DOI:
10.1039/C0CE00111B
(Paper)
CrystEngComm, 2011,
13, 118-123
Synthesis, structural characterization and magnetic behavior of a 2D bi-layer MOF, {[Ni2(C5O5)2(bpe)2]·H2O}n (bpe = 1,2-bis(4-pyridyl)ethylene)†
Received
19th April 2010
, Accepted 19th June 2010
First published on 26th August 2010
Abstract
Coordination polymers of metal-croconate (C5O52−) complex, {[Ni2(bpe)2(C5O5)2]·H2O}n (1) (bpe = 1,2-bis(-4-pyridyl)ethylene)) with two-dimensional (2D) metal–organic frameworks (MOF), have been synthesized under hydrothermal conditions and characterized by single-crystal X-ray diffraction studies. Structural determination reveals that compound 1 contains two crystallographically independent Ni(II) ions which both lie in a distorted octahedral environment bonded to two bpe nitrogen atoms and four oxygen donors from two croconates. The croconate acts as a bridging ligand with two different bridging modes, one presents a bis-bidentate adjacent μ3-coordination mode and the other a bis-bidentate μ4-coordination mode. A 2D brick-wall-like layered framework is formed by using a rectangle grid as a basic building unit through the connectivity of NiII with hybrid μ3-, μ4-croconates and bpe ligands. Two layers are then cross-linked by the other bpe ligands to complete a 2D bi-layered MOF. The TGA associated with in situXRD measurements demonstrated that 1 have quite high thermo-stability that keep their crystalline forms up to 350 °C. Magnetic measurements show the long range magnetic ordering and spin canting behavior in 1 below the critical temperature of 22 K with an approximate canting angle of 0.79°. A large coercivity of 3.5 kOe in the hysteresis loop is observed at 1.8 K, indicating that 1 is a weak hard magnet.
Introduction
Multidimensional molecular-based magnets with metal coordination frameworks (MOFs) are the focus of considerable current research effort from both fundamental and device-related perspectives.1–3 Two- or three-dimensional (2D, 3D) homometallic magnetic systems can behave as ferromagnetic, antiferromagnetic, or as metamagnetic materials, depending on the nature of the spin coupling.1,2 The synthesis of mixed-ligands metal–organic frameworks (MOFs) containing two different types of ligand allows for greater flexibility in the design of different structural topology.4 The croconate (C5O52−, dianion of 4,5-dihydroxycyclo-pent-4-1,2,3-trione) bridge has been widely utilized because its diverse binding modes lead to an antiferromagnetic interaction.5 Recently, four bridging modes of croconate as a tridentate ligand,6a tetradentate ligand,6a bidentate/modentate,6b and tetradentate,6c have been used on the construction of 2D or 3D MOFs. We are currently studying the synthetic approaches to mixed-ligands homometallic coordination polymers of different dimensionalities with croconate ligand.7 Two types of mixed-ligands coordination polymers, with 2D MOFs are constructed by the connectivity between the metal ions and croconate with two different bridging modes and related pyridyl-based ligands such as 4,4′-bipy (4,4′-bipyridine), dpe (1,2-bis(4-pyridyl)ethane) or bpe (1,2-bis(4-pyridyl)ethylene),7 where both of their 3D extended supramolecular architecture are further stabilized by the inter-layer π–π interaction between the five-membered cyclic ring of croconate and pyridyl rings of 4,4′-bipy, or dpe. The magnetic studies unveil antiferromagnetic coupling within the metal centres viaμ3-croconate bridges.7e With our continuous effort on the study of metal-croconate coordination polymers, we report here the synthesis, crystal structure, and spin canting magnetic behavior of a 2D bilayered homometallic network, {[Ni2(C5O5)2(bpe)2]·H2O}n (1) that orders magnetically at 22 K.
Experimental
Materials and physical techniques
General: all chemicals were of reagent grade and were used as commercially obtained without further purification. Elementary analyses (carbon, hydrogen and nitrogen) were performed using a Perkin-Elmer 2400 elemental analyzer. IR spectra were recorded on a Nicolet Fourier Transform IR, MAGNA-IR 500 spectrometer in the range of 500–4000 cm−1 using the KBr disc technique. Thermogravimetric analyses (TGA) of the title compound were performed on a computer-controlled Perkin-Elmer 7 Series/UNIX TGA7 analyzer. Single-phased powder samples of 1 (2.083 mg) were loaded into alumina pans and heated with a ramp rate of 5 °C min−1 from room temperature to 800 °C under a nitrogen atmosphere. The temperature-dependent magnetic susceptibility was measured on the SQUID system with 2 kOe external magnetic field. The in situX-ray powder diffraction was performed at the beamline BL01C2 of the National Synchrotron Radiation Research Center (NSRRC) Taiwan. The synchrotron X-ray radiation was generated from the superconducting wavelength shifted magnet of 5.0 Tesla with ring energy of 1.5 GeV typical ring current of 200 to 120 mA. The X-ray wavelength was 0.5166 Å which was delivered by a double crystal monochromator with two Si(111) crystals. Samples of 1 were sealed in 1 mm-quartz capillary and heated under hot air stream from room temperature up to 650 °C during the XRD measurement. Two dimensional powder X-ray diffraction patterns were recorded by using Mar345 imaging plate detector with the pixel size of 100 μm and the typical exposure time of 5 min. The sample to imaging plate distance was ca. 260 mm and the diffraction angle was calibrated by the standard powders of Ag-Behenate and Si powder (NBS640b). The one dimensional XRD profile was converted using FIT2D program of a cake type integration.
Synthesis of {[Ni2(bpe)2(C5O5)2]·H2O}n (1)
In a typical experiment, Na2C5O5 (0.0373 g, 0.20 mmol) in an aqueous solution (2 mL) was added to an aqueous solution (4 mL) of 1,2-bis(4-pyridyl)ethylene (bpe) (0.0205 g, 0.11 mmol) and NiCl2 (0.0268 g, 0.20 mmol). The new mixture was placed in a 23 mL Teflon-lined autoclave and heated at 180 °C for three days. The autoclave was then cooled over a period of 26 h at a rate of 6 K h−1. Green block crystals of 1 were collected by filtration, washed with water, and dried in air. Phase-pure 1 was obtained by manual separation (final yield: 0.017 mg, 37.9% based on NiII). IR (KBr pellet): ν = 3635 (s, br), 3505 (s, br), 1684 (m, sharp), 1612 (s, sharp), 1520 (vs, sharp), 1476 (vs, sharp), 1420 (s, sharp), 1216 (m, sharp) cm−1. Anal. Calc. for C34H22N4Ni2O11 (Mr = 779.98): C, 53.98; H, 2.91; N, 7.41. Found: C, 53.58; H, 2.41; N, 7.30.
Single-crystal structure analyses of compound 1 was performed on a Siemens SMART diffractometer with a CCD detector with Mo radiation (λ = 0.71073 Å) at room temperature. A preliminary orientation matrix and unit cell parameters were determined from 3 runs of 15 frames each, each frame corresponds to a 0.3° scan in 10 s, followed by spot integration and least-squares refinement. For each structure, data were measured using ω scans of 0.3° per frame for 20 s until a complete hemisphere had been collected. Cell parameters were retrieved using SMART8 software and refined with SAINT9 on all observed reflections. Data reduction was performed with the SAINT9 software and corrected for Lorentz and polarization effects. Absorption corrections were applied with the program SADABS.10 Direct phase determination and subsequent difference Fourier map synthesis yielded the positions of all non-hydrogen atoms, which were subjected to anisotropic refinements. All hydrogen atoms were generated geometrically (C–H = 0.93) with the exception of the hydrogen atoms of the coordinated water molecules, which were located in the difference Fourier map with the corresponding positions and isotropic displacement parameters being refined. The final full-matrix, least-squares refinement on F2 was applied for all observed reflections [I > 2σ(I)]. All calculations were performed using the SHELXTL-PC V 5.03 software package.11Crystallographic data and details of data collections and structure refinements of compound 1 are listed in Table 1.
Table 1
Crystal data and refinement details of {[Ni2(bpe)2(C5O5)2]·H2O}∞1a
|
R
F = ∑‖Fo − Fc‖/∑|Fo|; Rw(F2) = [∑w|Fo2 − Fc2|2/∑w(Fo4)]1/2.
|
| Chemical formula |
C34H22N4Ni2O11 |
Formula mass |
779.98 |
| Crystal system |
Monoclinic |
Space group |
P21/c |
|
a/Å |
7.9639(5) |
α/° |
90.0 |
|
b/Å |
24.326(2) |
β/° |
90.412(2) |
|
c/Å |
16.065(1) |
γ/° |
90.0 |
|
V/Å3 |
3112.1(3) |
Z
|
4 |
|
μ/mm−1 |
1.283 |
θ range/° |
2.56–24.00 |
| Total no. of data collected |
25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 212 212 |
No. of obsd data (I > 2σ(I)) |
5389 |
|
R
1, wR2 (I > 2σ(I)) |
0.0651, 0.1223 |
R
1, wR2 (all data) |
0.0942, 0.1325 |
| GOF |
1.116 |
No. of variable |
460 |
Results and discussion
Structure description of {[Ni2(bpe)2(C5O5)2]·H2O}n (1)
Hydrothermal treatment of NiCl2 with disodium croconate and bpe in aqueous solution at 180 °C leads to the formation of the coordination polymer, {[Ni2(C5O5)2(bpe)2]n·(H2O)}n (1). The X-ray single-crystal analysis shows that compound 1 is isostructural with {[Zn2(bpe)2(C5O5)2]·H2O}n,7a in which each of the two NiII ions (Ni(1) and Ni(2)) in the asymmetric unit is located in a distorted octahedron, bonded with two nitrogen donors of two bpe ligands at the cis position and with four oxygen donors of two croconate ligands (shown in ESI, Fig. S1(a) and S1(b)†). Selected bond lengths and angles around the Ni centers are listed in Table 2. Both of the bpe and croconate ligands are used in the construction of the 2D bi-layer MOF. The bpe ligand acts as a bis-monodentate bridging ligand to link Ni(1) and Ni(2) ions with the Ni⋯Ni separations of 13.52 and 13.41 Å. The croconate ligand acts as a bridging ligand, adopting two different bridging bis-bidentate modes. The first one is a bridging bis-bidentate mode3,7 through three-adjacent (μ3) croconate oxygen atoms to link Ni ions with the Ni⋯Ni separation of 4.47 Å. The bond analysis of the C–C and C–O bond lengths of croconate (see ESI, Table S1†) reveals that two uncoordinated oxygen atoms (O(4) and O(5)) show a ketonic form with shorter bond lengths compared to the C–O bond lengths of the metal-coordinated oxygen atoms. Notably, the C–C bond lengths between two non-bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups are close to being single bond in character, and they are also longer than other C–C bond lengths, indicating a partial π-electron localization form of croconate in the five-membered ring. Such partial π-electron localization molecular geometry of C5O52− seems to be in accordance with charge-localized form of croconate in a hydrogen-bonded host lattice,12alkali metal croconate salts13 and theoretical predictions.14,15 The other one also adopts a bridging bis-bidentate mode,3b,7,16 but through four-adjacent croconate oxygen atoms (μ4), to link Ni ions with the Ni⋯Ni separation of 6.954(6) Å. The uncoordinated oxygen atom (O(10)) also shows a ketonic form with shorter bond lengths compared to other C–O bond lengths of the metal-coordinated oxygen atoms. The differences among five C–C bond lengths (see ESI, Table S1†) of μ4-croconate are not so obvious, indicating an intermediate pattern between the charge-localized (C2v) and charge-delocalized (D5h) forms4 of croconate upon complexation.
O groups are close to being single bond in character, and they are also longer than other C–C bond lengths, indicating a partial π-electron localization form of croconate in the five-membered ring. Such partial π-electron localization molecular geometry of C5O52− seems to be in accordance with charge-localized form of croconate in a hydrogen-bonded host lattice,12alkali metal croconate salts13 and theoretical predictions.14,15 The other one also adopts a bridging bis-bidentate mode,3b,7,16 but through four-adjacent croconate oxygen atoms (μ4), to link Ni ions with the Ni⋯Ni separation of 6.954(6) Å. The uncoordinated oxygen atom (O(10)) also shows a ketonic form with shorter bond lengths compared to other C–O bond lengths of the metal-coordinated oxygen atoms. The differences among five C–C bond lengths (see ESI, Table S1†) of μ4-croconate are not so obvious, indicating an intermediate pattern between the charge-localized (C2v) and charge-delocalized (D5h) forms4 of croconate upon complexation.
Table 2 Bond lengths (Å) and angles (°) around Ni(1) and Ni(2) ions in 1a
|
Symmetry transformations used to generate equivalent atoms: i = x − 1, y, z.
|
| Ni(1)–O(1) |
2.072(3) |
Ni(2)–O(3) |
2.073(3) |
| Ni(1)–O(2) |
2.267(3) |
Ni(2)–O(2) |
2.269(3) |
| Ni(1)–O(7) |
2.102(2) |
Ni(2)–O(8)i |
2.102(3) |
| Ni(1)–O(6) |
2.129(3) |
Ni(2)–O(9)i |
2.125(3) |
| Ni(1)–N(3) |
2.030(3) |
Ni(2)–N(4) |
2.030(3) |
| Ni(1)–N(1) |
2.065(3) |
Ni(2)–N(2) |
2.070(3) |
| N(3)–Ni(1)–O(7) |
169.2(1) |
N(4)–Ni(2)–O(3) |
102.0(1) |
| N(3)–Ni(1)–O(2) |
89.4(1) |
N(4)–Ni(2)–O(8)i |
169.4(1) |
| N(3)–Ni(1)–O(6) |
90.6(1) |
N(4)–Ni(2)–O(9)i |
92.0(1) |
| N(3)–Ni(1)–O(1) |
102.5(1) |
N(4)–Ni(2)–O(2) |
87.3(1) |
| N(3)–Ni(1)–N(1) |
94.0(1) |
N(4)–Ni(2)–N(2) |
93.6(1) |
| N(1)–Ni(1)–O(2) |
170.1(1) |
N(2)–Ni(2)–O(3) |
87.4(1) |
| N(1)–Ni(1)–O(6) |
93.9(1) |
N(2)–Ni(2)–O(8)i |
95.0(1) |
| O(7)–Ni(1)–O(1) |
85.4(1) |
O(3)–Ni(2)–O(8)i |
84.7(1) |
| O(7)–Ni(1)–O(2) |
84.4(1) |
O(3)–Ni(2)–O(9)i |
165.8(1) |
| O(7)–Ni(1)–O(6) |
81.2(1) |
O(8)–Ni(2)–O(9)i |
81.1(1) |
| O(7)–Ni(1)–N(1) |
93.6(1) |
N(2)–Ni(2)–O(9)i |
94.3(1) |
| O(1)–Ni(1)–O(2) |
81.6(1) |
N(2)–Ni(2)–O(2) |
169.2(1) |
| O(1)–Ni(1)–O(6) |
166.4(1) |
O(3)–Ni(2)–O(2) |
81.9(1) |
| O(1)–Ni(1)–N(1) |
88.6(1) |
O(8)–Ni(2)–O(2)i |
85.5(1) |
| O(2)–Ni(1)–O(6) |
95.3(1) |
O(9)–Ni(2)–O(2)i |
96.5(1) |
The 2D brick-wall-like layer shown in Fig. 1(a) is formed with a rectangle-grid as the basic building unit, which consists of six Ni(II) ions bridged by two μ3-croconate, two μ4-croconate, and two bis-monodentate bpe, giving rise to 12-membered rectangular apertures (Fig. 1(a)) with sizes of approximate 17.98 × 6.95 Å (Ni⋯Ni distances). Two layers are cross-linked by an out-of-plane bpe ligand with a bis-monodentate fashion to give a 2D bi-layers framework, as illustrated in Fig. 1(b). This result demonstrates a coordination polymer with its 2D bi-layers MOF being constructed by using bpe ligand and croconate with a hybrid bis-bidentate through four-adjacent (μ4) and three-adjacent (μ3) bridging modes. It is also interesting to note that a 1D alternatively trapezoid, inverse-trapezoid chain of the bi-layers MOF is formed viewing along the direction of the a-axis, as shown in Fig. 1(c). Adjacent independent 2D bi-layered frameworks with identical connectivity are also mutually interlocked by an inter-layer “sandwiched” π–π interaction6,17 between the μ3-croconate and pyridyl rings of bpe (see ESI, Fig. S2, Table S2†) and C–H⋯O interactions18 between the croconates and bpe ligands (Table 3), resulting in a stable 3D supramolecular architecture. The lattice water molecules (O(11)) are encapsulated by 1D channels formed from the bi-layer MOF (Fig. 1(c)) and are stabilized by the O–H⋯O hydrogen bond interactions between the lattice water and croconate ligands (Table 3). The pore volume (201.7 Å3) occupied by the lattice water molecules is calculated to be 6.5% of the unit cell volume (3112.1 Å3).
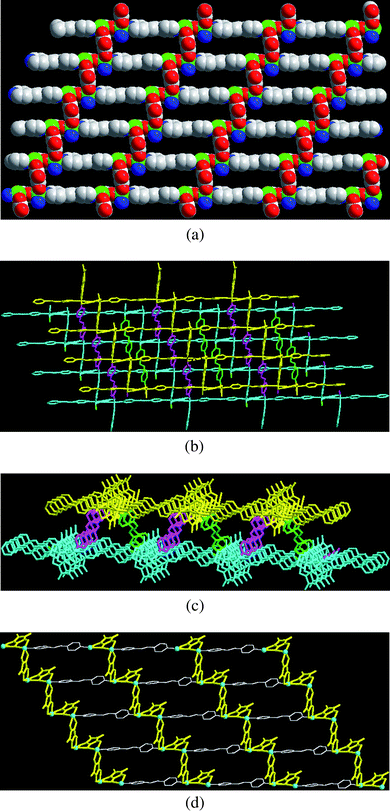 |
| | Fig. 1 (a) A 2D brick-wall-like layer MOF of 1 with the rectangle grid as the basic building block. (b) Clear view of the 2D bi-layer MOF with two layers (yellow, upper layer and blue, lower layer) cross-linkage by bpe ligands (pink and green). (c) 1D alternative trapezoid, inverse-trapezoid chain in the bi-layer viewing along a-axis. (d) 2D layer attributed to bpe ligands (white) linking the 1D zig-zag alternating magnetic chains by croconate ligands (yellow). | |
Table 3 The O–H⋯O H-bonds and C–H⋯O interaction for 1a
| D–H⋯A/Å |
D–H/Å |
H⋯A/Å |
D⋯A/Å |
∠ D–H⋯A/° |
|
Symmetry code: (i) = 1 − x, −y, −z; (ii) = x − 1, y, 1 + z; (iii) = 1 + x, y, z (iv) = 2 + x, −y + 1/2, z + 1/2.
|
|
O(11)–H(11C)⋯O(4)i |
0.99 |
2.58 |
3.061 |
110 |
|
C(11)–H(11A)⋯O(1) |
0.93 |
2.39 |
2.915 |
116 |
|
C(22)–H(22A)⋯O(3)ii |
0.93 |
2.52 |
2.972 |
110 |
|
C(30)–H(30A)⋯O(11)iii |
0.93 |
2.60 |
3.331 |
136 |
|
C(30)–H(30A)⋯O(9)iv |
0.93 |
2.50 |
3.011 |
115 |
To assess the thermal stability and its structural variation as a function of temperature, thermogravimetric analysis (TGA) was performed on single-phase polycrystalline samples. During the heating process (see ESI, Fig. S3†), the TGA analysis shows that the first weight loss of 3.6% (calc 2.4%), corresponding to the loss of crystallized water molecules, occurred in the range of approximate 25–383 °C. On further heating, these samples decomposed at approximately 383–460 °C, with a loss of 76.2%. The variable temperature XRD patterns are shown in Fig. 2. The crystallinity of compound 1 was thermally stable up to 400 °C, evidenced from the diffraction data. Above that temperature, pure Ni metal was formed (PDF #040850, cubic). At that temperature, the first weight loss observed by TGA was considered as dehydration of crystallized water. We observed only some weak reflection changes during the dehydration process. The diffraction pattern showed that the framework was stable as a crystalline. After complete removal of water at 350 °C, the sample was cooled to room temperature again and opened to air at room humidity (60% RH) for 12 h, and the sample returned to its hydration form (Fig. 2). This crystalline compound is not only thermally stable but also reversible upon its de-hydrated and re-hydrated. These excellent properties may play an important role in potential applications. The XRD measurements are also in accordance with the TG analysis and both results evidence that compound 1 has quite high thermo-stability that keep their crystalline forms up to at least 350 °C. The great thermal stability of the 3D supramolecular architecture could be mainly attributed to mutual interlocking among the 2D bi-layer frameworks.
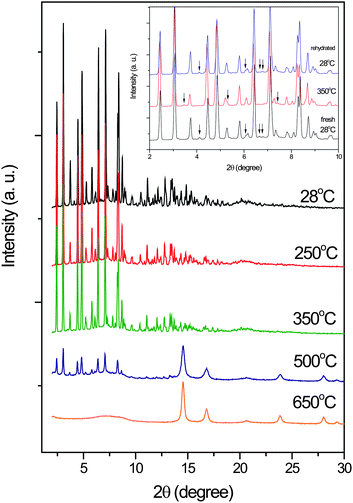 |
| | Fig. 2 Temperature-dependent powder X-ray diffraction patterns of 1 from room temperature to 650 °C. The temperature ramps to 350 °C and back to 28 °C for dehydration and rehydration are shown in the inset. The baselines for each temperature were shifted for clarity. | |
Magnetic properties of 1
The magnetic properties of 1 were measured from 300 to 1.8 K in the forms of χM and χMT vs. T shown in Fig. 3. At room temperature, χMT is equal to 2.03 emu K mol−1, which is close to the spin-only value 2.0 emu K mol−1 for a dinuclear non-interacting NiII unit with g = 2 and S = 1. When the temperature is lowered, χMT continuously decreases, reaching a minimum of 0.781 emu K mol−1 at 30 K, further cooling the system, χMT sharply increases to a maximum of 1.06 emu K mol−1 at 22 K and then down to 0.093 emu K mol−1 at 1.8 K. This magnetic behavior is the obvious nature of a spin-canting antiferromagnet. The decreasing region of χMT indicates the antiferromagnetic coupling interaction between NiII ions. Below 30 K, there is a sharp increase in the χMT value. After the maximum at 22 K, it dramatically decreased. The χMT value of 1.06 emu K mol−1 at 22 K corresponds to a spin state of S = 1, which is less than the spin value of two paramagnetic NiII ions. The sharp peak at low temperature indicates that the spins in 1 are in a canting state. It is confirmed with the variable-field magnetization, as shown in Fig. 4. At 1.8 K, the magnetization successively increases with the increasing applied field and reaches 0.055 Nβ·mol−1 at 7 T, which is much less than the saturation state of 4 Nβ·mol−1 for NiII2 unit. However, a hysteresis loop with coercivity about 3.5 kOe at 1.8 K was observed, indicating that 1 is a weak ferromagnet. The spin canting angle can be estimated by the magnetization value at 7 T, which is about 0.79°. When the temperature increases, the hysteresis loop becomes smaller and finally disappears above 20 K, suggesting a magnetic ordering temperature in the range of 20 to 25 K. The zero-field cooled magnetization (zfcm) and field cooling magnetization (fcm) at 10 Oe (Fig. 5) show the non-reversibility and bifurcation below 21.5 K, implying the spontaneous magnetization due to the long-range ordering transition. Fig. 6 gives the ac magnetic susceptibility measurements. An obvious peak was observed at 21 K for both in-phase and out-of-phase signals. Examined by different frequencies, we see no frequency dependent phenomenon for complex 1. This measurement further confirmed that a long-range order transition in this compound.
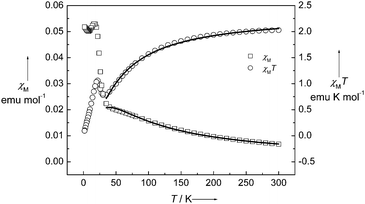 |
| | Fig. 3 Temperature dependence of magnetic susceptibilities of 1 from 300 to 1.8 K in the form of χM (□) and χMT (○) vs T. The solid lines are the best fitting, as described in the text. | |
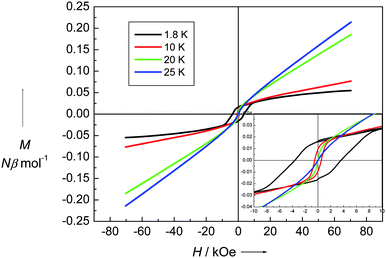 |
| | Fig. 4 Field dependence of magnetization of 1 at different temperatures. | |
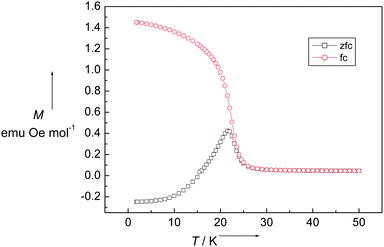 |
| | Fig. 5
ZFCM and FCM of 1 at an applied field of 10 Oe. | |
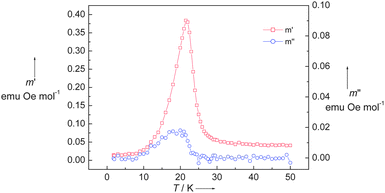 |
| | Fig. 6 AC magnetization of 1 at Hdc = 0, Hac = 2 Oe and f = 10 Hz. | |
In the crystal structure of 1, both ligands of C5O52− and bpe bridge between NiII ions to form a 2D double layer MOF, but only C5O52− is suggested to mediate the magnetic coupling between NiII ions due to the bpe groups' connecting magnetic centers over a long distance. Thus, the MOF of 1 can be considered as a 2D structure attributed to bpe groups linking the 1D zig-zag alternating magnetic chains (Fig. 1(d)). Furthermore, a Heisenberg exchange alternating chain model19 with spin Hamiltonian H = −J[∑S2i·S2i + 1 + ∑S2i·S2i−1] (where J and αJ1 are the two nearest neighbor antiferromagnetic exchange interactions) can be used here and
| | | χM = (2Ng2β2/3kT)(χrTr) | (1) |
in which
χr = [
ATr2 +
BTr +
C]/[
Tr3 +
DTr2 +
ETr +
F],
Tr =
kT/|
J| and the
A ∼
F expression with alternating parameter
α given as
X(
α) =
x0 +
x1α +
x2α (0.5 ≤
α ≤ 1.0). Below the ordering temperature, the magnetic properties being out of the paramagnetic region cannot be fitted by van Vleck equation; however, above 30 K, the interchain interaction is relatively weak and can be neglected at higher temperature. Thus, the best fitting results above 30 K (
Fig. 4) give a set of parameters describing the intrachain magnetic properties:
g = 2.17(2), |
J| = 25(6) cm
−1, |
αJ| = 0.6(6) × 25 = 15 cm
−1 and
R = 4.6 × 10
−4 (
R = ∑[(
χMT)
calcd − (
χMT)
obs]
2/∑(
χMT)
2obs). The fitting results indicate that μ
3-croconate bridges mediate the intermediately strong coupling between Ni
II ions in
1. The spin canting behavior is attributed to the superexchange interaction of antisymmetric components (Dzyaloshinsky-Moriya interaction, DMI),
20 which minimizes the coupling energy when two spins are perpendicular to one another. The mechanism for spin canting requires that the canted spins in the solid state are not related by a center of inversion.
21 In
1, there exists two sublattices corresponding to Ni1 and Ni2, respectively, therefore, it is possible that the spin canting occurs between different sublattices.
Conclusions
In this paper, we have presented the synthesis, crystal structure of a mixed-ligands coordination polymer, {[Ni2(bpe)2(C5O5)2]·H2O}n (1), with interesting structural topologies and high thermal stability. A 2D bi-layer framework is formed via two 2D layers cross-linkage by out-of-plane bpe ligands. Each layer is constructed by using a rectangle-grid as the basic building unit through the connectivity between the NiII ions and bpe, croconate (μ3- and μ4-bridging modes) ligands. The magnetic measurement shows the long range magnetic ordering behavior as well as spin canting in 1 below the critical temperature of 22 K with an approximate canting angle of 0.79°. A large hysteresis loop of coercivity 3.5 was observed at 1.8 K, indicating that 1 is a weak hard magnet. To the best of our knowledge, 1 is the first example of croconate-based Ni(II) compounds exhibiting the interesting magnetic properties such as spin-canting behavior.
Notes and references
- For example, see
(a) M. Ohba, N. N. Usuki, N. N. Fukita and H. Okawa, Angew. Chem., Int. Ed., 1999, 38, 1795–1798 CrossRef CAS;
(b) X. Hao, Y. Wei and S. Zhang, Chem. Commun., 2000, 2271–2273 RSC;
(c) P. S. Mukherjee, S. Dalai, G. Mostafa, E. Zangrando, T. H. Lu, G. Rogez, T. Mallah and N. R. Chaudhuri, Chem. Commun., 2001, 1346–1348 RSC;
(d) S. Konar, P. S. Mukherjee, E. Zangrando, F. Lloret and N. R. Chaudhuri, Angew. Chem., Int. Ed., 2002, 41, 1561–1563 CrossRef CAS;
(e) M. H. Zeng, W. X. Zhang, X. Z. Sun and X. M. Chen, Angew. Chem., Int. Ed., 2005, 44, 3079–3082 CrossRef CAS;
(f) T. E. Vos and J. S. Miller., Angew. Chem., Int. Ed., 2005, 44, 2416–2419 CrossRef CAS;
(g) D. Maspoch, D. Ruiz-Molina and J. Veciana, Chem. Soc. Rev., 2007, 36, 770–818 RSC;
(h) D. Maspoch, D. Ruiz-Molina and J. Veciana, J. Mater. Chem., 2004, 14, 2713–2723 RSC;
(i) S. R. Batten and K. S. Murray, Coord. Chem. Rev., 2003, 246, 103–130 CrossRef CAS.
-
(a)
O. Kahn
Molecular Magnetism, VCH, Weinheim, 1993, chap. 11 Search PubMed;
(b) P. Day, J. Chem. Soc., Dalton Trans., 1997, 701–707 RSC.
-
(a) H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 4877–4884 RSC;
(b) K. Drabent, Z. Ciunik and A. Ozarowski, Inorg. Chem., 2008, 47, 3358–3365 CrossRef CAS;
(c) H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 1734–1744 RSC;
(d) D. G. Branzea, L. Sorace, C. Maxim, M. Andruh and A. Caneschi, Inorg. Chem., 2008, 47, 6590–6592 CrossRef CAS;
(e) Y.-Z. Zheng, W. Xue, M.-L. Tong, X.-M. Chen, F. Grandjean and G. J. Long, Inorg. Chem., 2008, 47, 4077–4087 CrossRef CAS;
(f) G. Agustí, M. C. Muñoz, A. B. Gaspar and J. A. Real, Inorg. Chem., 2008, 47, 2552–2561 CrossRef CAS;
(g) X.-Y. Wang and S. C. Sevov, Inorg. Chem., 2008, 47, 1037–1043 CrossRef CAS;
(h) A. M. Kirillov, Y. Y. Karabach, M. Haukka, M. F. C. G. da Silva, J. Sanchiz, M. N. Kopylovich and A. J. L. Pombeiro, Inorg. Chem., 2008, 47, 162–175 CrossRef CAS;
(i) H.-L. Sun, Z.-M. Wang and S. Gao, Inorg. Chem., 2005, 44, 2169–2176 CrossRef CAS;
(j) R. Sessoli, Inorg. Chim. Acta, 2008, 361, 3356–3364 CrossRef CAS;
(k) E. Rentschler and C. von Malotki, Inorg. Chim. Acta, 2008, 361, 3646–3653 CrossRef CAS;
(l) R. A. Mole, S. P. Cottrell, J. A. Stride and P. T. Wood, Inorg. Chim. Acta, 2008, 361, 3718–3722 CrossRef CAS;
(m) M. A. M. Abu-Youssef, F. A. Mautner and R. Vicente, Inorg. Chim. Acta, 2008, 361, 2895–2900 CrossRef CAS;
(n) H.-R. Wen, C.-F. Wang, Z. Y. Du and J.-L. Zuo, Inorg. Chim. Acta, 2008, 361, 2901–2908 CrossRef CAS;
(o) A. Ray, J. Chakraborty, B. Samanta, S. Thakurta, C. Marschner, M. S. El Fallah and S. Mitra, Inorg. Chim. Acta, 2008, 361, 1850–1860 CrossRef CAS;
(p) G. S. Matouzenko, M. Perrin, B. Le Guennic, C. Genre, G. Molnar, A. Bousseksou and S. A. Borshch, Dalton Trans., 2007, 934–942 RSC;
(q) J. Chakraborty, M. Nandi, H. Mayer-Figge, W. S. Sheldrick, L. Sorace, A. Bhaumik and P. Banerjee, Eur. J. Inorg. Chem., 2007, 5033–5044 CrossRef CAS;
(r) M. Seredyuk, A. B. Gaspar, M. C. Munoz, M. Verdaguer, F. Villain and P. Guetlich, Eur. J. Inorg. Chem., 2007, 4481–4491 CrossRef CAS.
-
(a) B. Wisser, Yirong Lu and C. Janiak, Z. Anorg. Allg. Chem., 2007, 633, 1189–1192 CrossRef CAS;
(b) S. C. Manna, K.-I. Okamoto, E. Zangrando and N. R. Chaudhuri, CrystEngComm, 2007, 9, 199–202 RSC;
(c) Z.-F. Chen, S.-F. Zhang, H.-S. Luo, B. F. Abrahams and H. Liang, CrystEngComm, 2007, 9, 27–29 RSC;
(d) A. Pichon, C. M. Fierro, M. Nieuwenhuyzen and S. James, CrystEngComm, 2007, 9, 449–451 RSC;
(e) J. Pasán, J. Sanchiz, F. Lloret, M. Julve and C. Ruiz-Pérez, CrystEngComm, 2007, 9, 478–487 RSC;
(f) M. D. Stephenson and M. J. Hardie, Dalton Trans., 2006, 3407–3417 RSC;
(g) R. Carballo, B. Covelo, E. M. Vázquez-López, E. García-Martínez, A. Castiñeiras and C. Janiak, Z. Anorg. Allg. Chem., 2005, 631, 2006–2010 CrossRef CAS.
-
(a) I. Castro, M. L. Calatayud, F. Lloret, J. Sletten and M. Julve, J. Chem. Soc., Dalton Trans., 2002, 2397–2403 RSC;
(b) I. Castro, J. Sletten, J. Faus, M. Julve, Y. Journaux, F. Lloret and S. Alvarez, Inorg. Chem., 1992, 31, 1889–1894 CrossRef CAS.
-
(a) T. K. Maji, S. Konar, G. Mostafa, E. Zangrando, T. H. Lu and N. R. Chaudhuri, J. Chem. Soc., Dalton Trans., 2003, 171–175 RSC;
(b) T. K. Maji, D. Ghoshal, E. Zangrando, J. Ribas and N. R. Chaudhuri, CrystEngComm, 2004, 6, 623–626 RSC;
(c) D. Ghoshal, A. K. Ghosh, J. Ribas, G. Mostafa and N. R. Chaudhuri, CrystEngComm, 2005, 7, 616–620 RSC.
-
(a) C.-C. Wang, C.-H. Yang, S.-M. Tseng, G.-H. Lee, Y.-P. Chiang and H.-S. Sheu, Inorg. Chem., 2003, 42, 8294–8299 CrossRef CAS;
(b) C.-C. Wang, H.-W. Lin, C.-H. Yang, C.-H. Liao, I.-T. Lan and G.-H. Lee, New J. Chem., 2004, 28, 180–183 RSC;
(c) C. C. Wang, C. H. Yang and G. H. Lee, Inorg. Chem., 2002, 41, 1015–1018 CrossRef CAS;
(d) C. C. Wang, S. M. Tseng, S. Y. Lin, F. C. Liu, S. C. Dai, G. H. Lee, W. J. Shih and H. S. Sheu, Cryst. Growth Des., 2007, 7, 1783–1790 CrossRef CAS;
(e) C. C. Wang, S. C. Dai, H. W. Lin, G. H. Lee, H. S. Sheu, Y. H. Lin and H. L. Tsai, Inorg. Chim. Acta, 2007, 360, 4058–4067 CrossRef.
-
SMART V 4.043 Software for CCD Detector System; Siemens Analytical Instruments Division: Madison, WI, 1995 Search PubMed.
-
SAINT V 4.035 Software for CCD Detector System; Siemens Analytical Instruments Division: Madison, WI, 1995 Search PubMed.
-
G. M. Sheldrick. Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1993 Search PubMed.
-
SHELXTL 5.03 (PC-Version), Program Liberary for Structure Solution and Molecular Graphics; Siemens Analytical Instruments Division: Madison, WI, 1995 Search PubMed.
- C. K. Lam, M. F. Cheng., C. L. Li, J. P. Zhang, X. M. Chen, W. K. Li and T. C. K. Mak, Chem. Commun., 2004, 448–449 RSC.
-
(a) D. Braga, L. Maini and F. Grepioni, Chem.–Eur. J., 2002, 8, 1804–1812 CrossRef CAS;
(b) J. D. Duntiz, P. Seiler and W. Czechtizky, Angew. Chem., Int. Ed., 2001, 40, 1779–1780 CrossRef CAS.
- M. F. Cheng, C. L. Li and W. K. Li, Chem. Phys. Lett., 2004, 391, 157–164 CrossRef CAS.
- P.v. R. Schleyer, K. Najafian, B. Kiran and H. Jiao, J. Org. Chem., 2000, 65, 426–431 CrossRef.
- I. Castro, M. L. Calatayud, J. Sletten, M. Julve and F. Lloret, C.R. Acad. Sci. Paris, Chim., 2001, 4, 235 Search PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2002, 3885 Search PubMed.
-
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, in IUCr Monograph on Crystallography, 1999, 9, Oxford Science Search PubMed.
- J. J. Borrás-Almenar, E. Coronado, J. Curely and R. Georges, Inorg. Chem., 1995, 34, 2699–2704 CrossRef CAS.
-
(a) I. Dzyaloshinsky, J. Phys. Chem. Solids, 1958, 4, 241–255 CrossRef;
(b) T. Moriya, Phys. Rev., 1960, 120, 91 CrossRef.
-
(a)
R. L. Carling, Magnetochemistry, Springer, Berlin, 1989 Search PubMed;
(b) N. Robertson, C. Bergemann, H. Becker, P. Agarwal, S. R. Julian, R. H. Friend, N. J. Hatton, A. E. Underhill and A. Kobayashi, J. Mater. Chem., 1999, 9, 1713–1717 RSC;
(c) X. Ren., Y. Chen, C. He and S. Gao, J. Chem. Soc., Dalton Trans., 2002, 3915–3918 RSC.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.