DOI:
10.1039/C0CE00142B
(Paper)
CrystEngComm, 2011,
13, 293-305
N-Heterocyclic carbene
copper(I), mercury(II) and silver(I) complexes containing durene linker: synthesis and structural studies†
Received
28th April 2010
, Accepted 8th July 2010
First published on 6th September 2010
Abstract
The dibenzimidazolium salts bis[N-(alkyl)benzimidazoliumylmethyl]durene·2X (1a: alkyl = C2H5, X = Br; 1b: alkyl = C2H5, X = PF6; 1c: alkyl = n-C3H7, X = Cl; 1d: alkyl = n-C4H9, X = I; 1e: alkyl = 1-PyCH2, X = Br; durene = 1,2,4,5-tetramethylbenzene) and the diimidazolium salt bis[N-(butyl)imidazoliumylmethyl]durene hexafluorophosphate (1f), and their seven NHC copper(I), mercury(II) and silver(I) complexes, [durene(CH2bimyEt)2Cu2I2] (2b), [durene(CH2bimyEt)2HgBr]·0.5[HgBr4] (2c), [durene(CH2bimynBu)2Hg2(CHCN)][HgI4] (2d), [durene(CH2imynBu)2Ag2(CH2CN)2] (2e), [durene(CH2bimyPyCH2)2Ag2Br2] (2f), [durene(CH2bimyEt)2Ag][PF6] (2g) and [durene(CH2bimynBu)2Ag2(OH)2] (2h), as well as one anionic complex [durene(CH2benzimidazoliumylnPr)2][PdCl3(DMSO)]2 (2a) (bimy = benzimidazol-2-ylidene, imy = imidazol-2-ylidene), have been prepared and characterized. These compounds adopt two diverse conformations (i.e., cis- and trans-conformations). In 2d and 2e, an interesting phenomenon is that the α-carbon atoms of deprotonated acetonitrile ([CHCN]2− for 2d and [CH2CN]− for 2e) participate in coordination with metal ions. In the crystal packings of these complexes, 1D supramolecular chains or 2D supramolecular layers are formed via intermolecular weak interactions, including π–π interactions, hydrogen bonds and C–H⋯π contacts.
Introduction
N-Heterocyclic carbenes (NHCs) have received a burst of attention as new ligands since the isolation of the first free stable N-heterocyclic carbenes by Arduengo and coworkers.1 A variety of relevant metal complexes have been synthesized through deprotonation of N,N′-disubstituted benzimidazolium (or imidazolium) salts.2 Strong electron-donating ability of NHC ligands leads to their metal complexes of high stability to heat, moisture and air.3 NHC-metal complexes have shown to be efficient catalysts for some organic reactions, such as Heck, Suzuki and Kumada couplings, and olefin metathesis.4 Of known NHC-metal complexes, the NHC copper(I),5mercury(II)6 and silver(I)7 complexes have played important roles in the development of N-heterocyclic carbene chemistry. Particularly, N-heterocyclic carbene silver(I) complexes can be used as carbene transfer reagents for the synthesis of Ni, Pd, Pt, Cu, Au, Rh, Ir and Ru carbene complexes, which affords a convenient method for the preparation of these metal carbene complexes.8
In the family of NHCs, a number of functional bidentate bis-NHC ligands and their metal complexes have been studied. Among these bidentate bis-NHC ligands, a pair of NHC moieties are connected by a bridging linker, such as alkyl,9 aryl,10pyridinediyl11 and ether chain.12 This type of ligand along with metal ions readily form cyclic NHC-metal complexes, which may have potential application in organic reactions as catalyst and in molecular recognition as an acceptor. We are interested in bidentate bis-NHC ligands and their metal complexes. Recently, some silver(I) and mercury(II) complexes based on bidentate bis-NHC ligands with alkyl or oligoether linkers have been reported by us.13 We report herein the synthesis, structures and fluorescent emission spectra of NHC-metal complexes containing durene linker, [durene(CH2bimyEt)2Cu2I2] (2b), [durene(CH2bimyEt)2HgBr]·0.5[HgBr4] (2c), [durene(CH2bimynBu)2Hg2(CHCN)][HgI4] (2d), [durene(CH2imynBu)2Ag2(CH2CN)2] (2e), [durene(CH2bimyPyCH2)2Ag2Br2] (2f), [durene(CH2bimyEt)2Ag][PF6] (2g) and [durene(CH2bimynBu)2Ag2(OH)2] (2h), and one anionic complex [durene(CH2benzimidazoliumylnPr)2][PdCl3(DMSO)]2 (2a) (bimy = benzimidazol-2-ylidene, imy = imidazol-2-ylidene, durene = 1,2,4,5-tetramethylbenzene) is also described in the paper. In addition, we also report two diverse conformations (i.e., cis- and trans-conformations) of the dibenzimidazolium salts and their metal complexes. Moreover, the α-carbon atoms of deprotonated acetonitrile in 2d and 2e participate in coordination with metal ions, and this observation is also interesting in organometallic chemistry.
Results and discussion
Synthesis and general characterization of precursors 1a–1f and complexes 2a–2h
The dibenzimidazolium salts, bis[N-(alkyl)benzimidazoliumylmethyl]durene halogenide (1a: alkyl = Et, halogenide = Br; 1c: alkyl = nPr, halogenide = Cl; 1e: alkyl = 1-PyCH2, halogenide = Br) were prepared from benzimidazole by alkylation with 1-bromoethane or 2-chloromethylpyridine followed by quarterization with bis(bromomethyl)durene in sequence (method 1, Scheme 1). The bis[N-(ethyl)benzimidazoliumylmethyl]durene hexafluorophosphate (1b) and the bis[N-(n-butyl)benzimidazoliumylmethyl]durene iodide (1d) were obtained in two steps. The first step is in a manner similar to that of 1a, and the second step is through an anionic exchange with ammonium hexafluorophosphate in methanol for 1b and with sodium iodide in acetone for 1d (method 2, Scheme 1). The diimidazolium salt bis[N-(n-butyl)imidazoliumylmethyl]durene hexafluorophosphate (1f) was prepared in a manner similar to that for 1b (Scheme 2). Precursors 1a–1f are stable towards air and moisture, soluble in polar organic solvents such as dichloromethane, acetonitrile and methanol, and scarcely soluble in benzene, diethyl ether and petroleum ether. In the 1H NMR spectra of 1a–1f, the benzimidazolium (or imidazolium) proton signals (NCHN) appear at δ = 9.90–10.60 ppm, which are consistent with the chemical shifts of reported benzimidazolium (or imidazolium) salts.2
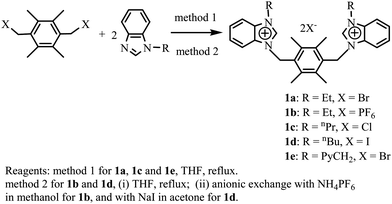 |
| | Scheme 1 Preparation of precursors 1a–1e. | |
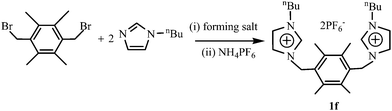 |
| | Scheme 2 Preparation of precursor 1f. | |
Synthetic methods of complexes 2a–2h are shown in Scheme 3. The reaction of 1c with [PdCl2(CH3CN)2] afford an anionic complex [durene(CH2benzimidazoliumylnPr)2][PdCl3(DMSO)]2 (2a) (Scheme 3(1)). 1b was treated with copper(I) iodide and silver oxide, respectively, to afford complexes [durene(CH2bimyEt)2Cu2I2] (2b) and [durene(CH2bimyEt)2Ag][PF6] (2g) (Scheme 3(2) and (7)). Complex [durene(CH2bimyEt)2HgBr]0.5[HgBr4] (2c) was prepared by the reaction of 1a with anhydrous mercury(II) bromide (Scheme 3(3)). 1d was treated with anhydrous mercury(II) iodide and silver acetate, respectively, to afford complexes [durene(CH2bimynBu)2Hg2(CHCN)][HgI4] (2d) and [durene(CH2bimynBu)2Ag2(OH)2] (2h) (Scheme 3(4) and (8)). Complex [durene(CH2imynBu)2Ag2(CH2CN)2] (2e) was prepared via reaction of 1f with silver oxide (Scheme 3(5)). Complex [durene(CH2bimyPyCH2)2Ag2Br2] (2f) was prepared via reaction of 1e with silver oxide (Scheme 3 (6)). Complexes 2a–2h are stable towards air and moisture, soluble in DMSO and insoluble in diethyl ether and hydrocarbon solvents. In the 1H NMR spectra of 2b–2h, the disappearance of the resonances for the benzimidazolium (or imidazolium) protons (NCHN) shows the formation of the expected metal carbene complexes, and the chemical shifts of other hydrogens are similar to those of corresponding precursors. The 1H NMR spectrum of anionic complex 2a is similar to that of corresponding precursor 1c. In 13C NMR spectra, the signals for the carbene carbon of 2b–2d and 2f–2h appear at 174.4–187.3 ppm, which are similar to known metal carbene complexes.5–7 The signal of carbene carbon in 2e was not observed. Similar results have been reported for some carbene-silver(I) complexes, which may result from the fluxional behavior of the NHC complexes.14 For complexes 2d and 2e containing deprotonated acetonitriles, the signals of carbon of cyano group (CN) appear at 113.4 ppm for 2d and 112.7 ppm for 2e, and the signals of α-carbon atom of acetonitrile appear at 30.3 ppm for 2d and 31.2 ppm for 2e. IR spectra contain the characteristic bands of cyano groups at 2191 cm−1 for 2d and 2166 cm−1 for 2e. These values are comparable to those of the reported complexes containing cyano groups.15
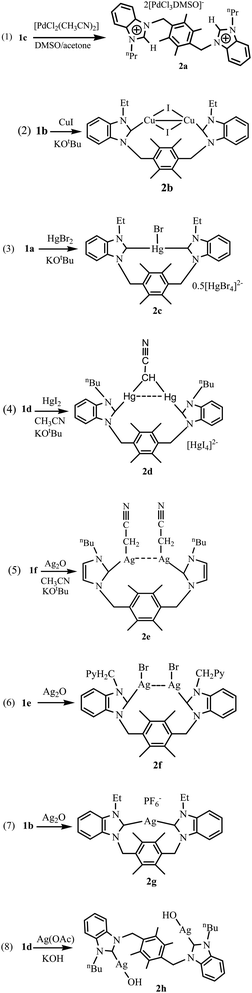 |
| | Scheme 3 Preparation of complexes 2a–2h. | |
Structures of dibenzimidazolium salt 1e and complexes 2a–2h
Molecular structures of precursor 1e and complexes 2a–2h were demonstrated by X-ray analysis. The crystals of 1e·CH3CN, 2a, 2b·0.5CH3CN, 2c·0.5DMSO, 2d·CH3CN, 2e, 2f·DMSO, 2g·0.5H2O and 2h suitable for X-ray diffraction were obtained by slow diffusion of diethyl ether into their DMSO/CH3CN solution. In complexes 2c and 2g, each cation contains a mononuclear macrometallocycle formed by one bidentate chelate carbene ligand and one metal atom, whereas complexes 2b, 2d, 2e and 2f possess dinuclear macrometallocyclic structures as shown in Fig. 1(a), Fig. 2(a) and Fig. 6(a)–Fig. 9(a). In each of 1e and 2b–2g, two Rbimi (or Rimi) arms lie in the same side of durene plane to adopt a cis-conformation. The two benzimidazole (or imidazole) rings within each molecule form the dihedral angles varying from 5.1° to 56.0°, and they form the dihedral angles in the ranges of 76.4–91.9° and 81.7–96.9° with the centric durene plane, respectively (the related data being given in Table 1). In contrast, the molecular structures of complexes 2a and 2h are different from those of above-mentioned compounds, and two Rbimi arms within each molecule in 2a and 2h lie in the opposite sides of durene plane to adopt an open trans-conformation as shown in Fig. 3(a) and Fig. 4(a). The X-ray crystal structural analyses of 2a and 2h show that there are inversion centers in the complexes. Additionally, two benzimidazole rings within each molecule are parallel, and they form the dihedral angles 82.3(5)° for 2a and 88.8(2)° for 2h with the centric durene plane, respectively. In 2b–2h, the internal ring angles (N–C–N) at the carbene centers are in the range of 102.8–107.5°, which are comparable to those of the known NHC-metal complexes,2 but these values are smaller than the corresponding values (110.9–111.1°) of precursor 1e and anionic complex 2a.
| Compounds |
A
|
B
|
|
1e
|
9.5(1) |
86.4(1), 81.8(1) |
|
2b
|
5.1(3) |
82.0(2), 86.4(1) |
|
2c
|
6.6(1) |
88.1(1), 84.3(4) |
|
2d
|
56.0(2) |
76.4(2), 81.7(1) |
|
2e
|
9.0(1) |
81.7(1), 90.5(1) |
|
2f
|
6.4(2) |
91.9(2), 92.9(3) |
|
2g
|
18.6(1) |
78.7(1), 96.9(2) |
 |
| | Fig. 1
(a) Perspective view of 2b and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms were omitted for clarity. Selected bond lengths (Å) and angles (°): Cu(1)–I(1) 2.660(1), Cu(1)–I(2) 2.600(1), Cu(1)–Cu(2) 2.668(1); I(1)–Cu(1)–Cu(2) 57.8(4), N(1)–C(7)–N(2) 106.7(7), N(3)–C(22)–N(4) 105.6(6). (b) The 2D supramolecular layers by the π–π interactions and C–H⋯π contacts in 2b. All hydrogen atoms except those participating in the C–H⋯π contacts were omitted for clarity. | |
 |
| | Fig. 2
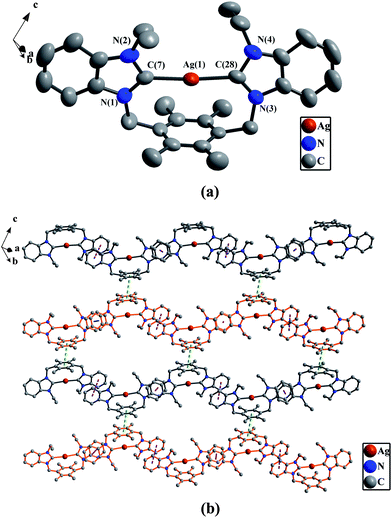
(a) Perspective view of 2g and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms and anions 2PF6− were omitted for clarity. Selected bond lengths (Å) and angles (°): Ag(1)–C(7) 2.121(2), Ag(1)–C(28) 2.100(2); N(1)–C(7)–N(2) 105.5(4), N(3)–C(28)–N(4) 106.1(4). (b) The 2D supramolecular layers by the both types of π–π interactions in 2g. All hydrogen atoms were omitted for clarity. | |
 |
| | Fig. 3
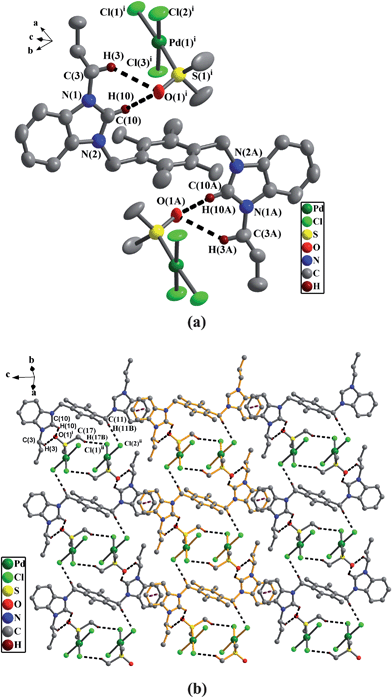
(a) Perspective view of 2a and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms except those participating in the C–H⋯O hydrogen bonds were omitted for clarity. Selected bond lengths (Å) and angles (°): N(1)–C(10) 1.332(8), Pd(1)–S(1) 2.239(1), Pd(1)–Cl(1) 2.341(1), Pd(1)–Cl(2) 2.328(1), Pd(1)–Cl(3) 2.348(1); Cl(1)–Pd(1)–Cl(2) 92.1(6), Cl(1)–Pd(1)–Cl(3) 89.0(6), Cl(2)–Pd(1)–Cl(3) 176.9 (7), S(1)–Pd(1)–Cl(2) 88.6(6), S(1)–Pd(1)–Cl(3) 90.2(6), N(1)–C(10)–N(2) 111.0(6). (b) The 2D supramolecular layers by the π–π interactions, C–H⋯O hydrogen bonds and C–H⋯Cl hydrogen bonds in 2a. All hydrogen atoms except those participating in the hydrogen bonds were omitted for clarity. | |
 |
| | Fig. 4
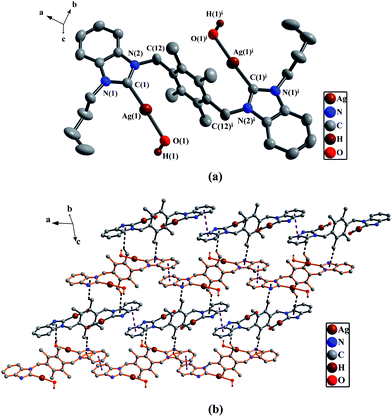
(a) Perspective view of 2h and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms except those in the OH were omitted for clarity. Selected bond lengths (Å) and angles (°): Ag(1A)–C(1A) 2.101(3), O(1A)–H(1A) 0.820(1); N(1A)–C(1A)–N(2A) 105.7(3). (b) The 2D supramolecular layers by the π–π interactions and C–H⋯π contacts in 2h. All hydrogen atoms except those participating in the C–H⋯π contacts and in the OH, and all butyl groups on the nitrogen atoms were omitted for clarity. | |
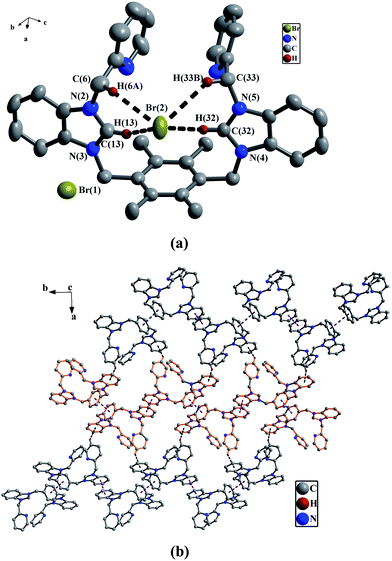
In precursor 1e, one bromine atom and four hydrogen atoms on two PyCH2bimi arms form four C–H⋯Br hydrogen bonds16 as shown in Fig. 5(a) (the data of hydrogen bonds being given in Table 2). Anionic complex 2a is different from precursor 1e, one oxygen atom from DMSO of anionic [PdCl3(DMSO)] and two hydrogen atoms on one nPrbimi arm in 2a form two C–H⋯O hydrogen bonds17 as shown in Fig. 3(a) (Table 2). In anionic unit [PdCl3(DMSO)], the center Pd is coordinated to three chlorine atoms and one sulfur atom of DMSO to adopt a planar square array. The bond distances of Pd–Cl are in the range of 2.328–2.348 Å.
Table 2 H-Bonding geometry (Å, °) for 1e, 2a, 2d and 2ea
| |
D–H⋯A |
D–H |
H⋯A |
D⋯A |
D–H⋯A |
|
Symmetry code: i = 1 − x, 1 − y, 1 − z; ii = −1 + x, 2 + y, 2 + z for 2a. i = x, −1 + y, z for 2d. i = 1 + x, y, z for 2e.
|
|
1e
|
C(6)–H(6A)⋯Br(2) |
0.97(4) |
2.781(5) |
3.654(3) |
150.0(2) |
| C(13)–H(13)⋯Br(2) |
0.93(3) |
2.814(5) |
3.572(4) |
139.4(2) |
|
C(33)–H(33B)⋯Br(2) |
0.97(4) |
3.135(6) |
3.974(4) |
145.5(2) |
| C(32)–H(32)⋯Br(2) |
0.93(3) |
3.155(7) |
3.723(4) |
121.2(4) |
|
2a
|
C(3)–H(3)⋯O(1)i |
0.97(8) |
2.668(8) |
3.463(1) |
139.4(4) |
| C(10)–H(10)⋯O(1)i |
0.93(7) |
2.679(8) |
3.227(1) |
118.4(4) |
|
C(11)–H(11B)⋯Cl(2)ii |
0.97(8) |
2.721(1) |
3.608(7) |
152.3(5) |
|
C(17)–H(17B)⋯Cl(1)ii |
0.96(9) |
2.829(2) |
3.518(1) |
129.5(6) |
|
2d
|
C(35)–H(35)⋯I(1)i |
0.98(1) |
3.189(1) |
3.923(3) |
133.1(1) |
| C(12)–H(12)⋯I(4) |
0.97(1) |
3.010(2) |
3.689(1) |
128.0(3) |
|
2e
|
C(21)–H(21)⋯N(6)i |
0.93(1) |
2.334(1) |
3.219(1) |
158.9(3) |
 |
| | Fig. 5
(a) Perspective view of 1e and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms except those participating in the C–H⋯Br hydrogen bonds were omitted for clarity. Selected bond lengths (Å) and angles (°): C(13)–N(2) 1.317(4); N(2)–C(13)–N(3) 111.1(3), N(4)–C(32)–N(5) 110.9(2). (b) The 2D supramolecular layers by the π–π interactions and C–H⋯π contacts in 1e. All hydrogen atoms except those participating in the C–H⋯π contacts, all methyl groups on the durenes and all bromine atoms were omitted for clarity. | |
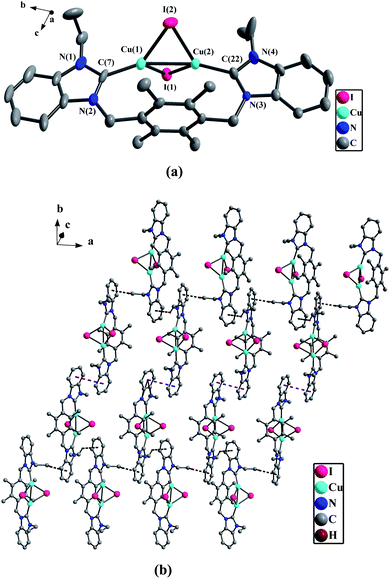
In NHC-copper(I) complex 2b (Fig. 1(a)), each copper(I) atom is tetra-coordinated with one carbene carbon atom (C(carbene)), two iodine atoms and another copper(I) atom. Two copper(I) atoms are linked by two bridging iodine atoms to form a distorted Cu2I2 quadrangular arrangement. In the Cu2I2 core, the dihedral angle between the Cu(1)–I(1)–Cu(2) plane and Cu(1)–I(2)–Cu(2) plane is 44.3(1)°. The Cu(1)–Cu(2) distance of 2.668(1) Å (van der Waals radii of copper = 1.86 Å) indicates a strong metal–metal interaction.18 The bond angles of two Cu–I–Cu are 61.3(4)° and 61.5(3)°. The Cu–I distance of 2.57–2.66 Å falls in the regular range.19 Additionally, the distances of two Cu–C bonds are 1.962(8) Å and 1.929(1) Å, and the bond angles of two C–Cu–Cu are 159.4(3)° and 161.5(1)°. These values are comparable to those of the reported NHC copper(I) complexes.8
In two NHC-mercury(II) complexes 2c and 2d (Fig. 6(a) and Fig. 7(a)), the bond distances of Hg–C(carbene) vary from 2.082(8) Å to 2.101(6) Å, which are similar to those of known NHC-mercury(II) complexes.6 The anionic units of 2c and 2d are tetrahedral [HgBr4]2− and [HgI4]2−, respectively, in which the values of bond lengths and bond angles fall in regular range.20 In 2c, the mercury(II) atom within the cationic macrometallocycle is tri-coordinated with two carbene carbons and one bromine atom. The C(1)–Hg(1)–C(22) array is nearly linear with the bond angle of 173.7(3)°. The two ethyl chains and one bromine atom in the cation are stretching in the same direction. In the cationic unit of 2d, the Hg(2) and Hg(3) are dicoordinated with one carbene carbon atom and one α-carbon atom (C(α)) of deprotonated acetonitrile. The bond distances of two Hg–C(carbene) (2.082(8) Å and 2.084(8) Å) are slightly shorter than those of Hg–C(α) (2.090(8) Å and 2.112(8) Å). The Hg(2)⋯Hg(3) separation of 3.338(5) Å (van der Waals Radii of mercury = 1.70 Å) indicates a weak metal–metal interaction.20 Two C(carbene)–Hg–C(α) arrays are nearly linear with the bond angles of 172.8(4)° and 176.9(3)°. The bond angles of two Hg–C(35)–C(36) are 113.5(6)° and 114.7(7)°, and the bond angle of Hg(2)–C(35)–Hg(3) is 105.2(3)°. Notably, the cyanomethyl moiety (C(35)–C(36)–N(5)) is nearly linear with a bond angle of 177.9(1)°. The C(35)–C(36) bond distance (1.398(1) Å) is shorter than that of the regular C–C single bond (1.54–1.59 Å), and it has partial double-bond character. The C(36)–N(5) bond distance (1.167(1) Å) is similar to that of other complexes containing cyano groups, and this value is expected for a triple bond with a small amount of double bond character.21 The two butyl chains on the nitrogen atoms are stretching in the opposite directions.
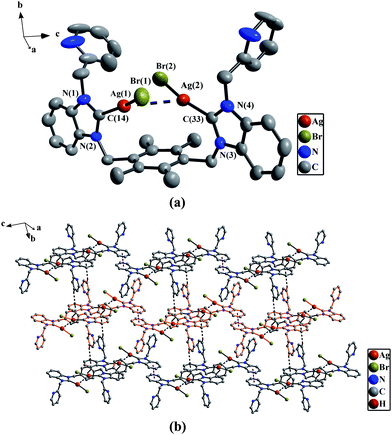
![(a) Perspective view of 2c and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms and anion 0.5[HgBr4]2− were omitted for clarity. Selected bond lengths (Å) and angles (°): Hg(1)–C(1) 2.097(6), Hg(1)–Br(1) 3.076(1); C(1)–Hg(1)–C(22) 173.7(3), C(1)–Hg(1)–Br(1) 90.4(2), N(1)–C(1)–N(2) 107.5(5), N(3)–C(22)–N(4) 108.2(6). (b) The 2D supramolecular layers by the weak Hg⋯Br bonds, π–π interactions and C–H⋯π contacts in 2c. All hydrogen atoms except those participating in the C–H⋯π contacts were omitted for clarity.](/image/article/2011/CE/c0ce00142b/c0ce00142b-f6.gif) |
| | Fig. 6
(a) Perspective view of 2c and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms and anion 0.5[HgBr4]2− were omitted for clarity. Selected bond lengths (Å) and angles (°): Hg(1)–C(1) 2.097(6), Hg(1)–Br(1) 3.076(1); C(1)–Hg(1)–C(22) 173.7(3), C(1)–Hg(1)–Br(1) 90.4(2), N(1)–C(1)–N(2) 107.5(5), N(3)–C(22)–N(4) 108.2(6). (b) The 2D supramolecular layers by the weak Hg⋯Br bonds, π–π interactions and C–H⋯π contacts in 2c. All hydrogen atoms except those participating in the C–H⋯π contacts were omitted for clarity. | |
![(a) Perspective view of 2d and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms except that in the [CHCN]2− were omitted for clarity. Selected bond lengths (Å) and angles (°): N(5)–C(36) 1.167(1), C(35)–C(36) 1.398(1), Hg(3)–C(11) 2.084(8); N(5)–C(36)–C(35) 177.9(1), C(30)–Hg(2)–C(35) 176.9(3), C(36)–C(35)–Hg(3) 113.5(6), C(36)–C(35)–Hg(2) 114.7(7), N(1)–C(11)–N(2) 107.5(7), N(3)–C(30)–N(4) 106.9(7). (b) The 1D supramolecular chains by the C–H⋯I hydrogen bonds in 2d. All hydrogen atoms except those participating in the C–H⋯I hydrogen bonds were omitted for clarity.](/image/article/2011/CE/c0ce00142b/c0ce00142b-f7.gif) |
| | Fig. 7
(a) Perspective view of 2d and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms except that in the [CHCN]2− were omitted for clarity. Selected bond lengths (Å) and angles (°): N(5)–C(36) 1.167(1), C(35)–C(36) 1.398(1), Hg(3)–C(11) 2.084(8); N(5)–C(36)–C(35) 177.9(1), C(30)–Hg(2)–C(35) 176.9(3), C(36)–C(35)–Hg(3) 113.5(6), C(36)–C(35)–Hg(2) 114.7(7), N(1)–C(11)–N(2) 107.5(7), N(3)–C(30)–N(4) 106.9(7). (b) The 1D supramolecular chains by the C–H⋯I hydrogen bonds in 2d. All hydrogen atoms except those participating in the C–H⋯I hydrogen bonds were omitted for clarity. | |
In four NHC-silver(I) complexes 2e–2h, each silver(I) atom possesses a dicoordinated geometry, and the bond distances of Ag–C(carbene) vary from 2.058(7) Å to 2.121(2) Å. These values are comparable to known NHC-silver(I) complexes.5 As for 2e, the data of all bond lengths and angles in the [CH2CN]− unit are comparable to the corresponding values in [CHCN]2− unit of 2d. The two C(carbene)–Ag–C(α) arrays are nearly linear with the bond angles of 173.0(3)° and 173.6(3)°. The bond distances of two Ag–C(carbene) (2.058(7) Å and 2.066(7) Å) are slightly longer than those of two Ag–C(α) (1.913(1) Å and 1.975(7) Å). The Ag⋯Ag separation of 3.304(1) Å (van der Waals Radii of silver = 1.72 Å) indicates a weak metal–metal interaction.22 The two n-butyl groups on the nitrogen atoms are stretching in the same direction, whereas the two [CH2CN]− units are stretching in the opposite directions.
The macrometallocycle of 2f is analogous to that of 2e, however, the Ag⋯Ag separation of 3.113(7) Å in 2f is shorter than that of 3.304(1) Å in 2e. The bond distances of two Ag–Br in 2f are 2.492(6) Å and 2.437(7) Å, and the bond angles of two C–Ag–Br are 159.6(1)° and 166.9(1)°. The two bromine atoms are stretching in the opposite directions. The dihedron angle between pyridine ring and adjacent benzimidazole is 88.2(1)° (73.3(1)° for the corresponding precursor 1e), and the dehedral angle between two pyridine rings is 64.3(1)° (37.5(2)° for 1e). Compared to 1e, some relevant bond lengths and angles of complex 2f occur relatively large changes due to the introduction of metal silver(I) atom. As for 2g, the C(7)–Ag(1)–C(28) array is almost linear with the bond angle of 175.8(6)°. The two ethyl chains on the nitrogen atoms are stretching in the opposite directions.
In the trans structure of 2h, the hydroxy groups linked on silver(I) atoms come from KOH. Two [nBubimyAg(OH)] units are placed in the opposite sides of the durene plane. An n-butyl group and a AgOH part on same benzimidazole ring are stretched along the benzimidazole ring plane in the same direction, but they point to the opposite directions as compared with the corresponding parts on another benzimidazole ring. The angles of two C–Ag–O (177.1(1)°) and the bond distances of two Ag–O (2.274(3) Å) are the same, respectively, and they fall into the regular range.23
The conformations of the dibenzimidazole salts and their metal complexes
As shown in Scheme 4, the dibenzimidazole salts containing durene linker adopt two diverse conformations (i.e., cis-conformation I and trans-conformation II) according to the different arrangements of two Rbimi arms. The conformations I and II can converse each other in the solution, and their ratio are mostly relative to two factors: (1) the weak interactions between anions and two Rbimi arms; (2) the steric hindrance between two Rbimi arms. The former may have larger effect on the ratio of conformation than the latter. When there exist strong interactions between one anion and two Rbimi arms at the same time in a compound, the two arms lie in same side of durene plane, and the compound adopts mainly cis-conformation (e.g., 1e). When there exist strong interactions between one anion and only one Rbimi arm in a compound, the two arms lie in opposite sides of durene plane due to steric hindrance, and the compound adopts mainly trans-conformation (e.g., 2a).
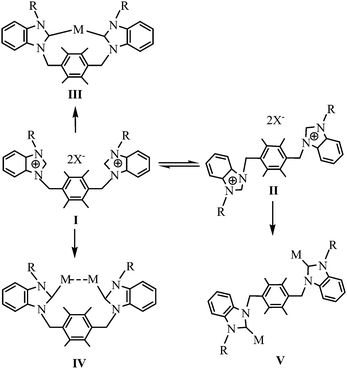 |
| | Scheme 4 The diverse conformations of dibenzimidazole and their metal complexes | |
Similarly, NHC metal complexes formed viadibenzimidazole salts and metal compounds possess both cis-conformations III or IV (e.g., 2b–2g) and trans-conformation V (e.g., 2h), and their conformations may depend mainly on the conformations of the corresponding precursors. Additionally, NHC metal complexes possessing cis-conformation have usually two different structures, namely, single nuclear macrocycle complexes III (e.g., 2c and 2g) and dinuclear macrocycle complexes IV (e.g., 2b, 2d, 2e and 2f). In dinuclear macrocycle complexes, metal–metal interactions are observed.
The different formulae of deprotonated acetonitrile
An interesting phenomenon during the preparation of 2d or 2e is that α-hydrogen atoms of acetonitrile are deprotonated due to the presence of strong base KOtBu to generate doubly deprotonated acetonitrile ([CHCN]2−) for 2d and singly deprotonated acetonitrile ([CH2CN]−) for 2e. The α-carbon atom (C(α)) of these deprotonated acetonitrile along with didentate carbene ligands are coordinated to metal atoms to form complexes 2d and 2e, respectively. A distinction is that the α-carbon atom of [CHCN]2− in 2d is bonded to two mercury(II), whereas the α-carbon atom of [CH2CN]− in 2e is bonded to one silver(I). From the structures of complexes 2d and 2e, as well as reported compounds containing deprotonated acetonitrile, such as (Me3Ge)2CHCN,21a[Pt(CH2CN)(PMe2Ph)3]PF6,21bPdCl(CH2CN)(PPh3)221c and Fe3(Cp)3(μ-CO)3[μ3-CCN],21d we know that three α-hydrogen atoms of acetonitrile may be deprotonated one by one in the presence of base to form singly, doubly and triply deprotonated acetonitrile (i.e., [CH2CN]−, [CHCN]2− and [CCN]3−), respectively. The different formulae of deprotonated acetonitriles probably are relative to the amount of base and the type of coordinated metal ions in the system of reaction.
The crystal packings of precursor 1e and complexes 2a–2h
Analyses of the crystal packings of 1e and 2a–2h reveal that 2D supramolecular layers of 1e, 2a, 2b, 2c and 2f–2h, as well as 1D supramolecular chains of 2d and 2e are formed via intermolecular weak interactions, including π–π stacking interactions, hydrogen bonds, C–H⋯π contacts and weak Hg⋯Br bonds (the data of hydrogen bonds being given in Table 2, and the related data of π–π interactions and C–H⋯π contacts being given in Table 3).
Table 3 Distances (Å) of π–π interactions, and distances (Å) and angles (°) of C–H⋯π contacts for 1e, 2a–2c and 2e–2h
In 1e, the intermolecular weak interactions involve the C–H⋯π contacts24 (the hydrogen atoms being from pyridine rings) and two types of aromatic π–π stacking interactions.25 In these two types of π–π interactions, one is from intermolecular benzene rings of durenes, and another is from intermolecular benzimidazole rings (Fig. 5(b)). An interesting feature in the crystal packing of 2a is that 1D supramolecular chains are formed by the π–π stacking interactions from intermolecular benzimidazole rings. In addition, an anionic dimer with a 12-membered ring [PdCl3(DMSO)]22− are formed by two anions [PdCl3(DMSO)]−via two C–H⋯Cl hydrogen bonds (C(17)–H(17B)⋯Cl(1)).26 These anionic dimeric units ([PdCl3(DMSO)]2) are packed between the 1D chains to extend 1D chains into 2D supramolecular layers via new C–H⋯Cl hydrogen bonds (C(11)–H(11B)⋯Cl(2)) as shown in Fig. 3(b).
In 2b, 2f and 2h, there exist the π–π stacking interactions from intermolecular benzimidazole rings and C–H⋯π contacts (the hydrogen atoms being from ethyl groups on nitrogen atoms for 2b, from pyridine rings for 2f, and from methyl groups of durenes for 2h) (Fig. 1(b) for 2b, Fig. 8(b) for 2f and Fig. 4(b) for 2h). In 2c, the intermolecular weak interactions include the π–π stacking interactions from intermolecular benzimidazole rings, C–H⋯π contacts (the hydrogen atoms being from methyl groups of durenes) and weak Hg⋯Br bonds (Hg⋯Br distance = 3.602(2) Å)20 (Fig. 6(b)). In 2g, both types of aromatic π–π stacking interactions are observed (Fig. 2(b)). One type is formed via intermolecular benzene rings of durenes, and another is formed via intermolecular benzimidazole rings.
 |
| | Fig. 8
(a) Perspective view of 2f and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms were omitted for clarity. Selected bond lengths (Å) and angles (°): Ag(1)–C(14) 2.115(4), Ag(2)–C(33) 2.081(4); C(14)–Ag(1)–Br(1) 159.6(1), C(33)–Ag(2)–Br(2) 166.9 (1), N(1)–C(14)–N(2) 105.2(3). (b) The 2D supramolecular layers by the π–π interactions and C–H⋯π contacts in 2f. All hydrogen atoms except those participating in the C–H⋯π contacts and all methyl groups on the durenes were omitted for clarity. | |
Different from the compounds above-mentioned, the adjacent cationic units in 2d are held together by anionic units [HgI4]2−via C–H⋯I hydrogen bonds27 to form 1D supramolecular chains (Fig. 7(b)). The double-strand 1D supramolecular chains in 2e are formed via C–H⋯N hydrogen bonds28 (the hydrogen atoms being from imidazole rings, and nitrogen atoms being from cyano groups) and π–π stacking interactions from intermolecular imidazole rings (Fig. 9(b)).
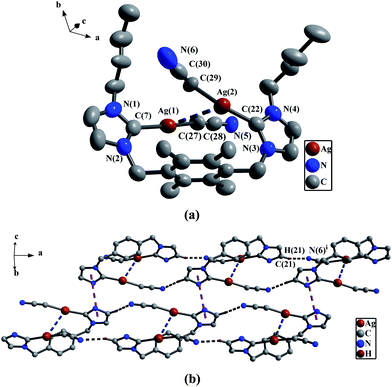 |
| | Fig. 9
(a) Perspective view of 2e and anisotropic displacement parameters depicting 30% probability. All hydrogen atoms were omitted for clarity. Selected bond lengths (Å) and angles (°): C(27)–C(28) 1.259(5), N(5)–C(28) 1.071(9), Ag(1)–C(7) 2.058(7), C(29)–C(30) 1.276(5), N(6)–C(30) 1.092(5); Ag(2)–C(22) 2.066(7); N(5)–C(28)–C(27) 178.2(1), C(28)–C(27)–Ag(1) 165.7(6), C(30)–C(29)–Ag(2) 158.9(1), N(3)–C(22)–N(4) 103.3(6), N(6)–C(30)–C(29) 169.1(1), N(1)–C(7)–N(2) 102.8(6). (b) The 1D double-strand supramolecular chains by the π–π interactions and C–H⋯N hydrogen bonds in 2e. All hydrogen atoms except those participating in the C–H⋯N hydrogen bonds, all methyl groups on durenes and all butyl groups on the nitrogen atoms were omitted for clarity. | |
Fluorescent emission spectra of precursors 1a, 1e and 1f, and complexes 2b and 2d–2f
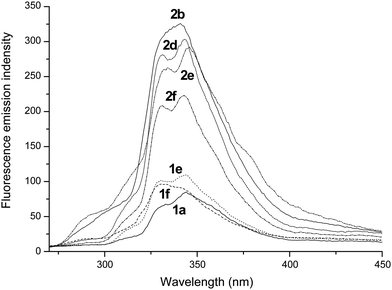
As shown in Fig. 10, the fluorescent emission spectra of precursors 1a, 1e and 1f and complexes 2b and 2d–2f in dichloromethane at room temperature are obtained upon excitation at 230 nm (the fluorescent emission spectra of 1b–1d and 2a are similar to that of 1a; the fluorescent emission spectra of 2c, 2g and 2h are similar to that of 2b). Precursors 1a, 1e and 1f exhibit double emission bands in the region of 330–345 nm, corresponding to intraligand transitions. Complex 2b exhibits a broad and intense emission band in the region of 325–350 nm. Complexes 2d–2f show double emission bands in the range of 330–345 nm. The fluorescence emissions of these complexes are stronger than those of their corresponding precursors, which may be assigned to the incorporation of metal–ligand coordination interactions.29 The results show that these metal complexes might be candidates for potential photoactive materials.
 |
| | Fig. 10
Emission spectra of 1a (—), 1e (⋯), 1f (---), 2b (⋯), 2d (⋯), 2e (---) and 2f (⋯) at 298 K in CH2Cl2 (5.0 × 10−6 M) solution. | |
Conclusions
In summary, a series of new NHC copper(I), mercury(II) and silver(I) complexes have been synthesized and characterized. Precursor 1e and complexes 2b–2g adopt cis-conformations, and complexes 2a and 2h adopt open trans-conformations. Additionally, complexes 2b–2g possess a macrometallocycle, respectively, formed by a didentate chelate carbene ligand and one/two metal ions. During the preparation of 2d or 2e, the acetonitrile are deprotonated in the presence of strong base KOtBu to form deprotonated acetonitrile ([CHCN]2− for 2d and [CH2CN]− for 2e). Then the α-carbon atom of deprotonated acetonitrile participates in coordination with metal atoms. In crystal packings, 2D supramolecular layers of 1e, 2b, 2c and 2f–2h, as well as 1D supramolecular chains of 2d and 2e are formed via intermolecular weak interactions, including π–π interactions, hydrogen bonds, C–H⋯π contacts and weak Hg⋯Br bonds. Further studies on new organometallic compounds from precursors 1a–1f and analogous ligands are under way.
Experimental
General procedures
Bis(bromomethyl)durene was prepared according to the literature method.30 All manipulations were performed using Schlenk techniques, and solvents were purified by standard procedures. All the reagents for synthesis and analyses were of analytical grade and used without further purification. Melting points were determined with a Boetius Block apparatus. IR spectra were measured on a Bruker IR Equinox-55 infrared spectrophotometer. 1H and 13C{1H} NMR spectra were recorded on a Varian Mercury Vx 400 spectrometer at 400 MHz and 100 MHz, respectively. Chemical shifts, δ, are reported in ppm relative to the internal standard TMS for both 1H and 13C NMR. J values are given in Hz. Elemental analyses were measured using a Perkin-Elmer 2400C Elemental Analyzer. The luminescent spectra were conducted on a Cary Eclipse fluorescence spectrophotometer.
Preparation of dibenzimidazolium (or diimidazolium) salts
Preparation of bis[N-(ethyl)benzimidazoliumylmethyl]durene bromide (1a) and bis[N-(ethyl)benzimidazoliumylmethyl]durene hexafluorophosphate (1b).
A THF solution of benzimidazole (2.000 g, 16.9 mmol) was added to a suspension of oil-free sodium hydride (0.480 g, 20.3 mmol) in THF (50 mL) and stirred for 1 h at 60 °C. Then THF (40 mL) solution of ethyl bromide (2.029 g, 18.6 mmol) was dropwise added to above solution. The mixture was continued to stir for 48 h at 60 °C, and a yellow solution was obtained. The solvent was removed with a rotary evaporator, and H2O (100 mL) was added to the residue. Then the solution was extracted with CH2Cl2 (3 × 30 mL), and the extracting solution was dried over anhydrous MgSO4. After removing CH2Cl2, a pale yellow liquid 1-ethylbenzimidazole was obtained. Yield: 2.220 g (90%).
A solution of 1-ethylbenzimidazole (2.013 g, 14.0 mmol) and bis(bromomethyl)durene (2.000 g, 6.25 mmol) in THF (150 mL) was stirred for three days under reflux, and a precipitate was formed. The product was filtered and washed with THF. The white powder of bis[N-(ethyl)benzimidazoliumylmethyl]durene bromide (1a) was obtained by recrystallization from methanol–diethyl ether. Yield: 3.300 g (86%). Mp: 170–172 °C. Anal. Calcd for C30H36N4Br2: C, 58.83; H, 5.92; N, 9.15%. Found: C, 58.66; H, 5.96; N, 9.43%. 1H NMR (400 MHz, DMSO-d6): δ 1.62 (t, J = 7.2, 6H, CH3), 2.31 (s, 12H, CH3), 4.84 (q, J = 7.2, 4H, CH2), 5.90 (s, 4H, CH2), 7.75 (m, 6H, PhH), 8.52 (d, J = 8.1, 2H, PhH), 10.40 (s, 2H, 2-bimiH) (bimi: benzimidazole).
NH4PF6 (1.171 g, 7.2 mmol) was added to a methanol solution (100 mL) of 1a (2.000 g, 3.3 mmol) whilst stirring and a white precipitate formed immediately. The product was collected by filtration, washed with small portions of cold methanol, and dried in vacuum to give 1b. Yield: 1.980 g (82%). Mp: 292–296 °C. Anal. Calcd for C30H36F12N4P2: C, 48.52; H, 4.89; N, 7.55%. Found: C, 48.85; H, 5.22; N, 7.81%. 1H NMR (400 MHz, DMSO-d6): δ 1.66 (t, J = 7.2, 6H, CH3), 2.44 (s, 12H, CH3), 4.89 (q, J = 7.2, 4H, CH2), 5.95 (s, 4H, CH2), 7.67 (m, 6H, PhH), 8.55 (d, J = 8.3, 2H, PhH), 10.60 (s, 2H, 2-bimiH).
The following dibenzimidazolium (or diimidazolium) salts 1c and 1e were prepared in a manner analogous to that for 1a, and 1d and 1f were prepared in a manner analogous to that for 1b.
Preparation of bis[N-(propyl)benzimidazoliumylmethyl]durene chloride (1c).
Yield: 4.009 g (84%). Mp: 282–284 °C. Anal. Calcd for C32H40N4Cl2: C, 69.26; H, 7.31; N, 10.16%. Found: C, 69.12; H, 7.44; N, 10.52%. 1H NMR (400 MHz, DMSO-d6): δ 1.65 (t, J = 7.2, 6H, CH3), 1.74 (m, 4H, CH2), 2.34 (s, 12H, CH3), 4.84 (t, J = 7.2, 4H, CH2), 5.93 (s, 4H, CH2), 7.75 (m, 6H, PhH), 8.57 (d, J = 8.1, 2H, PhH), 10.43 (s, 2H, 2-bimiH) (bimi: benzimidazole).
Preparation of bis[N-(butyl)benzimidazoliumylmethyl]durene iodide (1d).
Yield: 1.919 g (80%). Mp: 320–322 °C. Anal. Calcd for C34H44N4I2: C, 53.55; H, 5.82; N, 7.35%. Found: C, 53.84; H, 5.48; N, 7.64%. 1H NMR (400 MHz, DMSO-d6): δ 0.94 (t, J = 7.3, 6H, CH3), 1.44 (m, 4H, CH2), 1.95 (m, 4H, CH2), 2.31 (s, 12H, CH3), 4.85 (t, J = 7.3, 4H, CH2), 5.95 (s, 4H, CH2), 7.81 (m, 6H, PhH), 8.67 (d, J = 8.3, 2H, PhH), 10.41 (s, 2H, 2-bimiH).
Preparation of bis[N-(1-pyridinylmethyl)benzimidazoliumylmethyl]durene bromide (1e).
Yield: 3.200 g (73%). Mp: 324–326 °C. Anal. Calcd for C38H38N6Br2: C, 61.80; H, 5.19; N, 11.38%. Found: 61.44; H, 5.53; N, 11.65%. 1H NMR (400 MHz, DMSO-d6): δ 2.31 (s, 12H, CH3), 5.90 (s, 4H, CH2), 5.94 (s, 4H, CH2), 7.36 (m, 2H, PhH), 7.66 (m, 6H, PhH), 7.95 (m, 4H, PhH), 8.14 (m, 2H, PhH), 8.42 (d, J = 4.8, 2H, PhH), 9.98 (s, 2H, 2-bimiH).
Preparation of bis[N-(n-butyl)imidazoliumylmethyl]durene hexafluorophosphate (1f).
Yield: 2.100 g (86%). Mp: 286–288 °C. Anal. Calcd for C26H40F12N4P2: C, 44.70; H, 5.77; N, 8.02%. Found: C, 44.71; H, 6.23; N, 7.74%. 1H NMR (400 MHz, DMSO-d6): δ 0.91 (t, J = 5.3, 6H, CH3), 1.37 (m, 4H, CH2), 1.86 (t, J = 5.3, 4H, CH2), 2.15 (s, 12H, CH3), 4.40 (t, J = 5.3, 4H, CH2), 5.83 (s, 4H, CH2), 7.36 (s, 2H, imiH), 8.58 (s, 2H, imiH), 9.90 (s, 2H, 2-imiH) (imi: imidazole).
Preparation of [durene(CH2BimynPr)2][PdCl3(DMSO)]2 (2a).
A suspension of bis[N-(n-propyl)benzimidazoliumylmethyl]durene chloride (1c) (0.200 g, 0.36 mmol), PdCl2(CH3CN)2 (0.187 g, 0.72 mmol) in acetonitrile (10 mL) and DMSO (5 mL) was refluxed for 24 h. A brown solution was formed, and the acetonitrile were removed with a rotary evaporator. The water (30 mL) was added to the residue, and the solution extracted with CH2Cl2 (3 × 20 mL). The extracting solution was dried over anhydrous MgSO4, then the solution was concentrated to 10 mL and hexane (4 mL) was added. 2a was obtained as a pale yellow powder. Yield: 0.167 g (57%). Mp: > 320 °C. Anal. Calcd for C36H52Cl6N4O2Pd2S2: C, 40.69; H, 4.93; N, 5.27%. Found: C, 40.33; H, 4.72; N, 5.56%. 1H NMR (400 MHz, DMSO-d6): δ 1.67 (t, J = 6.8, 6H, CH3), 1.74 (m, 4H, CH2), 2.37 (s, 12H, CH3), 4.89 (t, J = 5.6, 4H, CH2), 5.96 (s, 4H, CH2), 7.78 (m, 6H, PhH), 8.60 (d, J = 8.1, 2H, PhH), 10.02 (s, 2H, 2-bimiH) (bimi: benzimidazole). 13C NMR (100 MHZ, DMSO-d6): δ 138.3, 136.2, 132.8, 132.1, 127.1, 126.0 and 125.1 (PhC or imiC), 46.9 (NCH2Ph), 31.8 (NCH2C), 17.3 (CCH2C), 16.8 (PhCH3), 12.0 (CH2CH3).
Preparation of [durene(CH2BimyEt)2Cu2I2] (2b).
A suspension of KOtBu (0.146 g, 1.3 mmol), bis[N-(ethyl)benzimidazoliumylmethyl]durene hexafluorophosphate (1b) (0.200 g, 0.3 mmol) and copper(I) iodide (0.100 g, 0.5 mmol) in acetonitrile (30 mL) was refluxed for 24 h. A brown solution was formed, and the solvent was removed with a rotary evaporator. The water (30 mL) was added to the residue, and the solution extracted with CH2Cl2 (3 × 20 mL). The extracting solution was dried over anhydrous MgSO4, then the solution was concentrated to 10 mL and hexane (4 mL) was added. As a result, a pale yellow powder was obtained. Yield: 0.126 g (56%). Mp: 352–354 °C. Anal. Calcd for C30H34Cu2I2N4: C, 43.33; H, 4.12; N, 6.74%. Found: C, 43.72; H, 4.50; N, 6.33%. 1H NMR (400 MHz, DMSO-d6): δ 1.41 (t, J = 5.1, 6H, CH3), 2.36 (s, 12H, CH3), 4.40 (q, J = 5.1, 4H, CH2), 5.49 (s, 4H, CH2), 7.49 (m, 4H, PhH), 7.80 (d, J = 5.4, 2H, PhH), 8.3 (d, J = 5.4, 2H, PhH). 13C NMR (100 MHZ, DMSO-d6): δ 174.4 (Ccarbene), 138.2, 135.3, 132.8, 132.0, 127.2, 126.1 and 124.3 (PhC), 47.4 (NCH2Ph), 32.5 (NCH2C), 17.1 (PhCH3), 12.1 (CCH3).
The following two NHC mercury(II) complexes 2c and 2d were prepared in a manner analogous to that for 2b.
Preparation of [durene(CH2BimyEt)2HgBr]·0.5[HgBr4] (2c).
Yield: 0.178 g (55%). Mp: 306–308 °C. Anal. Calcd for C30H34Br3Hg1.5N4: C, 36.35; H, 3.46; N, 5.65%. Found: C, 36.78; H, 3.74; N, 5.33%. 1H NMR (400 MHz, DMSO-d6): δ 1.59 (t, J = 7.2, 6H, CH3), 2.27 (s, 12H, CH3), 4.62 (q, J = 7.2, 4H, CH2), 5.82 (s, 4H, CH2), 7.72 (s, 4H, PhH), 8.02 (d, J = 6.2, 2H, PhH), 8.28 (d, J = 6.2, 2H, PhH). 13C NMR (100 MHZ, DMSO-d6): δ 180.1 (Ccarbene), 137.2, 135.1, 132.9, 132.0, 131.3, 126.3 and 125.8 (PhC), 46.8 (NCH2Ph), 33.6 (NCH2C), 15.1 (PhCH3), 10.0 (CCH3).
Preparation of [durene(CH2benzimynBu)2Hg2(CHCN)][HgI4] (2d).
Yield: 0.230 g (54%). Mp: 276–278 °C. Anal. Calcd for C36H43Hg3I4N5: C, 26.12; H, 2.62; N, 4.23%. Found: C, 26.57; H, 2.32; N, 4.65%. 1H NMR (400 MHz, DMSO-d6): δ 0.88 (t, J = 7.4, 6H, CH3), 1.29 (m, 4H, CH2), 1.83 (m, 4H, CH2), 2.07 (s, 1H, CH), 2.27 (s, 12H, CH3), 4.48 (t, J = 7.2, 2H, CH2), 5.81 (s, 2H, CH2), 7.76 (m, 4H, PhH), 8.12–8.19 (m, 4H, PhH). 13C NMR (100 MHZ, DMSO-d6): δ 187.3 (Ccarbene), 137.0, 136.2, 133.2, 132.7, 132.3, 126.3 and 126.0 (PhC), 113.4 (CN), 48.3 (NCH2Ph), 31.5 (NCH2C), 30.3 (HgCHHg), 19.2 (CCH2C), 17.3 (CCH2C), 16.2 (PhCH3), 13.4 (CCH3). IR (KBr): ν CN, 2191 cm−1.
Preparation of [durene(CH2imynBu)2Ag2(CH2CN)2] (2e).
Silver oxide (0.089 g, 0.4 mmol) and KOtBu (0.146 g, 1.3 mmol) was added to a solution of bis[N-(n-butyl)imidazoliumylmethyl]durene hexafluorophosphate (1f) (0.200 g, 0.3 mmol) in CH3CN–CH2Cl2 (30 mL), and the suspension solution was stirred for 24 h at refluxing. The resulting solution was filtered and concentrated to 10 mL, and Et2O (5 mL) was added to precipitate a white powder. Isolation by filtration yielded 2e. Yield: 0.101 g (50%), Mp: 164–166 °C. Anal. Calcd for C30H42Ag2N6: C, 51.30; H, 6.03; N, 11.96%. Found: C, 51.71; H, 6.46; N, 11.66%. 1H NMR (400 MHz, DMSO-d6): δ 0.87 (t, J = 7.2, 6H, CH3), 1.20 (m, 4H, CH2), 1.70 (m, 4H, CH2), 2.20 (s, 12H, CH3), 2.23 (s, 2H, CH2), 4.01 (t, J = 6.0, 4H, CH2), 5.35 (s, 4H, CH2), 7.39 (s, 2H, imiH), 7.48 (s, 2H, imiH) (imi: imidazole). 13C NMR (100 MHZ, DMSO-d6): δ 135.4, 133.0, 123.1 and 122.1 (PhC or imiC), 112.7 (CN), 52.0 (NCH2CH2), 50.0 (NCH2Ph), 33.6 (AgCH2C), 31.2 (CCH2C), 19.7 (CCH2C), 17.4 (PhCH3), 14.2 (CCH3). IR (KBr): ν CN, 2166 cm−1. The signal of carbene carbon was not observed.
Preparation of [durene(CH2BimyPymethyl)2Ag2Br2] (2f).
Silver oxide (0.070 g, 0.3 mmol) was added to a solution of bis[N-(1-pyridinylmethly)imidazoliumylmethyl]durene bromide (1e) (0.200 g, 0.3 mmol) in dichloromethane (30 mL) and the suspension solution was stirred for 24 h at refluxing. The resulting solution was filtered and concentrated to 10 mL, and Et2O (5 mL) was added to precipitate a white powder. Isolation by filtration yielded 2f. Yield: 0.156 g (60%), Mp: 288–290 °C. Anal. Calcd for C38H36Ag2Br2N6: C, 47.93; H, 3.81; N, 8.83%. Found: C, 47.58; H, 3.44; N, 8.62%. 1H NMR (400 MHz, DMSO-d6): δ 2.30 (s, 12H, CH3), 5.91 (s, 4H, CH2), 5.95 (s, 4H, CH2), 7.34 (m, 2H, PhH or PyH), 7.61 (m, 6H, PhH or PyH), 7.92 (m, 4H, PhH or PyH), 8.13 (m, 2H, PhH or PyH), 8.44 (d, J = 4.8, 2H, PhH or PyH). 13C NMR (100 MHZ, DMSO-d6): δ 182.0 (Ccarbene), 155.8, 150.2, 138.5, 136.2, 133.8, 132.8, 132.1, 124.1, 123.0, 118.9 and 116.8 (PhC or PyC), 50.4 (NCH2Py), 40.6 (NCH2Ph), 13.1 (PhCH3).
The following two NHC silver(I) complexes 2g and 2h were prepared in a manner analogous to that for 2f.
Preparation of [durene(CH2BimyEt)Ag](PF6) (2g).
Yield: 0.098 g (52%). Mp: 256–260 °C. Anal. Calcd for C30H34AgF6N4P: C, 51.22; H, 4.87; N, 7.96%. Found: C, 51.61; H, 4.48; N, 7.68%. 1H NMR (400 MHz, DMSO-d6): δ 1.50 (t, J = 5.2, 6H, CH3), 2.43 (s, 12H, CH3), 4.46 (q, J = 5.2, 4H, CH2), 5.53 (s, 4H, CH2), 7.51 (m, 4H, PhH), 7.84 (d, J = 5.5, 2H, PhH), 8.33 (d, J = 5.5, 2H, PhH). 13C NMR (100 MHZ, DMSO-d6): δ 175.0 (Ccarbene), 138.3, 133.1, 128.4, 118.1 and 112.2 (PhC), 50.1 (NCH2Ph), 48.2 (NCH2C), 14.8 (PhCH3), 10.8 (CCH3).
Preparation of [durene(CH2BimynBu)2Ag2(OH)2] (2h).
Yield: 0.146 g (50%). Mp: 150–152 °C. Anal. Calcd for C34H43.88I0.1N4O1.88Ag2: C, 53.23; H, 5.77; N, 7.30%. Found: C, 53.67; H, 5.32; N, 7.72%. 1H NMR (400 MHz, DMSO-d6): δ 0.92 (t, J = 7.0, 6H, CH3), 1.37 (m, 4H, CH2), 1.87 (m, 4H, CH2), 2.19 (s, 12H, CH3), 4.44 (t, J = 7.0, 4H, CH2), 5.57 (s, 4H, CH2), 7.56 (m, 4H, PhH), 7.86 (d, J = 7.8, 2H, PhH), 8.13 (d, J = 7.8, 2H, PhH). 13C NMR (100 MHZ, DMSO-d6): δ 181.3 (Ccarbene), 138.3, 133.1, 129.4, 119.9 and 115.3 (PhC), 53.2 (NCH2Ph), 48.3 (NCH2C), 33.7 (CCH2C), 25.6 (CCH2C), 14.2 (CCH3), 9.2 (PhCH3).
X-Ray structure determinations
†
For complexes 2a–2h, selected single crystals were mounted on a Bruker APEX II CCD diffractometer at 293(2) K with Mo-Kα radiation (λ = 0.71073 Å) by ω scan mode. Data collection and reduction were performed using the SMART and SAINT software31 with frames of 0.6° oscillation in the range of 1.8° < θ < 25°. An empirical absorption correction was applied using the SADABS program.32 The structures were solved by direct methods and all non-hydrogen atoms were subjected to anisotropic refinement by full-matrix least squares on F2 using the SHELXTL package.33 All hydrogen atoms were generated geometrically (C–H bond lengths fixed at 0.96 Å), assigned appropriated isotropic thermal parameters and included in the final calculations. Crystallographic data are summarized in Tables 4–Table 6 for 1e and 2a–2h.
| |
1e·CH3CN |
2a
|
2b·0.5CH3CN |
|
Goof = [∑w(Fo2 − Fc2)2/(n − p)]1/2, where n is the number of reflections and p is the number of parameters refined.
R1 = ∑(‖Fo| − |Fc‖)/∑|Fo|; wR2 = 1/[σ2(Fo2) + (0.0691P) + 1.4100P] where P = (Fo2 + 2Fc2)/3.
|
| Chemical formula |
C38H38N6·Br2·CH3CN |
[C32H40N4]·2[PdCl3DMSO] |
C30H34Cu2I2N4·0.5CH3CN |
|
Fw
|
779.62 |
1062.46 |
852.02 |
| Crystal system |
Monoclinic |
Triclinic |
Triclinic |
| Space group |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
P
|
|
a/Å |
14.781(7) |
8.885(1) |
9.709(1) |
|
b/Å |
14.678(7) |
10.627(1) |
17.216(3) |
|
c/Å |
18.540(9) |
13.042(1) |
19.642(4) |
|
α/° |
90 |
78.793(2) |
72.8 (3) |
|
β/° |
111.032(1) |
80.799(2) |
86.3 (3) |
|
γ/° |
90 |
66.276(2) |
88.6 (3) |
|
V/Å3 |
3754.4(3) |
1101.3(2) |
3131.2(1) |
|
Z
|
4 |
2 |
2 |
|
D
c/Mg m−3 |
1.379 |
1.602 |
1.807 |
|
μ/mm−1 |
2.197 |
1.311 |
3.358 |
|
F(000) |
1600 |
538 |
1668 |
| Crystal size/mm |
0.22 × 0.21 × 0.14 |
0.28 × 0.22 × 0.20 |
0.14 × 0.12 × 0.10 |
|
θ
min, θmax (°) |
1.82, 25.01 |
2.12, 25.02 |
1.39, 25.02 |
|
T/K |
296(2) |
296(2) |
113(2) |
| No. of data collected |
18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 776 776 |
5682 |
10999 |
| No. of unique data |
6616 |
3868 |
10999 |
| No. of refined params |
447 |
240 |
725 |
| Goodness-of-fit on F2a |
1.032 |
1.061 |
1.077 |
| Final R indicesb [I > 2σ(I)] |
|
R
1
|
0.0405 |
0.0559 |
0.0689 |
|
wR2 |
0.1205 |
0.1756 |
0.1383 |
|
R indices (all data) |
|
R
1
|
0.0675 |
0.0637 |
0.0921 |
|
wR2 |
0.1358 |
0.1845 |
0.1524 |
| |
2c·0.5DMSO |
2d·CH3CN |
2e
|
|
Goof = [∑w(Fo2 − Fc2)2/(n − p)]1/2, where n is the number of reflections and p is the number of parameters refined.
R
1 = ∑(‖Fo| − |Fc‖)/∑|Fo |; wR2 = 1/[σ2(Fo2) + (0.0691P) + 1.4100P] where P = (Fo2 + 2Fc2)/3.
|
| Chemical formula |
C30H34BrHgN4 ·0.5HgBr4·0.5DMSO |
C36H43Hg3I4N5·CH3CN |
C30H42Ag2N6 |
|
Fw
|
1030.29 |
1696.18 |
702.43 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P
|
P
|
P
|
|
a/Å |
14.017(1) |
12.212(8) |
10.950(6) |
|
b/Å |
16.221(2) |
13.311(8) |
14.822 (7) |
|
c/Å |
18.554(2) |
16.692(1) |
19.4 (9) |
|
α/° |
89.7 (2) |
71.1 (1) |
98.1 (1) |
|
β/° |
69.7 (2) |
75.7 (1) |
95.9 (1) |
|
γ/° |
72.1 (2) |
62.9 (1) |
107.0 (1) |
|
V/Å3 |
3745.0(8) |
2271.9(2) |
2955.6(3) |
|
Z
|
2 |
2 |
2 |
|
D
c/Mg m−3 |
1.826 |
2.479 |
1.579 |
|
μ/mm−1 |
9.403 |
12.862 |
1.355 |
|
F(000) |
1950 |
1536 |
1432 |
| Crystal size/mm |
0.24 × 0.20 × 0.16 |
0.24 × 0.20 × 0.18 |
0.28 × 0.26 × 0.20 |
|
θ
min, θmax (°) |
1.63, 26.00 |
1.77, 25.03 |
1.07, 25.03 |
|
T/K |
296(2) |
296(2) |
296(2) |
| No. of data collected |
14562 |
11724 |
15297 |
| No. of unique data |
14562 |
7972 |
10382 |
| No. of refined params |
783 |
466 |
690 |
| Goodness-of-fit on F2a |
1.029 |
1.030 |
1.020 |
| Final R indicesb [I > 2σ(I)] |
|
R
1
|
0.0412 |
0.0383 |
0.0488 |
|
wR2 |
0.1001 |
0.0978 |
0.1413 |
|
R indices (all data) |
|
R
1
|
0.0657 |
0.0481 |
0.0823 |
|
wR2 |
0.1063 |
0.1043 |
0.1698 |
| |
2f·DMSO |
2g·0.5H2O |
2h
|
|
Goof = [∑w(Fo2 − Fc2)2/(n − p)]1/2, where n is the number of reflections and p is the number of parameters refined.
R
1= ∑(‖Fo| − |Fc‖)/∑|Fo|; wR2 = 1/[σ2(Fo2) + (0.0691P) + 1.4100P] where P = (Fo2 + 2Fc2)/3.
|
| Chemical formula |
C38H36Ag2Br2N6·DMSO |
C30H34AgN4·PF6·0.5H2O |
C34H43.88Ag2I0.1N4O1.88 |
|
Fw
|
1030.42 |
712.46 |
768.56 |
| Crystal system |
Triclinic |
Triclinic |
Monoclinic |
| Space group |
P
|
P
|
C
2/c |
|
a/Å |
10.097(2) |
9.368(1) |
3.133(4) |
|
b/Å |
13.533(3) |
13.897(2) |
18.772(4) |
|
c/Å |
15.4 (3) |
14.595(2) |
20.002(2) |
|
α/° |
104.0 (3) |
103.6 (2) |
90 |
|
β/° |
97.6 (3) |
102.9 (2) |
15.8 (5) |
|
γ/° |
94.5 (4) |
104.9 (2) |
90 |
|
V/Å3 |
2010.4(7) |
1699.1(4) |
3784.7(2) |
|
Z
|
2 |
2 |
4 |
|
D
c/Mg m−3 |
1.699 |
1.393 |
1.349 |
|
μ/mm−1 |
3.051 |
0.699 |
1.155 |
|
F(000) |
1024 |
726 |
1563 |
| Crystal size/mm |
0.28 × 0.22 × 0.20 |
0.34 × 0.32 × 0.30 |
0.22 × 0.20 × 0.18 |
|
θ
min, θmax (°) |
1.56, 25.03 |
2.38, 25.01 |
1.94, 25.01 |
|
T/K |
296(2) |
296(2) |
296(2) |
| No. of data collected |
10282 |
8589 |
3344 |
| No. of unique data |
7035 |
5920 |
3344 |
| No. of refined params |
510 |
431 |
197 |
| Goodness-of-fit on F2a |
1.032 |
1.071 |
1.150 |
| Final R indicesb [I > 2σ(I)] |
|
R
1
|
0.0357 |
0.0473 |
0.0352 |
|
wR2 |
0.0962 |
0.1486 |
0.1164 |
|
R indices (all data) |
|
R
1
|
0.0525 |
0.0543 |
0.0400 |
|
wR2 |
0.1072 |
0.1573 |
0.1192 |
Acknowledgements
This work was financially supported by the National Science Foundation of China (Project Grant No. 20872111) and the Natural Science Foundation of Tianjin (07JCYBJC00300).
References
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef.
-
(a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS;
(b) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS;
(c) F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2007, 26, 150–154 CrossRef CAS.
-
(a) F. E. Hahn, L. Wittenbecher, D. Le Van and R. Frohlich, Angew. Chem., Int. Ed., 2000, 39, 541–544 CrossRef CAS;
(b) F. E. Hahn, T. von Fehren and T. Lügger, Inorg. Chim. Acta, 2005, 358, 4137–4144 CrossRef;
(c) F. E. Hahn and M. Foth, J. Organomet. Chem., 1999, 585, 241–245 CrossRef CAS;
(d) S. C. Zinner, C. F. Rentzsch, E. Herdtweck, W. A. Herrmann and F. E. Kühn, Dalton Trans., 2009, 7055–7062 RSC.
-
(a) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS;
(b) L. H. Gade and S. B. Laponnaz, Coord. Chem. Rev., 2007, 251, 718–725 CrossRef CAS;
(c) W. J. Sommer and M. Weck, Coord. Chem. Rev., 2007, 251, 860–873 CrossRef CAS.
-
(a) J. C. Y. Lin, R. T. W Huang, C. S. Lee, A. Bhattacharyya, W. S. Huang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS;
(b) S. Gischig and A. Togni, Organometallics, 2005, 24, 203–205 CrossRef CAS;
(c) B. S. Khalid, M. Yasumasa, Y. Ken-ichi and T. Kiyoshi, Angew. Chem., Int. Ed., 2009, 48, 8733–8735 CrossRef CAS.
-
(a) K. M. Lee, J. C. C. Chen, C. J. Huang and I. J. B. Lin, CrystEngComm, 2007, 9, 278–281 RSC;
(b) U. J. Scheele, S. Dechert and F. Meyer, Inorg. Chim. Acta, 2006, 359, 4891–4900 CrossRef CAS;
(c) Q. X. Liu, L. N. Yin and J. C. Feng, J. Organomet. Chem., 2007, 692, 3655–3663 CrossRef CAS.
-
(a) J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS;
(b) F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2006, 25, 5927–5936 CrossRef CAS;
(c) M. C. Jahnke, J. Paley, F. Hupka, J. J. Weigand and F. E. Hahn, Z. Naturforsch., 2009, 64b, 1458–1462 Search PubMed.
-
(a) J. J. Van Veldhuizen, J. E. Campbell, R. E. Giudici and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127, 6877–6882 CrossRef CAS;
(b) K. S. Coleman, H. T. Chamberlayne, S. Turberville, M. L. H. Green and A. R. Cowley, Dalton Trans., 2003, 2917–2922 RSC;
(c) W. A. Herrmann, S. K. Schneider, K. Öfele, M. Sakamoto and E. Herdtweck, J. Organomet. Chem., 2004, 689, 2441–2444 CrossRef CAS.
-
(a) M. C. Perry, X. H. Cui and K. Burgess, Tetrahedron: Asymmetry, 2002, 13, 1969–1972 CrossRef CAS;
(b) L. G. Bonnet, R. E. Douthwaite and R. Hodgson, Organometallics, 2003, 22, 4384–4386 CrossRef;
(c) C. Marshall, M. F. Ward and W. T. A. Harrison, J. Organomet. Chem., 2005, 690, 3970–3975 CrossRef CAS;
(d) H. Clavier, J. C. Guillemin and M. Mauduit, Chirality, 2007, 19, 471–476 CrossRef CAS;
(e) M. S. Jeletic, I. Ghiviriga, K. A. Abboud and A. S. Veige, Organometallics, 2007, 26, 5267–5270 CrossRef CAS.
-
(a) C. M. Zhang and M. L. Trudell, Tetrahedron Lett., 2000, 41, 595–598 CrossRef CAS;
(b) D. S. Clyne, J. Jin, E. Genest, J. C. Gallucci and T. V. RajanBabu, Org. Lett., 2000, 2, 1125–1128 CrossRef CAS;
(c) A. A. Danopoulos, A. A. D. Tulloch, S. Winston, G. Eastham and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 2003, 1009–1015 RSC;
(d) W. L. Duan, M. Shi and G. B. Rong, Chem. Commun., 2003, 2916–2917 RSC;
(e) A. R. Chianese and R. H. Crabtree, Organometallics, 2005, 24, 4432–4436 CrossRef;
(f) W. F. Wang, F. J. Wang and M. Shi, Organometallics, 2010, 29, 928–933 CrossRef CAS;
(g) B. P. Morgan, G. A. Galdamez, R. J. Gilliard Jr. and R. C. Smith, Dalton Trans., 2009, 2020–2028 RSC.
-
(a) D. H. Brown, G. L. Nealon, P. V. Simpson, B. W. Skelton and Z. Wang, Organometallics, 2009, 28, 1965–1968 CrossRef CAS;
(b) A. T. Normand and K. J. Cavell, Eur. J. Inorg. Chem., 2008, 2781–2800 CrossRef CAS;
(c) B. Liu, W. Chen and S. Jin, Organometallics, 2007, 26, 3660–3667 CrossRef CAS;
(d) D. Pugh, J. A. Wright, S. Freeman and A. A. Danopoulos, Dalton Trans., 2006, 775–782 RSC;
(e) F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2007, 26, 150–154 CrossRef CAS.
-
(a) D. J. Nielsen, K. J. Cavell, B. W. Skelton and A. H. White, Organometallics, 2006, 25, 4850–4856 CrossRef;
(b) A. A. D. Tulloch, A. A. Danopoulos, G. J. Tizzard, S. J. Coles, M. B. Hursthouse, R. S. Hay-Motherwell and W. B. Motherwell, Chem. Commun., 2001, 1270–1271 RSC;
(c) D. J. Nielsen, K. J. Cavell, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2002, 327, 116–125 CrossRef CAS;
(d) X. Q. Zhang, Y. P. Qiu, B. R and M. M. Luo, Organometallics, 2009, 28, 3093–3099 CrossRef CAS.
-
(a) Q. X. Liu, X. J. Zhao, X. M. Wu, J. H. Guo and X. G. Wang, J. Organomet. Chem., 2007, 692, 5671–5679 CrossRef CAS;
(b)
Q. X. Liu, X. Q. Yang, X. J. Zhao, S. S. Ge, S. W. Liu, Y. Zang, H. B. Song, J. H. Guo and X. G. Wang, CrystEngComm, 10.1039/b919007d.
- A. A. D. Tulloch, A. A. Danopoulos, S. Winston, S. Kleinhenz and G. Eastham, J. Chem. Soc., Dalton Trans., 2000, 4499–4506 RSC.
-
(a) I. B. Michael, T. W. Hambley, R. S. Michael and A. G. Swincer, Organometallics, 1985, 4, 494–500 CrossRef CAS;
(b) Y. Han and H. V. Huynh, Dalton Trans., 2009, 2201–2209 RSC.
- L. Rajput and K. Biradha, CrystEngComm, 2009, 11, 1220–1222 RSC.
- C. A. Ellis, M. A. Miller, J. Spencer, J. Z. Schpectorc and E. R. T. Tiekink, CrystEngComm, 2009, 11, 1352–1361 RSC.
- X. P. Zhou, S. H. Lin, D. Li and Y. G. Yin, CrystEngComm, 2009, 11, 1899–1903 RSC.
- J. Ramos, V. M. Yartsev, S. Golhen, L. Ouahab and P. Delhaes, J. Mater. Chem., 1997, 7, 1313–1319 RSC.
- G. Mahmoudi and A. Morsali, CrystEngComm, 2007, 9, 1062–1072 RSC.
-
(a) S. Inoue and Y. Sato, Organometallics, 1986, 5, 1197–1201 CrossRef CAS;
(b) P. S. Pregosin, Inorg. Chim. Acta, 1980, 45, L7–L9 CrossRef CAS;
(c) M. Robert, F. George, A. J. McAlees, M. Parvez and P. J. Roberts, J. Chem. Soc., Dalton Trans., 1982, 1699–1708 RSC;
(d) B. Luigi, B. Silvia, Z. Valerio, A. G. Vincenzo and B. Dario, New J. Chem., 1992, 16, 693–696 Search PubMed;
(e) H. William and G. O. Allen, Acta Cryst., 1999, 55, 1406–1408.
-
(a) F. E. Hahn, C. Radloff, T. Pape and A. Hepp, Chem.–Eur. J., 2008, 14, 10900–10904 CrossRef CAS;
(b) A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572–4573 CrossRef CAS.
- Z. X. Lian, J. W. Cai, C. H. Chen and H. B. Luo, CrystEngComm, 2007, 9, 319–327 RSC.
-
R. P. A. Bettens, D. Dakternieks, A. Duthie, F. S. Kuan and E. R. T. Tiekink, CrystEngComm., 2009, 11, pp. 1362–1372.
- L. H. Zhao, Y. P. Quan, A. H. Yang, J. Z. Cui, H. L. Gao, F. L. Lu, W. Shi and P. Cheng, CrystEngComm, 2009, 11, 1427–1432 RSC.
- M. L. Cole and P. C. Junk, CrystEngComm, 2004, 6, 173–176 RSC.
- F. Neve and A. Crispini, CrystEngComm, 2003, 5, 265–268 RSC.
- S. F. Alshahateet, R. Bishop, D. C. Craig and M. L. Scudder, CrystEngComm, 2001, 48, 1–5 Search PubMed.
-
(a) F. J. B. dit Dominique, H. Gornitzka, A. Sournia-Saquet and C. Hemmert, Dalton Trans., 2009, 340–352 RSC;
(b) X. X. Zhao, J. P. Ma, D. Z. Shen, Y. B. Dong and R. Q. Huang, CrystEngComm, 2009, 11, 1281–1290 RSC.
- A. W. vander Made and R. H. vander Made, J. Org. Chem., 1993, 58, 1262–1263 CrossRef CAS.
-
SMART 5.0 and SAINT 4.0 for Windows NT, Area Detector Control and Integration Software, Bruker Analytical X-Ray Systems, Inc., Madison, WI, USA, 1998 Search PubMed.
-
G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, Univ. of Göttingen, Germany, 1996 Search PubMed.
-
G. M. Sheldrick, SHELXTL 5.10 for Windows NT, Structure Determination Software, Brucker Analytical X-Ray Systerms, Inc., Madison, WI, USA, 1997 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.