DOI:
10.1039/C0CE00258E
(Paper)
CrystEngComm, 2011,
13, 88-92
A supramolecular twist to the structures of bis(polyfluorophenyl)mercurials†
Received
2nd June 2010
, Accepted 11th August 2010
First published on 13th September 2010
Abstract
The crystal structure of Hg(C6F4-o-NO2)21 is planar with intramolecular Hg–O coordination whereas those of the polymorphs of Hg(C6F4-m-NO2)22a and 2b have the two aryl rings rotated relative to each other (87° and 59°). At a supramolecular level, both complexes exhibit weak Hg⋯O interactions. Theoretical calculations (B3LYP and MP2) for gas-phase Hg(C6F4-o-NO2)2 and Hg(C6F4-m-NO2)2 molecules predict non-planar conformations in both complexes corresponding to one polymorph of the latter (2a). However, when the supramolecular Hg⋯O interactions are included, using a dimeric model, the calculations more accurately predict the observed structures of 1 and 2b.
Introduction
In general, bis(polyfluorophenyl)mercury complexes range from those in which the aryl rings are coplanar e.g.Hg(C6F4-o-H)21a to those in which there is a substantial twist, e.g.Hg(C6F5)2 (dihedral angle 59.4°), Hg(C6F4-p-X)2 (dihedral angle X = Me 60.4°, X = OMe 52.9°, and X = NH2 62.2°).1b,c,d The transoid disposition of the orthofluorine atoms in Hg(C6F4-o-H)21a suggests that non-planar configurations may be in response to steric and/or electrostatic repulsion between the orthofluorine atoms of opposing polyfluoroaryl rings. However, in a recent study, the structures of [Hg(C6F4-m-H)2], [Hg(C6F4-p-H)2] and [Hg(C6H2-2,4,6-F3)2] were all shown to be planar despite the presence of four orthofluorine atoms in each compound, yet contrastingly [Hg(C6H3-2,6-F2)2] was twisted with a dihedral angle of 88°.2a Similarly, the mixed aryl/fluoroaryl compound, [Hg(C6F5)(C6H4-p-OMe)], was shown to be twisted (dihedral angle 87°) rather than planar despite having opposing H and F atoms in the ortho positions.2b Furthermore, calculations for gas-phase Hg(C6F5)2 (B3LYP)3a and HgPh2 (MP2)3b predict orthogonal structures for both molecules as the minimum-energy conformation (cf.HgPh2 is planar in the solid state2c). Clearly, other factors such as crystal packing and supramolecular interactions have a significant structural effect.
In this contribution, we report the crystal structures and theoretical calculations for a pair of isomeric mercurials [Hg(C6F4-o-NO2)2 (1) and Hg(C6F4-m-NO2)2 (2)] (Scheme 1). The presence of the bulky electron withdrawing nitro groups allows us to study potential steric as well as Hg–O coordination effects, both in an intramolecular and supramolecular framework. In addition, we have been able to isolate two polymorphs (rotamers) of 2 (2a and 2b) through crystallization under marginally different conditions, an observation highly relevant to the issue of varying twist angles for closely related bis(polyfluorophenyl)mercurials. Earlier we have reported on the synthesis, structures and supramolecular interactions of the complexes between these mercurials and a number of arenes.4
 |
| | Scheme 1 | |
Results and discussion
The X-ray crystal structure of [Hg(C6F4-o-NO2)2] (1) shows an essentially planar molecule (Fig. 1) and one oxygen atom from each of the nitro groups forms an intramolecular Hg–O interaction. The o-NO2 groups are in a transoid disposition, with the O–Hg–O angle near linear, and the ‘bite’ angles of the two C6F4-o-NO2 ligands are 67.4(2)° and 68.3(2)°. The o-NO2 groups are only slightly twisted out of the aryl ring plane (dihedral angles 5.5(6)° and 17.6(5)°) and clearly the steric bulk of the o-NO2 substituent can be accommodated in a planar structure. The m-NO2 complex [Hg(C6F4-m-NO2)2] (2) has been crystallised in two polymorphs (one from a room temperature crystallisation, 2a, and one from a crystallisation at −10 °C, 2b). The HgR2 structural unit is similar for both polymorphs differing predominantly in the relative orientation of the two C6F4-m-NO2 rings. The structure of 2a (Fig. 2a) shows a twisted HgR2 unit (dihedral angle 87.3(1)°) with the m-NO2 groups in a cisoid orientation. For 2b, the corresponding twist angle is 59.9(1)° similar to that of Hg(C6F5)2 and a range of Hg(C6F4-p-X)2 compounds (above), and the m-NO2 groups are transoid (Fig. 2b). Isolation of two polymorphs 2a and 2b with quite different twist angles under reasonably similar crystallization conditions is indicative of a small energy difference between them and suggests polymorphs may be also accessible for other bis(polyfluorophenyl)mercurials. Indeed, two forms of Hg(C6F5)2 with different melting points have been reported, but without crystallographic validation of a second form.5 In both structures 2a and 2b, the oxygen atoms of the m-NO2 groups are not in a position to form intramolecular Hg–O contacts, as is evident in 1, and are twisted out of the fluoroarene plane (dihedral angles 39.8(4)° and 47.8(2)°, 2a, and 33.6(5)° and 42.1(4)°, 2b). The Hg–C distances (Table 1) are typical for Hg(ArF)2 compounds,1,2 but show slightly longer bonds for 1 which may reflect the higher coordination number due to the o-NO2 coordination. The latter interaction has a Hg–O bond distance (Table 1) marginally shorter than that of [Hg(C6H4-o-P(O)Ph2)2] (2.874(5) Å).6a Shorter Hg–O distances are observed for [Hg(C6F5)Cl(L)] (L = dmso, 2.542(4) Å),6b a consequence of the greater Lewis acidity of mercury halide compounds.
Table 1 Selected bond distances (Å) and angles (°) in [Hg(C6F4-o-NO2)2] and [Hg(C6F4-m-NO2)2]
| |
1
|
2a
a
|
2b
b
|
|
Room temperature crystallisation.
Low temperature crystallisation.
|
| Hg–C |
2.081(6), 2.106(6) |
2.061(4), 2.069(4) |
2.037(6), 2.052(5) |
| Hg–O |
2.729(4), 2.784(4) |
|
|
| C–Hg–C |
176.6(2) |
178.1(1) |
174.5(2) |
| O–Hg–O |
178.1(1) |
|
|
| Hg⋯O |
3.007(4) |
2.973(3) |
2.987(4) |
| |
3.027(4) |
3.224(3) |
3.114(4) |
| |
3.132(4) |
|
3.268(4) |
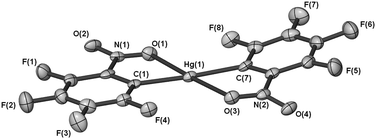 |
| | Fig. 1 Molecular diagram of 1 shown with 50% thermal ellipsoids. | |
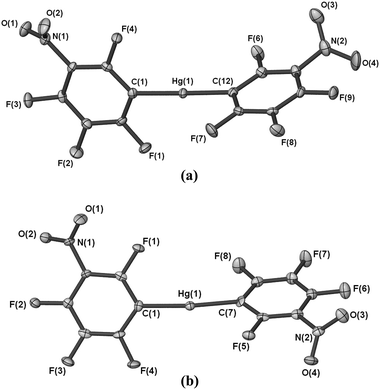 |
| | Fig. 2 (a) Molecular diagram of 2a shown with 50% thermal ellipsoids. (b) Molecular diagram of 2b shown with 50% thermal ellipsoids. | |
At the intermolecular level, the structure of 1 has three plausible Hg⋯O interactions (Table 1) which are within the sum of the van der Waals radii of oxygen (1.54 Å)7a,b and mercury (1.73–2.2 Å).7a,c,d Supramolecular interactions of Hg with nitro moieties have been observed in [{Hg(o-C6F4)}3(O2NPh)] with comparable Hg⋯O bond lengths (2.858(6)–3.254(5) Å),8 whereas Hg⋯O(ether) contacts tend to be longer, e.g. [Hg(C6F4-o-NO2)2(TMO)2] (TMO = 1,2,4-trimethoxybenzene) (Hg⋯O 3.203(3) Å)4 and [Hg(C6F4-p-OMe)2] (3.313 Å).1d The two shorter Hg⋯O bond lengths in 1 are approximately perpendicular to the HgR2 plane and link successive HgR2 units into a stepped linear chain (Fig. 3). The O⋯Hg⋯O angle is near linear giving a skewed octahedral mercury environment. Octahedral mercury has been observed in [Hg(OH2)2(O3SCF3)2]∞, which has short axial Hg–OH2 and longer equatorial Hg–O3SCF3 bonds,9a whilst more regular octahedra are evident for the fully solvated Hg2+ cations e.g.[Hg(S)6][X]2 (S = dmso, X = ClO4 and O3SCF3).9b,c The longer Hg⋯O distance in 1 is from a NO2 group to the mercury atom of a neighbouring chain resulting in a 2D network.
![Ball and stick representation of 1 showing a stepped linear chain of [Hg(C6F4-o-NO2)2] molecules with supramolecular Hg⋯O contacts as dotted lines. Symmetry operators: (i) 1 + x, y, z and (ii) x − 1, y, z.](/image/article/2011/CE/c0ce00258e/c0ce00258e-f3.gif) |
| | Fig. 3 Ball and stick representation of 1 showing a stepped linear chain of [Hg(C6F4-o-NO2)2] molecules with supramolecular Hg⋯O contacts as dotted lines. Symmetry operators: (i) 1 + x, y, z and (ii) x − 1, y, z. | |
The structure of 2a has two intermolecular Hg⋯O contacts within the sum of the respective van der Waals radii (Table 1) and involving only one of the m-NO2 groups. Two such groups form bidentate bridges between two neighbouring and approximately parallel HgR2 units (Fig. 4). The O⋯Hg⋯O coordination is cisoid with an angle of 113.51(8)°. The opposing face of the mercury centre has no analogous Hg⋯O contacts, with the closest approach being a fluorine atom (ca. 3.310 Å). Propagation of the bridging Hg–(ON(R)O)2–Hg moiety results in a porous 2D sheet structure containing rectangular holes into which protrude C6F4-m-NO2 groups (those not involved in Hg⋯O interactions) from a neighbouring 2D sheet. The structure of 2b has three Hg⋯O contacts (Table 1) in a T-shaped geometry; the next nearest contact is to a distant fluorine atom (Hg⋯F ca. 3.295 Å). The two shorter interactions are cisoid with O⋯Hg⋯O 106.8(1)° and form a bidentate nitro bridged core similar to that of 2a. The third Hg⋯O interaction (transoid to O(2), O⋯Hg⋯O 156.9(1)°) involves the other nitro group and participates in a head to tail dimer structure (Fig. 5) resulting, overall, in a more compact 2D sheet structure.
![Ball and stick representation of 2a showing bidentate m-NO2 bridging of two [Hg(C6F4-m-NO2)2] molecules with supramolecular Hg⋯O contacts as dotted lines. Symmetry operators: (i) x − ½, ½ − y, z − ½; (ii) 1½ − x, ½ − y, 2½ − z; and (iii) 1 − x, −y, 2 − z.](/image/article/2011/CE/c0ce00258e/c0ce00258e-f4.gif) |
| | Fig. 4 Ball and stick representation of 2a showing bidentate m-NO2 bridging of two [Hg(C6F4-m-NO2)2] molecules with supramolecular Hg⋯O contacts as dotted lines. Symmetry operators: (i) x − ½, ½ − y, z − ½; (ii) 1½ − x, ½ − y, 2½ − z; and (iii) 1 − x, −y, 2 − z. | |
![Ball and stick representation of 2b showing head to tail m-NO2 bridging of [Hg(C6F4-m-NO2)2] molecules with supramolecular Hg⋯O contacts as dotted lines. Symmetry operators: (i) ½ + x, ½ − y, ½ + z and (ii) x − ½, ½ − y, z − ½.](/image/article/2011/CE/c0ce00258e/c0ce00258e-f5.gif) |
| | Fig. 5 Ball and stick representation of 2b showing head to tail m-NO2 bridging of [Hg(C6F4-m-NO2)2] molecules with supramolecular Hg⋯O contacts as dotted lines. Symmetry operators: (i) ½ + x, ½ − y, ½ + z and (ii) x − ½, ½ − y, z − ½. | |
Calculated structures of [Hg(C6F4-o-NO2)2] and [Hg(C6F4-m-NO2)2]
The optimized bond distances and angles in minimum-energy structures of the HgR2 molecules and corresponding dimeric models are given in Table 2. The optimized minimum-energy geometry of an isolated (gas phase) [Hg(C6F4-o-NO2)2] molecule has the aryl rings almost perpendicular to one another (Fig. 6a), and differs strikingly from that observed for 1. The minimum-energy structure for [Hg(C6F4-m-NO2)2] (Fig. 6b) is similarly twisted, as observed in 2a. However, the energy difference calculated at the MP2 level of theory between the non-planar (minimum-energy) and planar (calculated in the Ci symmetry) geometries is only 2 kJ mol−1 for the o-NO2 complex, whereas for the m-NO2 complex there is an even more negligible difference (<1 kJ mol−1). The planar configurations represent saddle points with one imaginary frequency and the o-NO2 and m-NO2 groups are tilted away from planarity by 20° and 54°, respectively.
Table 2 Gas-phase optimized bond distances (Å) and angles (°) of the lowest energy structures of HgAr2 molecules and their dimersa
| |
[Hg(C6F4-o-NO2)2] |
[Hg(C6F4-m-NO2)2] |
| Monomer |
Dimer |
Monomer |
Dimer |
|
In the case of the [Hg(C6F4-o-NO2)2] dimer the planar structure (Ci symmetry) is presented.
|
| Hg–C |
2.147 |
2.149 |
2.124 |
2.125 |
| |
|
|
2.136 |
| Hg–O |
2.652 |
2.758 |
— |
— |
| |
2.770 |
|
|
| C–Hg–C |
175 |
177 |
180 |
177 |
| O–Hg–O |
95 |
169 |
— |
— |
| Ar twist |
97 |
0 |
92 |
33 |
|
NO2 twist |
1 |
20 |
54 |
53 |
| Hg⋯O |
— |
2.980 |
— |
2.983 |
![Gas-phase optimized minimum-energy structures of [Hg(C6F4-o-NO2)2] and [Hg(C6F4-m-NO2)2].](/image/article/2011/CE/c0ce00258e/c0ce00258e-f6.gif) |
| | Fig. 6 Gas-phase optimized minimum-energy structures of [Hg(C6F4-o-NO2)2] and [Hg(C6F4-m-NO2)2]. | |
To take into account the intermolecular Hg⋯O interactions observed in the crystal structures, we optimised the geometries of dimeric models (Fig. 7-1 and 7-2). For [Hg(C6F4-o-NO2)2], two structures were found with the through space interaction between the Hg atom and the oxygen atoms of the o-NO2 group resulting in a planar and a non-planar configuration. Out of these structures the non-planar configuration is a true minimum, whereas the planar configuration represents a saddle point with a negligible imaginary frequency. The structure with the non-planar configuration of [Hg(C6F4-o-NO2)2] has the interaction energy (calculated with respect to the monomer in its minimum-energy geometry) of 62 kJ mol−1, which is about 14 kJ mol−1 greater than that of the corresponding planar configuration. This suggests further that interactions over and above solely Hg⋯O contacts (optimised Hg⋯O distance 3.350 and 3.962 Å) contribute to stronger binding in the dimer. A comparison of these structures with the crystal structure in Fig. 3 indicates that the overall planar arrangement of the dimeric [Hg(C6F4-o-NO2)2] model is in very good agreement with the extended [Hg(C6F4-o-NO2)2] structure (see Fig. 3), with the optimized Hg⋯O distance of 2.980 Å being only marginally shorter than those observed in 1. The fact that the more energetically preferable non-planar arrangement of [Hg(C6F4-o-NO2)2] has not been observed in the crystal structure of 1 suggests that the formation of a highly ordered lattice with the planar configuration of monomers is more likely to be attributed to a significant change in entropy going from a disordered to highly ordered lattice, thus leading a stronger lattice energy overall.
![Gas-phase optimized structures of the dimeric models of [Hg(C6F4-o-NO2)2] (1 and 2) and [Hg(C6F4-m-NO2)2] (3).](/image/article/2011/CE/c0ce00258e/c0ce00258e-f7.gif) |
| | Fig. 7 Gas-phase optimized structures of the dimeric models of [Hg(C6F4-o-NO2)2] (1 and 2) and [Hg(C6F4-m-NO2)2] (3). | |
The dimer of [Hg(C6F4-m-NO2)2] represents a different case in which the analogous interaction between the Hg atom and the m-NO2 group can only occur with the aryl rings twisted out of the plane by 53° (see Fig. 7-3). In the dimeric [Hg(C6F4-m-NO2)2] model, the optimized Hg⋯O distance, 2.983 Å, is in good agreement with the shorter of the observed values in 2b. Furthermore, the calculated dimeric arrangement is similar to a portion of the extended structure of 2b (see Fig. 5b). The interaction energy of this twisted arrangement is only 3 kJ mol−1 lower compared to that of the planar [Hg(C6F4-o-NO2)2] dimer. The analysis of these interaction energies for both complexes revealed that the interaction was strongly dominated by dispersion-type interactions, with the electron correlation energy contributing up to 93% and 98% for the dimers of [Hg(C6F4-o-NO2)2] and [Hg(C6F4-m-NO2)2], respectively.
There are relevant studies bearing on the strength/existence of intermolecular Hg⋯O interactions in solution. Stability constants (from molecular weight determinations) show that solid complexes of Hg(C6F5)2 with 1,10-phenanthroline, 2,2′-bipyridyl, and, relevant to the current study, Ph3PO persist to varying extents in benzene.10 Further, phenyl(8-quinolinolato)mercury(II) has a polymeric crystal structure through intermolecular Hg⋯O contacts of 3.3–3.4 Å11 at or over the limit of the sum of the van der Waals radii of oxygen7b and a conservative 1.73 Å7c for mercury, yet significant dimer formation was detected in benzene, chloroform and carbon tetrachloride.12 Likewise, of some relevance to gas-phase calculations, dimercury species were detected in the vapour state by mass spectrometry.12 These data indicate that intermolecular Hg⋯O interactions play a significant role.
Conclusions
The relative twist angle of the aryl rings for two isomeric bis(tetrafluorophenyl)mercurials appears to be determined by supramolecular Hg⋯O interactions rather than intramolecular steric or electronic factors. Thus, [Hg(C6F4-o-NO2)2] 1 is planar, whereas [Hg(C6F4-m-NO2)2] is twisted in both polymorphs (2a and 2b). Isolation of two polymorphs under only slightly different crystallization conditions highlights that the differing aryl ring twist angles arise from very small energy differences, and account for the unpredictability of reported twist angles. These observations are consistent with theoretical predictions, which show an existence of a planar configuration for [Hg(C6F4-o-NO2)2], only when Hg⋯O bonding is included. The less energetically preferable planar arrangement of [Hg(C6F4-o-NO2)2] in the crystal structure is likely to form due to entropic contributions resulting in a highly ordered lattice. Polymorph 2a corresponds to the minimum-energy monomeric structure calculated for [Hg(C6F4-m-NO2)2] whereas the calculated dimeric model represents the major features of 2b.
Experimental
The diorganomercurials [Hg(C6F4-o-NO2)2] and [Hg(C6F4-m-NO2)2] were prepared by the literature methods.13 Crystals of 1 were obtained by slow evaporation of a CH2Cl2 solution whereas 2 was crystallised in two polymorphs from either a concentrated CH2Cl2/hexane solution at room temperature (2a) or cooling a CH2Cl2/hexane solution to −10 °C (2b).
X-Ray structure determinations
Single crystals suitable for X-ray analysis were covered in viscous oil and mounted on a glass fibre. Data (2θmax 55°) were collected at 123(1) K using a Enraf Nonius KAPPA or Bruker X8 Apex CCD and MoKα (λ 0.71073 Å) radiation. After integration and scaling, datasets were merged (Rint as quoted) and the structures were solved using conventional methods and refined by full matrix least squares using the SHELX-97 software,14a in conjunction with the X-Seed interface.14bNon-hydrogen atoms were refined with anisotropic thermal parameters. Data were corrected for absorption using SORTAV14c or SADABS.14d
Crystal and refinement data
[Hg(C6F4-o-NO2)2] (1).
C12F8HgN2O4 FW 588.73. Monoclinic, P21/c. a = 6.1736(2), b = 9.1500(2), c = 24.7578(6) Å, β = 92.857(1)°. V = 1396.79(6) Å3. Z = 4. Dcalcd = 2.800 g cm−3. μ = 11.15 mm−1, Nt = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 080, N = 3169 (Rint = 0.052). R1 = 0.030 (I > 2σI), wR2 = 0.066 (all data).
080, N = 3169 (Rint = 0.052). R1 = 0.030 (I > 2σI), wR2 = 0.066 (all data).
[Hg(C6F4-m-NO2)2] (2a).
C12F8HgN2O4 FW 588.73. Monoclinic, P21/n. a = 9.2625(4), b = 13.1795(5), c = 11.7280(5) Å, β = 97.562(2)°. V = 1419.25(10) Å3. Z = 4. Dcalcd = 2.755 g cm−3. μ = 10.97 mm−1, Nt = 16244, N = 3252 (Rint = 0.046). R1 = 0.022 (I > 2σI), wR2 = 0.044 (all data) (room temperature crystallisation).
[Hg(C6F4-m-NO2)2] (2b).
C12F8HgN2O4 FW 588.73. Monoclinic, P21/n. a = 12.7462(3), b = 8.5095(2), c = 13.1712(3) Å, β = 105.339(1)°. V = 1377.71(6) Å3. Z = 4. Dcalcd = 2.838 g cm−3. μ = 11.30 mm−1, Nt = 12971, N = 3155 (Rint = 0.050). R1 = 0.036 (I > 2σI), wR2 = 0.092 (all data) (low temperature crystallisation).
Theoretical procedures
The optimizations were performed at B3LYP/6-31 + G(d) using the Gaussian 03 package.15a In order to describe the core electrons of Hg we employed the Stuttgart/Dresden effective core potential (ECP) with a Dirac–Fock relativistic correction, abbreviated MDF, as implemented in Gaussian 03. The number of electrons included in the core was 60. For the valence electrons on Hg we used a double-ζ basis set optimized for the chosen ECP. Improved electronic energies were calculated at a correlated level of theory, MP2/cc-pVTZ, using the B3LYP optimized geometries. For Hg the relativistic cc-pVTZ-PP basis set of Peterson et al.15b was employed. Interaction energies of the dimeric models studied were calculated as the difference in the improved electronic energies of the dimer and the corresponding monomers taken in their minimum geometries. The interaction energies were also counter-poised corrected using the Boys algorithm as implemented in Gaussian 03.
Acknowledgements
We gratefully acknowledge generous allocations of computing time from the National Facility of the National Computational Infrastructure and the Monash Sun Grid Cluster at the e-research centre of Monash University. We thank the Australian Research Council for financial support and for a postdoctoral fellowship (EII).
References
-
(a) D. S. Brown, A. G. Massey and D. A. Wickens, J. Organomet. Chem., 1980, 194, 131–135 CrossRef CAS;
(b) N. R. Kuchner and M. Mathew, J. Chem. Soc., Chem. Commun., 1966, 71–73 RSC;
(c) D. L. Wilkinson, J. Riede and G. Müller, Z. Naturforsch., B: Chem. Sci., 1991, 46, 285–288 CAS;
(d) G. B. Deacon, C. M. Forsyth, D. M. M. Freckmann, G. Meyer and D. Stellfeldt, Z. Anorg. Allg. Chem., 2000, 626, 540–546 CrossRef CAS.
-
(a) D. Naumann and F. Schulz, Z. Anorg. Allg. Chem., 2005, 631, 122–125 CrossRef CAS;
(b) G. B. Deacon and P. C. Junk, J. Chem. Crystallogr., 2003, 33, 605–607 CrossRef CAS;
(c) C. Glidewell, J. N. Low and J. L. Wardell, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, m107–m108 CrossRef.
-
(a) C. N. Buress, M. I. Bodine, Q. Elbjeirami, J. H. Reibenspies, M. A. Omary and F. C. Gabbaï, Inorg. Chem., 2007, 46, 1388–1395 CrossRef CAS;
(b) I. Antes and G. Frenking, Organometallics, 1995, 14, 4263–4268 CrossRef CAS.
- G. B. Deacon, C. M. Forsyth, P. C. Junk, T. J. Ness, E. Izgorodina, J. Baldamus, G. Meyer, I. Pantenburg, J. Hitzbleck and K. Ruhlandt-Senge, Eur. J. Inorg. Chem., 2008, 4770–4780 CrossRef CAS.
- S. C. Cohen and A. G. Massey, Adv. Fluorine Chem., 1970, 6, 83–285 Search PubMed.
-
(a) M. A. Bennett, M. Contel, D. C. R. Hockless, L. L. Welling and A. C. Willis, Inorg. Chem., 2002, 41, 844–855 CrossRef CAS;
(b) M. Tschinkl, A. Schier, J. Riede and F. P. Gabbaï, Organometallics, 1999, 18, 2040–2042 CrossRef CAS.
-
(a) A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS;
(b) S. C. Nyburg and C. H. Faerman, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 274–279 CrossRef;
(c) A. J. Canty and G. B. Deacon, Inorg. Chim. Acta, 1980, 45, L225–L227 CrossRef CAS;
(d) S. S. Batsanov, J. Chem. Soc., Dalton Trans., 1998, 1541–1546 RSC.
- I. A. Tikhonova, K. I. Tugashov, F. M. Dolgushin, A. A. Yakovenko, P. V. Petrovskii, G. G. Furin, A. P. Zaraisky and V. B. Shur, J. Organomet. Chem., 2006, 692, 953–962.
-
(a) A. Molla-Abbassi, L. Eriksson, J. Mink, I. Persson, M. Sandström, M. Skripkin, A.-S. Ullström and P. Lindqvist-Reis, J. Chem. Soc., Dalton Trans., 2002, 4357–4364 RSC;
(b) M. Sandström and I. Persson, Acta Chem. Scand., Ser. A, 1978, 32, 95–100;
(c) J. M. Hook, P. A. W. Dean and D. C. R. Hockless, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1547–1549 CrossRef.
- A. J. Canty and G. B. Deacon, Aust. J. Chem., 1971, 24, 489 CAS.
- C. L. Raston, B. W. Skelton and A. H. White, Aust. J. Chem., 1978, 31, 537–545 CAS.
- R. J. Bertino, G. B. Deacon and J. M. Miller, Aust. J. Chem., 1978, 31, 527–535 CrossRef CAS.
-
(a) H. B. Albrecht and G. B. Deacon, J. Organomet. Chem., 1973, 57, 77–86 CrossRef CAS;
(b) G. B. Deacon and D. Tunaley, J. Organomet. Chem., 1978, 156, 403–426 CrossRef CAS.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122;
(b) L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS;
(c) R. H. Blessing, J. Appl. Crystallogr., 1997, 30, 421–426 CrossRef CAS;
(d)
G. M. Sheldrick, SADABS: Program for Scaling and Absorption Correction of Area Detector Data, Universität Göttingen, 1997 Search PubMed.
-
(a)
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, A. L. G. Liu, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed;
(b) K. A. Peterson, D. Figgen, M. Dolg and H. Stoll, J. Chem. Phys., 2007, 126, 124101–124112 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.