DOI:
10.1039/C0CE00098A
(Paper)
CrystEngComm, 2011,
13, 279-286
Synthesis, crystal structures, and luminescent properties of Cd(II) coordination polymers assembled from asymmetric semi-rigid V-shaped multicarboxylate ligands†
Received
15th April 2010
, Accepted 18th July 2010
First published on 7th September 2010
Abstract
A series of seven Cd(II) coordination polymers, namely [Cd3(L1)2(phen)3]n·(H2O)2n (1), [Cd3(L1)2(2,2′-bpy)3(H2O)2]n·(H2O)3n (2), [Cd3(L1)2(4,4′-bpy)2]n·(H2O)4n (3), [Cd3(L2)2(phen)2]n (4), [Cd3(L2)2(2,2′-bpy)2]n (5), [Cd3(L3)2(phen)2(H2O)]n·(H2O)2n (6), and [Cd3(L4)2(phen)2(H2O)2]n·(H2O)3n (7), have been prepared on the basis of four asymmetric semi-rigid V-shaped multicarboxylate ligands including 4-(2-carboxyphenoxy)phthalic acid (H3L1), 3-(2-carboxyphenoxy)phthalic acid (H3L2), 3-(4-carboxyphenoxy)phthalic acid (H3L3), and 4-(4-carboxyphenoxy)phthalic acid (H3L4) with the help of 1,10-phenanthroline (phen), 2,2′-bipyridine (2,2′-bpy), or 4,4′-bipyridine (4,4′-bpy) as secondary ligand. Single-crystal X-ray diffraction analysis reveals that compounds 1, 4, 5, and 6 exhibit one-dimensional (1D) double chain structures constructed from trinuclear Cd(II) clusters, which further forms 2D supramolecular architecture via intermolecular π–π interaction. Complex 2 possesses a 3D supramolecular network assembled from 2D layered architecture composed of dinucelar cadmium subunits and isolated Cd(II) ions via intermolecular π–π interaction. Compound 3 shows a 3D framework structure which is generated from 2D layered motif comprised of trinuclear cadmium subunits pillared by 4,4′-bpy ligands, while the 3D framework of 7 is constructed from two different directional ribbon chains consisting of dinuclear cadmium clusters and isolated Cd(II) ions as node. The different molecular structure for compounds 1–3 formed from the same L1 ligand reveals the spacer effect of secondary ligands on tuning the structure of coordination polymers, while the structural difference among 1, 4, 6, and 7 built from different asymmetric semi-rigid V-shaped multicarboxylate ligand (L1, L2, L3, or L4) but employing the same secondary ligand of phen reveals the important role of the positions of carboxylic substituents at the asymmetric multicarboxylate ligand in the formation of coordination compounds. Photophysical properties over the series of complexes 1–7 have been systematically and comparatively investigated.
Introduction
In the field of supramolecular chemistry and crystal engineering, the design and synthesis of coordination polymers have been emerging as an ongoing field owing to their structural aesthetics and topologies as well as diverse functional properties.1–3 Among various kinds of complexes fabricated, those with intriguing fluorescent property attract great attention due to their potential applications in light-emitting diodes (LEDs).4 Thus far, significant advance achieved in this field has led to a lot of promising materials through the self-assembly of organic ligands and metal ions.5 Nevertheless, it still remains a great and long-term challenge to exactly predict the molecular structure and luminescent properties of coordination polymers because of many subtle factors involved in the crystallization process.
In comparison with the rigid aromatic multicarboxylate ligands such as 1,4-benzenedicarboxylate, 1,3,5-benzenetricarboxylate, and 1,2,4,5-benzenetetracarboxylate with one benzene ring as central molecular framework, the so-called semi-rigid V-shaped multicarboxylate ligands with two benzene rings bridged by an nonmetallic atom (C, O, S, or N atom) as central molecular framework are of increasing flexibility and therefore able to lead to metal complexes with diverse structures because of the free rotation of two benzene rings around the bridged non-metallic atom.6,7 By using such semi-rigid V-shaped ligands, different coordination polymers with diverse molecular structures and potential applications in the field of separation, absorption, catalyst, and sensor have been prepared.8 It is worth noting that symmetric semi-rigid V-shaped multidentate O-donor ligands with two or four carboxylic substituents attached at the symmetric positions of semi-rigid V-shaped central molecular framework usually generate coordination polymers with discrete metal ions as node, leading to the limitation in tuning the structure and functionality of metal–organic coordination frameworks (MOFs). As a result, effort has been started recently to be devoted to the construction of MOFs using asymmetric semi-rigid V-shaped multidentate O-donor ligands with carboxylic substituents attached at asymmetric positions of central V-shaped molecular framework, resulting in coordination polymers composed of polymetallic subunits through tuning the positions of asymmetrically attached carboxylic groups at the central framework.9 For the purpose of further clarifying the effect of positions of asymmetrically attached carboxylic groups on the molecular structure and functional properties of MOFs, in the present work four asymmetric semi-rigid V-shaped multicarboxylate ligands H3L1-H3L4, Scheme 1, were selected and employed to assemble a series of seven new coordination complexes including [Cd3(L1)2(phen)3]n·(H2O)2n (1), [Cd3(L1)2(2,2′-bpy)3(H2O)2]n·(H2O)3n (2), [Cd3(L1)2(4,4′-bpy)2]n·(H2O)4n (3), [Cd3(L2)2(phen)2]n (4), [Cd3(L2)2(2,2′-bpy)2]n (5), [Cd3(L3)2(phen)2(H2O)]n·(H2O)2n (6), and [Cd3(L4)2(phen)2(H2O)2]n·(H2O)3n (7) with the help of phen, 2,2′-bpy, or 4,4′-bpy as secondary ligand. Their single crystal structures and luminescent properties were also systematically and comparatively investigated.
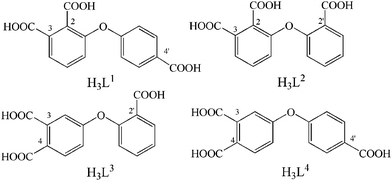 |
| | Scheme 1 Schematic molecular structures of H3L1–H3L4. | |
Experimental
All reagents and solvents employed in the present work were obtained from commercial source and used directly without further purification. The four ligands H3L1-H3L4 were synthesized according to the reported procedure.9b
General synthesis procedure for complexes 1–7
In the general synthesis, the target complexes were obtained by using hydrothermal method with the same stoichiometric proportion for the starting materials in the presence of KOH. Teflon-lined stainless steel container (25 mL) was employed as reaction vessel containing all starting materials, which was heated to appropriate temperature and held for 72 h, then cooled to 50 °C at a descent rate of 10 °C h−1. Finally, the oven was cut off and kept for another 10 h, perfect crystals were isolated.
[Cd3(L1)2(phen)3]n·(H2O)2n (1)
The mixture of Cd(OAc)2·2H2O (0.0533 g, 0.2 mmol), phen (0.0360 g, 0.2 mmol), H3L1 (0.0302 g, 0.1 mmol), KOH (0.0168 g, 0.3 mmol), and H2O (15 mL) was sealed in 25 mL Teflon-lined stainless steel reactor, which was heated to 130 °C. Colourless block-shaped crystals suitable for X-ray diffraction analysis were separated by filtration with the yield of 0.0325 g, 43% (based on L1 ligand). Anal. calcd for C66H42Cd3N6O16: C 52.42, H 2.80, N 5.56. Found: C 52.45, H 2.82, N 5.49. IR/cm−1 (KBr): 3064 (m), 1589 (s), 1553 (s), 1406 (s), 1370 (s), 1224 (s), 860 (s), 726 (m).
[Cd3(L1)2(2,2′-bpy)3(H2O)2]n·(H2O)3n (2)
The mixture of Cd(OAc)2·2H2O (0.0533 g, 0.2 mmol), 2,2′-bpy (0.0312 g, 0.2 mmol), H3L1 (0.0302 g, 0.1 mmol), KOH (0.0168 g, 0.3 mmol), and H2O (15 mL) H3L1 was sealed in 25 mL Teflon-lined stainless steel reactor, which was heated to 150 °C. Colourless block-shaped crystals suitable for X-ray diffraction analysis were separated by filtration with the yield of 0.0366 g, 49% (based on L1 ligand). Anal. calcd for C60H48Cd3N6O19: C 48.22, H 3.23, N 5.62. Found: C 48.01, H 3.32, N 5.48. IR/cm−1 (KBr): 3069 (m), 1586 (s), 1553 (s), 1400 (s), 1367 (s), 1223 (s), 870 (m), 733 (m).
[Cd3(L1)2(4,4′-bpy)2]n·(H2O)4n (3)
By employing the above-described procedure for preparing 2 with 4,4′-bpy (0.0312 g, 0.2 mmol) instead of 2,2′-bpy (0.0312 g, 0.2 mmol) as starting material, colourless block-shaped crystals suitable for X-ray diffraction analysis were obtained after the reactor was cooled to room temperature from 150 °C with the yield of 0.0369 g, 56% (based on L1 ligand). Anal. calcd for C50H38Cd3N4O18: C 45.49, H 2.90, N 4.24. Found: C 45.41, H 2.92, N 4.19. IR/cm−1 (KBr): 3071 (m), 1606 (s), 1540 (m), 1390 (s), 1236 (s), 865 (m), 762 (m).
[Cd3(L2)2(phen)2]n (4)
Compound 4 was synthesized in a similar manner to 1 by using H3L2 (0.0302 g, 0.1 mmol) instead of H3L1 as starting material. Colourless blocked crystals suitable for X-ray diffraction analysis were obtained after the reactor was cooled to room temperature from 130 °C with the yield of 0.0473 g, 73% (based on L2 ligand). Anal. calcd for C54H30Cd3N4O14: C 50.04, H 2.33, N 4.32. Found: C 50.06, H 2.28, N 4.19. IR/cm−1 (KBr): 3064 (m), 1581 (s), 1552 (s), 1390 (s), 1233 (s), 851 (m), 726 (m).
[Cd3(L2)2(2,2′-bpy)2]n (5)
By using the same procedure used to prepare 2 with H3L2 (0.0302 g, 0.1 mmol) instead of H3L1 as starting material, colourless block-shaped crystals suitable for X-ray diffraction analysis were separated by filtration with the yield of 0.0381 g, 61% (based on L2 ligand). Anal. calcd for C50H30Cd3N4O14: C 48.03, H 2.29, N 2.21. Found: C 48.11, H 2.39, N 2.29. IR/cm−1 (KBr): 3072 (m), 1591 (s), 1562 (s), 1382 (s), 1233 (s), 857 (m), 761 (m).
[Cd3(L3)2(phen)2(H2O)]n·(H2O)2n (6)
By using the same procedure used to prepare 1 with H3L3 (0.0302 g, 0.1 mmol) instead of H3L1 (0.0302 g, 0.1 mmol) as starting material, colourless blocked crystals suitable for X-ray diffraction analysis were obtained after the reactor was cooled to room temperature with the yield of 0.0324 g, 48% (based on L3 ligand). Anal. calcd for C54H36Cd3N4O17: C 48.04, H 2.69, N 4.15. Found: C 48.01, H 2.67, N 4.13. IR/cm−1 (KBr): 3057 (m), 1599 (s), 1552 (s), 1387 (s), 1241 (s), 856 (m), 776 (m).
[Cd3(L4)2(phen)2(H2O)2]n·(H2O)3n (7)
By employing the above-described procedure of 1 with H3L4 (0.0302 g, 0.1 mmol) instead of H3L1 (0.0302 g, 0.1 mmol) as starting material, colourless blocked crystals suitable for X-ray diffraction analysis were obtained after the reactor was cooled to room temperature with the yield of 0.0454 g, 58% (based on L4 ligand). Anal. calcd for C66H47Cd3N6O19: C 50.64, H 3.03, N 5.37. Found: C 50.61, H 3.09, N 5.33. IR/cm−1 (KBr): 3071 (m), 1589 (s), 1559 (s), 1384 (s), 1236 (s), 858 (m), 763 (m).
Physical measurements
Elemental analyses were carried out with an Elementary Vario El. The infrared spectroscopy on KBr pellets was performed on a Magna-IR 750 spectrophotometer in the region of 4000–400 cm−1. TGA was performed on a Perkin-Elmer TG-7 analyzer heated from 30–600 °C under nitrogen. The fluorescence spectra were measured on a multifrequency phase and modulation fluorometer.
Crystal data for all the seven complexes were collected on a Bruker SMART APEXII CCD diffractometer with graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å) using the SMART and SAINT programs at 298 K, and the structures were solved by the direct method (SHELXS-97) and refined by full-matrix least-squares (SHELXL-97) on F2. Anisotropic thermal parameters were used for the non-hydrogen atoms and isotropic parameters for the hydrogen atoms. Hydrogen atoms were added geometrically and refined using a riding model. Crystallographic data and other pertinent information for all the complexes are summarized in Table 1 and 2. Selected bond distances and bond angles with their estimated standard deviation are listed in Table S1 (ESI).†
Table 1 Details of the crystal parameters, data collection and refinement for complexes 1–3
| Complex |
1
|
2
|
3
|
| Formula. |
C66H42Cd3N6O16 |
C60H48Cd3N6O19 |
C50H38Cd3N4O18 |
|
FW/g mol−1 |
1512.29 |
1494.24 |
1320.09 |
| System |
Monoclinic |
Triclinic |
Monoclinic |
| Space group |
C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
C2/c |
|
a/Å |
15.159(3) |
9.5260(5) |
20.8380(15) |
|
b/Å |
18.821(3) |
15.0310(8) |
10.485(2) |
|
c/Å |
19.798(3) |
22.0200(11) |
22.5800(10) |
|
α/° |
90 |
70.5440(10) |
90 |
|
β/° |
92.382(3) |
78.3470(10) |
102.9680(10) |
|
γ/° |
90 |
84.9060(10) |
90 |
|
Z
|
4 |
2 |
4 |
|
V/Å3 |
5643.4(16) |
2910.8(3) |
4807.6(10) |
|
D
calcd/g cm−3 |
1.780 |
1.705 |
1.824 |
|
μ/mm−1 |
1.202 |
1.168 |
1.398 |
|
F000
|
3008 |
1492 |
2616 |
|
R
int
I > 2θ |
0.0521 |
0.0419 |
0.0234 |
|
R
w2
I > 2θ |
0.1383 |
0.1143 |
0.0619 |
|
R
int all |
0.0609 |
0.0547 |
0.0264 |
|
R
w2 all |
0.1435 |
0.1211 |
0.0635 |
|
S
|
1.064 |
1.054 |
1.046 |
Table 2 Details of the crystal parameters, data collection and refinement for complexes 4–7
| Complex |
4
|
5
|
6
|
7
|
| Formula |
C54H30Cd3N4O14 |
C50H30Cd3N4O14 |
C54H36Cd3N4O17 |
C66H47Cd3N6O19 |
|
FW/g mol−1 |
1296.02 |
1247.98 |
1350.07 |
1565.30 |
| System |
Triclinic |
Triclinic |
Monoclinic |
Triclinic |
| Space group |
P
|
P
|
Cc
|
P
|
|
a/Å |
11.6201(5) |
11.2000(9) |
14.0039(5) |
13.511(3) |
|
b/Å |
11.9085(5) |
11.5369(9) |
28.8677(11) |
14.864(3) |
|
c/Å |
18.7329(8) |
18.3362(14) |
13.6293(5) |
17.527(3) |
|
α/° |
84.387(10) |
88.2210(10) |
90 |
66.948(2) |
|
β/° |
86.887(10) |
81.4750(10) |
118.5460(10) |
86.831(2) |
|
γ/° |
66.265(10) |
70.8300(10) |
90 |
67.297(3) |
|
Z
|
2 |
2 |
4 |
2 |
|
V/Å3 |
2361.27(2) |
2212.8(3) |
4840.0(3) |
2968.8(1) |
|
D
calcd/g cm−3 |
1.823 |
1.873 |
1.853 |
1.751 |
|
μ/mm−1 |
1.416 |
1.506 |
1.390 |
1.150 |
|
F000
|
1276 |
1228 |
2672 |
1562 |
|
R
int
I > 2θ |
0.0305 |
0.0246 |
0.0172 |
0.0439 |
|
R
w2
I > 2θ |
0.0962 |
0.0607 |
0.0462 |
0.1175 |
|
R
int all |
0.0380 |
0.0300 |
0.0184 |
0.0626 |
|
R
w2 all |
0.1120 |
0.0629 |
0.0555 |
0.1296 |
|
S
|
1.134 |
1.047 |
1.150 |
1.106 |
Results and discussion
Preparation of the complexes 1–7
In the present study, complexes 1–7 were prepared from the hydrothermal reaction between corresponding semi-rigid V-shaped multicarboxylate ligand and Cd(OAc)2·2H2O with the help of assistant ligand including phen, 2,2′-bpy, and 4,4′-bpy ligands, Scheme 2. It was just reported that a similar hydrothermal reaction between Co(OAc)2·2H2O and semi-rigid V-shaped multicarboxylate ligand in the presence of 4,4′-bpy ligand led to the formation of single crystals suitable for X-ray analysis without the necessity using base to adjust the pH value of reaction mixture.9b However, in the present case the reaction between Cd(OAc)2·2H2O and H3L(L = L1, L2, L3, and L4) with the help of secondary ligand without adding base gave only some white precipitate instead of single crystals of coordination polymers. In good contrast, when solid KOH with the molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to the tricarboxylate ligand was added to completely deprotonate the ligand, good single crystals of seven coordination polymers were obtained. In addition, the reaction temperature was also revealed to play an important role in the formation of corresponding complexes as exemplified for the isolation of 2, 3, and 5 only at higher temperature of 150 °C but isolation of 1, 4, 6, and 7 at lower temperature of 130 °C.
:1 to the tricarboxylate ligand was added to completely deprotonate the ligand, good single crystals of seven coordination polymers were obtained. In addition, the reaction temperature was also revealed to play an important role in the formation of corresponding complexes as exemplified for the isolation of 2, 3, and 5 only at higher temperature of 150 °C but isolation of 1, 4, 6, and 7 at lower temperature of 130 °C.
 |
| | Scheme 2 Schematic synthesis procedure of compounds 1–7. | |
Thermal analysis
The thermal behaviour for compounds 1–7 was studied to reveal their thermal stability. TGA experiments were performed on pure single crystal sample of complexes 1–7 under N2 atmosphere with a heating rate of 10 °C min−1 in the range of 30–600 °C.
The thermal curves are exhibited in Fig. S1 (ESI).† Complex 1 displays the weight loss by stage owing to the release of water molecules in the range from room temperature to 144 °C (obsd 2.18%, calcd 2.38%). The organic spacers then decomposes from 287–387 °C. For 2, the TGA curve shows that the water molecules are lost from room temperature to 127 °C, obsd 3.58%, calcd 3.62%. The decomposition temperature of this compound spans the range of 384–485 °C. According to the TGA curve of 3, the water molecules are lost in the range of 30–243 °C, obsd 5.14%, calcd 5.46%. Both 4 and 5 show similar TGA curve due to their similar molecular architecture with only different secondary ligand. The weight loss is observed owing to the direct decomposition of organic ligands in the range of 330–442 °C for 4 and 334–440 °C for 5. The release of water molecules is observed in 6 in the range from room temperature to 242 °C (obsd 13.05%, calcd 13.38%). The curve of 7 reveals that the lattice water molecules are lost from room temperature to 214 °C, obsd 3.20%, calcd 3.45%. The residual composition of this complex is then decomposed in the range of 284–481 °C.
Crystal structures of complexes 1, 4, 5, and 6
The crystal structures of 1, 4, 5, and 6 have been shown in Fig. 1–3, and selected bond lengths and angles compiled in Table S1 (ESI).† Despite the different asymmetric semi-rigid V-shaped ligand or secondary ligand, single-crystal X-ray diffraction analysis reveals that all these four complexes feature a 2D layered supramolecular structure assembled from 1D chain composed of trinuclear subunits via intermolecular π–π interaction. As shown in Fig. 1A, compound 1 is composed of two kinds of crystallographically independent Cd(II) ions, of which both Cd1 and Cd2 ions locate in a slightly distorted octahedral coordination sphere formed by two nitrogen atoms from chelating phen ligand and four carboxylic oxygen atoms of L1 ligand in cis-typed conformation with μ4-bridging mode binding four Cd(II) ions. The Cd–O and Cd–N bond lengths are in the range of 2.197(5)–2.466(6) and 2.327(5)–2.386(5) Å, respectively. These data are comparable with those of reported compounds containing O–Cd–N segments.10 As can be seen, the 3- and 4-carboxylic groups in 1 as linkers assemble three Cd(II) ions into trinuclear aggregate, and the 2′-carboxylic group in monodentate mode connects Cd1 ion of the neighbouring trinuclear Cd(II) cluster, forming a 1D infinite chain, Fig.1B. These chains are further packed into a 2D supramolecular network via intermolecular π–π interaction (face–face distance amounting to 3.535(4) Å) between phen ligands of neighbouring chains, Fig. 1C, which consolidates the structure of 1 in cooperation with the coordination interaction.
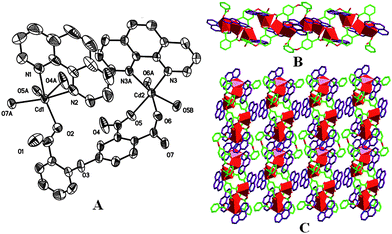 |
| | Fig. 1 (A) The ORTEP diagram of coordination environment for Cd(II) atoms in 1 (the 30% probability level), all hydrogen atoms and water molecules have been omitted for clarity. (B) An infinite 1D chain composed of trinuclear subunits. (C) 2D supramolecular network via intermolecular π–π interaction. | |
 |
| | Fig. 2 (A) The ORTEP diagram of coordination sphere for Cd(II) atoms in 4 with the 30% probability level, all hydrogen atoms and water molecules have been omitted for clarity. (B) A infinite 1D molecular structure of 4. (C) 2D supramolecular network. | |
 |
| | Fig. 3 The coordination geometry of 6 (A), 1D wave ribbon chain along the c axis (B), and the 2D supramolecular structures of 6via intermolecular interactions (C). | |
The detail of molecular structures of isostructural 4 and 5 is elucidated by taking 4 as a typical representative, Fig. 2. Compound 4 is constructed by three kinds of Cd(II) ions, Fig. 2A. Cd1 and Cd3 ions are coordinated by two nitrogen atoms of phen ligand and five oxygen atoms from three L2 ligands. Cd2 ion is hexa-coordinated by six oxygen atoms. 2- and 3-carboxylic groups of two kinds of L2 ligands with μ2-η2-η1 coordination mode bind to three Cd(II) ions, leading to a trinuclear cluster. These trinuclear subunits are further linked by 2′-carboxylic groups from L2 ligand, generating a 1D infinitely double chain, Fig. 2B. The neighbouring chains are packed in a × c plane, giving a 2D supramolecular structure via intermolecular π–π interaction (face–face distance amounting to 3.394(7) and 3.361(3) Å for 4 and 5, respectively).
In complex 6, a much wavier chain than in 1, 4, and 5 containing two kinds of L3 ligands and three types of Cd(II) ions can be found, Fig. 3A. Cd1 and Cd3 ions are hepta-coordinated. For Cd1 ion, the coordination environment is composed of two nitrogen atoms, four oxygen atoms from two carboxylic groups in μ2-η2-η1 mode, and one oxygen atom of water molecules. Cd2 ion locates in a distorted octahedral coordination geometry completed by four oxygen atoms and two nitrogen atoms of phen ligand. The coordination geometry around Cd3 ion is constructed by seven oxygen atoms of five different carboxylic groups. The Cd1, Cd2, and Cd3 ions are connected by carboxylic groups of L3 ligand to form a trinuclear Cd(II) cluster, which further expands to give a wavy double chain, Fig. 3B. The characteristic wavy chains composed of Cd(II) trimers are further assembled into a 2D supramolecular structure via the π–π interaction (face to face distance amounting to 3.379(3) Å) between the phen ligands of different chains, Fig. 3C. At the end of this paragraph, it is worth noting that the molecular structure of 1, 4, 5, and 6 support the fact that the chelating bidentate ligands phen and 2,2′-bpy favour the formation of 1D coordinative chain by restricting the available metal coordination sites.11
Crystal structure of complex 2
Compound 2 was obtained only by employing 2,2′-bpy instead of phen for preparing 1 as secondary ligand. As can be found in Fig. 4A, 2 contains three kinds of Cd(II) ions, three kinds of chelating 2,2′-bpy ligand, two types of L1 ligands, and three coordination water molecule in the asymmetric unit. Cd1 ion is hepta-coordinated by two nitrogen atoms of 2,2′-bpy ligands, four oxygen atoms from two bidentate chelating carboxylic groups, and one oxygen atom of water molecules. Both Cd2 and Cd3 ions possess a distorted octahedral coordination geometry completed by two nitrogen atoms and four oxygen atoms. L1 ligands of 2 in trans-typed conformation with μ3- and μ4-bridging modes link Cd1 and Cd2 ions into a dinuclear subunit. These dinuclear subunits and isolated Cd3 ions are bridged by L1 ligands to further generate a 2D covalent network. In the case that L1 ligand, Cd1 ion, and dinuclear subunit built from Cd2 and Cd3 ions are considered as 3-connected node, the molecular structure of 2 could be classified as a (6,3) herringbone network, Fig. S2 (ESI).† The neighbouring 2D covalent structures further form a 3D supramolecular framework via the π–π interaction (face to face distance amounting to 3.412(3) Å) between 2,2′-bpy ligands. The different structure between 1 and 2 indicates the effect of N-donor size in tuning the structure of coordination polymer.8f,12
 |
| | Fig. 4 (A) The ORTEP diagram of coordination geometry for Cd(II) atoms in 2 (the 30% probability level), all hydrogen atoms and water molecules have been omitted for clarity. (B) 2D covalent molecular structure of 2. (C) 3D supramolecular network via intermolecular π–π interaction. | |
Crystal structure of complex 3
Complex 3 was synthesized by introducing rod-like N-donor (4,4′-bpy) secondary ligand instead of the chelating N-donor (phen and 2,2′-bpy ligands), which exhibits a complicated 3D molecular structure as expected. There are two kinds of different Cd(II) ions in 3, both Cd(II) ions are in octahedral coordination environment, Fig. 5A. However, the octahedral coordination sphere around Cd1 ion is completed by six oxygen atoms from four L1 ligands in trans-typed configuration, and Cd2 ion is hexa-coordinated with the equatorial plane formed by four oxygen atoms and the axis positions occupied by two nitrogen atoms of 4,4′-bpy ligands. One Cd1 ion in the middle position and two Cd2 ions in the terminal positions form a trinuclear cluster, and the coordination polyhedrons of adjacent Cd(II) ions share the same selvedge. The neighbouring trinuclear subunits are linked by 4-carboxylic groups in syn–anti coordination mode to form an infinite 1D metal chain. The adjacent chains are further connected by the L1 ligand to form a 2D covalent network, Fig. 5B. Then 4,4′-bpy ligands connect the Cd2 ions of neighbouring 2D layered networks as pillar, forming a 3D molecular structure, Fig. 5C.
 |
| | Fig. 5 (A) The ORTEP diagram of coordination geometry for Cd(II) atoms in 3 (the 30% probability level), all hydrogen atoms and water molecules have been omitted for clarity. (B) A 2D covalent molecular structure of 3. (C) 3D network pillared by 4,4′-bpy ligands. | |
 |
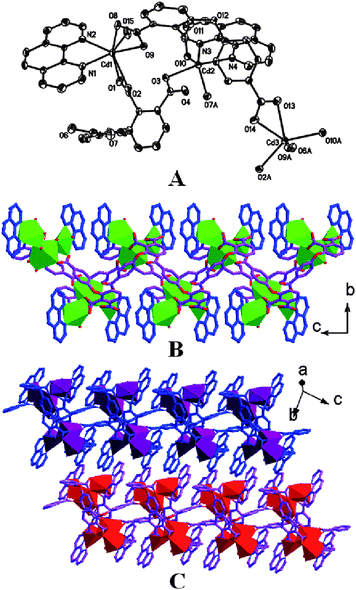
| | Fig. 6 ORTEP drawing of coordination sphere in the complex 7 (A), an infinite 1D ribbon chain composed of Cd1 and Cd2 ions (B), a infinite 1D ribbon chain composed of Cd1 and Cd3 ions (C), and a 2D molecular structure of 7 (D). | |
Crystal structure of complex 7
In the structure of 7, there exist three types of Cd(II) ions in Figure 6. Cd1 ion exhibits a hepta-coordinated geometry built from two nitrogen atoms, one oxygen atom from carboxylic group in monodentate mode, and two oxygen atoms from one carboxylic group in bidentate chelating mode, and two oxygen atoms from one carboxylic group in μ2-η2-η1 coordination mode. Cd2 ion possesses an octahedral coordination sphere formed by two nitrogen atoms of phen ligand, two oxygen atoms from one bidentate chelating carboxylic group, and one oxygen atom of water molecule, and one oxygen atom from one carboxylic group in μ2-η2-η1 coordination mode. For Cd3 ion, hepta-coordinated geometry is completed by two nitrogen atoms and five oxygen atoms (four from two bidentate chelating carboxylic groups and one from water molecule). As can be seen, the Cd1 and Cd2 ions share one carboxylic group of the L4 ligand in μ2-η2-η1 coordination mode to form a dinuclear subunit, which is linked by L4 ligands to generate a 1D ribbon chain along the a axis. The isolated Cd1 and Cd3 ions are assembled into a 1D double chain along the b axis. These two kinds of different directional double chains are compiled through the collective Cd1 ions as node, leading to a 2D covalent framework in a × b plane. The single crystal structures of 2 and 7 reveal that multicarboxylate ligands with semirigid central framework lead a 2D instead of 1D structure for the resulting complexes in cooperation with the 2,2′-bpy ligand.
Luminescent property of ligands L1–L4 and complexes 1–7
The solid state emission spectra of complexes 1–7 together with the four precursors H3L1–H3L4 have been recorded at room temperature and the related data complied in Table 3. As shown in Fig. S3 (ESI),† the solid emission spectra of ligands H3L1–H3L4 are very similar with the emission band observed at 395, 397, 399, and 408 nm, respectively, which is assigned to the typical intraligand π–π* or π–n electronic transition.13 The slight difference in the emission peak for H3L1–H3L4 is probably due to the slightly different electron-withdrawing effect resulted from different positions of asymmetrically attached carboxylic groups for these four ligands. Formation of Cd(II) complexes with either di- or trinuclear subunits from metal free L1–L4 ligands with the help of secondary ligand does not induce change in the luminescent emission of 1–7 (except 3) in comparison with corresponding metal free ligand, and the corresponding intraligand emission bands of H3L1–H3L4 are in the range of 381–394 nm in the luminescent emission spectra of 1–7 (except 3), Fig. S4 (ESI).† The emission band at 450 nm observed for 3 with a red shift of 55 nm relative to that of metal free ligand is assigned to LMCT on the basis of previous experimental result together with molecular orbital calculation on the Cd(II) coordination polymers.4b,8e,14
Table 3 The data of emissions in solid state of ligands H3L1–H3L4 and complexes 1–7
| Compound |
λ
ex/nm |
λ
em/nm |
| L1 |
246 |
395 |
| L2 |
246 |
397 |
| L3 |
246 |
399 |
| L4 |
246 |
408 |
|
1
|
246 |
394 |
|
2
|
246 |
389 |
|
3
|
320 |
450 |
|
4
|
320 |
381 |
|
5
|
320 |
383 |
|
6
|
246 |
390 |
|
7
|
246 |
391 |
The effect of secondary N-donor ligands and tricarboxylate ligands on the structures of 1–7
As can be seen in Fig. 7, nine types of different coordination modes and two kinds of coordination configurations exist in 1–7 for the four ligands L1–L4. For the three complexes 1–3 built from L1 ligand, the difference in their molecular structures reveals the significant spacer effect of secondary N-donor ligands in tuning the molecular structures of coordination polymers as well as the bridging modes and conformations of L1 ligand, Fig. 7A–7D. In particular, most carboxylate ligands have been revealed to form isostructures of coordination polymers for most transition metals with the help of phen and 2,2′-bpy as secondary ligand due to their similar coordination modes of the later two ligands.15 However, different framework structure observed between 1 and 2 in the present case clearly indicates the size effect of the secondary N-donor ligand on the complex structure. It is worth noting that the size effect between phen and 2,2′-bpy secondary ligands on tuning the molecular structure of 4 and 5 assembled from L2 ligand is not observed, indicating that L1 ligand is more sensitive to the size of secondary ligand during the formation of its coordination polymers than L2.
 |
| | Fig. 7 The coordination modes of the four ligands L1–L4 in 1–7 (A for 1, B and C for 2, D for 3, E for 4 and 5, F and G for 6, and H and I for 7). | |
Despite the fact that exact coordination mode and conformation of the four ligands in every complex have been detailed in the above-described molecular structure section, it still appears necessary to reveal and understand the relationship between the coordination mode of carboxylic groups as well as conformation of ligands and the molecular structure of complex for the purpose of rational design and synthesis of functional coordination polymers. In 1–3, the 4-carboxylic group of L1 in μ2-η2-η1 linking mode as main factor connects the Cd(II) ions into a di- or trinuclear subunit. For 4 and 5, L2 ligand exhibits a μ5-bridging mode with the 2-, 2′-, and 3-carboxylic groups possessing μ2-η2-η1, μ2-η2-η1, and μ2-η1-η1 coordination mode, respectively, inducing the formation of trinuclear subunit in these two complexes. In addition, both cis- and trans-typed configurations have been observed for symmetric semi-rigid V-shaped ligands.7,16 This is also true for the asymmetric L1 and L2 ligands in 1 and 4. As for the other two complexes 6 and 7 assembled from L3 and L4, respectively, also with the same phen secondary ligand, L3 ligand exhibits a much bender conformation than L1 and L2, resulting in the formation of a much wavier 1D double chain in 6 than in 1 and 4. This indicates the effect of the positions of three asymmetric carboxylic substituents at the semi-rigid V-shaped central molecular framework on the structure of MOFs. For L4 ligand in 7, the long trans-coordination conformation plays an important role in the formation of a 2D covalent network. Comparison in the molecular structure of 7 with 1, 4, and 6 also reveals the effect of the asymmetric positions of three carboxylic groups on tuning the structure of coordination polymers.
Conclusion
In summary, four asymmetric semi-rigid V-shaped tricarboxylate ligands have been employed for the first time to construct Cd(II) coordination polymers with di- or trinuclear subunit. Structural investigation reveals that the subunits could be successfully tuned by tuning the positions of asymmetrically attached carboxylic groups at the V-shaped central molecular framework in the tricarboxylate ligand and size of secondary ligand.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 20931001).
References
-
(a) O. M. Yaghi, H. L. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474 CrossRef CAS;
(b) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS;
(c) K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552 CrossRef CAS;
(d) M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725 CrossRef CAS;
(e) S. I. Noro, S. Kitagawa, M. Kondo and K. Seki, Angew. Chem., Int. Ed., 2000, 39, 2081 CrossRef.
-
(a) L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247 CrossRef CAS;
(b) S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef CAS;
(c) D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273 CrossRef CAS;
(d) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef;
(e) S. R. Batten, CrystEngComm, 2001, 3, 67 RSC;
(f) P. J. Steel, Acc. Chem. Res., 2005, 38, 243 CrossRef CAS;
(g) A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127 CrossRef CAS;
(h) S. A. Barnett and N. R. Champness, Coord. Chem. Rev., 2003, 246, 145 CrossRef CAS.
-
(a) J.-H. Chou, M. E. Kosal, S. Nakagaki, D. W. Smithenry and S. R. Wilson, Acc. Chem. Res., 2005, 38, 283 CrossRef CAS;
(b) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS;
(c) R. J. Hill, D. L. Long, N. R. Champness, P. Hubberstey and M. Schroöder, Acc. Chem. Res., 2005, 38, 335 CrossRef CAS;
(d) G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217 CrossRef CAS.
-
(a) M. Li, J. F. Xiang, L. J. Yuan, S. M. Wu, S. P. Chen and J. T. Sun, Cryst. Growth Des., 2006, 6, 2036 CrossRef CAS;
(b) S. L. Zheng, J. H. Yang, X. L. Yu, X. M. Chen and W. T. Wong, Inorg. Chem., 2004, 43, 830 CrossRef CAS;
(c) J. H. He, J. H. Yu, Y. T. Zhang, Q. H. Pan and R. R. Xu, Inorg. Chem., 2005, 44, 9279 CrossRef CAS;
(d) C. A. Bauer, T. V. Timofeeva, T. B. Settersten, B. D. Patterson, V. H. Liu, B. A. Simmons and M. D. Allendorf, J. Am. Chem. Soc., 2007, 129, 7136 CrossRef CAS;
(e) Q. R. Fang, G. S. Zhu, Z. Jin, Y. Y. Ji, J. W. Ye, M. Xue, H. Yang, Y. Wang and S. L. Qiu, Angew. Chem., Int. Ed., 2007, 46, 6638 CrossRef CAS.
-
(a) O. R. Evans, R. Xiong, Z. Wang, G. K. Wong and W. Lin, Angew. Chem., Int. Ed., 1999, 38, 536 CrossRef CAS;
(b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS;
(c) M. D. Hollingsworth, Science, 2002, 295, 2410 CAS;
(d) G. Férey, Chem. Mater., 2001, 13, 3084 CrossRef CAS.
-
(a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keefe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef;
(b) C. Serre, F. Millange, J. Marrot and G. Férey, Chem. Mater., 2002, 14, 2409 CrossRef CAS;
(c) H. Chun, H. Jung, G. Koo, H. Jeong and D.-K. Kim, Inorg. Chem., 2008, 47, 5355 CrossRef CAS;
(d) L. Xu, E.-Y. Choi and Y.-U. Kwon, Inorg. Chem., 2007, 46, 10670 CrossRef CAS;
(e) J.-W. Ye, J. Wang, J. Y. Zhang, P. Zhang and Y. Wang, CrystEngComm, 2007, 9, 515 RSC;
(f) D. Bradshaw, T. J. Prior, E. Cussen, J. B. Claridge and M. J. Rosseinsky, J. Am. Chem. Soc., 2004, 126, 6106 CrossRef CAS;
(g) Z. Lin, D. S. Wragg, J. E. Warren and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 10334 CrossRef CAS.
- D.-R. Xiao, E.-B. Wang, H.-Y. An, Y.-G. Li, Z.-M. Su and C.-Y. Sun, Chem.–Eur. J., 2006, 12, 6528 CrossRef CAS.
-
(a) X.-L. Wang, C. Qin, E.-B. Wang, Y.-G. Li, Z.-M. Su, L. Xu and L. Carlucci, Angew. Chem., Int. Ed., 2005, 44, 5824 CrossRef CAS;
(b) P. Mahata, G. Madras and S. Natarajan, J. Phys. Chem. B, 2006, 110, 13759 CrossRef CAS;
(c) S.-L. Li, Y.-Q. Lan, J.-F. Ma, J. Yang, G.-H. Wei, L.-P. Zhang and Z.-M. Su, Cryst. Growth Des., 2008, 8, 1610 CrossRef CAS;
(d) Y.-Q. Lan, S.-L. Li, K.-Z. Shao, X.-L. Wang, D.-Y. Du, Z.-M. Su and D.-J. Wang, Cryst. Growth Des., 2009, 9, 1353 CrossRef CAS;
(e) X.-L. Chen, B. Zhang, H.-M. Hu, F. Fu, X.-L. Wu, T. Qin, M.-L. Yang, G.-L. Xue and J.-W. Wang, Cryst. Growth Des., 2008, 8, 3706 CrossRef CAS;
(f) Q. Chu, G.-X. Liu, Y.-Q. Huang, X.-F. Wang and W.-Y. Sun, Dalton Trans., 2007, 4302 RSC.
-
(a) Y. Su, S. Zang, Y. Li, H. Zhu and Q. Meng, Cryst. Growth Des., 2007, 7, 1277 CrossRef CAS;
(b) H. Wang, D. Zhang, D. Sun, Y. Chen, L.-F. Zhang, L. Tian, J. Jiang and Z.-H. Ni, Cryst. Growth Des., 2009, 9, 5273 CrossRef CAS;
(c) W. Li, H.-P. Jia, Z.-F. Ju and J. Zhang, Dalton Trans., 2008, 5350 RSC;
(d) H. Wang, D. Zhang, D. Sun, Y. Chen, K. Wang, Z.-H. Ni, L. Tian and J. Jiang, CrystEngComm, 2010, 12, 1096 RSC.
-
(a) D. P. Martin, M. R. Montney, R. M. Supkowski and R. L. LaDuca, Cryst. Growth Des., 2008, 8, 3091 CrossRef CAS;
(b) L.-L. Wen, D.-B. Dang, C.-Y. Duan, Y.-Z. Li, Z.-F. Tian and Q.-J. Meng, Inorg. Chem., 2005, 44, 7161 CrossRef CAS;
(c) J.-D. Lin, J.-W. Cheng and S.-W. Du, Cryst. Growth Des., 2008, 8, 3345 CrossRef CAS;
(d) C.-S. Liu, X.-S. Shi, J.-R. Li, J.-J. Wang and X.-H. Bu, Cryst. Growth Des., 2006, 6, 656 CrossRef CAS.
-
(a) M. J. Plater, M. R. St. J. Foreman, E. Coronado, C. J. Gómez-García and A. M. Z. Slawin, J. Chem. Soc., Dalton Trans., 1999, 4209 RSC;
(b) M. J. Plater, M. R. St J. Foreman, R. A. Howie, J. M. S. Skakle, E. Coronado, C. J. Gómez-García, T. Gelbrich and M. B. Hursthouse, Inorg. Chim. Acta, 2001, 319, 159 CrossRef CAS.
- Z.-B. Han, X.-N. Cheng and X.-M. Chen, Cryst. Growth Des., 2005, 5, 695 CrossRef CAS.
-
(a) S. Jin, W. Chen and H. Qiu, Cryst. Growth Des., 2007, 7, 2071 CrossRef CAS;
(b) Y. Z-Du, X.-L. Li, Q.-Y. Liu and J.-G. Mao, Cryst. Growth Des., 2007, 7, 1051.
-
(a) J.-C. Dai, X.-T. Wu, Z.-Y. Fu, C.-P. Cui, S.-M. Hu, W.-X. Du, L.-M. Wu, H.-H. Zhang and R.-Q. Sun, Inorg. Chem., 2002, 41, 1391 CrossRef CAS;
(b) S. V. Ganesan and S. Natarajan, Inorg. Chem., 2004, 43, 198 CrossRef CAS;
(c) X.-L. Wang, C. Qin, E.-B. Wang, Y.-G. Li, N. Hao, C.-W. Hu and L. Xu, Inorg. Chem., 2004, 43, 1850 CrossRef CAS.
-
(a) F. Costantino, A. Lenco, S. Midollini, A. Orlandini, L. Sorace and A. Vacca, Eur. J. Inorg. Chem., 2008, 19, 3046 CrossRef;
(b) Y.-F. Yue, J. Liang, E.-Q. Gao, C.-J. Fang, Z.-G. Yan and C.-H. Yan, Inorg. Chem., 2008, 47, 6115 CrossRef CAS;
(c) A. K. Ghosh, D. Ghoshal, E. Zangrando, J. Ribas and N. R. Chaudhuri, Inorg. Chem., 2007, 46, 3057 CrossRef CAS.
- S. Zang, Y. Su, Y. Li, H. Zhu and Q. Meng, Inorg. Chem., 2006, 45, 2972 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-Ray crystallographic files (CIF), diagrams of the structures, selected bond distances, bond angles and TGA curves of compounds 1–7. CCDC reference numbers 746377–746383. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0ce00098a |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.