binding features exhibited by the lanthanoid ions. Compounds 3 and 4 are characterized by the same type of 14-connected uninodal net with the Schlafli point symbol (345.444.52). Their corresponding vertex symbol is [3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.4.4.4.4.4.4.4.4.4(2).4(2).4(2).4(2).4(2).4(2).5.5.5.5.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞]. Compounds 5, 9, 10 and 11 have similar type of 11-connected uninodal net, point (Schlafli) symbol (327.426.52), and vertex symbol [3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.4.4.4.4.5.5.5.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞]. Compounds 6 and 7 represent 10-connected uninodal nets with point symbol (315.422.58) and FeB topology. The extended point symbol is [3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4(2).4(2).4(2).4(2).4(2).4(2).5(2).5(3).5(3).5(3).5(3).5(3).5(3).5(3)], and the vertex symbol is [3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.4.4.4.4.4.4.4.4.5.5.5.5.5.5.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞]. Finally compound 8 is characterized by Ca0.3 Sn1.7-type inter-metallic topology with one kind of 14-connected uninodal net, point symbol (333.454.54), and vertex symbol [3.3.3.3.3.3. 3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.3.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.4.5.5.5.5.5.5. ∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞.∞].
Thermogravimetric analyses were carried out under argon in the range from room temperature to about 400 °C at heating rate of 10 K min−1. Features of the thermal stability of compounds 3, 5, 6, 8 and 9 are illustrated in Fig. 9. Compound 3 shows weight loss of 4.0% between 151 to 184 °C, corresponding to the loss of one molecule of water (calcd: 3.4%). Upon further heating, the anhydrous compound is stable up to 410 °C, and then decomposes above this temperature. Compound 6 exhibits weight loss of 14.8% in the 91–264 °C temperature range, which accounts for the total removal of the two metal-coordinated and three non-coordinated water molecules (calcd: 14.4%). The remaining material shows no further weight loss up to 400 °C. Compound 8 looses 12.7% of its weight within 128–177 °C, most likely due to the de-sorption of the four water molecules (calcd: 12.0%), the dehydrated residue remains stable up to about 395 °C, and starts to decompose above this temperature. Compound 9 shows weight loss of 14.3% within 45–299 °C, and then remains stable up to 400 °C. The observed weight loss corresponds to the five water molecules (calculated weight loss: 14.3%) in this compound. The only apparent discrepancy between the crystallographic (at 110 K) and TGA data relates to compound 5. In spite of the relatively high water content in compound 5 (see above), it shows weight loss of only 4.7% within the 100–255 °C temperature range, due the apparent loss of two water molecules (calcd: 5.3%). It is possible that in this case the five non-coordinated water species accommodated in the wide channel voids of the polymeric lattice diffused out of the crystalline material during the drying process of the analyzed sample. In such case the observed weight loss can be attributed to the two metal coordinated molecules. As expected, relatively high temperatures are required to release the metal-coordinated water ligands. After completion of the dehydration processes, no other phase transitions have been observed in DTA and DSC diagrams prior to decomposition of the metal–ligand coordination frameworks near 400 °C.
 | ||
| Fig. 9 Thermogravimetric analysis in compounds 3, 5, 6, 8 and 9, showing weight loss of the tested samples during the dehydration processes. | ||
Concluding remarks
We report herein on the synthesis and structure of two novel bis(oxy)isophthalic acid ligands with CH2– and (CH2)2– spacers, which impart features of flexibility to the tetracarboxylic acid moiety. These ligands, H4L′ and H4L″, have been then successfully utilized in the construction of two-dimensional and three-dimensional coordination polymers by reacting them with a variety of the oxophilic lanthanoid metal ions. The latter have multiple coordination capacity (observed coordination numbers are either 8 or 9, in their most common 3+ oxidation state) and serve as connectors in tessellating the supramolecular hybrid organic–inorganic assemblies (compounds 3–11). The structures of all the new compounds 1–11 have been precisely characterized by X-ray diffraction. The conformational degrees of freedom imparted to the two ligands evidently facilitate the formation of polymeric assemblies, by adjusting the shape of the ligand to the spatial coordination requirements of the metal bridging unit. However, due to this flexibility factor, as well as the high affinity of the lanthanoid ions to supplement their first coordination sphere with an unpredictable number of water ligands from the aqueous reaction mixture, it is practically impossible to control the preferential formation of a given coordination scheme. Compounds 3–5 and 9–11 contain coordination networks of 2D connectivity, and loose crystallinity on removal of the crystallization solvent. Compounds 6–8 are characterized by open architectures of 3D connectivity, yet they also represent soft materials. Their structures collapse upon heating and dehydration into either polycrystalline or amorphous phases. Current findings are in some contrast with our earlier observations on the coordination polymerization features of the more rigid tetra(4-carboxyphenyl)porphyrin and tetra(3-carboxy)phenylporphyrin moieties with lanthanoid ions.6f,6g In that case most of the formed porphyrin–lanthanoid polymers represented open 3D-framework materials, where the metal–carboxylate interaction schemes provided robust construction pillars to sustain the porphyrin-layered (dictated by the large aromatic frameworks of these building blocks) multi-level architectures. It is evident that the delicate interplay between molecular rigidity vs. flexibility is one of the major factors to consider in targeted formulations of metal–organic framework coordination polymers with the tetraacid ligands.Acknowledgements
This research was supported by The Israel Science Foundation (Grant No. 502/08).References
- (a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460–1494 CrossRef; (b) M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151–1152 CrossRef CAS; (c) G. B. Gardner, D. Venkataraman, J. S. Moore and S. Lee, Nature, 1995, 374, 792–795 CrossRef CAS; (d) C. Piguet and J. C. G. Bünzli, Chem. Soc. Rev., 1999, 28, 347–358 RSC; (e) L. H. Gade, Acc. Chem. Res., 2002, 35, 575–582 CrossRef CAS; (f) L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247–289 CrossRef CAS; (g) A. N. Khlobystov, A. J. Blake, N. R. Champness, D. A. Lemenovskii, A. G. Majouga, N. V. Zyk and M. Schröder, Coord. Chem. Rev., 2001, 222, 155–192 CrossRef CAS; (h) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS; (i) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS; (j) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS; (k) S. Subramanian and M. J. Zaworotko, Angew. Chem., Int. Ed. Engl., 1995, 34, 2127–2129 CrossRef CAS; (l) S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS; (m) D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273–282 CrossRef CAS; (n) V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 378–395 RSC; (o) Y. Z. Zhang, S. Gao, H. L. Sun, G. Su, Z. M. Wang and S. W. Zhang, Chem. Commun., 2004, 1906–1907 RSC; (p) S. R. Batten, B. F. Hoskins and R. Robson, Angew. Chem., Int. Ed. Engl., 1997, 36, 636–637 CrossRef CAS.
- (a) G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surble, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef CAS; (b) P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638–2684 CrossRef; (c) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 466–468; (d) Y. B. Dong, M. D. Smith and H.-C. Zur Loye, Angew. Chem., Int. Ed., 2000, 39, 4271–4273 CrossRef CAS; (e) B. Zhao, P. Cheng, Y. Dai, C. Cheng, D. Z. Liao, S. P. Yan, Z. H. Jiang and G. L. Wang, Angew. Chem., Int. Ed., 2003, 42, 934–936 CrossRef CAS; (f) B. Zhao, P. Cheng, X. Y. Chen, C. Cheng, W. Shi, D. Z. Liao, S. P. Yan and Z. H. Jiang, J. Am. Chem. Soc., 2004, 126, 3012–3013 CrossRef CAS.
- (a) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS; (b) W. Mori and S. Takamizawa, J. Solid State Chem., 2000, 152, 120–129 CrossRef CAS.
- (a) B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390–1393 CrossRef CAS; (b) C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 119, 1093–1096 CrossRef; (c) S. Horike, M. Dincă, K. Tamaki and J. R. Long, J. Am. Chem. Soc., 2008, 130, 5854–5855 CrossRef CAS; (d) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- (a) R. Cao, D. Sun, Y. Liang, M. Hong, K. Tatsumi and Q. Shi, Inorg. Chem., 2002, 41, 2087–2094 CrossRef CAS; (b) L. Pan, N. Zheng, Y. Wu, S. Han, R. Yang, X. Huang and J. Li, Inorg. Chem., 2001, 40, 828–830 CrossRef CAS; (c) T. M. Reineke, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 1999, 38, 2590–2594 CrossRef CAS; (d) T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651–1657 CrossRef CAS; (e) L. Pan, E. B. Woodlock and X. Wang, Inorg. Chem., 2000, 39, 4174–4178 CrossRef; (f) V. Kiritsis, A. Michaelides, S. Skoulika, S. Golhen and L. Ouahab, Inorg. Chem., 1998, 37, 3407–3410 CrossRef CAS; (g) F. Serpaggi and G. Férey, J. Mater. Chem., 1998, 8, 2737–2741 RSC.
- (a) H. Tsukube and S. Shinoda, Chem. Rev., 2002, 102, 2389–2404 CrossRef CAS; (b) S. K. Ghosh and P. K. Bharadwaj, Inorg. Chem., 2003, 42, 8250–8254 CrossRef CAS; (c) X. Li, Q. Shi, D. Sun, W. Bi and R. Cao, Eur. J. Inorg. Chem., 2004, 2747–2753 CrossRef; (d) C.-Y. Su, M. D. Smith, A. M. Goforth and H.-C. zum Loye, Inorg. Chem., 2004, 43, 6881–6883 CrossRef CAS; (e) A. Thirumurugan and S. Natarajan, Dalton Trans., 2004, 2923–2928 RSC; (f) S. Lipstman, S. Muniappan, S. George and I. Goldberg, Dalton Trans., 2007, 3273–3281 RSC; (g) S. Muniappan, S. Lipstman, S. George and I. Goldberg, Inorg. Chem., 2007, 46, 5544–5554 CrossRef CAS.
- (a) O. Kahn, Acc. Chem. Res., 2000, 33, 647–657 CrossRef CAS; (b) W. S. Liu, T. Q. Jiao, Y. Z. Li, Q. Z. Liu, M. Y. Tan, H. Wang and L. F. Wang, J. Am. Chem. Soc., 2004, 126, 2280–2281 CrossRef CAS; (c) B. Q. Ma, D. S. Zhang, S. Gao, T. Z. Jin, C. H. Yan and G. X. Xu, Angew. Chem., Int. Ed., 2002, 39, 3644–3662; (d) C. Serre, N. Stock, T. Bein and G. Férey, Inorg. Chem., 2004, 43, 3159–3163 CrossRef CAS; (e) G. Mancino, A. J. Ferguson, A. Beeby, N. J. Long and T. S. Jones, J. Am. Chem. Soc., 2005, 127, 524–525 CrossRef CAS.
- (a) D. M. Shin, I. S. Lee, Y. A. Lee and Y. K. Chung, Inorg. Chem., 2003, 42, 2977–2982 CrossRef CAS; (b) M. A. Withersby, A. J. Blake, N. R. Champness, P. Hubberstey, W. S. Li and M. Schröder, Angew. Chem., Int. Ed. Engl., 1997, 36, 2327–2329 CrossRef CAS; (c) A. S. Batsanov, M. J. Begley, P. Hubberstey and J. Stroud, J. Chem. Soc., Dalton Trans., 1996, 1947–1957 RSC.
- (a) M. A. Withersby, A. J. Blake, N. R. Champness, P. A. Cooke, P. Hubberstey, W. S. Li and M. Schröder, Inorg. Chem., 1999, 38, 2259–2266 CrossRef CAS; (b) K. Nomiya and H. Yokoyama, J. Chem. Soc., Dalton Trans., 2002, 2483–2490 RSC; (c) R. Koner and I. Goldberg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m160–m164 CrossRef.
- (a) D. M. Shin, I. S. Lee, Y. K. Chung and M. S. Lah, Inorg. Chem., 2003, 42, 5459–5461 CrossRef CAS; (b) K. Mitsurs, M. Shimamura, S. I. Noro, S. Minakoshi, A. Asami, K. Seki and S. Kitagawa, Chem. Mater., 2000, 12, 1288–1299 CrossRef CAS; (c) L. Carlucci, G. Ciani and D. M. Proserpio, J. Chem. Soc., Dalton Trans., 1999, 1799–1804 RSC; (d) C. He, B. G. Zhang, C. Y. Duan, J. H. Li and Q. J. Meng, Eur. J. Inorg. Chem., 2000, 2549–2554 CrossRef CAS; (e) R. Koner and I. Goldberg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m149–m151 CrossRef.
- (a) N. Sabbatini, M. Guardigi, F. Bolletta, I. Manet and R. Ziessel, Angew. Chem., Int. Ed. Engl., 1994, 33, 1501–1503 CrossRef; (b) X.-P. Yang and R. A. Jones, J. Am. Chem. Soc., 2005, 127, 7686–7687 CrossRef CAS.
- B. D. Chandler, D. T. Cramb and G. K. H. Shimizu, J. Am. Chem. Soc., 2006, 128, 10403–10412 CrossRef CAS.
- (a) Y. J. Kim, M. Suh and D. Y. Jung, Inorg. Chem., 2004, 43, 245–250 CrossRef CAS; (b) H. F. Zhu, Z. H. Zhang, T. A. Okamura, W. Y. Sun and N. Ueyama, Cryst. Growth Des., 2005, 5, 177–182 CrossRef CAS.
- (a) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 5th edn, Wiley & Sons, New York, 1997 Search PubMed; (b) G. Socrates, Infrared Characteristic Group Frequencies, Wiley, New York, 1980 Search PubMed.
- (a) A. Altomare, M. C. Burla, M. Camalli, M. Cascarano, C. Giacovazzo, A. Guagliardi and G. Polidori, J. Appl. Crystallogr., 1994, 27, 435–436 CrossRef; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- (a) X.-J. Zhang, Y.-H. Xing, J. Han, X.-Q. Zeng, M.-F. Ge and S.-Y. Niu, Cryst. Growth Des., 2008, 8, 3680–3688 CrossRef CAS; (b) D. Weng, X. Zheng, L. Li, W. Yanga and L. Jin, Dalton Trans., 2007, 4822–4828 RSC; (c) J.-Y. Wu, T.-T. Yeh, Y.-S. Wen, J. Twu and K.-L. Lu, Cryst. Growth Des., 2006, 6, 467–473 CrossRef CAS; (d) Z. Li, G. Zhu, X. Guo, X. Zhao, Z. Jin and S. Qiu, Inorg. Chem., 2007, 46, 5174–5178 CrossRef CAS.
- S. M. Hawxwell, G. M. Espallargas, D. Bradshaw, M. J. Rosseinsky, T. J. Prior, A. J. Florence, J. van de Streeke and L. Brammer, Chem. Commun., 2007, 1532–1534 RSC.
- (a) J. J. Perry, V. Ch. Kravtsov, G. J. McManus and M. J. Zaworotko, J. Am. Chem. Soc., 2007, 129, 10076–10077 CrossRef CAS; (b) Z. Pan, H. Zheng, T. Wang, Y. Song, Y. Li, Z. Guo and S. R. Batten, Inorg. Chem., 2008, 47, 9528–9536 CrossRef CAS; (c) A. Zafar, J. Yang, S. J. Geib and A. D. Hamilton, Tetrahedron Lett., 1996, 37, 2327–2330 CrossRef CAS.
- R. P. C. R. Soares-Santos, L. Cunha-Silva, F. A. Almeida Paz, R. A. Sá Ferreira, J. Rocha, T. Trindade, L. D. Carlos and H. I. S. Nogueira, Cryst. Growth Des., 2008, 8, 2505–2514 CrossRef CAS.
- TOPOS freeware: http://www.topos.ssu.samara.ru/.
- (a) B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693–1696 CrossRef CAS; (b) C. Daiguebonne, O. Guillou and K. Boubekeur, Inorg. Chim. Acta, 2000, 304, 161–169 CrossRef CAS; (c) X. Guo, G. Zhu, Z. Li, F. Sun, Z. Yang and S. Qiu, Chem. Commun., 2006, 3172–3174 RSC; (d) Z.-H. Zhang, Z.-L. Shen, T. Okamura, H.-F. Zhu, W.-Y. Sun and N. Ueyama, Cryst. Growth Des., 2005, 5, 1191–1197 CrossRef CAS.
- C. Daiguebonne, O. Guillou, Y. Gérault and K. Boubekeur, J. Alloys Compd., 2001, 323–324, 199–203 CrossRef CAS.
- (a) D. Sun, R. Cao, Y. Liang, Q. Shi and M. Hong, J. Chem. Soc., Dalton Trans., 2002, 1847–1851 RSC; (b) C. Daiguebonne, A. Deluzet, M. Camara, K. Boubekeur, N. Audebrand, Y. Gérault, C. Baux and O. Guillou, Cryst. Growth Des., 2003, 3, 1015–1020 CrossRef CAS; (c) Y.-B. Wang, W.-J. Zhuang, L.-P. Jin and S.-Z. Lu, J. Mol. Struct., 2005, 737, 165–172 CrossRef CAS.
- (a) K. M. L. Taylor, A. Jin and W. Lin, Angew. Chem., Int. Ed., 2008, 47, 7722–7725 CrossRef CAS; (b) L. P. Wu, M. Munakata, T. Kuroda-Sowa, M. Maekawa and Y. Suenaga, Inorg. Chim. Acta, 1996, 249, 183–189 CrossRef CAS; (c) A. Deluzet and O. Guillou, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m277–m279 CrossRef.
- (a) N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS; (b) S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814–3815 CrossRef CAS.
- (a) Y.-F. Han, X.-H. Zhou, Y.-X. Zheng, Z. Shen, Y. Song and X.-Z. You, CrystEngComm, 2008, 10, 1237–1242 RSC; (b) X. Guo, G. Zhu, Q. Fang, M. Xue, G. Tian, J. Sun, X. Li and S. Qiu, Inorg. Chem., 2005, 44, 3850–3855 CrossRef CAS; (c) Y.-B. Wang, W.-J. Zhuang, L.-P. Jin and S.-Z. Lu, J. Mol. Struct., 2004, 705, 21–27 CrossRef CAS.
- (a) X. Zheng, C. Sun, S. Lu, F. Liao, S. Gao and L. Jin, Eur. J. Inorg. Chem., 2004, 3262–3268 CrossRef CAS; (b) C.-S. Lim, A. C. Van Noord, J. W. Kampf and V. L. Pecoraro, Eur. J. Inorg. Chem., 2007, 134–1350; (c) A. Deluzet, W. Maudez, C. Daiguebonne and O. Guillou, Cryst. Growth Des., 2003, 3, 475–479 CrossRef CAS; (d) P.-K. Chen, S. R. Batten, Y. Qi and J.-M. Zheng, Cryst. Growth Des., 2009, 9, 2756–2761 CrossRef CAS.
- Y. Cui, H. L. Ngo, P. S. White and W. Lin, Chem. Commun., 2002, 1666–1667 RSC.
- L. Yan, Q. Yue, Q.-X. Jia, G. Lemercier and E.-Q. Gao, Cryst. Growth Des., 2009, 9, 2984–2987 CrossRef CAS.
- T. Devic, C. Serre, N. Audebrand, J. Marrot and G. Férey, J. Am. Chem. Soc., 2005, 127, 12788–12789 CrossRef CAS.
Footnote |
| † CCDC reference numbers 772531–772541 (for structures 1–11, respectively) For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ce00474j |
| This journal is © The Royal Society of Chemistry 2011 |