Horning-crown diamine complexes and salts: proton transfer mediated by solid-state intermolecular hydrogen bonding†
Fera
Luciawati
a,
Luke T.
Higham
a,
Christopher R.
Strauss
b and
Janet L.
Scott‡
*a
aSchool of Chemistry and Centre for Green Chemistry, Monash University, Wellington Rd, Clayton, Victoria 3800, Australia. E-mail: Janet.Scott@sci.monash.edu.au
bQUILL Centre, The Queen's University of Belfast, David Keir Building, Stranmillis Rd, Belfast BT9 5AG, Northern Ireland, UK. E-mail: Chris.Strauss@qub.ac.uk
First published on 27th August 2010
Abstract
Horning-crown macrocycles form sheath/core inclusion complexes or salts with alkyldiamines. The degree of proton transfer appears to be mediated by intermolecular hydrogen bonding interactions. Among seven crystal structures examined, neutral complexes, hemi-salts and salts were revealed. With crystalline materials, the extent of proton transfer appeared to reflect the degree of stabilisation of phenolates and ammonium groups by hydrogen bond donors and acceptors rather than merely differences in solution phase pKa.
Introduction
Horning-crown macrocycles (HCs) are hybrid molecules of calixarenes and crown ethers, introduced by our group in 2004.1 As illustrated in Scheme 1, their preparation involved controlled Claisen–Schmidt condensation of aryldialdehyde linkers I with cyclohexanones II to produce dienone ether macrocycles (DEMs) Va,2 followed by a multi-isoaromatisation step analogous to the process introduced by Horning in 1945.1 This direct, green approach to the synthesis of such macrocycles provides opportunities for tuning of molecular structure by judicious selection of building blocks from both commercially available, and readily prepared, starting materials.3 The use of a multi-tasking solvent and catalyst, DIMCARB (an adduct of dimethylamine and carbon dioxide),4,5 enables control over the sequence of condensations as well as construction of DEMs Vb and HCs VIb with different length linkers on either side.6,7 Thus, HCs of types VIa and VIb are synthetically accessible via short reaction sequences. | ||
| Scheme 1 DEM macrocycles may be prepared by direct Claisen–Schmidt condensation of I and II to yield Va, or as trapezoidal, unsymmetrical forms, by stepwise DIMCARB mediated reaction of I and II to yield the intermediate III, which following subsequent condensation provides access to Vb. DEMs formed with cyclohexanone derivatives II may be isoaromatised to provide HCs VIa or VIb. | ||
HCs are flexible macrocycles, which, depending on the length of the flexible linker, may self-associate to form clasped dimers in both solution and the solid state.8 For the ethylene-linked HC 1 these dimers are highly conserved in the presence of various guest molecules,9 but are switchable to monomeric forms by the introduction of polar hydrogen-bond disrupting solvents8 or by guests possessing terminal amino functional groups.1

While investigating the formation of inclusion complexes of ethylene-linked HC 1, and propylene-linked HC 2, with diamine guests: ethylenediamine 3, propylenediamine 4, and butylenediamine 5, we noted an interesting phenomenon in the crystalline complexes. While pKa for a given HC must be constant (in the same solvent or solvent/guest mixture) the crystalline complexes exhibited a range of structural types including salts (complete host to guest proton transfer), neutral inclusion complexes (no proton transfer) and mixed hemi-salts (proton transfer from half of the phenolic groups to acceptor amines).
Herein we describe these different structural types and analyse the resultant hydrogen bonding motifs, intermolecular interactions and crystal packing. In the light of recent interest in co-crystals (particularly of active pharmaceutical ingredients)10 and discussion concerning differentiation between co-crystals, salts and solvates11–13 as well as a recent report of a mixed salt/neutral co-crystal of Dianin's compound,14 this paper presents further examples highlighting difficulties in the assignment of rigid nomenclature for such complexes.15
Results and discussion
Crystalline complexes of HCs 1 and 2 and diamines 3–5 were prepared (Table 1) and several single crystal structures examined. The structure of complex 1a was published in a preliminary report pertaining to synthesis of HCs.1| 3 | 4 | 5 | |
|---|---|---|---|
a Complexes are numbered as if the “missing” complexes exist.
b No crystals suitable for X-ray analysis were isolated.
c 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex detected by mass spectrometric and 1H NMR spectroscopic analysis. :1 complex detected by mass spectrometric and 1H NMR spectroscopic analysis.
|
|||
| 1 | 1·3·CH2Cl2 complex 1aa | 1·4·CHCl3 complex 1b | 1·5·CHCl3 complex 1c |
| —b | 1·42 complex 1e | —b | |
| 2 | 2·3b,c | 2·4 complex 2b | 2·5 complex 2c |
| 2·32 complex 2d | —b | —b |
In the majority of cases, 1:1 HC:diamine stoichiometry implied matching of NH2 and OH groups, but two 1:2 complexes were also isolated: 1·42 and 2·32.
The structures of complexes 1a, 1b, 1c, 2b and 2c exhibited similar packing of HCs and amines: each had two crystallographically independent ½ HC macrocycles centred on an inversion centre (thus generating the complete macrocycle) and one diamine molecule in the asymmetric unit (ASU). The HCs were in an open conformation and diamines were largely surrounded or enclosed by folded HCs. The sheath/core structures are non-covalent (or supramolecular), 1D polymers that resemble coated wires with the HC acting as “insulation” and diamines as “wire”, Fig. 1.
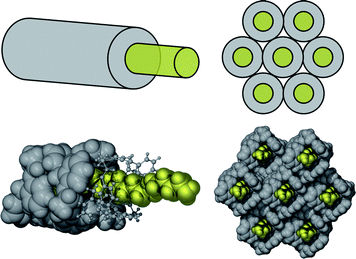 | ||
| Fig. 1 Top: schematic of sheath core structures typical of most of these HC diamine complexes. The “insulated wires” form close packed bundles, although, as these sheath core assemblies exhibit 2-fold symmetry in cross-section, these are not hexagonal close-packed structures. Bottom: illustrated by space filling diagrams of complex 2b. | ||
In addition, the complexes 1a, 1b and 1c each contained one solvent molecule in the ASU. The solvent molecules served to fill gaps in the sheath of the complexes with 1, while propylene-linked 2, bearing a longer flexible linker, was able to fold more effectively, completely sheathing the diamine guests, Fig. 2. Attempts to prepare complexes of HC 1 lacking included solvent molecules led to isolation of complex 1e, where a diamine molecule replaces solvent in the sheath.
![One sheath/core assembly each of complex 1a (top) and complex 2c (bottom), both viewed down [100]. Diamine guests are represented with atoms in space fill mode in blue, while the two crystallographically independent HCs are shown in pale and dark grey, with the solvent in complex 1a in yellow.](/image/article/2011/CE/c003571h/c003571h-f2.gif) | ||
| Fig. 2 One sheath/core assembly each of complex 1a (top) and complex 2c (bottom), both viewed down [100]. Diamine guests are represented with atoms in space fill mode in blue, while the two crystallographically independent HCs are shown in pale and dark grey, with the solvent in complex 1a in yellow. | ||
Complex 1e, with one HC 1 and two propylenediamine molecules in the ASU, was prepared from direct crystallisation of HC 1 dissolved directly in propylenediamine. Thus, it did not contain occluded solvent molecules, but the additional propylenediamine molecule served the same role as the chlorinated solvents in complexes 1a, 1b and 1c, viz it acted to complete the sheath surrounding the diamine core, Fig. 3. Thus the sheath/core form was retained and the extra diamine guest molecule (here it truly was a guest molecule) plugged a gap in the sheath. The unprotonated amine groups acted as hydrogen bond donors and acceptors, as illustrated in Fig. 3a. As a result, polar amine groups were buried in the core of the sheath/core assembly while the linking alkyl chains formed part of the sheath. This is clearly illustrated in the Hirshfeld surface16,17 representation of the assembly generated using the programme CrystalExplorer18 and depicted in Fig. 3b.
 | ||
| Fig. 3 Complex 1e (a) hydrogen bonding scheme showing the two independent cyclic hydrogen-bonding motifs (in this case both are hydrogen bonded salts) and the extra included diamine guest which forms part of the sheath (both HC 1 molecules are shown in thin stick form, all heteroatoms are depicted as spheres and hydrogen bonds as dotted lines). The “extra” uncharged propylenediamine molecule is nonetheless hydrogen bonded to each of the cyclic hydrogen bond systems. (b) Hirshfeld surface of the HC sheath (treated as a single surface and depicted in grey) with embedded alkyl chains of propylenediamine shown as coloured surfaces. | ||
Inspection of the intermolecular interactions of solvent molecules in complexes 1a, 1b and 1c revealed that these also participated in hydrogen bonding type interactions. Thus, these solvent molecules formed an integral part of the sheath/core assembly, Fig. 4. Hydrogen bond motifs and salt formation versus neutral hydrogen bonded complementary donors and acceptors are discussed below.
 | ||
| Fig. 4 Hydrogen bonding of solvent molecules in sheath/core assemblies of (a) complex 1a, with the dichloromethane solvent forming a weak hydrogen bond to the phenol group oxygen atom (d H⋯O = 2.31 Å) and participating in a C–H⋯π interaction (d H⋯centroid = 2.46 Å); (b) complex 1b with the chloroform solvent participating in a C–H⋯π interaction with a “corner” aromatic group (d H⋯centroid = 2.39 Å); and (c) complex 1c with the disordered chloroform solvent molecule participating in a C–H⋯π interaction with a phenolate Ar group (d H⋯centroid = 2.91 and 2.80 Å for each disorder component). | ||
Similarly to 1e, two crystallographically independent ½ HC macrocycles and two diamines appeared in the ASU of 2c, however, in contrast to 1e, the HCs in 2c were arranged as discrete ball-shaped assemblies of an HC situated on a centre of symmetry with two attendant diamine molecules. In each case, only one amine group of the ethylenediamine molecules interacted with the HC phenolic group, Fig. 5. Such structures are similar to those noted before with mono-amine guest molecules.1 One ethylenediamine amine group, not involved in ammonium/phenolate hydrogen bonding, is disordered over two positions.
 | ||
| Fig. 5 Ball shaped HC 2·(ethylenediamine)2assemblies of complex 2d (one ethylenediamine molecule is partially disordered). Top: ASU atoms represented as ellipsoids, non-ASU atoms in stick mode with thin bonds and hydrogen bonds as dotted red lines (all hydrogen atoms except those of the ammonium groups are removed for clarity). Bottom: with all atoms represented in space fill mode. | ||
Close examination of the phenolic groups of the HCs and amine groups of the diamines revealed some surprising differences between superficially similar complexes. In some cases, complete proton transfer from phenol to amine had occurred, resulting in a phenolate/ammonium salt, while in others, hydrogen bonded phenol/amine assemblies occurred, with no evidence of proton transfer (or even significant proton sharing). As described in the Experimental section, hydrogen atoms of potential salt-forming or hydrogen bond donor groups were located in electron density difference maps and refined. Owing to the poor scattering power of the hydrogen atom and hence the unreliability of its location and refinement from X-ray data (even good quality, low temperature data) as well as the apparent shortening of the D–H bond or H–A+ bond, we include information about relative D–H, H⋯A and H–A+ for comparative purposes only. Deprotonation of phenols is confirmed by consideration of C–O bond lengths and, in all complexes, shorter C–O distances correspond to the occurrence of an extra peak in the electron density map at a position, relative to the amine functionality, consistent with amine protonation. Metrics describing the H-bond geometries and the H-bond types are presented in Table 2.
| D–H⋯Aa | Type | d D⋯A/Å | d D–H/Å | d H⋯A/Å | ∠ DHA/° | d C–O/Å | Comment | |
|---|---|---|---|---|---|---|---|---|
| a The parent atom bearing the hydrogen atom at covalent bonding distance is designated as the “donor” i.e. we do not follow the convention that the moiety derived from the neutral acid is designated as the donor, even where complete proton transfer has occurred. b Complex 2d is not a sheath/core structure. | ||||||||
| 1a | O1–H1O⋯N21−x,2−y,−z | OHB (OH⋯N) | 2.643(2) | 1.05(3) (OH) | 1.66(3) (H⋯N) | 154(3) | 1.360(3) | Concerted H-bond cycle 1 |
| N2–H2NA⋯O1 | OHB (NH⋯O) | 3.095(2) | 1.05(3) (NH) | 2.24(3) (H⋯O) | 138(2) | 1.360(3) | ||
| N1–H1NB⋯O1′1−x,2−y,1−z | OHB (NH⋯O) | 3.107(2) | 0.94(3) (NH) | 2.40(3) (H⋯O) | 132(2) | 1.380(2) | Concerted H-bond cycle 2 | |
| O1′–H1′O⋯N1 | OHB (OH⋯N) | 2.620(2) | 1.00(3) (OH) | 1.64(3) (H⋯N) | 169(3) | 1.380(2) | ||
| N1–H1NA⋯Cl2x,y+1,z | 3.617(2) | 1.05(3) (NH) | 2.64(3) (H⋯Cl) | 155(2) | NH2 to solvent | |||
| 1b | O1–H1O⋯N1x,y+1,z | OHB (OH⋯N) | 2.639(3) | 0.85(3) (OH) | 1.84(3) (H⋯N) | 156(2) | 1.357(3) | Concerted H-bond cycle 1 |
| N1–H1NB⋯O11−x,1−y,1−z | OHB (NH⋯O) | 3.051(3) | 0.97(4) (NH) | 2.30(3) (H⋯O) | 134(2) | 1.357(3) | ||
| O1′–H1′O⋯N21−x,1−y,1−z | OHB (OH⋯N) | 2.597(3) | 0.93(4) (OH) | 1.70(3) (H⋯N) | 161(3) | 1.362(2) | Concerted H-bond cycle 2 | |
| N2–H2NB⋯O1′ | OHB (NH⋯O) | 3.107(3) | 0.87(3) (NH) | 2.33(3) (H⋯O) | 149(2) | 1.362(2) | ||
| 1c | O1–H1O⋯N1 | OHB (OH⋯N) | 2.552(4) | 1.09(5) (OH) | 1.51(4) (H⋯N) | 160(4) | 1.363(4) | Concerted H-bond cycle 1 |
| N1–H1NA⋯O1−x,1−y,1−z | OHB (NH⋯O) | 3.010(4) | 0.93(4) (NH) | 2.12(4) (H⋯O) | 161(3) | 1.363(4) | ||
| N2–H2NC⋯O1′ | (±)CAHB | 2.614(4) | 1.12(5) (NH)a | 1.51(4) (H⋯O) | 168(4) | 1.323(4) | Concerted H-bond cycle 2 | |
| N2–H2NA⋯O1′−x,−y,1−z | (±)CAHB | 2.701(4) | 0.98(5) (NH)a | 1.89(3) (H⋯O) | 159(4) | 1.323(4) | ||
| N2–H2NB⋯O3′ | OHB (NH⋯O) | 3.452(4) | 0.77(4) (NH) | 2.72(4) (H⋯O) | 160(4) | NH3+ to ether linker of 1 | ||
| 1e | N2–H2NB⋯O1′−x,2−y,−z | (±)CAHB | 2.587(3) | 1.09(4) (NH)a | 1.51(4) (H⋯O) | 167(3) | 1.323(3) | Concerted H-bond cycle 1 |
| N2–H2NA⋯O1′ | (±)CAHB | 2.739(3) | 0.90(3) (NH)a | 1.89(3) (H⋯O) | 157(3) | 1.323(3) | ||
| N1–H1NA⋯O1−x,2−y,1−z | (±)CAHB | 2.648(3) | 1.08(4) (NH)a | 1.58(4) (H⋯O) | 167(3) | 1.332(3) | Concerted H-bond cycle 2 | |
| N1–H1NB⋯O1 | (±)CAHB | 2.649(3) | 1.15(4) (NH)a | 1.51(4) (H⋯O) | 169(3) | 1.332(3) | ||
| N2–H2NC⋯N4 | OHB (NH⋯N) | 2.964(4) | 0.97(3) (NH) | 2.01(3) (H⋯N) | 167(3) | NH3+ to diamine “solvate” | ||
| N1–H1NC⋯O3 | OHB (NH⋯O) | 3.010(3) | 1.01(4) (NH) | 2.12(4) (H⋯O) | 147(3) | NH3+ to ether linker of 1 | ||
| N3–H3NA⋯O1′−x,2−y,1−z | OHB (NH⋯O) | 3.350(4) | 1.24(4) (NH) | 2.17(4) (H⋯O) | 158(3) | NH2 (diamine “solvate”) to phenolate O of 1 | ||
| 2b | N2–H2NB⋯O1 | (±)CAHB | 2.603(5) | 1.11(7) (NH) | 1.53(7) (H⋯O) | 160(5) | 1.330(4) | Concerted H-bond cycle 1 |
| N2–H2NC⋯O12−x,−y,−z | (±)CAHB | 2.621(5) | 1.06(7) (NH) | 1.61(7) (H⋯O) | 157(5) | 1.330(4) | ||
| N1–H1NB⋯O1′3−x,1−y,1−z | OHB (NH⋯O) | 2.966(5) | 0.96(6) (NH) | 2.07(6) (H⋯O) | 154(5) | 1.362(4) | Concerted H-bond cycle 2 | |
| O1′–H1′O⋯N1 | OHB (OH⋯N) | 2.640(5) | 0.95(6) (OH) | 1.70(6) (H⋯N) | 166(5) | 1.362(4) | ||
| 2c | N1–H1NB⋯O11−x,2−y,−z | (±)CAHB | 2.614(2) | 1.04(3) (NH)a | 1.59(3) (H⋯O) | 168(2) | 1.321(2) | Concerted H-bond cycle 1 |
| N1–H1NA⋯O1 | (±)CAHB | 2.650(2) | 1.08(3) (NH)a | 1.59(3) (H⋯O) | 165(2) | 1.321(2) | ||
| O1′–H1′O⋯N2 | OHB (OH⋯N) | 2.640(2) | 0.87(2) (OH) | 1.80(3) (H⋯N) | 164(2) | 1.358(2) | Concerted H-bond cycle 1 | |
| N2–H2NB⋯O1′1−x,1−y,1−z | OHB (NH⋯O) | 3.012(2) | 1.09(4) (NH) | 1.93(3) (H⋯O) | 168(2) | 1.358(2) | ||
| N1–H1NC⋯O31−x,2−y,−z | OHB (NH⋯O) | 3.409(2) | 0.98(2) (NH) | 2.53(2) (H⋯O) | 149(2) | NH3+ to ether linker of 2 | ||
| 2d b | N3–H3NA⋯O1′1−x,2−y,1−z | (±)CAHB | 2.656(3) | 1.28(6) (NH)a | 1.41(6) (H⋯O) | 161(5) | 1.3141(3) | Concerted H-bond cycle 1 |
| N3–H3NC⋯O1′ | (±)CAHB | 2.650(3) | 1.14(4) (NH)a | 1.56(4) (H⋯O) | 157(3) | 1.3141(3) | ||
| N4–H4NB⋯O1 | (±)CAHB | 2.614(3) | 1.09(4) (NH)a | 1.54(4) (H⋯O) | 168(3) | 1.3265(3) | Concerted H-bond cycle 2 | |
| N4–H4NC⋯O1−x,1−y,−z | (±)CAHB | 2.663(3) | 0.96(3) (NH) | 1.73(3) (H⋯O) | 164(3) | 1.3265(3) | ||
| N3–H3NB⋯O3′1−x,2−y,1−z | OHB (NH⋯O) | 3.332(4) | 1.12(4) (NH)a | 2.30(4) (H⋯O) | 153(3) | NH3+ to ether linker of 2 | ||
| N2–H2NA⋯O31−x,1−y,1−z | OHB (NH⋯O) | 3.142(4) | 0.88(7) (NH) | 2.27(7) (H⋯O) | 170(6) | NH2 (disordered) to ether linker of 2 |
The types of hydrogen bonds have been defined following Gilli, Pretto, Bertolasi and Gilli.19 Cyclic H-bonded systems with alternating phenol (ArOH)/phenolate (ArO−) and amine (NH2)/ammonium (NH3+) groups allow for a number of possibilities, as depicted in Fig. 6. Ordinary Hydrogen Bonds (OHBs, following the classification of Gilli et al.19) result from the interaction D–H⋯A where each OH and NH2 group acts as both donor and acceptor (Fig. 6a) or two types of double Charge Assisted Hydrogen Bonds [(±)CAHBs] resulting from proton “sharing” (Fig. 6b) or proton transfer (Fig. 6c). The two extremes thus represent a completely neutral cyclic hydrogen bonded system and a double salt hydrogen bonded system. The (±)CAHB is strongest when the differences between acid dissociation constants of donor and protonated acceptor are minimised such that the expression ΔpKa = pKa(D–H) − pKa(A–H+) tends to 0.20 Values of pKa for the HCs have not been measured, but if that of dibenzylphenol is expected to lie between pKa of 2,6-dimethylphenol (10.5921) and 2,6-tert-butylphenol (11.7021), this would provide a good first approximation of pKa1 for the HCs. The values for the diamines are collected in Table 3. The ΔpKa values for both HCs 1 and 2 and all of the diamines fall well within the value of ±2.5 pKa units compatible with strong H-bond formation according to Gilli et al.20
| 3 | 4 | 5 | |
|---|---|---|---|
| R | C2H4 | C3H6 | C4H8 |
| pKa1 | 9.92 | 10.47 | 10.82 |
| pKa2 | 6.85 | 8.49 | 9.35 |
 | ||
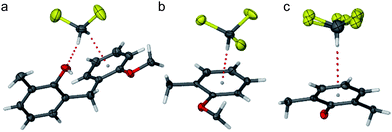
| Fig. 6 Interactions between two phenol and two amine groups may result in the formation of various circular hydrogen bond motifs following the descriptions of Gilli et al.:19 (a) all uncharged with O–H⋯N and N–H⋯O hydrogen bonds (OHB); (b) partial proton transfer O½−⋯H⋯N½+ in which the H-atom is equally “shared” between O and N atoms resulting in formal partial negative and positive charges ((±)CAHB); or (c) O−⋯H–N+ in which complete proton transfer has occurred, resulting in a salt which is also hydrogen bonded ((±)CAHB). | ||
Notwithstanding this, while some long N–H or O–H distances are detected, no truly equally shared hydrogen atoms were noted in these structures, but both the “weak” OHB, with uncharged ArOH and R–NH2 groups (Fig. 6a) and fully charged (±)CAHB salt type systems (Fig. 6c) appeared. As the sheath/core structures are 1D polymers formed by diamines interacting with sequential HCs, this yields three possible motifs, as illustrated in Fig. 7: all neutral (type I), all charged (type II) and mixed neutral and charged (type III). Type IV, representing the ball structures exhibited in complex 2d, is included for completeness (uncharged versions of this have been detected in other complexes with monoamines).1 Each of the complexes may be classified according to the hydrogen bond system types illustrated in Fig. 7 as reflected in Table 4 and Fig. 8.
| 3 | 4 | 5 | |
|---|---|---|---|
| 1 | 1·3·CH2Cl2 complex 1aType I | 1·4·CHCl3 complex 1bType I | 1·5·CHCl3 complex 1cType III |
| — | 1·42 complex 1eType II | — | |
| 2 | — | 2·4 complex 2bType III | 2·5 complex 2cType III |
| 2·32 complex 2dType IV | — | — |
 | ||
| Fig. 7 When the complex is an HC: diamine 1D polymer, as here, three possible motifs result: type I, all uncharged OHB (neutral); type II, all charged (±)CAHB (salt); and type III, alternating charged (±)CAHB and uncharged (hemi-salt). Type IV represents the 1:2H:G ratio assemblies found in some HC amine inclusion complexes such as 2d. The cartoons at the bottom are schematic representations of theses types. | ||
 | ||
| Fig. 8
Hydrogen bond schemes of 1:1 HC:diamine 1D polymers in the sheath/core structures. Complexes 1a and 1b exhibit all uncharged hydrogen bond cycles (type 1); complex 1e exhibits all fully charged hydrogen bond cycles (type II); and complexes 1c, 2b and 2c show alternating neutral and charged hydrogen bonded cycles (type III). (Both disorder components are illustrated for complex 1c, phenol/phenolate oxygen atoms and nitrogen atoms are shown as spheres and all macrocycles are shown as thin lines with Ar carbon atoms, not forming part of the cycle, removed for clarity. The solvent-like propylenediamine molecule of complex 1e is included, but the included solvent molecules of 1a, 1b and 1c are not shown.) | ||
The differences in hydrogen bond interactions are particularly easy to discern in decomposed Hirshfeld fingerprint plots,23 where the specific types of close contacts are highlighted, Fig. 9. Thus, in type I hydrogen bonded complex 1a (all neutral), the ethylenediamine fingerprint exhibits a prominent feature denoting N to H close contacts, Fig. 9a, while the propylenediamine fingerprint in the type III hydrogen bonded complex 2b (hemi-salt) exhibits features due to both N to H and H to O close contacts, pointing to its roles as both a strong hydrogen bond donor (NH3+ group) and acceptor (NH2 group). As both crystallographically independent HC 1 molecules in complex 1a are present as uncharged phenols, their fingerprint plots (Fig. 9b and c) are similar and exhibit strong features denoting H to N close contacts, reflecting the phenol OH to diamine NH2hydrogen bonds. Clearly, the fingerprint plot of the uncharged HC 2 in complex 2b (Fig. 9e) is similar to these, with a strong feature denoting H to N hydrogen bonding. Notably, however, the hydrogen bond O to H acceptor feature is also quite visible, reflecting the shorter D⋯A distance in the OHB of complex 2b compared with complex 1a. The fingerprint of the doubly charged phenolate HC 2 of complex 2b, Fig. 9f, bears no clear donor features, but, as expected, it has a strong O to H acceptor feature reflecting the (±)CAHBs resulting from interaction of phenolate O− with the NH3+ group of protonated propylenediamine. Both HC 1 molecules of the fully charged type II (salt) complex 1e exhibit fingerprint plots (not shown) similar to that depicted in Fig. 9f, although subtle differences due to other intermolecular interactions do arise. Concern that missed symmetry might give rise to the crystallographically independent ½ HCs in structures that are either type I or type II is allayed by the significant differences in their fingerprint plots, indicative of dissimilar intermolecular contacts. This illustrates another useful aspect of these plots: a quick visual check for missed symmetry or pseudosymmetry in structures with Z′ < 1.
 | ||
| Fig. 9 Hirshfeld surface fingerprint plots generated using CrystalExplorer18 for (a) ethylenediamine in complex 1a; (b and c) each of the crystallographically independent HC 1 molecules in complex 1a; (d) propylenediamine in complex 2b; and (e and f) each of the crystallographically independent HC 2 molecules in complex 2b. (The quantity de is the distance from the generated Hirshfeld surface to the closest atom outside the surface, while di is the distance from the generated Hirshfeld surface to the closest atom inside the surface, thus de > di values are consistent with donor type interactions, while points in the de < di region are due to acceptor type interactions.) | ||
Many other features of molecular packing and interactions are readily discerned from close inspection of fingerprint plots. For example, small features in the donor region of the diamine fingerprint plots, Fig. 9a and c, may be ascribed to H to Cl and H to C contacts respectively, i.e. quite different types of interactions. The former is due to relatively close approaches between amine hydrogen atoms and chlorine atoms of the included solvent (H to Cl), while the latter results from interactions between hydrogen atoms of CH2 groupsα to the NH2 groups of diamine molecule and π systems of the HCs. These interactions, ascribed to the relative acidity of the hydrogen atoms of this methylene group, are to be found in many of the complexes, as are π⋯π and C–H⋯π interactions of HC aromatic rings and linkers.
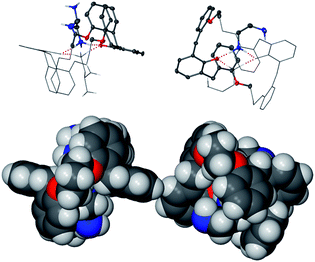
Careful consideration of the types of hydrogen bond systems formed and hence the degree of proton transfer in each structure might lead one to suggest that HC 2 is a stronger acid than HC 1 based on the formation of a type III hemi-salt of the former with propylenediamine (complex 2b) versus the type I neutral compound of the latter with ethylenediamine (complex 1a). This would immediately raise the question “why does the 1:2 complex 1c form a type II complex where complete proton transfer occurs, leading to an all salt system?” Closer inspection of hydrogen bonding interactions reveals a possible cause, other than pKa differences for the stabilisation of the charged systems. The “extra” unprotonated butylenediamine, as well as acting as part of the sheath structure, participates in a number of hydrogen bond interactions with the two cyclic four hydrogen bonded (±)CAHB systems, Fig. 3a. In addition, further interactions of NH3+ groups with ether linker oxygen atoms ensure that the hydrogen bond potential of the protonated NH3+ groups is completely fulfilled, Fig. 10.
 | ||
| Fig. 10 Complete hydrogen bonding scheme of the 1:2 complex 1e. Hydrogen bonded cycles of phenolate/ammonium groups are each composed of four (±)CAHBs, but are further stabilised by a series of extra hydrogen bond interactions. For cycle A: one (+)CAHB (ammonium to “extra” butylenediamine amine), and for cycle B: one OHB (“extra” butylenediamine amine group to phenolate) and one (+)CAHB (ammonium to HC ether oxygen atom). In addition, one amine group of the “extra” butylenediamine participates in NH⋯π interactions. | ||
Thus, all ammonium hydrogen atoms participate as donors and the B system phenolate is a triple acceptor. Each of these interactions would be expected to stabilise the charged systems. It would seem therefore that the degree of proton transfer is not exclusively a function of relative solution phase pKas of acid and base and that other hydrogen bond type interactions may serve to stabilise salt formation. This emphasises the need to take a holistic view of a crystalline system, something that we find to be facilitated by the use of CrystalExplorer to reveal otherwise unnoticed close contacts.
If one accepts that these interactions are stabilising factors in the proton transfer and salt formation, as frozen out in the crystalline materials, then it would be reasonable to suggest that the type III hemi-salts would show some clear differences between inter or intramolecular stabilisation of the hydrogen bonded cyclic systems that have undergone proton transferversus those that have not. Indeed, this appears to be the case. Thus, the ammonium group in complex 1c approaches to within 2.65 Å of an ether oxygen atom and that in complex 2c, to within 2.55 Å. The non-hydrogen bonded hydrogen atom of the ammonium group in complex 2b participates in a close contact with a π system (2.47 Å = closest H⋯C(Ar) contact). In contrast, neutral complexes 1a and 1b not only show no notable close contacts between amine and other acceptor groups, but they exhibit rather long N–H⋯O D⋯A distances of >3 Å. Thus proton transfer is not stabilised, in the solid state, by either concerted strong hydrogen bond cycles or by any extra hydrogen bond type interactions.
Experimental
HCs 1 (3,4:9,10:18,19:24,25-tetrabenzo-5,8,20,23-tetraoxa-tricyclo[25.3.1.112,16]dotriaconta-1(30),3,9,12,14,16(32),18, 24,27(31),28-decaene-31,32-diol) and 2 (3,4:10,11:19,20:26,27-tetrabenzo-5,9,21,25-tetraoxatricyclo [27.3.1.113,17]tetratriaconta1(32),3,10,13,15,17(34),19,26,29(33),30-decaene-33,34-diol) were prepared as described in ref. 1 and 6.Crystallography
Crystals suitable for single crystal X-ray diffraction experiments were grown either by slow evaporation of solutions of the HC in chloroform or dichloromethane and the relevant diamine (complexes 1a, 1b and 1c), or by slow cooling of solutions of the HC in the relevant diamine (complexes 1e, 2b, 2c and 2d). Data were collected on an Enraf-Nonius Kappa CCD or a Bruker Kappa Apex CCD diffractometer at 123 K using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å, φ and ω scans in 1° or 0.5° steps as appropriate). Structures were solved by direct methods using the program SHELXS-97 and refined by full matrix least squares refinement on F2 using the programs SHELXS-97 and SHELXL-9724 with gui X-Seed.25 Generally, non-hydrogen atoms of the hosts were refined anisotropically and hydrogen atoms inserted in geometrically determined positions with temperature factors fixed at 1.2 times that of the parent atom. Phenol OH group, amine and ammonium hydrogen atoms were located from electron density difference maps and refined. Specific details pertaining to each crystal structure solution and refinement are summarized below and crystal and refinement data are provided in Table 5.| a The structure of complex 1a was published in a preliminary report1 and cell parameters are included here for comparison. b Residual electron density is associated with the disordered diamine molecule. | |||||||
|---|---|---|---|---|---|---|---|
| Code | Complex 1aa | Complex 1b | Complex 1c | Complex 1e | Complex 2b | Complex 2c | Complex 2d |
| CCDC number | 254491a |
767013 |
767014 |
641527 |
641524 |
641526 |
641525 |
| H·G·solvent (ASU) | 1·3·CH2Cl2 | 1·4·CHCl3 | 1·5·CHCl3 | 1·42 | 2·4 | 2·5 | 2·32 |
| Formula | C47H50Cl2N2O6 | C48H51Cl3N2O6 | C49H53Cl3N2O6 | C50H60N4O6 | C49H54N2O6 | C50H56N2O6 | C50H60N4O6 |
| M r | 858.26 | 872.28 | 813.02 | 766.94 | 780.97 | 813.02 | |
| Crystal size/mm−3 | 0.40 × 0.17 × 0.10 | 0.48 × 0.35 × 0.25 | 0.19 × 0.15 × 0.12 | 0.30 × 0.10 × 0.10 | 0.35 × 0.12 × 0.10 | 0.35 × 0.20 × 0.10 | |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
P
|
P
|
P
|
P
|
P
|
P
|
| a/Å | 12.3043(3) | 12.6805(2) | 12.8654(2) | 12.6048(7) | 12.6886(3) | 12.7812(5) | 11.610(2) |
| b/Å | 12.9833(3) | 13.8085(3) | 14.0595(2) | 13.1155(8) | 14.0567(4) | 14.0559(5) | 11.669(2) |
| c/Å | 15.1589(4) | 14.8731(3) | 14.5748(3) | 15.0029(7) | 14.1856(6) | 14.2325(5) | 18.732(4) |
| α/° | 78.598(1) | 113.964(1) | 114.863(1) | 82.348(3) | 96.182(1) | 64.129(2) | 78.05(3) |
| β/° | 71.580(1) | 93.672(1) | 91.742(1) | 75.666(3) | 110.043(1) | 69.975(2) | 82.69(3) |
| γ/° | 64.698(1) | 108.788(1) | 109.327(1) | 63.130(3) | 116.463(2) | 64.398(2) | 60.57(3) |
| V/Å3 | 2071.61(9) | 2195.08(7) | 2212.46(7) | 2142.9(2) | 2038.3(1) | 2037.1(1) | 2161.5(7) |
| Z | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| D c/g cm−3 | 1.299 | 1.309 | 1.260 | 1.259 | 1.273 | 1.249 | |
| μ/mm−1 | 0.260 | 0.259 | 0.083 | 0.082 | 0.083 | 0.082 | |
| Refl. unique | 10317 |
10438 |
10212 |
7520 | 11755 |
11323 |
|
| Refl. I > 2σ(I) | 5653 | 4440 | 7001 | 4163 | 6989 | 10149 | |
| R 1/wR2 [I > 2σ(I)] | 0.0555/0.1105 | 0.0772/0.1640 | 0.0846/0.1399 | 0.0685/0.1529 | 0.0579/0.1182 | 0.0881/0.1657 | |
| R 1/wR2 [all data] | 0.1350/0.1340 | 0.2151/0.2137 | 0.1266/0.1581 | 0.1494/0.1820 | 0.1132/0.1420 | 0.0980/0.1704 | |
| GoF on F2 | 1.017 | 1.023 | 1.113 | 1.037 | 1.016 | 1.252 | |
| Parameters/restraints | 556/0 | 585/20 | 582/0 | 538/0 | 547/2 | 588/0 | |
| Δρmax/e Å3 | 0.62 | 0.62 | 0.56 | 0.66 | 0.97 | 1.34b | |
| Δρmin/e Å3 | −0.67 | −0.81 | −0.30 | −0.37 | −0.35 | −0.39 |
Hirshfeld surface analysis
Hirshfeld surface analysis and construction of fingerprint plots were achieved using CrystalExplorer 2.118 and decomposed fingerprint plots were used to discern the relationship between observed features and close contacts. The Hirshfeld surface is a 3D isosurface, which effectively defines the molecular volume in the crystal, as the surface is defined so that for each point on the surface 1/2 of the electron density is due to the spherically averaged noninteracting atoms comprising the molecule, and the other half is due to those comprising the rest of the crystal.26 The development of the Hirshfeld surface concept for molecular visualization in a crystal environment has been fully described by Spackman and co-workers27 and the use of fingerprint plots and decomposed fingerprint plots.Conclusions
The seven closely related crystal structures of Horning-crowns with alkyldiamines presented herein serve to illustrate an important difference between the solid and solution states with respect to relative acidities (reflected in proton transfer) and salt formation. We have shown that hydrogen bonding interactions serve to stabilise charged phenolate and ammonium salt assemblies and that absolute pKa or pKa differences (between acid and base) play a lesser role than in solution. Thus, in this case, proton transfer, resulting in salt formation in the solid state, reflects crystal packing and charge stabilisation by donor/acceptor interactions resulting in complexes ranging from all neutral to all charged for acid/base pairs within a very narrow range of pKa values. This serves to emphasise both the care that must be taken when applying “rules” such as those developed by Gilli et al. with respect to hydrogen bonding and pKa values. Hydrogen bonds may be considered as a form of acid/base interaction and thus careful analysis of these and consideration of the effect of a summation of weak interactions is important, as emphasised by Gilli and co-workers. The use of Hirshfeld surface analysis to discern otherwise difficult to detect interactions is shown to be of value in analysis of superficially similar crystal structures.Notes and references
- L. T. Higham, U. P. Kreher, C. L. Raston, J. L. Scott and C. R. Strauss, Org. Lett., 2004, 6, 3261 CrossRef CAS.
- L. T. Higham, U. P. Kreher, C. L. Raston, J. L. Scott and C. R. Strauss, Org. Lett., 2004, 6, 3257 CrossRef CAS.
- J. L. Scott, C. R. Strauss, in Eco-Friendly Processes for the Synthesis of Fine Chemicals, ed. R. Ballini, RSC Publishing, Cambridge, UK, 2009, p. 220 Search PubMed.
- U. P. Kreher, A. E. Rosamilia, C. L. Raston, J. L. Scott and C. R. Strauss, Org. Lett., 2003, 5, 3107 CrossRef CAS.
- A. E. Rosamilia, M. A. Giarrusso, J. L. Scott and C. R. Strauss, Green Chem., 2006, 8, 1042 RSC.
- M. A. Giarrusso, L. T. Higham, U. P. Kreher, R. S. Mohan, A. E. Rosamilia, J. L. Scott and C. R. Strauss, Green Chem., 2008, 10, 842 RSC.
- L. T. Higham, J. L. Scott and C. R. Strauss, Cryst. Growth Des., 2010, 10, 2409–2420 CrossRef CAS.
- L. T. Higham, U. P. Kreher, R. J. Mulder, C. R. Strauss and J. L. Scott, Chem. Commun., 2004, 2264 RSC.
- L. T. Higham, U. P. Kreher, J. L. Scott and C. R. Strauss, CrystEngComm, 2004, 6, 484 Search PubMed.
- M. J. Zaworotko, Cryst. Growth Des., 2007, 7, 4 CrossRef CAS.
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439 RSC.
- G. R. Desiraju, CrystEngComm, 2003, 5, 466 RSC.
- J. D. Dunitz, CrystEngComm, 2003, 5, 506 RSC.
- T. Jacobs, G. O. Lloyd, M. W. Bredenkamp and L. J. Barbour, Cryst. Growth Des., 2009, 9, 1284–1286 CrossRef CAS.
- A. D. Bond, CrystEngComm, 2007, 9, 833 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, CrystalExplorer 2.1, University of Western Australia, Perth, 2007, http://hirshfeldsurface.net/CrystalExplorer Search PubMed.
- P. Gilli, L. Pretto and G. Gilli, J. Mol. Struct., 2007, 844–845, 328 CrossRef.
- P. Gilli, L. Pretto, V. Bertolasi and G. Gilli, Acc. Chem. Res., 2009, 42, 33 CrossRef CAS.
- Dean's Handbook of Organic Chemistry, ed. G. W. Gokel, McGraw-Hill, New York, Chicago, San Francisco, Lisbon, London, Madrid, Mexico City, Milan, New Delhi, San Juan, Seoul, Singapore, Sydney, Toronto, 2nd edn, 2004 Search PubMed.
- Lange's Handbook of Chemistry, ed. J. A. Dean, McGraw-Hill, New York, St Louis, San Francisco, Auckland, Bogotá, Caracus, Lisbon, London, Madrid, Mexico, Milan, Montreal, New Delhi, Paris, San Juan, São Paulo, Singapore, Sydney, Tokyo, Toronto, 15th edn, 1999 Search PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC.
- J. J. McKinnon, A. S. Mitchell and M. A. Spackman, Chem.–Eur. J., 1998, 4, 2136 CrossRef CAS.
Footnotes |
| † CCDC reference numbers 767013, 767014, and 641524–641527. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c003571h |
| ‡ Current address: Centre for Sustainable Chemical Technologies, University of Bath, BA2 7AY, UK.E-mail: j.l.scott@bath.ac.uk |
| This journal is © The Royal Society of Chemistry 2011 |