Coordination polymers of flexible tetracarboxylic acids with metal ions. II. Supramolecular assemblies of 5,5′-methylene- and 5,5′-(ethane-1,2-diyl)-bis(oxy)diisophthalic acid ligands with d-transition metals†
Anirban
Karmakar
and
Israel
Goldberg
*
School of Chemistry, Sackler Faculty of Exact Sciences, Tel-Aviv University, Ramat-Aviv, 69978, Tel-Aviv, Israel. E-mail: anirban@iitg.ernet.in; goldberg@post.tau.ac.il
First published on 11th October 2010
Abstract
This report evaluates the coordination patterns of the 5,5′-methylene-bis(oxy)diisophthalic acid and 5,5′-(ethane-1,2-diyl)-bis(oxy)diisophthalic acid ligands, bearing flexible spacers between the two isophthalic acid fragments, with a series of d-metal ions. Thirteen new binary and ternary (with additional bipyridyl components) hybrid organic–inorganic coordination-driven assemblies were investigated in detail by X-ray crystallography. In most cases they exhibit polymeric architectures of three-dimensional connectivity, the formation of which is facilitated by the conformational flexibility of the ligands.
Introduction
The prospective of the molecular building block approach for the development of new functional solid state porous materials has been well documented.1 This approach offers a promising pathway toward the design and construction of hybrid functional materials, due to the ability to tune their properties in a modular fashion. The metal–ligand directed assembly approach can yield a new generation of multi-dimensional networks, which contain channels, or cavities of various sizes and shapes.2 Due to the porous features of such metal–organic frameworks (MOFs) a wide range of applications can be envisioned, e.g. in nonlinear optics,3 gas storage,4 catalysis5 and host–guest induced separation.6 Polycarboxylic acids are among the most attractive building blocks (usually in their de-protonated carboxylate form) for the construction of metal–organic frameworks, due to their versatile binding features, and several challenging efforts were made in the past towards the formulation in a predictable manner of different metal–carboxylate motifs,7 and the design of new open framework structures.8 Some earlier reports relate to the use of dicarboxylate,9 tricarboxylate,10 and tetracarboxylate11 ligands, which are inter-bridged by mono- or multi-nuclear metal nodes, leading to stable MOFs with permanent porosity. For example, highly porous MOFs have been built with the aid of tricarboxylate linkers as 4,4′,4″-s-triazine-2,4,6-triyltribenzoate,12a benzene-1,3,5-tribenzoate,12b–h 1,3,5-tris[4′-carboxy(1,1′-biphenyl-4-yl)]benzene12i and tetracarboxylate linkers such as 3,3′,5,5′-azobenzenetetracarboxylate,13a terphenyl-3,3″,5,5″-tetracarboxylate,13b quaterphenyl-3,3′,5,5′-tetracarboxylate,13c biphenyl tetracarboxylate,13d–i 4,4′,4″,4′-benzene-1,2,4,5-tetrayltetrabenzate,13j,k and binaphthyl carboxylate.13l–p MOFs constructed from tetracarboxylates with rigid spacer groups such as poly(phenylene)14 or 4,4′-bipyridyl units15 are well documented in the literature, but tetracarboxylates with flexible spacer units are far less prevalent.16In this study we have applied a different type of approach to the MOF synthesis, with a view to construct more reactive and adaptable ligands and the resulting organic–inorganic hybrid materials. It involves creating non-rigid ligands with four diverging tetracarboxylic acid groups by appending two dicarboxylic acid aromatic species through a flexible spacer entity. These were synthesized by inter-connecting between two oxyisophthalic acid moieties, through their ethereal O-sites by methane or ethane fragment units, to yield the 5,5′-methylene-bis(oxy)diisophthalic acid, H4L′, and 5,5′-(ethane-1,2-diyl)-bis(oxy)diisophthalic acid, H4L″ (Scheme 1).17 It has been anticipated that such molecular design of the organic component with diverging flexible and poly-dentate coordination sites may generate, in combination with metal ion connectors, multidimensional architectures. We described earlier the syntheses of H4L′ and H4L″, and their coordination polymerization features with lanthanoid metal ions.17 Here we report on the metal–organic assemblies obtained by reacting these ligands with a series of d-metal ions. The H4L′ and H4L″ ligands can be singly, doubly, triply or quadruply deprotonated to the corresponding carboxylate species (H3L−, H2L2−, H1L3−, and L4−) to balance the charge of the inorganic metal connectors without the need to incorporate foreign counter ions in the formed structures. Additional experiments were carried with the various metal ions and mixtures of the H4L′ or H4L″ ligands with either the 4,4′-bipyridine (bpy) or 1,2-di(4-pyridyl)ethane (dpye) moieties. The latter are known to reveal high coordination affinity towards d-metal ions, which may improve the odds of obtaining three-component (a given metal ion with two different bridging ligands) framework coordination polymers.
 | ||
| Scheme 1 Molecular structures of the H4L′ and H4L″ ligands. | ||
We report here on the structural features of those compounds that could be obtained in the form of X-ray diffraction quality single crystals and subjected to a complete crystallographic analysis. Their empirical formulae (excluding the disordered solvent which could not be reliably modeled) are:
1 [Zn2+·3(H2O)·(H2L′)2−]·3H2O
2 [(Zn2+)2·3(H2O)·(py)·(L′)4−]·2H2O
3 [(Mn2+)1½·(MeOH)·(CH3CN)·(HL')3−]
4 [(Co2+)2·(O2−)·(H2O)·(H2L′)2−]
5 [(Ag+)2·(H2O)·(H2L′)2−]
6 [(Cd2+)2·(L″)4−]
7 [(Zn2+)2·3(DMF)·(L″)4−]·DMF
8 [(Ni2+)2·(O2−)·(H2O)·(H2L″)2−]
9 [(Ag+)4·(H2O)·(H2L″)24−]·H2O
10 [(Zn2+)2·(H2O)·(L′)4−·2(bpy)]·DMSO·H2O
11 [(Ag+)2·(H2L′)2−·½(bpy)]
12 [(Co2+)·(H2L″)2−·(bpy)]
13 [(Co2+)2·2(H2O)·(L″)4−·2(dpye)]
Experimental
All reagents and solvents were purchased from commercial sources and used without further purification. FT-IR spectra were recorded in the range of 400–4000 cm−1 from KBr pellets (Aldrich 99+%, FT-IR grade) using a BRUKER PS15 spectrophotometer. Compounds 1–13 were synthesized via the following (mostly in hydrothermal conditions) procedures:Compound 1
Ligand H4L′ (37.5 mg, 0.1 mmol) and ZnCl2 (27.4 mg, 0.2 mmol) were mixed and dissolved in 3 ml of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMSO:H2O. The mixture was stirred at room temperature for 30 min to yield a white precipitate, which was then dissolved in 0.5 ml of NH4OH. This solution was placed in a capped glass vessel and gradually heated to, and kept for 2 days at, 100 °C. The resulting colorless mixture was cooled to room temperature (at 0.2 °C min−1 rate) and kept for crystallization. After 10 days crystalline product was obtained. It was separated by filtration and then dried briefly in air. Yield: 43% (based on Zn). FT-IR (KBr, cm−1): 3152 (bs), 1624 (s, νC
:1 DMSO:H2O. The mixture was stirred at room temperature for 30 min to yield a white precipitate, which was then dissolved in 0.5 ml of NH4OH. This solution was placed in a capped glass vessel and gradually heated to, and kept for 2 days at, 100 °C. The resulting colorless mixture was cooled to room temperature (at 0.2 °C min−1 rate) and kept for crystallization. After 10 days crystalline product was obtained. It was separated by filtration and then dried briefly in air. Yield: 43% (based on Zn). FT-IR (KBr, cm−1): 3152 (bs), 1624 (s, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1574 (s, νCO asymmetric), 1463 (m, νCC), 1372 (s, νCO symmetric), 1246 (m, νC–O), 1158 (w), 1111 (w), 1030 (s), 779 (s), 720 (s).
C), 1574 (s, νCO asymmetric), 1463 (m, νCC), 1372 (s, νCO symmetric), 1246 (m, νC–O), 1158 (w), 1111 (w), 1030 (s), 779 (s), 720 (s).
Compound 2
The mixture of Zn(NO3)2·6H2O (59.4 mg, 0.2 mmol), H4L′ (37.5 mg, 0.1 mmol), and triethylamine (0.021 g, 0.2 mmol) was dissolved in 5 ml of MeOH and water (1:1). White precipitate was obtained when the mixture was stirred at room temperature for 1 h. The precipitate was dissolved in 0.5 ml of NH4OH and 0.5 ml of pyridine. Then, the resulting mixture was sealed in an 8 ml glass vessel and heated at 60 °C for 48 h. It was subsequently cooled to room temperature (0.2 °C min−1), affording after 2 weeks prism-like colorless crystals. Yield: 51% (based on Zn). FT-IR (KBr, cm−1): 3336 (bs, νO–H coordinated water), 2921 (w, νC–H), 1623 (s, νCC), 1575 (s, νCO asymmetric), 1497 (w), 1451 (w, νCC), 1374 (s, νCO symmetric), 1237 (m, νC–O), 1138 (w), 1026 (s), 921 (w), 777 (m), 695 (m).
Compound 3
Mn(ClO4)2·6H2O (9.4 mg, 0.026 mmol), H4L′ (5 mg, 0.013 mmol) were mixed and dissolved in 5 ml of MeOH:CH3CN (1:1 by volume), then sealed in a capped glass vessel, which was heated to 100 °C for 48 h and then cooled to RT (0.2 °C min−1). Colorless crystals suitable for X-ray diffraction analysis were obtained in 41% yield (based on Mn). FT-IR (KBr, cm−1): 3444 (bs, νO–H coordinated water), 2922 (w, νC–H), 1694 (s, νCO), 1621 (s, νCO asymmetric), 1542 (s), 1459 (m, νCC), 1384 (s, νCO symmetric), 1237 (m, νC–O), 1027 (s), 945 (w), 777 (s), 700 (m).
Compound 4
Co(NO3)2·6H2O (7.2 mg, 0.026 mmol) and H4L′ (5 mg, 0.013 mmol) were mixed and dissolved in 6 ml of MeOH:DMF:H2O (1:1:1 by volume), then sealed in a capped glass vessel, and heated to 85 °C for 48 h and then 100 °C for 24 h. After cooling to room temperature (0.2 °C min−1), pink crystals were obtained in ca. 21% yield (based on Co). FT-IR (KBr, cm−1): 3420 (bs, νO–H coordinated water), 2923 (m, νC–H), 1622 (s, νCC), 1573 (s, νCO asymmetric), 1456 (w, νCC), 1384 (s, νCO symmetric), 1285, 1246 (m, νC–O), 1051 (s), 916 (w), 778 (w), 715 (m), 645 (m).
Compound 5
A mixture of AgNO3 (4.4 mg, 0.026 mmol), H4L′ (5 mg, 0.013 mmol) and H2O (6 ml) was placed in a capped glass vessel and heated at 85 °C for 48 h. After the sample was cooled to room temperature (0.2 °C min−1), block-shaped colorless crystals were obtained, which were washed with water and dried in air. Yield: 19% (based on Ag). FT-IR (KBr, cm−1): 3453 (bs), 2925 (bs, νC–H), 1659 (s, νCC), 1537 (s, νCO asymmetric), 1454 (m, νCC), 1389 (s, νCO symmetric), 1270 (m, νC–O), 1136 (w), 1092 (m), 993 (m), 912 (w), 767 (s), 731 (s), 692 (s).
Compound 6
A mixture of Cd(NO3)2·4H2O (30.8 mg, 0.1 mmol) and H4L″ (20 mg, 0.05 mmol) in 4.5 ml of MeOH/DMF/H2O (3:1:½ by volume) was placed in a capped glass vessel and heated to 100 °C for 48 h in a bath reactor. After cooling to room temperature (0.2 °C min−1), colorless crystals were obtained, separated by filtration and then dried in air. Yield: 76% (based on Cd). FT-IR (KBr, cm−1): 3434 (bs), 3133 (bs, νC–H), 1662 (s, νCC), 1567 (s, νCO asymmetric), 1451 (m, νCC), 1370 (s, νCO symmetric), 1257 (m, νC–O), 1124 (w), 1071 (m), 1011 (m), 894 (w), 787 (s), 731 (s), 455 (w).
Compound 7
A mixture of Zn(NO3)2·6H2O (30.8 mg, 0.1 mmol), H4L″ (20 mg, 0.05 mmol)) and DMF (5 ml) was placed in a capped glass vessel and heated at 100 °C for 2 days. After the sample was cooled to room temperature (0.2 °C min−1), colorless crystals of 7 were obtained. Yield: 38% (based on Zn). FT-IR (KBr, cm−1): 3396 (bs), 2925 (m, νC–H), 1658 (s, νCO DMF), 1620 (s, νCC), 1574 (s, νCO asymmetric), 1454 (s, νCC), 1377 (s, νCO symmetric), 1259 (m, νC–O), 1131 (w), 1069 (w), 1011 (w), 777 (m), 729 (w), 476 (w).
Compound 8
Ni(ClO4)2·6H2O (18.2 mg, 0.05 mmol) and H4L″ (10 mg, 0.025 mmol) were mixed and dissolved in 5.3 ml of EtOH:DMF:H2O (3:3:2 by volume), then sealed in a capped glass vessel, which was heated to 100 °C for 48 h. After cooling to room temperature (0.2 °C min−1), green block type crystals were obtained which were separated by filtration and then dried in air. Yield: 19% (based on Ni). FT-IR (KBr, cm−1): 3418 (bs), 2929 (w, νC–H), 1653 (s, νCC), 1568 (s, νCO asymmetric), 1451 (m, νCC), 1370 (s, νCO symmetric), 1260 (m, νC–O), 1118 (w), 1066 (w), 1005 (w), 891 (w), 782 (m), 719 (m), 677 (m).
Compound 9
A mixture of AgNO3 (13 mg, 0.076 mmol), H4L″ (5 mg, 0.013 mmol) and H2O (6 ml) was placed in a capped glass vessel and heated at 100 °C for 48 h, yielding after a few days colorless crystals. Yield: 17% (based on Ag). FT-IR (KBr, cm−1): 3444 (bs), 2920 (w, νC–H), 1666 (s, νCC), 1536 (s, νCO asymmetric), 1456 (m, νCC), 1380 (s, νCO symmetric), 1268 (s, νC–O), 1128 (w), 1057 (w), 980 (w), 879 (w), 763 (s), 686 (m), 613 (w).
Compound 10
Zn(NO3)2·6H2O (30 mg, 0.1 mmol), H4L′ (20 mg, 0.05 mmol) and 4,4′-bipyridine (bpy) (8 mg, 0.05 mmol) were mixed in a capped glass vessel and dispersed in 5 ml of dimethyl sulfoxide. The vessel was sealed and heated at 100 °C for 48 hours without stirring. Two months after cooling to room temperature (0.2 °C min−1), prism-like colorless crystal was obtained. Yield: 10% (based on Zn). FT-IR (KBr, cm−1): 3390 (bs, νO–H coordinated water), 2925 (w, νC–H), 1611 (s, νCC), 1557 (s, νCO asymmetric), 1424 (m, νCC), 1371 (s, νCO symmetric), 1232 (m, νC–O), 1148 (w), 1023 (s), 814 (w), 779 (w), 722 (m), 634 (m).
Compound 11
A mixture of AgNO3 (4.4 mg, 0.026 mmol), H4L′ (5 mg, 0.013 mmol) and 4,4′-bipyridine (bpy) (2 mg, 0.013 mmol) was placed in a capped glass vessel and heated at 100 °C for 48 h in 6 ml of water. After cooling to room temperature (0.2 °C min−1), colorless crystals were obtained, washed with methanol and dried in air. Yield: 15% (based on Ag). FT-IR (KBr, cm−1): 3568 (bs), 2935 (bs, νC–H), 1700 (s, νCO), 1554 (s, νCO asymmetric), 1461 (m, νCC), 1384 (s, νCO symmetric), 1287 (m, νC–O), 1132 (w), 1030 (m), 995 (m), 763 (w), 683 (s).
Compound 12
Co(NO3)2·6H2O (29.1 mg, 0.1 mmol), H4L″ (20 mg, 0.05 mmol) and 4,4′-bipyridine (bpy) (8 mg, 0.05 mmol) were mixed in a capped glass vessel and dispersed in 4.5 ml of MeOH:DMF:H2O (3:1:0.5 by volume). The vessel was sealed and heated at 100 °C for 48 hours. After cooling to room temperature (0.2 °C min−1), the pink crystals were collected by filtration. Yield: 26% (based on Co). FT-IR (KBr, cm−1): 3437 (bs), 2926 (w, νC–H), 1710 (m, νCO), 1611 (s, νCC), 1554 (s, νCO asymmetric), 1454 (m, νCC), 1398 (s, νCO symmetric), 1278 (m, νC–O), 1225 (s), 1115 (w), 1071 (m), 1013 (m), 770 (m), 722 (m), 682 (s).
Compound 13
A mixture of Co(NO3)2·6H2O (29.1 mg, 0.1 mmol), H4L″ (20 mg, 0.05 mmol) and 4-{2-(pyridine-4-yl)ethyl}pyridine (dpye) (9 mg, 0.05 mmol) was placed in a capped glass vessel which contained 4.5 ml of MeOH:DMF:H2O (3:1:0.5 by volume). The vessel was sealed and heated at 100 °C for 48 hours, and then cooled gradually to RT (0.2 °C min−1). Dark pink crystals were obtained and collected by filtration. Yield: 52% (based on Co). FT-IR (KBr, cm−1): 3420 (bs, νO–H coordinated water), 2941 (w, νC–H), 1621 (s, νCC), 1553 (s, νCO asymmetric), 1458 (m, νCC), 1377 (s, νCO symmetric), 1267 (m, νC–O), 1131 (w), 1069 (m), 1014 (m), 893 (w), 780 (s), 721 (s), 471 (w).
Compounds 1, 2, 3, 4, 6, 7, 10, 11, and 13 were obtained as pure crystalline materials either directly from the reactor or after evaporation of solvent, and the yields were calculated considering the amount of obtained product. Compounds 5, 8, 9, and 12 were obtained as crystalline materials, which precipitated in the reaction vessel along with some unidentified amorphous substance. The crystals were collected by filtration, properly washed with DMF and water to remove the amorphous residue, and dried in air. The calculated yields of 5, 8, 9, and 12 were thus based on the net amounts of the corresponding crystalline products.
Crystal structure determinations
The X-ray measurements (Nonius KappaCCD diffractometer, MoKα radiation) were carried out either at 110(2) K (for 1–10, 12 and 13) or at 200(2) K (for 11) on crystals coated with a thin layer of amorphous oil to minimize crystal deterioration, possible structural disorder and related thermal motion effects, and to optimise the precision of the structural results. These structures were solved by direct methods (SIR-97) and refined by full-matrix least-squares (SHELXL-97).18 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms attached to carbon were located in idealized/calculated positions and were refined using a riding model, with Uiso = 1.2Ueq of the parent atom. Those attached to O and N atoms, which are involved in hydrogen bonding (except for the disordered species) were located in difference-Fourier maps; then their O–H and N–H distances were restrained to common values. Crystallographic refinements of all the structures converged to acceptable R-values, representing precisely determined structural models of compounds 1–13 and allowing reliable characterizations of the molecular structures and supramolecular binding motifs. Structural disorder in some components of the polymeric lattices of 11 (Ag ions), 13 (Co ions and dpye ligands) was resolved successfully. However, crystals of 1 and of the polymeric assemblies 3, 4, 6, 8, 10 and 13, contained additional severely disordered non-coordinated molecules of crystallization solvent (water, methanol, and N,N-dimethylformamide), which could not be modeled reliably by discrete atoms (as detailed in the corresponding CIFs). Correspondingly, the contribution of the disordered solvent moieties was subtracted from the diffraction pattern by the SQUEEZE procedure and PLATON software.19 The crystallographic and experimental data for 1–13 are given in Table 1. The uniform identity of the formed crystal lattices in a given reaction was confirmed in each case by repeated measurements of the unit-cell dimensions from different single crystallites.| Compound | 1 a | 2 | 3 a | 4 a | 5 |
|---|---|---|---|---|---|
| a Data refer to crystallographic refinements after subtracting the contribution of the disordered solvent to the diffraction data. | |||||
| Formula | C17H20O16Zn | C22H23NO15Zn2 | C40H24Mn3N2O22 | C17H8Co2O12 | C17H12Ag2O11 |
| F w | 545.70 | 672.15 | 1049.45 | 522.09 | 608.01 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group | C2/c | P21/c | C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å | 20.7034(4) | 10.8788(2) | 28.0246(6) | 10.2067(4) | 8.5594(1) |
| b/Å | 7.5718(2) | 30.1049(6) | 12.3747(3) | 11.1297(6) | 9.7574(2) |
| c/Å | 30.7679(9) | 8.5449(2) | 14.1946(3) | 13.9251(5) | 11.5046(2) |
| α/° | 90.00 | 90.00 | 90.00 | 112.663(3) | 67.701(1) |
| β/° | 92.749(1) | 111.360(4) | 109.489(1) | 104.574(3) | 81.920(1) |
| γ/° | 90.00 | 90.00 | 90.00 | 94.796(2) | 71.414(1) |
| V/Å3 | 4817.7(2) | 2606.3(9) | 4640.6(2) | 1383.6(1) | 842.41(2) |
| Z | 8 | 4 | 4 | 2 | 2 |
| ρ calcd/Mg m−3 | 1.505 | 1.713 | 1.502 | 1.253 | 2.397 |
| µ/mm−1 | 1.094 | 1.918 | 0.877 | 1.245 | 2.393 |
| F(000) | 2240 | 1368 | 2116 | 520 | 592 |
| θ max/° | 27.86 | 27.89 | 27.86 | 27.20 | 27.82 |
| Refl. collected | 16675 |
19477 |
17293 |
16280 |
9318 |
| Refl. unique | 5583 | 6089 | 5508 | 5917 | 3942 |
| R(int) | 0.052 | 0.067 | 0.062 | 0.066 | 0.033 |
| Completeness | 97% | 98% | 100% | 96% | 99% |
| Refl. with I > 2σ(I) | 3499 | 4258 | 3309 | 3463 | 3329 |
| Refined param. | 308 | 361 | 306 | 280 | 271 |
| R1 [I > 2σ(I)] | 0.055 | 0.047 | 0.054 | 0.049 | 0.031 |
| wR2 [I > 2σ(I)] | 0.142 | 0.122 | 0.134 | 0.110 | 0.079 |
| R1 [all data] | 0.090 | 0.079 | 0.098 | 0.082 | 0.042 |
| wR2 [all data] | 0.156 | 0.135 | 0.148 | 0.116 | 0.085 |
| ±Δρmax/e Å−3 | +1.16, −0.86 | +1.38, −0.71 | +0.58, −0.54 | +0.65, −0.63 | +1.27, −1.22 |
| 6 a | 7 | 8 a | 9 | |
|---|---|---|---|---|
| Formula | C18H12CdO10 | C30H38N4O14Zn | C18H12Ni2O12 | C36H28Ag4O22 |
| F w | 500.68 | 612.76 | 537.70 | 1244.06 |
| Crystal system | Monoclinic | Orthorhombic | Triclinic | Monoclinic |
| Space group | C2/c | P212121 |
P |
P21/c |
| a/Å | 13.8667(4) | 11.1099(1) | 10.0777(2) | 15.4803(2) |
| b/Å | 14.8068(4) | 13.8909(1) | 11.0012(2) | 17.8109(2) |
| c/Å | 14.6050(4) | 22.4850(4) | 15.0573(3) | 13.2541(2) |
| α/° | 90.00 | 90.00 | 72.949(1) | 90.00 |
| β/° | 114.176(2) | 90.00 | 77.047(1) | 104.7111(6) |
| γ/° | 90.00 | 90.00 | 81.370(1) | 90.00 |
| V/Å3 | 2735.7(1) | 3470.03(7) | 1548.98(5) | 3534.60(8) |
| Z | 4 | 4 | 2 | 4 |
| ρ calcd/Mg m−3 | 1.216 | 1.549 | 1.153 | 2.338 |
| µ (MoKα)/mm−1 | 0.836 | 1.455 | 1.258 | 2.284 |
| F(000) | 992 | 1672 | 544 | 2432 |
| θ max/° | 27.87 | 27.87 | 27.85 | 27.87 |
| Refl. collected | 23187 |
24555 |
16652 |
32567 |
| Refl. unique | 3256 | 8172 | 7277 | 8351 |
| R(int) | 0.039 | 0.063 | 0.040 | 0.046 |
| Completeness | 100% | 99% | 99% | 99% |
| Refl. with I > 2σ(I) | 2925 | 6691 | 5054 | 6330 |
| Refined param. | 135 | 459 | 289 | 563 |
| R1 [I > 2σ(I)] | 0.034 | 0.038 | 0.038 | 0.039 |
| wR2 [I > 2σ(I)] | 0.095 | 0.082 | 0.096 | 0.089 |
| R1 [all data] | 0.037 | 0.056 | 0.055 | 0.058 |
| wR2 [all data] | 0.097 | 0.088 | 0.101 | 0.098 |
| ± Δρmax/e Å−3 | +1.10, −1.03 | +0.65, −0.61 | +0.64, −0.59 | +2.10, −1.73 |
| 10 a | 11 | 12 | 13 a | |
|---|---|---|---|---|
| Formula | C39H34N4O13SZn2 | C22H14Ag2NO10 | C28H20CoN2O10 | C30H26Co2N2O12 |
| F w | 929.50 | 668.08 | 603.39 | 724.39 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/c |
P |
C2/c | Cm |
| a/Å | 16.4370(2) | 7.7679(2) | 20.2908(13) | 13.5405(3) |
| b/Å | 14.6990(2) | 10.6578(4) | 9.9513(6) | 21.5306(5) |
| c/Å | 17.5061(2) | 12.8283(3) | 14.5720(9) | 13.5016(3) |
| α/° | 90.00 | 78.834(2) | 90.00 | 90.00 |
| β/° | 109.379(1) | 73.855(2) | 116.450(3) | 105.778(1) |
| γ/° | 90.00 | 80.300(1) | 90.00 | 90.00 |
| V/Å3 | 3990.0(1) | 993.41(5) | 2634.4(3) | 3787.9(2) |
| Z | 4 | 2 | 4 | 4 |
| ρ calcd/Mg m−3 | 1.547 | 2.233 | 1.521 | 1.270 |
| µ (MoKα)/mm−1 | 1.326 | 2.039 | 0.715 | 0.930 |
| F(000) | 1904 | 654 | 1236 | 1480 |
| θ max/° | 27.88 | 27.88 | 26.32 | 27.90 |
| Refl. collected | 34714 |
18547 |
16150 |
16647 |
| Refl. unique | 9442 | 4699 | 2672 | 7833 |
| R(int) | 0.053 | 0.041 | 0.067 | 0.049 |
| Completeness | 99% | 99% | 100% | 99% |
| Refl. with I > 2σ(I) | 5828 | 3680 | 2123 | 6458 |
| Refined param. | 539 | 338 | 186 | 484 |
| R1 [I > 2σ(I)] | 0.049 | 0.038 | 0.053 | 0.047 |
| wR2 [I > 2σ(I)] | 0.128 | 0.088 | 0.104 | 0.114 |
| R1 [all data] | 0.088 | 0.054 | 0.075 | 0.060 |
| wR2 [all data] | 0.142 | 0.097 | 0.112 | 0.118 |
| ±Δρmax/e Å−3 | +1.01, −1.01 | +0.61, −0.98 | +0.30, −0.42 | +0.92, −0.58 |
Results and discussion
The main conformational degrees of freedom in H4L′ and H4L″ involve possible rotation of the two isophthalic acid fragments with respect to the central aliphatic spacers and about the O–Cphenyl bonds. Two parameters characterize best the resulting molecular shape of the ligands: the dihedral angle δ between the two phenyl rings in a given molecule and the torsion angle τ (Cphenyl–O⋯O–Cphenyl in H4L′, and O–CH2–CH2–O in H4L″). The observed values in compounds 1–13 are summarized in Table 2. The terminal carboxylic/carboxylate groups may also rotate with respect to the phenyl groups they are attached too, in order to optimize ligation to the metal ion linkers or intermolecular hydrogen-bonding, though due to π-electron delocalization they tend to be nearly coplanar with the proximal phenyl rings.| Compound (metal type) | δ′/δ″/° | τ′/τ″/° | Metal–O/N bonds range/Å | Nuclearity of metal cluster | Metal–metal distance(s)/Å | Coordination dimensionality | Ligand conformation |
|---|---|---|---|---|---|---|---|
| a The 3D diamondoid arrays doubly interpenetrate in the crystal lattice. b The silver ions in this structure reveal twofold positional disorder. c In 12, the 2D coordination networks are sixfold interpenetrated. | |||||||
| 1 (Zn) | 39.5(1) | 142.3(3) | 1.976(2)–2.151(2) | 1 | — | 0D | Flattened V-conformation of L′ |
| 2 (Zn) | 52.6(1) | 132.1(3) | 1.979(2)–2.015(3), 1.942(2)–2.039(3) | 1 | — | 1D | Flattened V-conformation of L′ |
| 3 (Mn) | 64.0(1) | 127.9(4) | 2.078(2)–2.270(3), 2.447(3) | 3 | 3.941(1) | 3D | V-Shaped twisted conformation of L′ |
| 4 (Co) | 73.4(1) | 141.0(5) | 2.051(2)–2.206(3) | 4 | 3.149(1), 3.370(1) | 3D | V-Shaped twisted conformation of L′ |
| 5 (Ag) | 12.6(2) | 86.7(4) | 2.181(2)–2.276(2), 2.399(2)–2.478(2) | ∞ | 2.8588(5), 2.7929(5), 3.1778(4) | 3D | U-Shape conformation of L′ |
| 6 (Cd) | 84.3(5) | 80.2(4) | 2.274(2)–2.628(2) | 1 | — | 3Da | Tetrahedral conformation of L″ |
| 7 (Zn) | 78.4(1) | 66.3(3) | 1.963(2)–2.109(2) | 2 | 3.723(1) | 3D | V-Shape bent conformation of L″ |
| 8 (Ni) | 84.2(1) | 69.8(2) | 1.987(1)–2.164(1) | 4 | 3.069(1), 3.366(1) | 3D | Tetrahedral conformation of L″ |
| 9 (Ag) | 25.8(1), 28.9(1) | 64.8(4), 67.8(4) | 2.179(3)–2.255(3), 2.448(2)–2.510(3) | ∞ | 2.8303(5)–2.8919(6), 3.224(4), 3.2693(4) | 3D | U-Shape flat conformation of L″ |
| 10 (Zn) | 47.2(1) | 131.6(2) | 2.033(2)–2.432(2) | 1 | — | 3Da | V-Shaped twisted conformation of L′ |
| 11 (Ag) | 21.7(2) | 95.2(4) | 2.098(2)–2.455(5) | 4 | 2.9–3.1b | 3D | U-Shape conformation of L′ |
| 12 (Co) | 88.9(1) | 79.7(4) | 1.984(2)–2.036(2) | 1 | — | 2Dc | Tetrahedral conformation of L″ |
| 13 (Co) | 83.2(1) | 73.5(4) | 2.012(3)–2.132(3) | 1 | — | 3D | Tetrahedral conformation of L″ |
Coordination patterns in metal complexes with H4L′
The correlation between the molecular conformation of this ligand and the supramolecular architecture which forms is demonstrated by the following examples. In compounds 1 and 2 the organic component adopts a flattened V-type geometry, and the resulting coordination motifs are characterized by low dimensionality. On the other hand, in compounds 3 and 4 the L′-species has a more pronouncedly bent V-shape, which is associated with the formation of three-dimensional polymeric metal–ligand assemblies. The coordination pattern in 1 is shown in Fig. 1a, representing a 2:2 Zn2+·(H2L′)2− discrete macrocyclic complex, assembled about the axis of twofold rotation, with the metal linkers located on opposite sides of the formed macrocycle. Two of the four carboxylic groups of a given ligand are involved in metal coordination, while the other two groups directed outward are open for intermolecular interactions. Each zinc ion is coordinated to three water molecules and two carboxylic groups (in a monodentate fashion) of two neighboring ligands. In the crystal, the open space in the center of the macrocyclic dimer is filled by disordered solvent species (DMSO and water). The macrocyclic entities are arranged in the crystal in a layered manner. The peripherally oriented carboxylic groups of one macrocycle are hydrogen bonded to the metal–ligated water molecules of adjacent macrocycles (Fig. 1b). The latter are also bridged by extensive intermolecular hydrogen bonding through the interstitial molecules of water trapped in the lattice as crystallization solvent. As the hydrogen bonding interactions are of secondary importance in the discussion of the coordination chemistry of H4L′ and H4L″ in this paper, a complete list of the hydrogen bonds in structures 1–13 is given only in the CIFs†. The metal–ligand coordination distances in these hybrid coordination compounds are presented in Table 2.
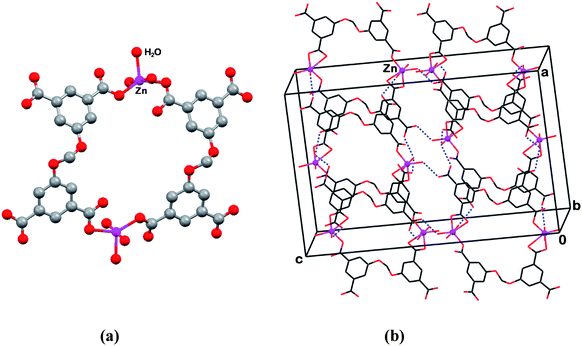 | ||
| Fig. 1 Compound 1. (a) Cyclic dimer of the zinc ion complex. Note the flattened conformation of the organic ligand. (b) View of the crystal structure and the intermolecular hydrogen bonding (dotted lines). The non-coordinated water molecules and their hydrogen bonding are omitted for clarity. | ||
Compound 2 represents a 2:1 zinc:(L′)4− complexation, with a complete deprotonation of the ligand, which allows expansion of the macrocyclic assembly into an extended one-dimensional coordination polymerization. This is associated with a slightly larger bending of the organic component (Table 2). In thus formed ladder-type chain (Fig. 2), the two zinc centers are four-coordinate with a tetrahedral geometry. They bind to the carboxylate groups of two neighboring (L′)4− units in a mono-dentate fashion. The coordination sphere of one zinc ion includes also two water molecules, while the other zinc ion links further to water and pyridyl species. The pyridyl group in oriented inward, filling the space within the macrocyclic segments of the polymeric array. The intermolecular organization in the crystal is characterized by multiple hydrogen bonding between neighboring polymeric chains (involving their carboxylate groups and metal ligated water molecules), either directly, or through additional molecules of the non-coordinated water crystallization solvent.
 | ||
| Fig. 2 The ladder-type coordination motif in 2. Note the pyridyl (py) moieties that fill the space between subsequent units of the flattened ligand. | ||
Single-framework metal–ligand coordination polymerization characterizes compounds 3–5. The coordination pattern in manganese polymer 3 is shown in Fig. 3. The V-shaped ligand coordinates simultaneously to seven different metal ions, in between them three carboxylate groups are bound to the metal centers via bridging bidentate fashion, and the remaining carboxylic acid function links to a single metal ion. The two crystallographically independent Mn-ions are arranged in tri-nuclear clusters and are hexa-coordinate. The metal core located on crystallographic inversion (Mn2) connects to six different carboxylate groups, and the second ion located in the inversion-related general positions (Mn1) each links with four different carboxylates, a molecule of methanol and acetonitrile. The Mn1⋯Mn2 distances within the tri-nuclear clusters are 3.941(1) Å. Every pair of the metal ions within the Mn1⋯Mn2⋯Mn1 trio is bridged by three carboxylate entities, in a µ2–η1–η1-bridging mode (in which the two O-sites coordinate to two different metal centers). The octahedral coordination environment around each of the manganese ions imparts three-dimensional connectivity pattern in this structure.
 | ||
| Fig. 3 The coordination scheme in 3. The Mn2 ion resides on crystallographic inversion binding to the shown O-sites of six different ligands. The Mn1 ion binds to the O-sites of four different ligands to MeOH and to MeCN, as shown. | ||
The inorganic connectors in 4 take the form of tetranuclear µ-dioxo-type clusters located on centers of inversion (Fig. 4a). Every tetranuclear connector is associated with eight different ligands, two molecules of water, and two triply bridged oxo anions. The cobalt centers are six-coordinate. The Co⋯Co distances in the cluster are 3.149(1) Å between the two inversion related central Co1 ions and 3.370(1) Å between the Co1 and the peripheral Co2 ion. On each side of the cluster, Co1 and Co2 are linked with one another by three µ2–η1–η1-bridged carboxylates of different ligands. Co2 is further coordinated to a carboxylic group of yet another ligand in a chelating bidentate mode. The ligand moiety is bent to a significant extent, with its phenyl rings oriented nearly perpendicular to one another. Association of its eight carboxylic/carboxylate groups to different (Co4O2)4+ connectors, results in the formation of single-framework polymeric arrays with open channels (approximately 7 Å × 8 Å) propagating through the polymer along the a-axis of the crystal (Fig. 4b). The latter are accommodated by disordered solvent (a mixture of water and methanol).
 | ||
| Fig. 4 (a) The coordination scheme in 4. The dioxo-bridged Co-tetramer, (Co4O2)4+, located on inversion is coordinated to eight organic ligands and two molecules of water. (b) Crystal structure of 4, viewed approximately down the a-axis, and down the solvent (not shown) channels. The Co-ions and the µ-oxo bridges are depicted by small spheres. | ||
The use of silver ions for coordination polymerization with H4L′ has led to a unique structure 5 in which the Ag+ ions aggregate into zigzag one-dimensional chains/backbones, which are crosslinked by the multiply coordinated doubly deprotonated ligands (adopting a U-type conformation) into a three-dimensional framework (Fig. 5). Dinuclear and polynuclear aggregation of the d10 silver ions, characterized by inter-ionic distances comparable to the Ag–Ag separation in metallic silver (2.889 Å), is known but only when stabilized by bridging anionic ligands.20 In 5, there are two crystallographically independent ions in the asymmetric unit Ag1 and Ag2. The observed interatomic distances between the proxime silver ions located across centers of inversion along the polynuclear cluster are Ag1⋯Ag1 = 2.8588(5) Å and Ag2⋯Ag2 = 2.7929(5) Å, the Ag1⋯Ag2 distance being slightly longer 3.1778(4) Å. Neighboring inversion-related Ag-pairs are µ2–η1–η1-bridged by the carboxylate groups of two different ligands, every ligand binding to differently shifted Ag-pairs along the chain. Then, Ag1 and Ag2 are each coordinated also to a carboxylic group of another ligand, Ag1 being further associated to a water molecule. Each doubly deprotonated ligand is interconnected with six different silver ions. Its carboxylate arms are coordinated with two of each Ag1 and Ag2 centers via a µ2–η1–η1-bridging mode, while the protonated parts of the ligand are connected with other two silver ions in a monodentate fashion. The two carboxylate groups of a given ligand bind to the same silver ion chain, and its two carboxylic acid functions, which diverge in opposite directions, coordinate to two other Ag-backbones. The latter function as construction pillars of the three-dimensional architecture.
 | ||
| Fig. 5 (a) Crystal structure of 5, showing the silver-chain pillars cross-linked by the organic ligand (note its U-shaped conformation with parallel phenyl rings). (b) Illustration of the bridging mode of the ligands to different silver chains. The silver ions are depicted as small spheres; the primed and non-primed labels denote ions related by inversion. | ||
Polymeric arrays were obtained also by reacting H4L′ with zinc and cadmium metal ions in hydrothermal conditions. The zinc material, of [(Zn2+)2(H2O)2(DMF)(L′)4−] composition (orthorhombic, space group P212121, a = 11.5219(1), b = 13.3523(1), c = 19.5742(2) Å, and Z = 4), exhibits di-nuclear metal ion connectors and three dimensional connectivity scheme. The cadmium material {[(Cd2+)(H2O)3]2(L′)4−} (monoclinic, space group P21/n, a = 10.1438(2), b = 22.8095(4), c = 14.4702(3) Å, β = 93.191(1)°, and Z = 4) reveals single metal-ion connectors coordinated to the four ends of the ligand species, forming two dimensional arrays (as three of its coordination sites are occupied by water molecules). These polymeric schemes are associated with significant V-type bending of the ligand at the central C-atom and a nearly perpendicular orientation of their phenyl rings (δ′ = 79.5(1)° and τ′ = 108.4(4)° in the Zn-compound, and δ′ = 83.6(2)° and τ′ = 101.7(4)° in the Cd-compound). Crystals of these two compounds were, however, of rather poor quality due to severely disordered crystallization solvent and fragments of the ligands, and they could not be analyzed with adequate precision.
Coordination patterns in metal complexes with H4L″
The ligand H4L″ has an extended spacer unit between the two oxo-isophthalic acids (Scheme 1), and additional degrees of conformational freedom. Its high potential for formulating robust metal–organic frameworks when reacted with metal ions is best illustrated by compound 6 where Cd2+ ions act as mononuclear connectors between the organic moieties. The Cd2+ ion reveals high propensity for coordination environment of tetrahedral geometry. This is readily matched by the conformation of the (L″)4− residue, the geometry of which being characterized by a nearly tetrahedral disposition of the carboxylate functions on the molecular skeleton. The observed polymer has, correspondingly, a diamondoid polymeric structure, in which every carboxylate group of the ligand coordinates in a bidentate-chelating fashion to a different metal ion, and every metal ion coordinates to four carboxylates of different (L″)4− ions (Fig. 6a), and exhibits a Cd(COO)4 coordination motif. The entire structure is composed of twofold interpenetrated polymeric diamondoid nets, with DMF-containing intra-lattice channels (approximately 10 Å by 10 Å), which extend parallel to the a-axis of the crystal (Fig. 6b). Efforts to replace the DMF in this material by another component, or to grow X-ray quality single crystals of this polymeric framework, but with other guest species, have not been successful thus far.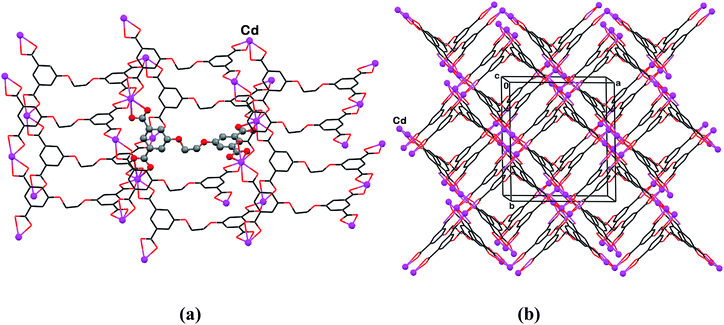 | ||
| Fig. 6 (a) The diamondoid-type coordination scheme in 6, revealing tetrahedral binding geometries around the Cd2+ and (L″)4− components of the polymer. The ligand in the center and all Cd-ions are denoted by small spheres. (b) The doubly interpenetrated architecture of the polymeric assemblies, with channel voids propagating through it parallel to the a-axis of the crystal. The disordered DMF species, which accommodate these channels, are not shown. | ||
Coordination polymers 7, 8 and 9, involving (H2L″)2−/(L″)4− and zinc, nickel or silver ions, respectively, also yielded three-dimensional single-framework assemblies. Polymer 7 is tessellated together by di-nuclear Zn-connectors (Fig. 7). One of the zinc ions has an octahedral environment, while the other ion adopts tetrahedral coordination geometry. The two metal ions are triply bridged in a µ2–η1–η1-mode by three carboxylate groups of different ligand species of bent shape. Then, the octahedral ion is further coordinated to three DMF ligands, while the tetrahedral ion is coordinated in a monodentate fashion to another (L″)4− ligand. Additional non-coordinated molecules of the DMF solvent occupy the interstitial voids.
![(a) Binding scheme in the [(Zn2+)2·(L″)4−] polymeric aggregate 7, via dinuclear metal ion connectors, tetrahedral Zn1 and octahedral Zn2, µ2–η1–η1-bridged by the carboxylate groups of three different ligands. Another carboxylate bound to Zn1 and three DMF ligands bound to Zn2 are also shown. (b) A perspective view of the polymeric structure of 7, showing also the non-coordinated DMF species (space-filling model) accommodated in the interlattice voids.](/image/article/2011/CE/c0ce00268b/c0ce00268b-f7.gif) | ||
| Fig. 7 (a) Binding scheme in the [(Zn2+)2·(L″)4−] polymeric aggregate 7, via dinuclear metal ion connectors, tetrahedral Zn1 and octahedral Zn2, µ2–η1–η1-bridged by the carboxylate groups of three different ligands. Another carboxylate bound to Zn1 and three DMF ligands bound to Zn2 are also shown. (b) A perspective view of the polymeric structure of 7, showing also the non-coordinated DMF species (space-filling model) accommodated in the interlattice voids. | ||
Compound 8 is stabilized by tetranuclear µ-dioxo-type clusters of (Ni4O2)4+ located on centers of inversion (Fig. 8), which resemble in their structure those observed in polymer 4. The (Ni4O2)4+ cluster is associated with eight different ligands and two molecules of water, each of the nickel ions being six-coordinate. On each side of the cluster, adjacent Ni-ions are bridged by either one or two carboxylic functions of different ligands by µ2–η1–η1-coordination mode. The peripheral ions of the cluster are further coordinated to a carboxylic group of yet another ligand in a chelating bidentate mode. The ligand itself is bent, with its phenyl rings oriented nearly perpendicular to one another (Table 2). The (Ni4O2)4+ clusters are arranged in layers. Three of the carboxylic/carboxylate groups of a given ligand associate with the metal connectors in one layer, while the fourth carboxylate group pillars at a perpendicular direction and binds to the metal cluster located in a neighboring layer either above or below. The resulting polymeric architecture is characterized by open channels (approximately 6 Å by 9 Å) aligned parallel to the b-axis of the crystals and centered at ½,y,0, which are accommodated by disordered solvent (a mixture of water and methanol).
![(a) Coordination pattern in the [(Ni2+)2·(O2−)·(H2L″)2−] polymer 8, sustained by dioxo-tetranickel connectors. All ions in the (Ni4O2)4+ clusters (located on inversion centers) are depicted as small spheres. (b) Molecular conformation of the (H2L″)2− species, with tetrahedral disposition of the COOH/COO− functions.](/image/article/2011/CE/c0ce00268b/c0ce00268b-f8.gif) | ||
| Fig. 8 (a) Coordination pattern in the [(Ni2+)2·(O2−)·(H2L″)2−] polymer 8, sustained by dioxo-tetranickel connectors. All ions in the (Ni4O2)4+ clusters (located on inversion centers) are depicted as small spheres. (b) Molecular conformation of the (H2L″)2− species, with tetrahedral disposition of the COOH/COO− functions. | ||
Polymerization of silver ions with the H4L″ ligand (compound 9) reveals a considerable similarity with the corresponding reaction product 5 between silver ions and H4L′. The asymmetric unit of 9 consists of four Ag+ and two (H2L″)2− ions. The ligand species are folded in such a way that their two phenyl rings are close to coplanar (Fig. 9). Linear polynuclear backbones of silver ions, arranged in pairs, inter-connect between the organic species to yield a single-framework polymer. As indicated above, the close separation between the silver ions of 2.83–2.89 Å, comparable to the Ag–Ag separation in metallic silver,20 is stabilized by the bridging anionic organic species. The three independent Ag-pairs are µ2–η1–η1-bridged by the carboxylate groups of two different ligands. Every silver ion is also singly coordinated to a carboxylic group of another ligand, one of the ions being further associated to a water molecule. While the two carboxylate groups of a given ligand bind to the same silver ion chain, its two carboxylic acid functions, which diverge in opposite directions, coordinate to two other Ag-backbones (Fig. 9).
 | ||
| Fig. 9 Crystal packing and the coordination pattern in the polymeric assembly of 9, sustained by polynuclear pillars of silver ions (shown as small spheres). Along the silver pillars, the alternating short and long Ag–Ag bonds (Table 2) are denoted by thick and dotted lines, respectively. | ||
Coordination patterns in ternary polymers of metal ions with combinations of the carboxylic acid (H4L′ or H4L″) and bipyridyl (bpy or dpye) ligands
As many of the d-transition metal ions reveal remarkable coordination affinity to both O- and N-type ligands, formation of polymeric arrays with ternary composition is feasible as well. The tetradentate nature of the flexible H4L′ and H4L″ molecules, and the bi-dentate binding capacity of bpy and dpye entities facilitate their polymerization through the metal connectors in three dimensions. The first two examples refer to H4L′-based ternary framework architectures with bpy, and either zinc (compound 10) or silver (compound 11) ions. Fig. 10 illustrates the coordination scheme in 10, which involves a V-shaped tetracarboxylate moiety (L′)4− and linear bpy components. There are two distinct zinc ions in the structure, Zn1 and Zn2, of different coordination geometries. One ion exhibits a distorted octahedral environment, linking to two bpy units, two carboxylate groups of different (L′)4− entities (in a chelating bi-dentate and mono-dentate modes, respectively), and one water molecule. The second zinc ion is characterized by a distorted square-pyramidal geometry with two different bpy molecules, and two carboxylate ligands coordinated to the metal center by a chelating bidentate and monodentate linkages. The bipyridyls bridge between the two types of the zinc ions. The tetracarboxylate ligands connect to two four different ions, with one isophthalic residue bound to two Zn1 centers and the other isophthalic arm to two Zn2 ions. This 3D framework represents twofold interpenetrated nets. The coordination polymer that formed has an open structure, being perforated by interstitial channels that propagate along the c-axis of the crystal. These channels are accommodated by non-coordinated molecules of water and DMSO. | ||
| Fig. 10 Crystal structure of polymer 10, sustained by mononuclear zinc ion connectors. The metal ions and water molecules coordinated to them are shown as small spheres. The non-coordinated solvent molecules of water and DMSO are omitted. | ||
As shown above in structures 5 and 9, coordination polymerization of H4L′ and H4L″ with silver ions is associated with flattened hemi-circular (U-type) conformations of the two ligands to conform with the linear/planar coordination preference of the Ag+ ions. This is valid also in the ternary compound 11, where the (H2L′)2− ligand species are characterized by a U-type conformation (Table 2). Every ligand coordinates to four different silver ions, each one of the latter being part of a different tetra-nuclear silver ion cluster. One carboxylate group coordinates in a µ2–η1–η1-mode to two inversion-related silver ions (within a given tetra-nuclear cluster), and the other carboxylate group coordinates in a chelating bidentate mode to a third metal ion. One of the carboxylic acid functions coordinates to a fourth ion in a monodentate fashion, while the second COOH group is not metal-coordinated—but it is involved in intermolecular hydrogen bonding. The tetra-nuclear silver ion clusters are composed of two inversion-related pairs of ions on each side which connect by a central bond, forming a Ag1–Ag2–°–Ag2′–Ag1' (where symbol ° represents crystallographic center of inversion) zigzag motif. The silver ions in this structure reveal a twofold positional disorder, yet all the relevant silver–silver distances are relatively short, varying within 2.9–3.1 Å. Within a given cluster, Ag2 and Ag2′ are each coordinated by the carboxylate groups of two different ligands in a chelating bidentate fashion. Then, Ag1 and Ag2, and similarly Ag1′ and Ag2′, are µ2–η1–η1-bridged by other carboxylate groups. Finally, each of the Ag1 and Ag1′ terminal ions coordinates in a monodentate mode to another carboxylic group and a bpy ligand (located on inversion), which in turn bridges between two silver ions of neighboring clusters. The resulting voids-free three-dimensional polymeric architecture tessellated by the tetranuclear silver ion connectors (and by additional intermolecular hydrogen bonding with the COOH groups) is shown in Fig. 11.
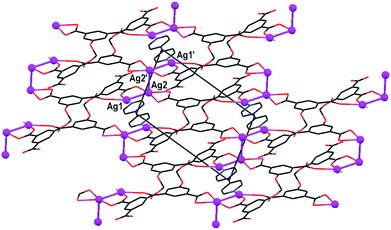 | ||
| Fig. 11 Crystal structure of framework polymer 11, viewed down the b-axis of the crystal. Note the tetra-nuclear silver ion connectors (spheres) that crosslink between the (H2L′)2− and bpy species. Only one set of the atomic positions of the disordered silver ions is shown. | ||
Irrespective of the presence of bpy ligands, the formation of compound 12 (Fig. 12) is driven by the complimentary tetrahedral coordination environment of the Co-ion connectors and the ability of the flexible H4L″ ligand to adopt a conformation characterized by tetrahedral disposition of the carboxylic groups. As in compound 6, the tetrahedral-type conformation of (H2L″)2− is characterized by nearly perpendicular orientation of its two phenyl arms (Table 2). At every site the cobalt ions are coordinated to two (H2L″)2− units in a monodentate mode and two bpy species. The Co ions and the (H2L″)2− ligands are located on axes of twofold rotation, while the bpy moieties reside on centers of inversion. In this structure, each of the two organic moieties bridges between two adjacent cobalt centers. The component Co, (H2L″)2− and bpy species in 12 assemble into two-dimensional polymeric arrays (Fig. 12), which are sixfold interpenetrated to yield a tightly packed structure. These polymers are lined on their upper and lower surfaces with the non-coordinated carboxylic groups, embedding the bipyridyl ligands in the central section of the polymer. The interpenetrated networks interact with each other by COOH⋯−OOC hydrogen bonding, as well as by π-stacking interactions between the aromatic pyridyl and phenyl fragments.
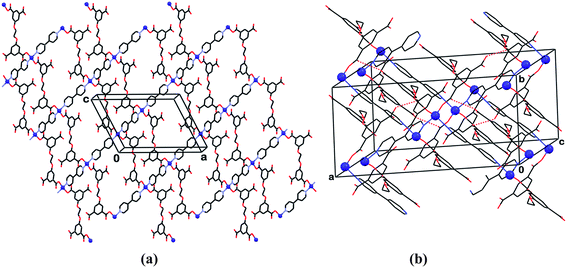 | ||
| Fig. 12 Compound 12. (a) The two dimensional polymeric array with tetrahedral cobalt nodes (small spheres) parallel to the ac plane of the crystal. (b) View of the structure showing interpenetrating nets, the packing of which is stabilized also by hydrogen bonds (dotted lines) and π-stacking of the aromatic fragments between the nets. | ||
A considerably more complex structure 13 was obtained when bpy was replaced by the more flexible dpye component in the reaction mixture with cobalt ions and H4L″ (Fig. 13). The dpye molecule can adopt both a linear as well as a bent conformation, which provides additional degrees of freedom in the polymeric assembly of the three reagents in this case. The 1:1:1 metal:L″:bipyridyl stoichiometry in 12 has changed to a 2:1:2 ratio between these components in 13. The cobalt ions in 13 are characterized by a pseudo-octahedral coordination environment. The fully de-protonated (L″)4− species adopts here a most common “tetrahedral” shape (Table 2). Two of the carboxylate functions coordinate at either end to twofold disordered Co1 ions, while the two other carboxylates link to Co2 ions, either in a chelating bidentate or a monodentate fashion. Paddle-wheel-type coordination characterize the supramolecular assembly around the disordered Co1 sites, where the two Co1 positions coordinate to the carboxylate groups of four different (L″)4− ligands. Neighboring Co(COO)4 paddle-wheel sites, located in sections parallel to the ac plane of the crystal (approximately at y = 0 and ½), are further bridged from both sides by the “linear” dpye bridges. The Co2 sites (gathered also in sections parallel to the ac plane, centered at y = ¼ and ¾) are coordinated to two carboxylates of two neighboring (L″)4− species, two water ligands, and one pyridyl site of the second and bent dpye ligand, the latter bridging by coordination between two different Co2 sites. The resulting polymer (Fig. 13) contains intra-lattice voids which are accommodated by disordered molecules of non-coordinated water.
 | ||
| Fig. 13 (a) Coordination mode in the polymeric framework 13. Only half of the (L″)4− and dpye ligands are shown. Co1a and Co1b ions have 50% occupancy. Note the paddle-wheel arrangement of the carboxylate groups around the Co1a–Co1b pair. (b) Crystal structure of 13. The cobalt ions and the coordinated water molecules are depicted as small spheres; the Co1a and Co1b pairs are shaded. Note the twofold orientational disorder of one of the dpye moieties, only the major orientation of the bent dpye moiety is shown. | ||
The above coordination features of H4L′ and H4L″ are in line with recent findings by other groups. Brammer et al. synthesized a similar type of torsionally flexible tetradentate ligands with diisophthalic acid moieties interconnected by butyl and butenyl spacer units. These were applied in the construction of thermally stable porous MOFs with zinc ions, which upon thermal de-solvation show rapid uptake of CO2 gas at low pressures.16a The two ligands adopted a planar conformation and yielded a square-grid-type two-dimensional coordination networks. The tetraacid with the butenyl spacer unit adopted also a twisted conformation, leading to the formation of a three-dimensional diamondoid network. Batten et al. evaluated the coordination chemistry of the 5,5′-(1,4-phenylenebis(methylene))- and 5,5′-(1,3-phenylene-bis(methylene))-derivatives of bis-(oxy)diisophthalic acid with aryloxy spacers. The reactions of these moieties with cobalt, manganese and zinc ions, in the presence or absence of auxiliary ligand, afforded three-dimensional metal–organic frameworks.21 In the zinc case, the framework structure is perforated by intra-lattice nano-channels (approximately 10 Å × 15 Å). A two-dimensionally intercoordinated product of ternary composition was observed from the reaction of the tetra-acid and bpy ligands with copper ions. It has a sheet-type structure, which in the crystal is further inter-connected by hydrogen bonds into a three-dimensional supramolecular architecture. Finally, the self-assembly of copper ions with 5,5′-(1,3-phenylenebis(methylene))-bis(oxy)diisophthalic acid by Zaworotko et al. yielded crystals of covalently crosslinked nanoball frameworks.16b The latter are characterized by a large internal cavity (1.8 × 1.8 × 1.4 nm3), and exhibit twofold interpenetration into one another.
Better understanding of the coordination networks 2–13 can be achieved by topological analysis, i.e. by reducing multidimensional structures to simple node-and-linker nets where the metallic nodes and the organic linkers represent secondary building units (SBUs).22 Thus, in 2 the zinc ion connected to two ligand moieties represents a 2-connected node, while the L′ ligand connected to four different metal ions acts as a 4-connected node. The polymeric chain structure is a binodal (2,4)-connected net. Framework 3 can be represented as a complex (5,6,7)-connected trinodal net. A related 3D framework, structurally similar to 3, in which [Mn3(COO)6(DMSO)4] units are linked via 1,5-dinitronaphthalene-dicarboxylates has been reported recently.23 In the latter, the central Mn(II) ion coordinated octahedrally to six carboxylate O-atoms, and each of the two outer Mn(II) ions, are coordinated to four carboxylate O-atoms and two DMSO molecules in a distorted octahedral geometry. Similar trinuclear Mn(II) motifs have been observed in other extended frameworks with dicarboxylate ligands.24 Coordination polymers 4 and 8 have similar topologies, representing 4-nodal (3,5,5,7)-connected nets. Due to the different lengths of the organic linkers the two frameworks differ in their potential porosity. Solvent accessible voids were assessed by PLATON19 to be 611 Å3 (44.2% of the crystal volume) in 4 and 750 Å3 (48% of the crystal volume) in 8. Two-dimensional frameworks of Co(II) and Ni(II) ions with the 5-oxyacetateisophthalate ligand, which exhibit a similar tetranuclear [M4(µ3-OH)2] core SBUs have been reported earlier.25
The three Ag-compounds 5, 9 and 11 have different topologies. Framework 5 can be represented as a (6,6,8)-connected trinodal net. Framework 9 is a 6-nodal (5,6,6,7,7,9)-connected nets. Compound 11 exhibits a trinodal (6, 7, 7)-connected net. The relatively short Ag⋯Ag distances in the poly-nuclear silver aggregates (within 2.8–3.2 Å, less than the 3.44 Å sum of the van der Waals radii of two Ag ions) provide supporting evidence for the significance of argentophilicity.26 Similarly short Ag⋯Ag bonds have been observed in a series of other compound with carboxylate and sulfonate ligands.27 Metal carboxylate frameworks having tetranuclear silver(I) cluster as secondary building block are rare.28 The 3D framework obtained by reaction of benzene-1,3,5-tricarboxylic acid with Ag2O under hydrothermal condition has close structural resemblance with that observed in 9.27b
Compound 6 with Cd-connectors of tetrahedral coordination environment represents a uninodal 4-connected net with diamondoid topology. The complimentary tetrahedral conformation of ligand L″ leads to a twofold interpenetrated diamondoid framework having square grid type of voids. The solvent-accessible void volume is 1193.1 Å3 in the unit-cell, comprising 43.6% of the crystal volume. Similar types of polymeric architectures of the Cd(II) ions with 3,3′-azodibenzoate and other carboxylate ligands have been reported.29 Framework 7 is a new trinodal (3, 4, 7)-connected net. The use of {Zn2(CO2)3}-type SBU present in 7 for the construction of MOFs is not abundant due to the distorted nature of the cluster, and only a few reports to this end have been published.30 This includes a 3D framework of this dinuclear cluster with adamantane tetracarboxylic acid as a linker,30f which has a structural resemblance to that in 7. However, while the former structure exhibits appreciable porosity, the bent conformation of L″ in 7 induces a more densely packed lattice. One the other hand, the more symmetric {Zn2(CO2)4}-type paddlewheel clusters have been more widely used for the construction of porous frameworks.31
The two different zinc ions and the L′ ligands in 10 can be regarded as 4-connected nodes, for the topological description. Correspondingly, structure 10 is a binodal (4,4)-connected net. Zinc-based interpenetrated MOFs with a variety of carboxylate linkers are known.32 Framework 12 has a binodal net, while compound 13 represents a trinodal (3,6,6)-connected net. Numerous polymeric compounds based on carboxylate linkers and either mononuclear or dinuclear paddle-wheel-type cobalt nodes have been reported in the literature, some with structural similarity to the polymeric networks 12 and 13.33,34
Concluding remarks
Studies of the coordination chemistry of the newly synthesized flexible H4L′ and H4L″ tetra-acid ligands have demonstrated their high reactivity towards transition metal ions in formulations of extended coordination polymers. In a separate publication (part I of this study)17 we reported on successful utilization of H4L′ and H4L″ in the construction of two-dimensional and three-dimensional hybrid coordination polymers with a variety of the oxophilic lanthanoid f-metal ions. In this part we illustrate the high propensity of these ligands to form framework coordination polymers with d-metals as well (compounds 3–9). This coordination affinity is preserved in the presence of competing bipyridyl ligands too, as demonstrated in compounds 10–13. All the structures of the new materials have been precisely characterized by X-ray diffraction, and our current and previous findings are summarized in Fig. 14.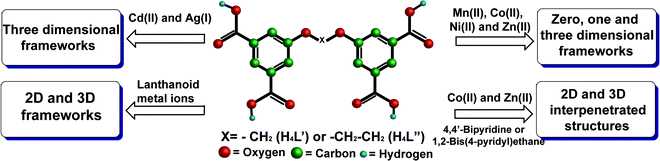 | ||
| Fig. 14 Coordination chemistry of H4L′ and H4L″ towards d- and f-transition metal ions. | ||
Evidently, the flexible molecular design of two new ligands allows for diverse coordination modes to metal ion connectors, by adjusting the shape of the ligand to the spatial coordination requirements of the inorganic centers (whether mono-, di- or poly-nuclear), in the formation of network coordination polymers (Fig. 15).
 | ||
| Fig. 15 Molecular flexibility of the L′ and L″ ligands. (a) Flattened V-conformation of L′ in 1 and 2. (b) V-shaped twisted conformation of L′ in 3, 4 and 10. (c) U-shape conformation of L′ in 5 and 11. (d) Tetrahedral conformation of L″ in 6, 8, 12 and 13. (e) V-shape bent conformation of L″ in 7. (f) U-shape flat conformation of L″ in 9. | ||
The construction of multidimensional structures is facilitated not only by the conformational flexibility of the ligands, but also by the presence of multiple diverging coordination sites, and their ability to balance the charge of the inorganic metal ion partner by facile deprotonation, without the need to incorporate into the polymeric lattice foreign counter ions. Compounds 3–13 exhibit single framework architectures of either three-dimensional polymeric assemblies, or interpenetrating two-dimensional polymers. The H4L″ derivative is particularly attractive in the above context. It is characterized by a preferred nearly gauche conformation about the central CH2–CH2 bond, and its four carboxylic acid functions can be readily oriented in tetrahedral directions (Fig. 8b). Its reaction with complimentary metal centers of tetrahedral coordination geometry may thus lead to porous frameworks, as in compounds 6 and 8.
There is little control over the topology of the different structures that form. Precise conditions have not yet been established to control the nuclearity of the inorganic connectors, predict a priori the metal–H4L ratio in the intercoordinated ensemble, and avoid interfering association of foreign moieties from the reaction mixture (as water, DMF or pyridine) to the metal coordination sites on account of the tetraacid ligand. Still some general observations can be drawn from the current study. Driven by thermodynamic considerations the supramolecular reaction is prone to yield extended polymeric architectures, mostly of 3D connectivity features, associated with optimal gain of binding energy. This is facilitated by complimentary coordination environments of the component organic and inorganic species. Correspondingly, in reactions with metal ion or metal ion cluster connectors (Co, Cd, Mn3, Ni4O2, Co4O2, and Zn2) of tetrahedral or octahedral geometry (see above) the H4L′ and H4L″ ligands adopt a strongly bent conformations with pseudo-tetrahedral disposition of their carboxylic functions to this end. In structures 3, 4, 6, 7, 8, 12 and 13 the δ-dihedral angles between the two aryl substituents range from 64 to 89° (Table 2 and Fig. 15). On the other hand, in reactions with silver ions (compounds 5, 9 and 11) the two ligands adopt a flattened U-shape geometry to match best the coordination preferences of the silver cations, which in the presence of bridging polydentate anionic moieties tend to arrange in linear polynuclear chains. Structures 1 and 2 with mononuclear zinc ion nodes, having some of their tetrahedral coordination sites blocked by water and pyridine ligands, provide exceptions to his general trend. They represent 0D and 1D coordination compounds in which the title ligands are characterized by flattened geometries.
Acknowledgements
This research was supported by The Israel Science Foundation (Grant No. 502/08).References
- (a) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS; (b) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS; (c) L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS; (d) A. K. Cheetham, G. Férey and T. Loiseau, Angew. Chem., 1999, 111, 3466–3492 ( Angew. Chem., Int. Ed. , 1999 , 38 , 3268–3292 ) CrossRef; (e) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS; (f) S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS; (g) R. J. Kuppler, D. J. Timmons, Q.-R. Fang, J.-R. Li, T. A. Makal, M. D. Young, D. Yuan, D. Zhao, W. Zhuang and H.-C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS.
- (a) P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638–2684 CrossRef; (b) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460–1494 CrossRef.
- (a) O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511–522 CrossRef CAS; (b) W. B. Lin, Z. Y. Wang and L. Ma, J. Am. Chem. Soc., 1999, 121, 11249–11250 CrossRef CAS.
- (a) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS; (b) B. Kesanli, Y. Cui, M. R. Smith, E. W. Bittner, B. C. Bockrath and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 72–75 CrossRef CAS; (c) M. Dincă, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904–8913 CrossRef CAS; (d) S. Ma, D. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H. Zhou, J. Am. Chem. Soc., 2007, 129, 1858–1859 CrossRef CAS; (e) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS; (f) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS.
- (a) C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075–1078 CrossRef CAS; (b) D. N. Dybtsev, A. L. Nuzhdin, H. Chun, K. P. Bryliakov, E. P. Talsi, V. P. Fedin and K. Kim, Angew. Chem., Int. Ed., 2006, 45, 916–920 CrossRef CAS; (c) W. Lin, J. Solid State Chem., 2005, 178, 2486–2490 CrossRef CAS; (d) C. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS; (e) A. Hu, H. L. Ngo and W. Lin, J. Am. Chem. Soc., 2003, 125, 11490–11491 CrossRef CAS.
- (a) A. J. Fletcher, E. J. Cussen, D. Bradshaw, M. J. Rosseinsky and K. M. Thomas, J. Am. Chem. Soc., 2004, 126, 9750–9759 CrossRef CAS; (b) L. Alaerts, C. E. A. Kirschhock, M. Maes, M. A. van der Veen, V. Finsy, A. Depla, J. A. Martens, G. V. Baron, P. A. Jacobs, J. E. M. Denayer and D. E. De Vos, Angew. Chem., Int. Ed., 2007, 46, 4293–4297 CrossRef CAS.
- (a) F. A. Cotton, C. Lin and C. A. Murillo, Acc. Chem. Res., 2001, 34, 759–771 CrossRef CAS; (b) R. Murugavel, M. G. Walawalkar, M. Dan, H. W. Roesky and C. N. R. Rao, Acc. Chem. Res., 2004, 37, 763–774 CrossRef CAS.
- (a) L. Brammer, M. D. Burgard, M. D. Eddleston, C. S. Rodger, N. P. Rath and H. Adams, CrystEngComm, 2002, 4, 239–248 RSC; (b) L. Brammer, M. D. Burgard, C. S. Rodger, J. K. Swearingen and N. P. Rath, Chem. Commun., 2001, 2468–2469 RSC; (c) C. M. Rivas and L. Brammer, Coord. Chem. Rev., 1999, 183, 43–80 CrossRef CAS.
- (a) K. K. Tanabe, Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2008, 130, 8508–8517 CrossRef CAS; (b) A. Schaate, S. Klingelhoefer, P. Behrens and M. Wiebcke, Cryst. Growth Des., 2008, 8, 3200–3205 CrossRef CAS; (c) S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2008, 130, 806–807 CrossRef; (d) S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS; (e) Y. B. Go, X. Wang and A. J. Jacobson, Inorg. Chem., 2007, 46, 6594–6600 CrossRef CAS; (f) P.-K. Chen, Y.-X. Che, L. Xue and J.-M. Zheng, Cryst. Growth Des., 2006, 6, 2517–2522 CrossRef CAS; (g) B. Chen, Y. Yang, F. Zapata, G. Qian, Y. Luo, J. Zhang and E. B. Lobkovsky, Inorg. Chem., 2006, 45, 8882–8886 CrossRef CAS; (h) R. Koner and I. Goldberg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m62–65 CrossRef; (i) R. Koner and I. Goldberg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m185–m189 CrossRef.
- (a) J. Luo, H. Xu, Y. Liu, Y. Zhao, L. L. Daemen, C. Brown, T. V. Timofeeva, S. Ma and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 9626–9627 CrossRef CAS; (b) Z. Lin, A. M. Z. Slawin and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 4880–4881 CrossRef CAS; (c) J.-H. Liao, P.-C. Wu and W.-C. Huang, Cryst. Growth Des., 2006, 6, 1062–1063 CrossRef CAS; (d) K. W. Chapman, G. J. Halder and P. J. Chupas, J. Am. Chem. Soc., 2008, 130, 10524–10526 CrossRef CAS; (e) L.-P. Zhang, J. Yang, J.-F. Ma, Z.-F. Jia, Y.-P. Xie and G.-H. Wei, CrystEngComm, 2008, 10, 1410–1420 RSC; (f) Z. Lin, F. Jiang, L. Chen, D. Yuan and M. Hong, Inorg. Chem., 2005, 44, 73–76 CrossRef CAS.
- (a) C. Ruiz-Perez, P. Lorenzo-Luis, M. Hernandez-Molina, M. M. Laz, F. S. Delgado, P. Gili and M. Julve, Eur. J. Inorg. Chem., 2004, 3873–3879 CrossRef CAS; (b) L.-J. Zhang, J.-Q. Xu, Z. Shi, W. Xu and T.-G. Wang, Dalton Trans., 2003, 1148–1152 RSC; (c) Y. Li, H. Zhang, E. Wang, N. Hao, C. Hu, Y. Yan and D. Hall, New J. Chem., 2002, 26, 1619–1623 RSC; (d) H.-P. Jia, W. Li, Z.-F. Ju and J. Zhang, Dalton Trans., 2007, 3699–3704 RSC; (e) D. Cheng, M. A. Khan and R. P. Houser, J. Chem. Soc., Dalton Trans., 2002, 4555–4560 RSC; (f) S. K. Ghosh and P. K. Bharadwaj, Inorg. Chem., 2004, 43, 5180–5182 CrossRef CAS; (g) O. Fabelo, J. Pasan, F. Lloret, M. Julve and C. Ruiz-Perez, Inorg. Chem., 2008, 47, 3568–3576 CrossRef CAS; (h) S. Ma, J. M. Simmons, D. Sun, D. Yuan and H.-C. Zhou, Inorg. Chem., 2009, 48, 5263–5268 CrossRef CAS; (i) R. Koner and I. Goldberg, CrystEngComm, 2009, 11, 367–374 RSC.
- (a) D. Sun, S. Ma, Y. Ke, D. J. Collins and H. Zhou, J. Am. Chem. Soc., 2006, 128, 3896–3897 CrossRef CAS; (b) L. Hou, J.-P. Zhang and X.-M. Chen, Cryst. Growth Des., 2009, 9, 2415–2419 CrossRef CAS; (c) S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, Inorg. Chem., 2008, 47, 7751–7756 CrossRef CAS; (d) B. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS; (e) L. Hou, J.-P. Zhang, X.-M. Chen and S. Weng Ng, Chem. Commun., 2008, 4019–4021 RSC; (f) K. Koh, A. G. Wong-Foy and A. J. Matzger, Angew. Chem., Int. Ed., 2008, 47, 677–680 CrossRef CAS; (g) H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. B. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS; (h) Z. Wang, K. K. Tanabe and S. M. Cohen, Inorg. Chem., 2009, 48, 296–306 CrossRef CAS; (i) D. Sun, Y. Ke, T. M. Mattox, S. Parkin and H. C. Zhou, Inorg. Chem., 2006, 45, 7566–7568 CrossRef CAS.
- (a) Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. C. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS; (b) X. Lin, J. Jia, X. Zhao, K. M. Thomas, A. J. Blake, G. S. Walker, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef; (c) X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159–2171 CrossRef CAS; (d) G.-P. Yang, Y.-Y. Wang, L.-F. Ma, J.-Q. Liu, Y.-P. Wu, W.-P. Wu and Q.-Z. Shi, Eur. J. Inorg. Chem., 2007, 3892–3898 CrossRef CAS; (e) X.-R. Hao, Z.-M. Su, Y.-H. Zhao, K.-Z. Shaoa and Y. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m2477–m2479 CrossRef; (f) C. Qin, X.-L. Wang and E.-B. Wang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m3073–m3074 CrossRef; (g) G.-P. Yang, Y.-Y. Wang, H. Wang, C.-J. Wang, G.-L. Wen, Q.-Z. Shi and S.-M. Peng, J. Mol. Struct., 2008, 888, 366–374 CrossRef CAS; (h) J.-J. Wang, L. Gou, H.-M. Hu, Z.-X. Han, D.-S. Li, G.-L. Xue, M.-L. Yang and Q.-Z. Shi, Cryst. Growth Des., 2007, 7, 1514–1521 CrossRef CAS; (i) T. Jia, S. Zhu, M. Shao, Y. Zhao and M. Li, Inorg. Chem. Commun., 2008, 11, 1221–1223 CrossRef CAS; (j) K. L. Mulfort, O. K. Farha, C. L. Stern, A. A. Sarjeant and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 3866–3868 CrossRef CAS; (k) O. K. Farha, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2008, 47, 10223–10225 CrossRef CAS; (l) L. Ma and W. Lin, J. Am. Chem. Soc., 2008, 130, 13834–13835 CrossRef CAS; (m) L. Ma and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 3637–3640 CrossRef CAS; (n) L. Ma, D. J. Mihalcik and W. Lin, J. Am. Chem. Soc., 2009, 131, 4610–4612 CrossRef CAS; (o) Y. Cui, O. R. Evans, H. L. Ngo, P. S. White and W. Lin, Angew. Chem., Int. Ed., 2002, 41, 1159–1162 CrossRef CAS; (p) B. Kesanli, Y. Cui, M. R. Smith, E. W. Bittner, B. C. Bockrath and W. Lin, Angew. Chem., Int. Ed., 2005, 44, 72–75 CrossRef CAS.
- (a) B. Chen, N. W. Ockwig, F. R. Fronczek, D. S. Contreras and O. M. Yaghi, Inorg. Chem., 2005, 44, 181–183 CrossRef CAS; (b) B. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745–4749 CrossRef CAS; (c) S. Yang, X. Lin, A. Dailly, A. J. Blake, P. Hubberstey, N. R. Champness and M. Schröder, Chem.–Eur. J., 2009, 15, 4829–4835 CrossRef CAS; (d) S. Ma, D. Sun, J. M. Simmons, C. D. Collier, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2008, 130, 1012–1016 CrossRef CAS; (e) H. Furukawa, J. Kim, N. W. Ockwig, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2008, 130, 11650–11661 CrossRef CAS; (f) N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS; (g) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef.
- X. Lin, A. J. Blake, C. Wilson, X. Z. Sun, N. R. Champness, M. W. George, P. Hubberstey, R. Mokaya and M. Schröder, J. Am. Chem. Soc., 2006, 128, 10745–10753 CrossRef CAS.
- (a) S. M. Hawxwell, G. M. Espallargas, D. Bradshaw, M. J. Rosseinsky, T. J. Prior, A. J. Florence, J. van de Streeke and L. Brammer, Chem. Commun., 2007, 1532–1534 RSC; (b) J. J. Perry, V. Ch. Kravtsov, G. J. McManus and M. J. Zaworotko, J. Am. Chem. Soc., 2007, 129, 10076–10077 CrossRef CAS.
- A. Karmakar and I. Goldberg, CrystEngComm, 2010 10.1039/c0ce00474j.
- (a) A. Altomare, M. C. Burla, M. Camalli, M. Cascarano, C. Giacovazzo, A. Guagliardi and G. Polidori, J. Appl. Crystallogr., 1994, 27, 435–436 CrossRef; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef.
- (a) O. Kristiansson, Inorg. Chem., 2001, 40, 5058 CrossRef CAS; (b) M.-L. Tong, X.-M. Chen and B.-H. Ye, Inorg. Chem., 1998, 37, 5278 CrossRef CAS; (c) K. Singh, J. R. Long and P. Stavropoulos, J. Am. Chem. Soc., 1997, 119, 2942 CrossRef CAS; (d) H. M. Titi and I. Goldberg, CrystEngComm, 2010 10.1039/c0ce00134a.
- Z. Pan, H. Zheng, T. Wang, Y. Song, Y. Li, Z. Guo and S. R. Batten, Inorg. Chem., 2008, 47, 9528–9536 CrossRef CAS.
- (a) M. O'Keeffe and O. M. Yaghi, Reticular Chemistry Structure Resource, Arizona State University, Tempe, AZ, 2005, http://rcsr.anu.edu.au/ Search PubMed; (b) V. A. Blatov, IUCr, Comput. Comm. Newslett., 2006, 7, 4, http://www.topos.ssu.samara.%20ru/ Search PubMed.
- H. Tian, Q.-X. Jia, J.-Y. Zhang and E.-Q. Gao, Inorg. Chim. Acta, 2010, 363, 2481–2487 CrossRef CAS.
- (a) B. Liu, R.-Q. Zou, R.-Q. Zhong, S. Han, H. Shioyama, T. Yamada, G. Maruta, S. Takeda and Q. Xu, Microporous Mesoporous Mater., 2008, 111, 470–477 CrossRef CAS; (b) T. Ladrak, S. Smulders, O. Roubeau, S. J. Teat, P. Gamez and J. Reedijk, Eur. J. Inorg. Chem., 2010, 3804–3812 CrossRef CAS; (c) R.-Q. Zhong, R.-Q. Zou, M. Du, T. Yamada, G. Maruta, S. Takeda, J. Lid and Q. Xu, CrystEngComm, 2010, 12, 677–681 RSC; (d) Z.-L. Chen, Y.-Z. Zhang, F.-P. Liang and Q. Wu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m2409–m2411 CrossRef.
- G.-H. Wang, Y.-Q. Lei, N. Wang, R.-L. He, H.-Q. Jia, N.-H. Hu and J.-W. Xu, Cryst. Growth Des., 2010, 10, 534–540 CrossRef CAS.
- (a) P. Pyykkö, Chem. Rev., 1997, 97, 597–636 CrossRef; (b) D. Rais, J. Yau, D. M. P. Mingos, R. Vilar, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 3464–3467 CrossRef CAS.
- (a) J. Wang, S. Hu and M. L. Tong, Eur. J. Inorg. Chem., 2006, 2069–2077 CrossRef CAS; (b) B. Ding, L. Yi, Y. Liu, P. Cheng, Y. B. Dong and J. P. Ma, Inorg. Chem. Commun., 2005, 8, 38–40 CrossRef CAS; (c) G. K. H. Shimizu, G. D. Enright, C. I. Ratcliffe, K. F. Preston, J. L. Reid and J. A. Ripmeester, Chem. Commun., 1999, 1485–1486 RSC.
- (a) Z.-F. Chen, L.-C. Yu, D.-C. Zhong, H. Liang, X.-H. Zhu and Z.-Y. Zhou, Inorg. Chem. Commun., 2006, 9, 839–843 CrossRef CAS; (b) A. Michaelides, V. Kiritsis, S. Skoulika and A. Aubry, Angew. Chem., Int. Ed. Engl., 1993, 32, 1495–1497 CrossRef.
- (a) Z.-F. Chen, R.-G. Xiong, B. F. Abrahams, X.-Z. You and C.-M. Che, J. Chem. Soc., Dalton Trans., 2001, 2453–2455 RSC; (b) Q.-X. Yao, Z.-F. Ju, X.-H. Jin and J. Zhang, Inorg. Chem., 2009, 48, 1266–1268 CrossRef CAS; (c) Q. Fang, G. Zhu, M. Xue, Z. Wang, J. Sun and S. Qiu, Cryst. Growth Des., 2008, 8, 319–329 CrossRef CAS; (d) K. O. Ashiry, Y.-H. Zhao, K.-Z. Shao, Z.-M. Su, Y.-M. Fu and X.-R. Hao, Inorg. Chem. Commun., 2008, 11, 1181–1183 CrossRef CAS.
- (a) M. E. Braun, C. D. Steffek, J. Kim, P. G. Rasmussen and O. M. Yaghi, Chem. Commun., 2001, 2532–2533 RSC; (b) O. M. Yaghi, C. E. Davis, G. Li and H. Li, J. Am. Chem. Soc., 1997, 119, 2861–2868 CrossRef CAS; (c) W. Clegg, D. R. Harbron, C. D. Homan, P. A. Hunt, I. R. Little and B. P. Straughan, Inorg. Chim. Acta, 1991, 186, 51–60 CrossRef CAS; (d) W. Clegg, I. R. Little and B. P. Straughan, J. Chem. Soc., Chem. Commun., 1985, 73–74 RSC; (e) W. Clegg, I. R. Little and B. P. Straughan, J. Chem. Soc., Dalton Trans., 1986, 1283–1288 RSC; (f) J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239–8247 CrossRef CAS.
- (a) T. Gadzikwa, B.-S. Zeng, J. T. Hupp and S. B. T. Nguyen, Chem. Commun., 2008, 3672–3674 RSC; (b) S. A. Bourne, J. Lu, A. Mondal, B. Moulton and M. J. Zaworotko, Angew. Chem., Int. Ed., 2001, 40, 2111–2113 CrossRef CAS; (c) M. Lv and S. Weng Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m3136 CrossRef; (d) S.-Y. Yang, L.-S. Long, R.-B. Huang, L.-S. Zheng and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m1671–m1673 CrossRef; (e) Y.-G. Lee, H. R. Moon, Y. E. Cheon and M. P. Suh, Angew. Chem., Int. Ed., 2008, 47, 7741–7745 CrossRef CAS.
- (a) X. Liu, M. Park, S. Hong, M. Oh, J. W. Yoon, J.-S. Chang and M. Soo Lah, Inorg. Chem., 2009, 48, 11507–11509 CrossRef CAS; (b) L.-F. Ma, L.-Y. Wang, J.-L. Hu, Y.-Y. Wang and G.-P. Yang, Cryst. Growth Des., 2009, 9, 5334–5342 CrossRef CAS; (c) S. Bureekaew, H. Sato, R. Matsuda, Y. Kubota, R. Hirose, J. Kim, K. Kato, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2010, 49, 1–5; (d) Z.-B. Han and G.-X. Zhang, CrystEngComm, 2010, 12, 348–351 RSC.
- (a) S. Wang, Cryst. Res. Technol., 2008, 43, 894–898 CrossRef CAS; (b) M. A. Braverman, R. M. Supkowski and R. L. LaDuca, Inorg. Chem. Commun., 2008, 11, 568–571 CrossRef CAS; (c) K. M. Blake, L. L. Johnston, M. A. Braverman, J. H. Nettleman, L. K. Sposato and R. L. LaDuca, Inorg. Chim. Acta, 2010, 363, 2233–2242 CrossRef CAS; (d) L.-F. Ma, L.-Y. Wang, Y.-Y. Wang, S. R. Batten and J.-G. Wang, Inorg. Chem., 2009, 48, 915–924 CrossRef CAS; (e) M. H. Mir, S. Kitagawa and J. J. Vittal, Inorg. Chem., 2008, 47, 7728–7733 CrossRef CAS.
- (a) H. Chun, H. Jung and J. Seo, Inorg. Chem., 2009, 48, 2043–2047 CrossRef CAS; (b) S. W. Lee, H. J. Kim, Y. K. Lee, K. Park, J.-H. Son and Y.-U. Kwon, Inorg. Chim. Acta, 2003, 353, 151–158 CrossRef CAS; (c) E.-Y. Choi, K. Park, C.-M. Yang, H. Kim, J.-H. Son, S. W. Lee, Y. H. Lee, D. Min and Y.-U. Kwon, Chem.–Eur. J., 2004, 10, 5535–5540 CrossRef CAS.
Footnote |
| † CCDC reference numbers 779782–779794. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ce00268b |
| This journal is © The Royal Society of Chemistry 2011 |