Identification, classification and relative stability of tautomers in the cambridge structural database†
Aurora J.
Cruz-Cabeza
*ab and
Colin R.
Groom
a
aThe Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, UK CB2 1EZ. E-mail: cruz@ccdc.cam.ac.uk.; Tel: +44 1223 336408
bThe Pfizer Institute for Pharmaceutical Materials Science, Department of Materials Science and Metallurgy, University of Cambridge, Pembroke Street, Cambridge, UK CB2 3QZ
First published on 27th September 2010
Abstract
Families of tautomers in the Cambridge Structural Database (CSD) have been identified, analysed and classified. We identified 108 molecules that crystallise in two different tautomeric forms. Most commonly, pairs of tautomers crystallise together in the same crystal structure. Tautomeric polymorphs—pairs of tautomers crystallising in different crystal structures with no other or identical components—are very rare. The calculation of the relative stabilities of the different pairs of tautomers mirrors the relative frequencies with which tautomeric forms are observed in the CSD. Our improved understanding of the factors influencing tautomeric preferences in crystal structures may allow the prediction and design of crystal structures containing tautomeric forms that are, as yet, unobserved in the solid state.
Introduction
Although prototropic tautomerism is a widely studied phenomenon in organic heterocyclic chemistry1 and has been the focus of renewed interest by computational chemists2–6 it appears to have largely been forgotten within a solid state context.7 Even though there are early reports on the isolation and observation of different tautomers in crystal structures, modern literature on the subject has been scarce.8 There have been, however, interesting studies aimed at understanding the often complex tautomeric behaviour of azoles in the solid state using NMR, X-ray crystallography and computational methods.8–12 The paucity of research in this area is probably attributable to the difficulties associated with the determination of hydrogen atom positions with X-ray diffraction techniques and the lack of routine use of complementary characterisation techniques such as solid-state NMR.In the context of pharmaceutical materials, the identification and characterisation of tautomers of drug molecules in the solid state could have important intellectual property and commercial implications. This has been illustrated by the late identification of new tautomers of well-known pharmaceutical molecules such as barbituric acid,13,14 omeprazole,15 ranitidine,16 sulfasalazine17 and irbesartan.18 Indeed the potential tautomeric complexity of drug molecules is well illustrated by the case of the anticoagulant drug warfarin, which can exist in approximately 40 distinct tautomeric forms.19 Although usually formulated as crystalline materials, ultimately drugs are dissolved and their tautomeric state might also change in solution.
In solution, tautomers are of course in equilibrium, with the nature of the solvent, the pH and temperature able to influence their relative population. In the solid state, the observed tautomeric form may differ from that which predominates in solution;20–22 temperature, proton transfer reactions23 and mechanical processes can all influence the tautomeric forms observed in crystal structures. In an interesting example of the latter, grinding commercially available barbituric acid yielded a new phase that contained the less stable trihydroxyl tautomer.13 Another study also reported that upon preparation of KBr-discs of barbituric acid for IR-spectra recording, variable amounts of a new tautomer were observed depending on the grinding treatment of the mixture.14
Tautomers in the solid state have been referred to as desmotrops. The term desmotropy was defined as “tautomerism in which both tautomeric forms have been isolated”.24 This term should not, therefore, be used in instances where two different tautomers are observed within the same crystal structure. The term desmotropy is not, however, widely known or used within the crystallographic community so it will not be adopted in this article, but there is a clear definition and nomenclature problem at the confluence of tautomerism and polymorphism.25 We refer to crystal structures of different tautomers as “tautomeric polymorphs”, “multicomponent crystals of tautomers” or “crystal structures containing both tautomeric forms” as appropriate; all are discussed further.
The aim of this study has been to identify, analyse and classify families of tautomers in crystal structures. We have, additionally, calculated the relative molecular energies of the different families of tautomers using density functional theory (DFT) calculations with a polarisable continuum model (PCM) to account for the crystal structure environment. We hope that this analysis might be used for future predictions of the phenomenom of tautomerism and tautomeric polymorphism26 in the solid state.
Methods
CSD dataset
Molecular data from the entire Cambridge Structural Database CSD27 was stored in an individual sdf file, created with the structures from the CSD version 5.31 (November 2009). Those CSD entries describing multicomponent crystals were separated into individual components, with each component stored as an individual sdf file entry. All molecular entries were required to have 3D coordinates for all non-hydrogen atoms, no disorder and more than 3 atoms. This resulted in a dataset containing 551![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 432 distinct molecular entities, from a total of 410779 CSD crystal structure entries which met the above criteria. The molecular entities were processed to give 357892 unique molecular structures, based on canonical SMILES strings calculated using the software Pipeline Pilot (Accelrys Inc.).28 Canonical SMILES strings are unique “simplified molecular input line entry specifications” for unambiguously describing the structure of chemical molecules using short ASCII strings.29 Processing and comparing molecular structures with SMILES strings is extremely fast and efficient.
432 distinct molecular entities, from a total of 410779 CSD crystal structure entries which met the above criteria. The molecular entities were processed to give 357892 unique molecular structures, based on canonical SMILES strings calculated using the software Pipeline Pilot (Accelrys Inc.).28 Canonical SMILES strings are unique “simplified molecular input line entry specifications” for unambiguously describing the structure of chemical molecules using short ASCII strings.29 Processing and comparing molecular structures with SMILES strings is extremely fast and efficient.
Tautomer generation
Tautomers for each of the unique molecules in the CSD were generated using the “Enumerate Tautomers” component in Pipeline Pilot. We have focussed on prototropic tautomerism. Only heteroatoms were used as hydrogen atom donors or acceptors during the generation of tautomers (keto–enol type of tautomers were not generated). A maximum of 15 tautomers per molecule was generated.Identification of tautomers in the CSD
The generated tautomers were compared to the extracted CSD entries using canonical SMILES strings. Those structures for which more than one tautomer was observed, were retained. An additional search of the CSD was performed using a text query of “tautomer” to allow the identification of tautomeric structures showing disorder using the software Conquest.30DFT Calculations
Initial molecular geometries were taken from the crystal structures. For those molecules in which hydrogen positions were not determined, the “auto-edit functionality” implemented in the program Mercury31 was used for the generation of hydrogen atom positions. Molecules were geometry optimised in gas phase using DFT, with the B3LYP functional and the 6-311++G** basis set with the program GAUSSIAN03.32 This minimisation process has no effect on the tautomeric form found in the crystal, as each tautomer is a different local energy minimum in the gas phase for the cases studied here. Single point energy calculations using a Polarisable Continuum Model (PCM model), and the B3LYP/6-311++G** level of theory, were then performed on the gas-phase optimised molecular geometries with a dielectric constant value of ε = 3 for neutral molecules (average dielectric constant in neutral organic crystals) or a dielectric value of ε = 11 for charged molecules (average dielectric constant in crystals of molecular salts).33Results and discussion
Generation and identification of tautomeric families in the CSD
Around 10% of the molecules in the CSD (35232 molecules of a total of 357892 unique molecules considered) have the potential for tautomerisation. In 50% of the cases only two different tautomers are possible, in 20% three tautomers, in 20% four tautomers and in the remaining 10% over four tautomers are possible (Fig. 1).
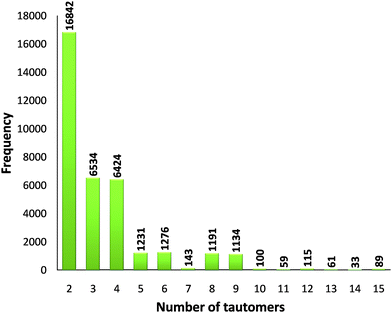 | ||
| Fig. 1 Frequency of the number of tautomers generated for a total of 35232 unique tautomerisable molecules in the CSD. | ||
Only 99 families of tautomers (a total of 198 different tautomers) were observed in the CSD, in a total of 324 crystal structures. In addition, 37 crystal structures containing molecules with considerable disorder contained multiple tautomers, belonging to 33 tautomeric families. In all cases, only two different tautomers were observed per family. It appears from our analysis that three or more tautomeric forms of any molecule, in the solid state, has not yet been reported.
Identification of structures with wrongly determined tautomers
The accuracy of the location of hydrogen atoms in crystal structures determined by X-ray diffraction can vary depending on the quality of data, the temperature of the study or the crystallographer! In most cases, the position of hydrogen atoms is derived from the geometry of the molecular heavy atoms. An analysis of such geometry usually leads to the correct determination of the hydrogen atom positions. However, when there is hydrogen atom disorder, or the quality of the data is poor, this can be a difficult process. In order to identify tautomers where hydrogen atoms may be wrongly placed, we applied a series of tests.For different tautomers of the same compound to be observed in the solid state, one might assume that their relative energy difference must be plausible. Tautomeric pairs with very large energy differences might therefore represent errors. After calculating the relative energies of all tautomers with a molecular weight of less than 300, we found that some pairs showed rather large tautomeric energy differences (> 50 kJ/mol) and unrealistic short contacts in the crystal structure. It seems clear that the least stable tautomer of these pairs represent incorrect determination of hydrogen atom positions, since such a large energy penalty is unlikely to be compensated for in order to generate a crystal. These large energy differences highlighted the presence of 12 incorrect tautomers in the CSD.‡
The COMPACK algorithm compares atomic positions in a molecule and its 15 closest neighbours in the crystal with other crystal structures.34 By ignoring hydrogen atom positions and bond types, we were able to use this algorithm to identify identical crystal structures containing different tautomers. Where we noted such cases, the molecular geometries of the pairs of molecules were studied using the CCDC software MOGUL.35 MOGUL can compare the bond lengths, bond angles, torsion angles and ring geometry of any molecule with all similar structural units in the CSD. We reasoned that incorrectly assigned tautomers may show molecular geometry that was unusual with respect to the distributions calculated by MOGUL. Fig. 2 illustrates one such example, where unusual bond lengths are seen for one tautomeric form of 3-chloro-1,2,4-triazole.This example happens to have been studied in depth, resulting in the re-determination of the crystal structure (CSD refcode: CLTRZL01).36
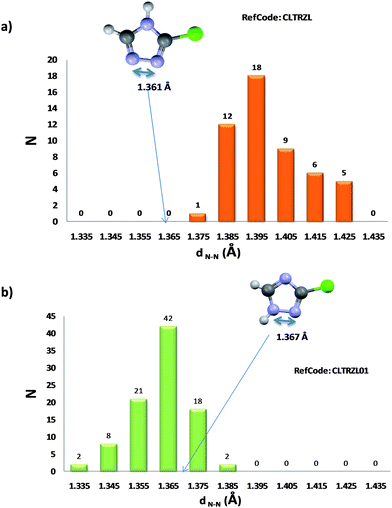 | ||
| Fig. 2 Example of the use of MOGUL geometry check to determine if a tautomeric form has been wrongly assigned. In (a), the N–N bond length is clearly outside the normal MOGUL distribution whereas for the correct tautomer assignment in (b), the N–N bond distance is within the normal ranges. | ||
Cases in which both tautomers show unusual values for bond lengths or angles were subject to further analysis, including assessment of the appropriate literature. After removing the incorrect tautomeric assignments identified by the tests described above, the remaining 108 tautomeric pairs, present in 277 distinct crystal structures were further analysed.
The nature and relative stability of tautomers
The energy difference between tautomeric pairs (ΔEt) of all molecules with molecular weights below 300 is plotted in Fig. 3. As the time to perform these calculations increases with molecular weight, the largest molecules were not analysed; however, we still optimised over 70% of the tautomers identified. As the larger molecules contain the same tautomerisable groups as this subset our conclusions should be valid for molecules of all sizes.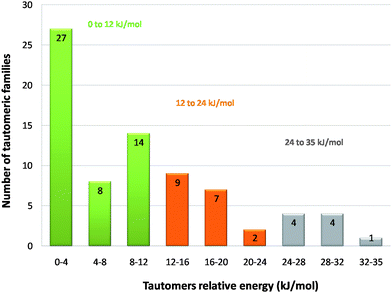 | ||
| Fig. 3 Histogram of the tautomers relative energies in kJ/mol. | ||
Pairs of tautomers were grouped in approximately equal energy ranges representing low, medium and high energy differences between tautomers: (i) 0 to 12 kJ/mol, (ii) 12 to 24 kJ/mol and (iii) 24 to 35 kJ/mol. 65% of the observed tautomeric pairs show a relative energy difference of less than 12 kJ/mol. In this energy range, small improvements in the intermolecular interactions may be sufficient to compensate the small energetic penalty of a molecule existing in a less favourable tautomeric form. 35% of all tautomeric pairs show relative energies of less than 4 kJ/mol.
We observe a great variety of tautomer types: from annular tautomers to functional tautomers and those enabled by 6-member ring resonance intramolecular hydrogen bonds.22
The molecular nature of the tautomers tends to correlate well with their relative stabilities. For example, most tautomers constituted by 6-member ring resonance intramolecular hydrogen bonds, show small relative energy differences and proton disorder (Fig. 4). Of course, although the resonance intramolecular hydrogen bonded pairs could be in dynamic equilibrium, the two states were optimised independently since they are both stationary points and local minima of the molecular energy surface, only the energy difference between the two ordered molecular states was calculated.
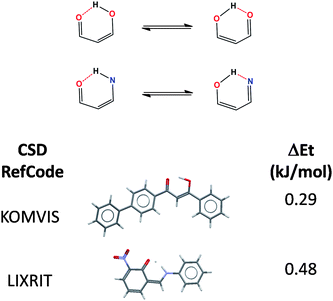 | ||
| Fig. 4 Examples of 6-ring intramolecular hydrogen bonded tautomers. | ||
The most abundant type of tautomers corresponds to derivatives of azoles (e.g. pyrazoles, indazoles, imidazoles, triazoles…). The majority of these tautomeric pairs (∼ 70%) show energy differences of <12 kJ/mol. However, we do observe higher energy differences for azolic tautomers in cases where one of the tautomers has a greater electronic resonance than the other, when the molecule is protonated or deprotonated (in salts) or when one of the tautomers shows stronger intramolecular interactions than the other.
Most of the molecules in the 20–35 kJ/mol energy range are derivatives of 4-pyrimidones (Fig. 5). As pair–pair hydrogen-bonding can be so complementary in the 2-amino derivatives both tautomers are often observed in the same crystal structure. Large energy differences might also be observed when a stable tautomer shows an intramolecular hydrogen bond but the higher energy tautomer does not. Fig. 6 illustrates this effect, the higher energy tautomer does not show an intramolecular hydrogen bond but this effect is compensated by forming extensive intermolecular hydrogen bonding in the crystal structure, as seen previously.1,37
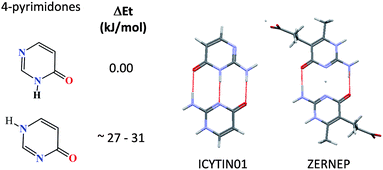 | ||
| Fig. 5 Energy differences in tautomers of 4-pyrimidones and hydrogen bonding complementarity in derivatives of 2-amino-4-pyrimidones. | ||
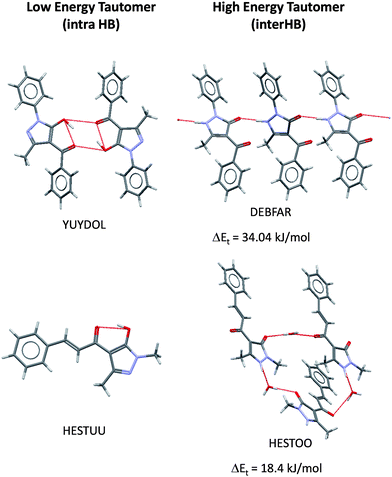 | ||
| Fig. 6 Intra- versus inter-molecular hydrogen bonding (HB) in two tautomeric pairs observed in the CSD. | ||
The nature of the crystal structures containing tautomers
Tautomers can crystallise as single or multicomponent crystals, as can any other organic molecule, but they can also crystallise with each other, as shown in Fig. 7. We use the term “tautomeric polymorph” to describe tautomers of the same compound crystallising in different crystal structures, i.e. both the tautomeric form and the crystal structure change, but only one tautomer is present in the crystal structure. Tautomeric polymorphs are the least common in the CSD, with only 16 pairs of tautomers showing tautomeric polymorphism (Fig. 8).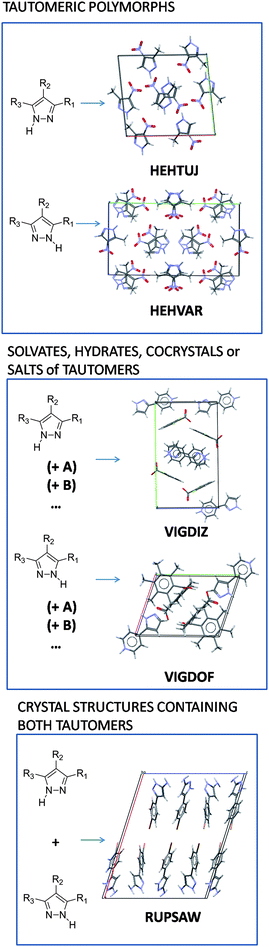 | ||
| Fig. 7 Classification of solid state occurring tautomers exemplified with pyrazole type molecules (where R1 ≠ R3). The CSD refcode of the crystal structures is indicated in the figure. | ||
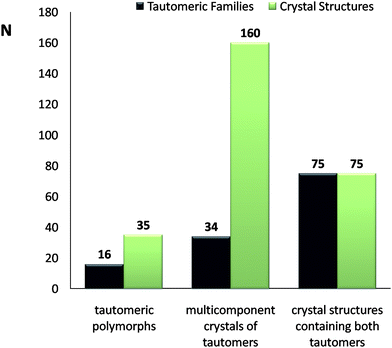 | ||
| Fig. 8 Frequency of observation of tautomeric polymorphs, multicomponent crystals of tautomers and crystal structures containing both tautomers in the CSD. The black bars indicate number of families of tautomers whereas the green bars indicate number of crystal structures. | ||
A second group of tautomers can be found in multicomponent crystals (solvates, hydrates, cocrystals or salts). In this group, we often observe that when a molecule crystallises with itself, it may do so in a less energetically favourable tautomer, in order to form a stable lattice. The presence of different components in the crystal means that this compromise may not be required, allowing it to adopt the most favourable tautomeric form. As one might expect a change of cocrystallisation component (or salt counter ion) might change the tautomeric form.
The final and largest group (Fig. 7 & 8) corresponds to crystal structures containing both tautomers. Here we find ordered crystal structures with varying stoichiometric ratios of tautomers and also structures showing disorder between the two tautomeric forms, be it static, dynamic or even dependent on temperature. We also find crystal structures in which the two tautomeric forms cocrystallise with a second or third additional component.
Discussion
The correct interpretation of tautomeric form from X-ray diffraction data can be challenging, particularly when the quality of the experimental data is low. This is confirmed by our molecular energy difference and molecular geometry tests, which suggest that almost 10% of our original dataset contained molecules with an incorrect tautomeric form.The use of complementary experimental techniques (such as solid state NMR) or theoretical models can help to clarify the nature of the tautomer in the crystal structure when needed. Whilst the use of DFT packages for this aim is more time consuming and might require the help of an expert user, straightforward geometrical analysis of the molecules, for example using the tool Mogul, is also effective.
The observation of different tautomers in the solid state is, however, a rare phenomenon. Only around 0.5% of molecules able to tautomerise in the CSD are observed in different tautomeric forms in the solid state. This represents just 0.05% of the molecules in the CSD. We identified no cases where more than two tautomeric forms per family were observed. Perhaps the generation of such systems represents an interesting challenge to the crystal engineer.
The majority of observed pairs of tautomers have small molecular energy differences of less than 12 kJ/mol. Annular tautomers and 6-member ring resonance intramolecular hydrogen bonds constitute a major proportion of the molecules crystallising in different tautomeric forms. Only derivatives of 2-pyrimidones and tautomeric pairs with intra- vs. inter-molecular hydrogen bond changes, show energy differences greater than 20 kJ/mol. Our observations allow us to make a general rule of thumb: “For different tautomers to be observed in the solid state, their relative energy must not exceed that of a strong hydrogen bond in an organic crystal”. The energy ranges presented for the different families of tautomers and their exceptions should be very useful for predicting the observation of one or more tautomers in the solid state based on ΔEt differences. They may also be useful in crystal structure prediction studies of molecules with the ability to form different tautomers.
Crystal structures containing two tautomers are the most abundant in terms of tautomeric diversity. Tautomeric polymorphs, on the other hand, are very rare. The use of different cocrystalisation components (or counter-ions) to isolate new tautomers, could perhaps be the only way of manipulating tautomeric preferences. It would be fascinating to see crystal engineering strategies developed to generate tautomers not yet seen in the solid state.
Conclusions
We have generated a database of tautomeric molecules from the Cambridge Structural Database, together with their calculated relative stabilities.§ We have made the important observation that for two tautomers to be observed in the solid state, their relative energy must not exceed that of a strong hydrogen bond typical of organic crystals. We also note that most commonly, tautomers tend to cocrystallise with each other, in one crystal structure, with or without disorder. The design of cocrystals for rare tautomeric forms could lead to the observation of different tautomers, for example by using coformers that would preferentially interact with them through strong synthon interactions. Perhaps, like polymorphism, the number of observed tautomers in the solid state is also proportional to the time and money spent on finding and designing them. Let's see.Acknowledgements
We should like to thank Prof. Jack Dunitz for very valuable discussions. AJCC thanks the Pfizer Institute for Pharmaceutical Materials Sciences for funding. The authors thank William R. Pitt for introducing them to the fascinating world of tautomers.Notes and references
- J. Elguero, A. R. Katritzky and O. V. Denisko, in Advances in Heterocyclic Chemistry, 2000, vol. 76, pp. 1–84 Search PubMed.
- T. Clark, J. Comput.-Aided Mol. Des., 2010 DOI:10.1007/s10822-010-9342-8.
- J. R. Greenwood, D. Calkins, A. P. Sullivan and J. C. Shelley, J. Comput.-Aided Mol. Des., 2010 Search PubMed.
- W. A. Warr, J. Comput.-Aided Mol. Des., 2010 DOI:10.1007/s10822-010-9338-4.
- R. A. Sayle, J. Comput.-Aided Mol. Des., 2010 DOI:10.1007/s10822-010-9329-5.
- A. J. Cruz-Cabeza, A. Schreyer and W. R. Pitt, J. Comput.-Aided Mol. Des., 2010 DOI:10.1007/s10822-010-9345-5.
- Y. C. Martin, J. Comput.-Aided Mol. Des., 2009, 23, 693–704 CrossRef CAS.
- W. Holzer, R. M. Claramunt, C. Lopez, I. Alkorta and J. Elguero, Solid State Nucl. Magn. Reson., 2008, 34, 68–76 CrossRef.
- W. Holzer, C. Kautsch, C. Laggner, R. M. Claramunt, M. Perez-Torralba, I. Alkorta and J. Elguero, Tetrahedron, 2004, 60, 6791–6805 CrossRef CAS.
- R. M. Claramunt, C. Lopez, M. A. Garcia, G. S. Denisov, I. Alkorta and J. Elguero, New J. Chem., 2003, 27, 734–742 RSC.
- S. Trofimenko, G. P. A. Yap, F. A. Jove, R. M. Claramunt, M. A. Garcia, M. D. S. Maria, I. Alkorta and J. Elguero, Tetrahedron, 2007, 63, 8104–8111 CrossRef CAS.
- O. Klein, F. Aguilar-Parrilla, J. M. Lopez, N. Jagerovic, J. Elguero and H. H. Limbach, J. Am. Chem. Soc., 2004, 126, 11718–11732 CrossRef CAS.
- M. R. Chierotti, R. Gobetto, L. Pellegrino, L. Milone and P. Venturello, Cryst. Growth Des., 2008, 8, 1454–1457 CrossRef CAS.
- N. Zencirci, E. Gstrein, C. Langes and U. J. Griesser, Thermochim. Acta, 2009, 485, 33–42 CrossRef CAS.
- P. M. Bhatt and G. R. Desiraju, Chem. Commun., 2007, 2057–2059 RSC.
- M. Mirmehrabi, S. Rohani, K. S. K. Murthy and B. Radatus, J. Cryst. Growth, 2004, 260, 517–526 CrossRef CAS.
- A. J. Blake, X. Lin, M. Schroder, C. Wilson and R. X. Yuan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, O226–O228 CrossRef.
- M. Bauer, R. K. Harris, R. C. Rao, D. C. Apperley and C. A. Rodger, J. Chem. Soc., Perkin Trans., 1998, 2, 475–481 Search PubMed.
- W. R. Porter, in Journal of Computer-Aided Molecular Design, 2010 DOI:10.1007/s10822-010-9335-7.
- Z. Q. Zhang, J. M. Njus, D. J. Sandman, C. Y. Guo, B. M. Foxman, P. Erk and R. van Gelder, Chem. Commun., 2004, 886–887 RSC.
- A. L. Llamas-Saiz, C. Foces-Foces, C. Fontenas, N. Jagerovic and J. Elguero, J. Mol. Struct., 1999, 484, 197–205 CrossRef CAS.
- T. Dziembowska, Pol. J. Chem., 1998, 72, 193–209 CAS.
- P. Gilli, V. Bertolasi, L. Pretto, L. Antonov and G. Gilli, J. Am. Chem. Soc., 2005, 127, 4943–4953 CrossRef CAS.
- M. A. Garcia, C. Lopez, R. M. Claramunt, A. Kenz, M. Pierrot and J. Elguero, Helv. Chim. Acta, 2002, 85, 2763–2776 CrossRef CAS.
- G. R. Desiraju, Cryst. Growth Des., 2008, 8, 3–5 CrossRef.
- S. L. Price, Phys. Chem. Chem. Phys., 2008, 10, 1996–2009 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- J. M. Stevenson and P. D. Mulready, J. Am. Chem. Soc., 2003, 125, 1437–1438 CrossRef CAS.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- T. G. Cooper, K. E. Hejczyk, W. Jones and G. M. Day, J. Chem. Theory Comput., 2008, 4, 1795–1805 CrossRef CAS.
- J. A. Chisholm and S. Motherwell, J. Appl. Crystallogr., 2005, 38, 228–231 CrossRef.
- I. J. Bruno, J. C. Cole, M. Kessler, J. Luo, W. D. S. Motherwell, L. H. Purkis, B. R. Smith, R. Taylor, R. I. Cooper, S. E. Harris and A. G. Orpen, J. Chem. Inf. Comput. Sci., 2004, 44, 2133–2144 CrossRef CAS.
- R. M. Claramunt, C. Lopez, M. A. Garcia, M. D. Otero, M. R. Torres, E. Pinilla, S. H. Alarcon, I. Akorta and J. Elguero, New J. Chem., 2001, 25, 1061–1068 RSC.
- C. Alvarez-Rua, S. Garcia-Granda, S. Goswani, R. Mukherjee, S. Dey, R. M. Claramunt, M. D. Santa Maria, I. Rozas, N. Jagerovic, I. Alkorta and J. Elguero, New J. Chem., 2004, 28, 700–707 RSC.
Footnotes |
| † Electronic Supplementary Information (ESI) available: CSD Refcodes and information on tautomers. See DOI: 10.1039/c0ce00123f/ |
| ‡ CSD refcodes: IMMTAZ, SOJNAG, MALHYZ10, ATTDAZ, VAHHAO, WEYROH, ILOQAB, ILOQEF, AHPAZP10, FOFBOS, LUNMPO10, SIHBOA |
| § Available by download from http://www.ccdc.cam.ac.uk |
| This journal is © The Royal Society of Chemistry 2011 |