Size does matter—the contribution of molecular volume, shape and flexibility to the formation of co-crystals and structures with Z′ > 1†
Kirsty M.
Anderson
*,
Michael R.
Probert
,
Andrés E.
Goeta
and
Jonathan W.
Steed
*
Department of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: jon.steed@durham.ac.uk
First published on 7th September 2010
Abstract
Systematic analysis of the Cambridge Structural Database (CSD) using parameters based on the shape, size and “awkwardness” of organic molecules shows that compounds which crystallise with Z′ > 1 are in general smaller (by around 50 Å3 on average) and less flexible (with ca. two fewer rotatable bonds) than molecules which crystallise with Z′ = 1. Molecules which are known to form co-crystals are, on average, even smaller and less flexible compared to molecules which crystallise with Z′ = 1. Thus formation of co-crystals or structures with Z′ > 1 is strongly linked to small, rigid, awkwardly shaped molecules that have more constraints on their crystal packing requirements. These results have some predictive utility in determining the likelihood of hydrate formation in pharmaceuticals, for example.
Introduction
Over the last 20 years crystal structures with Z′ > 1 (i.e. materials which crystallise with more than one molecule or formula unit in the asymmetric unit) have moved from a crystallographic curiosity to being a useful subset of structures which provide some insight into answering the age-old question of how molecules pack together to form a crystal.1 Recent studies on the phenomenon cover a wide range of topics including energy calculations,2 polymorphism3 and co-crystals4 as well as some healthy debates on the variety of underlying causes5–7 and nomenclature.8Recently we have shown4 that it is possible in some well-defined cases to successfully predict that a given compound is likely to pack with Z′ > 1. There are also categories of molecule with certain characteristics and functional groups which are known to have high incidences of Z′ > 1, such as mono-alcohols9 and chiral molecules containing a carboxylic acid or amide.10 Ourselves11 and others12 have noted that frustration between competing interactions also results in the formation of structures with Z′ > 1.
Investigations into the cause of these anomalies have mainly concentrated on frustration between intermolecular forces, for example hydrogen bonding13,14 and secondary interactions,15 and many structures which crystallise with high Z′ can be explained in this way. Other factors such as the size and shape of a given molecule are also known to play a large part in how molecules pack together16 but as yet the only link between shape and the propensity to pack with Z′ > 1 has been in some elegant research by Gavezzotti2 who calculated molecular volumes for a selection of structures with Z′ = 1 or 2 and found that the Z′ = 2 subset has a slightly smaller average size than similarly chosen structures with Z′ = 1. A more detailed description of molecular shape has been studied recently by Fayos, however, Z′ is not explicitly addressed.17 In this work we seek to examine the size, shape, flexibility and “awkwardness” of molecules using various parameters to determine if there are any important differences in the types of molecule which form structures with Z′ > 1 and Z′ = 1.
Results and discussion
A search of the Cambridge Structural Database18–20 (CSD) was carried out for organic crystal structures with Zr = 1 (i.e. one type of chemical residue4), three-dimensional coordinates present and with no errors or disorder. Our in-house program Mol2Man was used on the structures with Z′ > 1 to create a file with just one representative molecule for each structure, based on the first molecule recorded in the file. While there may be some conformational differences between independent molecules these are not on a scale where it becomes important to the final result in this work. This program was also used to output the total number of non-hydrogen atoms for each molecule in both the Z′ = 1 and Z′ > 1 subsets.The program MSROLL21 was used to calculate the molecular volume, as well as the surface area and the solvent accessible surface area (mapped using a probe sphere of radius = 1.5 Å) for a given molecule (Fig. 1). These data give an idea of the shape of the molecule, while the ratio of area![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :volume gives some idea of its “awkwardness”—i.e. how well it can fit together with other molecules. The use of area:volume ratio in defining molecular shape has been discussed previously by Gavezzotti who termed it the ‘molecular exposure ratio’.22 Here we propose that a high degree of molecular exposure may correlate with packing difficulties because highly surface-exposed molecules will contain concave surface features that will be unlikely to be fully shape complementary to the convex features on an adjacent molecule.
:volume gives some idea of its “awkwardness”—i.e. how well it can fit together with other molecules. The use of area:volume ratio in defining molecular shape has been discussed previously by Gavezzotti who termed it the ‘molecular exposure ratio’.22 Here we propose that a high degree of molecular exposure may correlate with packing difficulties because highly surface-exposed molecules will contain concave surface features that will be unlikely to be fully shape complementary to the convex features on an adjacent molecule.
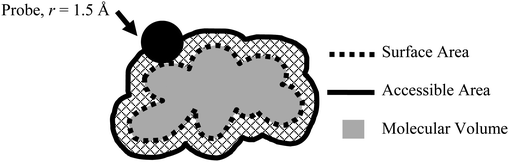 | ||
| Fig. 1 Areas and volume mapped for a given molecule. | ||
To complement these data RPLUTO23 was used to calculate the dimensions of a hypothetical box where the dimensions map out the boundaries of the molecule. The use of boxes to model molecular shape has been described previously.24 The volume of this box can be calculated and compared to the actual volume of the molecule in order to provide a guide to how well the molecule fits in the box. Molecules which fill the space in the box well will be those with no concave indentations and a more symmetrical shape and are closer to a cuboid shape which allows packing in three-dimensions without any empty space.
As well as geometric parameters we were also interested in the flexibility of the molecule and hence Marvin25 was used to calculate the total number of connections in a given compound as well as the number of rotatable bonds present. Bonds to hydrogen are ignored in these calculations. Tables 1–3 show the mean values for the calculated parameters, along with the median values. In this case it is more appropriate to compare median values due to the fact that the majority of data are distributed in a non-normal manner.‡
| Number | Volume (v)/Å3 | Number of non-hydrogen atoms | Molecular area (ma)/Å2 | Accessible area (aa)/Å2 | v/ma | v/aa | Box volume/Å3 | Box volume–volume/Å3 | Number of connections | Number of rotatable bonds | Ratio rotatable:connections |
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Z′ = 1 | 72757 |
Mean | 307.53 | 23.926 | 303.36 | 470.22 | 0.99084 | 0.63345 | 899.02 | 591.49 | 26.126 | 3.9434 | 0.14751 |
| Median | 283.71 | 22 | 287.74 | 451.31 | 0.97935 | 0.62376 | 790.08 | 501.61 | 24 | 3 | 0.13636 | ||
| Z′ > 1 | 10553 |
Mean | 289.44 | 22.575 | 288.54 | 450.56 | 0.98031 | 0.62191 | 827.9 | 538.46 | 24.638 | 3.5416 | 0.13832 |
| Median | 266.25 | 21 | 272.57 | 430.85 | 0.97102 | 0.61349 | 717.4 | 446.96 | 23 | 3 | 0.125 | ||
| Z′ > 2 | 1060 | Mean | 254.29 | 19.775 | 258.47 | 409.8 | 0.95663 | 0.59634 | 683.7 | 429.5 | 21.554 | 2.8759 | 0.12758 |
| Median | 233.67 | 18 | 242.4 | 389.85 | 0.94567 | 0.5878 | 593.95 | 353.7 | 20 | 2 | 0.11111 |
Table 1 shows values calculated described as above separated into organic molecules which crystallise with Z′ = 1, Z′ > 1 and Z′ > 2. The latter are included as a separate subset as we believe these represent the most extreme examples of type and hence the effects are expected to be considerably larger for this group. Looking at the molecular volumes for the three categories it is clear that, in agreement with the work of Gavezzotti2 which was done on a different subset of structures, compounds which crystallise with Z′ > 1 are comprised of entities with a smaller molecular volume. The difference in median molecular volumes between Z′ = 1 and Z′ > 2 molecules, significant to the 99.999% level according to calculated Mann–Whitney test values, is around 50 Å3 which is comparable to the volume of an isopropyl group (ca. 57 Å3). The Z′ > 1 subset also exhibits a smaller surface area and accessible area compared to the Z′ = 1 structures. In keeping with these observations the average number of non-hydrogen atoms also decreases from Z′ = 1 to Z′ > 1 structures. To represent “awkwardness” we have chosen the ratio of the surface area (or solvent accessible area) to the volume, where a smaller value represents a more awkward molecule. A molecule that has more concave indentations or convex protrusions that increase its surface area will be less likely to tessellate with its neighbours than a sphere, for example. The values for these ratios in Table 1 show that molecules which crystallise with Z′ > 1 are more awkward, that is they have a larger surface or solvent accessible area per unit volume.
In terms of box size the trend is repeated with Z′ > 1 structures represented by a smaller box size on average. We can also determine how well the molecule fills the box by calculating the free space remaining within the box (by subtracting the molecular volume from the box volume) and express this result as a percentage. The data show that on average the Z′ > 2 molecules do not fill the box as efficiently, having an occupied space percentage of 59.6% compared to 63.5% for the Z′ = 1 structures, indicating that the Z′ > 2 structures are less regular and hence more awkward, even despite obvious exceptions such as planar aromatics crystallising with Z′ > 1.
Looking at how the molecules themselves are constructed, the number of connections is larger for structures with Z′ > 1 despite having fewer atoms, with a median atom:connection ratio of 0.92 for Z′ = 1 and 0.90 for Z′ > 1 and Z′ > 2. Fig. 2 shows two structures (126 with Z′ = 1 and 227 with Z′ = 2) which have identical molecular formulae, C16H16O4 but different connectivities. The number of connections is 19 for 1 and 23 for 2 (bonds to hydrogen are ignored when calculating the total number of connections). An increase in the number of connections for molecules with Z′ > 1 indicates structures which have fewer terminal groups and are more rigid, as in 2. As hydrogen atoms are ignored when calculating the number of connections it is also possible to generate a structure with the same core non-hydrogen atoms as 1 and 2 but a differing number of hydrogen atoms, as in 328 which crystallises with Z′ = 2 and has the molecular formula C16H20O4. Here there is extra saturation (4 extra hydrogen atoms) and the same number of connections as 2. It should also be noted that by the definition used compound 2 has zero rotatable bonds, however, it does have some conformational freedom due to limited rotation about the rings which in this case leads to conformational polymorphism.27 Quantifying this sort of conformational freedom is not within the scope of this study.
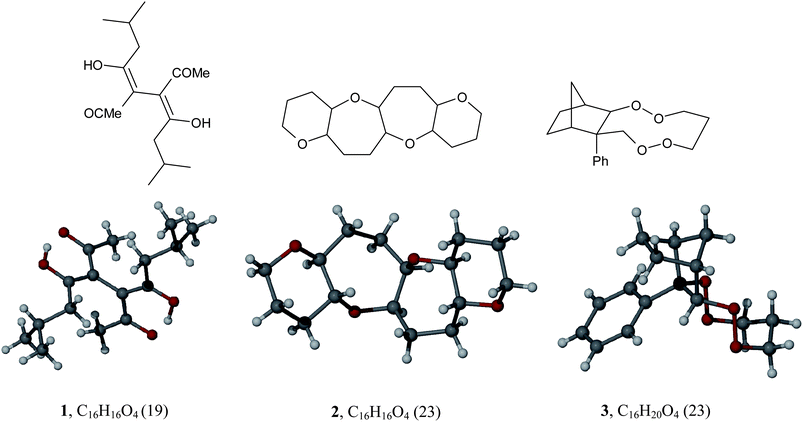 | ||
| Fig. 2 Structures with identical formulae but a different number of connections (1 and 2) and with the same number of non-hydrogen atoms but a different overall formula (3). The number of connections for each structure is shown in brackets. | ||
Perhaps more interestingly there are also fewer rotatable bonds for structures with Z′ > 1, and the ratio of rotatable bonds to connections is much less for Z′ > 1 and particularly Z′ > 2 structures than for those with Z′ = 1, indicating less flexibility in the Z′ > 1 structures. This observation holds true for molecules 1, 2 and 3 shown in Fig. 2 which have 7, 0 and 1 rotatable bonds respectively. This inability to adapt shape and/or conformation may well be very important when the system is exploring possible symmetry space during crystallisation and may be a key consideration in predicting whether a molecule will crystallise with Z′ = 1 or not.
In our previous work4 we showed that molecules which form co-crystals (dubbed “parent molecules”§) show a tendency to crystallise with Z′ > 1. Inclusion of a second, different molecule within the asymmetric unit is evidence for an inherent difficulty for the parent molecule in question to crystallise by itself (i.e. with Z′ = 1) and therefore it is more energetically favourable (in terms of packing efficiency) to include either a small unrelated molecule or a symmetry unrelated copy of the same molecule, to give co-crystals and structures with Z′ > 1, respectively. We were therefore interested to see if there were any significant differences in the size and shape parameters calculated for small organic molecules overall and for those designated as parent molecules. Table 2 shows the same set of calculated parameters discussed above as applied to parent molecules. Firstly we see that the difference in the median molecular volume between parents with Z′ = 1 and Z′ > 2 (53.0 Å3) is very similar in magnitude to the difference observed for the organic dataset, and is of a similar magnitude for the Z′ = 1 and Z′ > 1 difference (27.9 vs. 17.5 Å3 respectively). More interestingly though the data show that on average, the parent structures have a smaller molecular volume than the median values obtained for the organic dataset, a difference of 42.3, 52.7 and 45.3 Å3 for Z′ = 1, Z′ > 1 and Z′ > 1 sets respectively, all differences are significant to greater than the 98% level according to Mann–Whitney tests. Related differences are also observed for the other parameters, for example we observe fewer rotatable bonds and a smaller connections:rotatable bond ratio for the parents compared with the whole dataset. This indicates that parent molecules are more awkward to pack in general, and this may therefore be one of the reasons that these compounds form co-crystals rather than crystallising in “pure” form.
| Number | Volume (v)/Å3 | Number of non-hydrogen atoms | Molecular area (ma)/Å2 | Accessible area (aa)/Å2 | v/ma | v/aa | Box volume/Å3 | Box volume–volume/Å3 | Number of connections | Number of rotatable bonds | Ratio rotatable:connections |
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Z′ = 1 | 915 | Mean | 268.93 | 21.254 | 270.79 | 425.82 | 0.95658 | 0.59847 | 738.3 | 469.4 | 22.963 | 3.091 | 0.13799 |
| Median | 241.4 | 19 | 254.61 | 407.19 | 0.94038 | 0.58589 | 567.2 | 330.9 | 20 | 2 | 0.11628 | ||
| Z′ > 1 | 259 | Mean | 237.47 | 18.981 | 245.69 | 392.36 | 0.94186 | 0.58226 | 627.2 | 389.7 | 20.591 | 2.371 | 0.11777 |
| Median | 213.55 | 17 | 227.05 | 366.61 | 0.93183 | 0.57446 | 519.5 | 292.8 | 18 | 2 | 0.1 | ||
| Z′ > 2 | 47 | Mean | 214.2 | 17.32 | 225.9 | 365 | 0.919 | 0.5603 | 561.6 | 347.4 | 18.81 | 1.66 | 0.0919 |
| Median | 188.4 | 16 | 208.5 | 346 | 0.9183 | 0.5556 | 435.6 | 235.4 | 17 | 1 | 0.0667 |
Of particular interest in molecular solid state chemistry is the solid form of pharmaceuticals, particularly pharmaceutical co-crystals.29–31Table 3 shows the same parameters as discussed previously calculated for “bioactive” molecules in the Cambridge Structural Database (i.e. compounds which the author has indicated may be of biological interest). The same trends with smaller and more awkward Z′ > 1 and Z′ > 2 molecules are observed. In addition parameters were calculated for “parent” bioactive molecules with a view to the use of these data for predictive purposes in the formation of pharmaceutical co-crystals. The values in Table 3 show that bioactive parent molecules, i.e. molecules which are known to form co-crystals (including hydrates), are at the more extreme end of the awkwardness scale, with parameters similar to the overall Z′ > 2 bioactive species. Hence novel drug substances that are awkwardly shaped according to the parameters described here should be regarded as likely to form pharmaceutical co-crystals.
| Number | Volume (v)/Å3 | Number of non-hydrogen atoms | Molecular area (ma)/Å2 | Accessible area (aa)/Å2 | v/ma | v/aa | Box volume/Å3 | Box volume–volume/Å3 | Number of connections | Number of rotatable bonds | Ratio rotatable:connections |
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Z′ = 1 | 4499 | Mean | 293.24 | 23.433 | 295.98 | 462.88 | 0.97455 | 0.6192 | 856.7 | 563.46 | 25.525 | 3.6081 | 0.14215 |
| Median | 277.11 | 22 | 284.92 | 448.18 | 0.95939 | 0.60931 | 772.37 | 490.36 | 25 | 3 | 0.13043 | ||
| Z′ > 1 | 702 | Mean | 279.28 | 22.39 | 284.09 | 446.96 | 0.96875 | 0.61174 | 798.8 | 519.5 | 24.39 | 3.162 | 0.12992 |
| Median | 265.62 | 21 | 273.93 | 431.7 | 0.95334 | 0.60138 | 705.7 | 439.5 | 24 | 3 | 0.11396 | ||
| Z′ > 2 | 66 | Mean | 248.4 | 19.97 | 257 | 409.3 | 0.9496 | 0.59121 | 696.3 | 447.9 | 21.67 | 2.758 | 0.1177 |
| Median | 243.2 | 19 | 250.16 | 401.3 | 0.9436 | 0.58506 | 663.4 | 400.6 | 21.5 | 2 | 0.0931 | ||
| Parents | 265 | Mean | 243.67 | 19.589 | 250.57 | 399.61 | 0.93827 | 0.58054 | 664.2 | 420.5 | 21.155 | 2.423 | 0.11638 |
| Median | 215.85 | 18 | 231.71 | 377.93 | 0.92064 | 0.56872 | 557.5 | 339.2 | 19 | 2 | 0.1 |
Conclusions
We have shown that size and shape are important considerations in determining whether a molecule will pack with Z′ > 1 or not. A number of parameters have been determined based on molecular shape and size and we have shown that molecules which crystallise with Z′ > 1 are in general smaller (by around 50 Å3) and have fewer rotatable bonds than molecules which crystallise with Z′ = 1. We have also shown that molecules which are known to form co-crystals are even smaller and less flexible. Thus formation of co-crystals or structures with Z′ > 1 is strongly linked to small, rigid awkwardly shaped molecules that have more constraints on their crystal packing requirements. These results have some predictive utility in determining the likelihood of hydrate formation in pharmaceuticals, for example.Acknowledgements
We are very grateful for the assistance of the Cambridge Crystallographic Data Centre for their help and advice with the searches used in this work. We would also like to thank Dr Nicholas Green, Oxford University, for helpful discussions and the EPSRC for funding.References
- J. W. Steed, CrystEngComm, 2003, 5, 169–179 RSC.
- A. Gavezzotti, CrystEngComm, 2008, 10, 389–398 RSC.
- J. Bernstein, J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2008, 8, 2011–2018 CrossRef CAS.
- K. M. Anderson, M. R. Probert, C. N. Whiteley, A. M. Rowland, A. E. Goeta and J. W. Steed, Cryst. Growth Des., 2009, 9, 1082–1087 CrossRef CAS.
- G. R. Desiraju, CrystEngComm, 2007, 9, 91–92 RSC.
- K. M. Anderson and J. W. Steed, CrystEngComm, 2007, 9, 328–330 RSC.
- G. S. Nichol and W. Clegg, CrystEngComm, 2007, 9, 959–960 RSC.
- A. D. Bond, CrystEngComm, 2008, 10, 411–415 RSC.
- C. P. Brock and L. L. Duncan, Chem. Mater., 1994, 6, 1307–1312 CrossRef CAS.
- K. M. Anderson, K. Afarinkia, H. W. Yu, A. E. Goeta and J. W. Steed, Cryst. Growth Des., 2006, 6, 2109–2113 CrossRef CAS.
- K. M. Anderson, A. E. Goeta and J. W. Steed, Cryst. Growth Des., 2008, 8, 2517–2524 CrossRef CAS.
- G. S. Nichol and W. Clegg, Cryst. Growth Des., 2009, 9, 1844–1850 CrossRef CAS.
- N. J. Babu and A. Nangia, CrystEngComm, 2007, 9, 980–983 RSC.
- N. J. Babu and A. Nangia, Cryst. Growth Des., 2006, 6, 1995–1999 CrossRef CAS.
- K. M. Anderson, A. E. Goeta and J. W. Steed, Inorg. Chem., 2007, 46, 6444–6451 CrossRef CAS.
- A. I. Kitaigorodskii, Organic Chemical Crystallography, Iliffe, London, 1962 Search PubMed.
- J. Fayos, Cryst. Growth Des., 2009, 9, 3142–3153 CrossRef CAS.
- F. H. Allen and O. Kennard, Chem. Des. Autom. News, 1993, 8, 31–37 Search PubMed.
- Cambridge Structural Database Version 5.29 with 2 updates.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- M. L. Connolly, J. Mol. Graphics, 1993, 11, 139–143 CrossRef CAS.
- A. Gavezzotti, J. Am. Chem. Soc., 1985, 107, 962–967 CrossRef CAS.
- F. H. Allen, W. D. S. Motherwell, P. R. Raithby, G. P. Shields and R. Taylor, New J. Chem., 1999, 23, 25–34 RSC.
- E. Pidcock and W. D. S. Motherwell, Cryst. Growth Des., 2004, 4, 611–620 CrossRef CAS.
- Calculator Plugins, Marvin 4.1.12, ChemAxon, 2007, http://www.chemaxon.com/ Search PubMed.
- P. Groth and D. Semmingsen, Acta Chem. Scand., Ser. B, 1976, 30, 737.
- K. C. Nicolaou, C.-K. Hwang and D. A. Nugiel, J. Am. Chem. Soc., 1989, 111, 4136–4137 CrossRef CAS.
- K. J. McCullough, Y. Nonami, A. Masuyama, M. Nojima, H.-S. Kim and Y. Wataya, Tetrahedron Lett., 1999, 40, 9151–9155 CrossRef CAS.
- P. Vishweshwar, J. A. McMahon, J. A. Bis and M. J. Zaworotko, J. Pharm. Sci., 2006, 95, 499–516 CrossRef.
- O. Almarsson and M. J. Zaworotko, Chem. Commun., 2004, 1889–1896 RSC.
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439–448 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: A summary of all calculated parameters and statistical information. See DOI: 10.1039/c0ce00172d |
| ‡ For a full list of statistical parameters and explanation see the ESI†. |
| § “Parent” molecules in this context are analogous to the host in lattice host/guest compounds. |
| This journal is © The Royal Society of Chemistry 2011 |