 Open Access Article
Open Access ArticleProtein surface recognition using geometrically pure Ru(II) tris(bipyridine) derivatives†‡
Maria H.
Filby
ab,
James
Muldoon
ab,
Serin
Dabb
c,
Nicholas C.
Fletcher
c,
Alison E.
Ashcroft
b and
Andrew J.
Wilson
*ab
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK. E-mail: A.J.Wilson@leeds.ac.uk; Fax: +44 (0)113 3436565; Tel: +44 (0)113 3431409
bAstbury Centre for Structural Molecular Biology, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
cSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, UK
First published on 23rd November 2010
Abstract
This manuscript illustrates that the geometric arrangement of protein-binding groups around a ruthenium(II) core leads to dramatic differences in cytochrome c (cyt c) binding highlighting that it is possible to define synthetic receptors with shape complementarity to protein surfaces.
A major challenge in chemical biology is the competitive inhibition of protein-protein interactions (PPIs).1,2 To competitively inhibit PPIs, high affinity recogntion of one of the interfaces involved is required. Successful resolution of the challenge relies on being able to construct molecules capable of high affinity protein surface recognition by making discrete non-covalent contacts over a large (>800 Å2) surface with a less well-defined shape than the substrate binding cavities that have often served as targets for intervention.3 Over the past decade a number of groups have used various scaffolds to project functionality capable of making multivalent hydrophobic,4 ion-pairing5–17 and metal–ligand interactions18,19 over a protein surface to achieve high affinity binding. In certain cases inhibition of PPIs4,20–24 and selective recognition have been observed6,8,12,25,26 although the latter property has ordinarily derived from complementary matching of electrostatic interactions between protein and ligand. Thus far, these fundamental studies have focused on varying the dimensions and functional composition of molecules designed to recognise protein surfaces. In contrast, variation in the 3D projection of functional groups has not been exploited to modify binding properties. In the current manuscript we illustrate that the geometric arrangement of functional groups around a ruthenium(II) core such as compound 1 leads to differences in protein-binding affinity towards cytochrome c.
Hamachi and co-workers introduced ruthenium(II) trisbipyridine complexes as receptors for proteins in 1999.27 More recently Okhanda's group,28 followed by our own,26 illustrated such compounds act as fluorescent receptors for proteins. Compared with wholly organic receptors, these complexes are uniquely modular, allowing combinations of recognition elements to be assembled on a single rigid, three-dimensional scaffold with built-in sensing capability. Trischelate complexes of bipyridine ligands result in two optical isomers, Δ or Λ; and in the case of unsymmetrical bidentate ligands of Cs symmetry, geometrical facial (fac) and meridional (mer) isomers. This offers the opportunity to explore if different projections of binding functionality result in preferential binding of target proteins. Indeed, the binding properties of Δ or Λ optical isomers have been exploited for peptide assembly,29 lectin recognition30 and direct intercalation between the metal–ligand core and DNA.31 The isolation of geometrical isomers is less well established and so binding properties for these compounds have not been explored although simple anion recognition has been studied.32
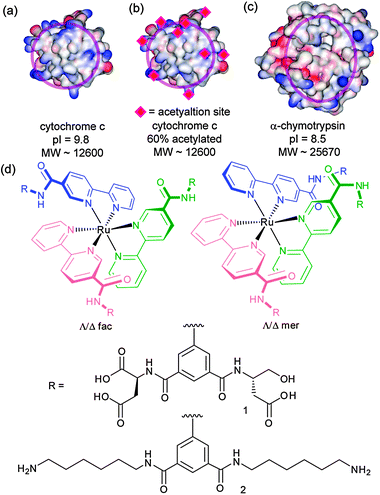 | ||
| Fig. 1 Structures and proteins used in this study (a) cyt c (b) acetylated cyt c (c) α-chymoytrypsin (d) compounds 1 and 2. | ||
Receptor 1 functionalized at the 5 position of bipyridine with an optically pure recognition motif incorporating two L-aspartyl groups was synthesized with tert-butyl ester protecting groups as described in the ESI.‡ We separated the tert-butyl protected fac/mer and Δ/Λ isomers using chromatography prior to deprotection. Our ability to achieve this difficult separation stems from the presence of optically pure L-aspartyl side chains in the ligand. Indeed, the diastereoisomeric nature of the Δ/Λ isomers is reflected in subtly different 1H NMR spectra for both fac and mer isomers. Shown in Fig. 2a are the 1H NMR spectra of all four fully protected isomers (noting the 1H NMR of the deprotected complexes exhibit broad poorly resolved resonances—see ESI‡). In addition to the subtle differences between Δ/Λ isomers, the lack of symmetry distinguishes the fac from mer isomers: the fac isomer is of C3 symmetry, whereas the mer isomer has a C1 symmetry, with all the ligands being inequivalent.33,34 The Δ/Λ isomers were assigned on the basis of circular dichroism spectra.35 Shown in Fig. 2b are the CD spectra of the fully deprotected complexes. We estimate on the basis of the CD spectra that the d.e. is ∼30% for Δ mer, 60% for Λ fac, >90% for both Λ mer and Δ fac however this property appears to have little effect on the binding affinity towards proteins (see below). The spectra of the fully protected complexes are similar (see ESI‡) indicating no loss in optical purity.
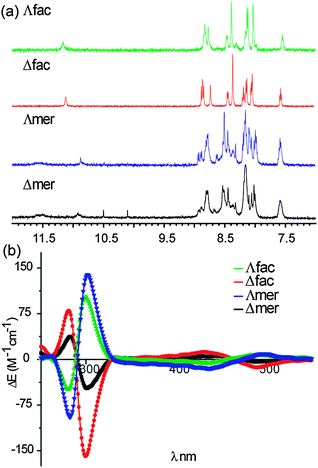 | ||
| Fig. 2 (a) 1H NMR Spectra (500 MHz, d6-acetone) of tbutyl protected fac/mer Λ/Δ 1 and (b) circular dichroism spectra (CD) of fac/mer Λ/Δ 1 (H2O, pH 7.0, 100 μM). | ||
Following synthesis of 1 we tested protein-surface recognition towards cytochrome c (cyt c), an ideal model protein with essential roles in electron-transfer and apoptosis pathways. Cyt c is positively charged at physiological pH (pI ∼ 10) and engages in PPIs with partners via a hydrophobic patch surrounded by lysine moieties (Fig. 1a).36 The ruthenium complexes 1 are fluorescent at 650 nm when excited at 467 nm.37Titration of cyt c into a solution of receptors (5 mM phosphate buffer, pH 7.4) resulted in efficient fluorescence quenching due to complex formation with the heme edge region of the protein (Fig. 3). The data were fit to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherm using non-linear regression to obtain dissociation constants, which for the highest affinity compound is in the low nM regime (Table 1). One-order of magnitude difference in the Kd between fac and the mer isomers upon binding to cyt c is observed (equivalent to 4 kJ mol−1 in free energy). The data, however, showed little discrimination between the Δ and Λ isomers for the pair of geometrical isomers. It is noteworthy that saturation is reached with only a 50% reduction in signal for mer1 and 25% for fac1—perhaps a manifestation of the different geometrical relationship that will exist between fluorophore and quencher as a consequence of geometrical selectivity. Ruthenium complex 2, which is functionalized on the isophthalamide motifs with hexylamino groups (see ESI‡), was synthesized as a negative control and as expected did not show any binding to cyt c. In addition, none of the compounds exhibited any quenching upon titration with 60% acetylated cyt c. Acetylation of surface exposed lysine residues prevents key electrostatic interactions involving these residues from taking place. The majority of solvent exposed lysine residues on cyt c surround the haem exposed edge so the result indicates that (a) the nature of recognition between cyt c and the isomers of 1 is electrostatic in nature (b) molecular recognition occurs at the haem exposed edge and (c) it is the geometrical placement of carboxylates that is the source of differential binding.
:1 binding isotherm using non-linear regression to obtain dissociation constants, which for the highest affinity compound is in the low nM regime (Table 1). One-order of magnitude difference in the Kd between fac and the mer isomers upon binding to cyt c is observed (equivalent to 4 kJ mol−1 in free energy). The data, however, showed little discrimination between the Δ and Λ isomers for the pair of geometrical isomers. It is noteworthy that saturation is reached with only a 50% reduction in signal for mer1 and 25% for fac1—perhaps a manifestation of the different geometrical relationship that will exist between fluorophore and quencher as a consequence of geometrical selectivity. Ruthenium complex 2, which is functionalized on the isophthalamide motifs with hexylamino groups (see ESI‡), was synthesized as a negative control and as expected did not show any binding to cyt c. In addition, none of the compounds exhibited any quenching upon titration with 60% acetylated cyt c. Acetylation of surface exposed lysine residues prevents key electrostatic interactions involving these residues from taking place. The majority of solvent exposed lysine residues on cyt c surround the haem exposed edge so the result indicates that (a) the nature of recognition between cyt c and the isomers of 1 is electrostatic in nature (b) molecular recognition occurs at the haem exposed edge and (c) it is the geometrical placement of carboxylates that is the source of differential binding.
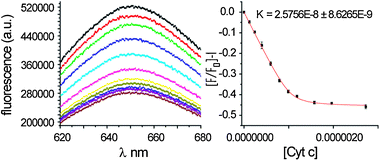 | ||
| Fig. 3 Raw fluorescence data and titration curve for binding of Δ-mer1 to cyt c. | ||
| Compound | K d/nM |
|---|---|
| a 1 μM 1, 5 mM sodium phosphate buffer, pH 7.4, ex 467 nm. | |
| Δ-mer1 | 25 ± 0.8 |
| Λ-mer1 | 29 ± 0.8 |
| Δ-fac1 | 172 ± 0.2 |
| Λ-fac1 | 130 ± 0.3 |
To provide further evidence that recognition occurs at the protein surface, all isomers of 1 were tested in a functional ascorbate reduction assay (see ESI‡).38 In the presence of the receptor 1, the reduction of cyt c by ascorbate occurs at a much slower rate, indicating binding of the receptors to the heme region. The degree of inhibition is dose dependent and although minor differences between fac and mer isomers are observed the experiment must be performed at concentrations well above Kd so a comparison with Kd determined by fluorescence is not possible.
To establish if the difference in binding between different isomers is protein-specific or an intrinsic difference between geometrical isomers, we performed titrations with α-chymotrypsin, which has been shown to be a good target for related ruthenium complexes by Okhanda.28 In our experiments, increases in fluorescence were observed for each isomer of 1, however these occurred over a concentration range much greater than the concentration of the ruthenium complex and did not reach saturation, which suggests ill defined binding (see ESI‡). Further studies with an expanded selection of funcitonalised ruthenium complexes will therefore be needed to probe this aspect.
In conclusion, we have illustrated that affinity of functionalized Ru(II) tris(bipyridine) complexes towards cyt c is dependent on their geometrical configuration. The complexes bind to cyt c in a specific and selective manner with differences in binding affinity attributable to the different geometrical placement of carboxylate groups that make contacts with surface exposed lysine residues on cyt c. Our future studies are directed towards a structural rationalisation of this result and testing the biological utility of these metal complexes.
This work was supported by the Wellcome Trust [WT 78112/Z/05/Z] and EPSRC [EP/F039069 & EP/F038712].
Notes and references
- A. J. Wilson, Chem. Soc. Rev., 2009, 38, 3289–3300 RSC.
- H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 4130–4163 CrossRef CAS.
- M. W. Peczuh and A. D. Hamilton, Chem. Rev., 2000, 100, 2479–2493 CrossRef CAS.
- D. K. Leung, Z. W. Yang and R. Breslow, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5050–5053 CrossRef CAS.
- Y. Hamuro, M. C. Calama, H.-S. Park and A. D. Hamilton, Angew. Chem., Int. Ed. Engl., 1997, 36, 2680–2683 CrossRef CAS.
- H. S. Park, Q. Lin and A. D. Hamilton, J. Am. Chem. Soc., 1999, 121, 8–13 CrossRef CAS.
- R. K. Jain and A. D. Hamilton, Org. Lett., 2000, 2, 1721–1723 CrossRef CAS.
- N. O. Fischer, C. M. McIntosh, J. M. Simard and V. M. Rotello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5018–5023 CrossRef CAS.
- A. J. Wilson, K. Groves, R. K. Jain, H. S. Park and A. D. Hamilton, J. Am. Chem. Soc., 2003, 125, 4420–4421 CrossRef CAS.
- M. Braun, S. Atalick, D. M. Guldi, H. Lanig, M. Brettreich, S. Burghardt, M. Hatzimarinaki, E. Ravanelli, M. Prato, R. v. Eldik and A. Hirsch, Chem.–Eur. J., 2003, 9, 3867–3875 CrossRef CAS.
- D. M. Tragore, K. I. Sprinz, S. Fletcher, J. Jayawickramarajah and A. D. Hamilton, Angew. Chem., Int. Ed., 2007, 46, 223–225 CrossRef CAS.
- D. Paul, H. Miyake, S. Shinoda and H. Tsukube, Chem.–Eur. J., 2006, 12, 1328–1338 CrossRef CAS.
- M. Arendt, W. Sun, J. Thomann, X. Xie and T. Schrader, Chem.–Asian J., 2006, 1, 544–554 CrossRef CAS.
- O. Hayashida, H. Ogawa and M. Uchiyama, J. Am. Chem. Soc., 2007, 129, 13698–13705 CrossRef CAS.
- F. Chiba, T.-C. Hu, L. J. Twyman and M. Wagstaff, Chem. Commun., 2008, 4351–4353 RSC.
- P. B. Crowley, P. Ganji and H. Ibrahim, ChemBioChem, 2008, 9, 1029–1033 CrossRef CAS.
- A. J. Wilson, J. Hong and A. D. Hamilton, Org. Biomol. Chem., 2007, 5, 276–285 RSC.
- A. Oida, Y. Mito-oka, M.-a. Inoue and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 6256–6257 CrossRef CAS.
- A. L. Banerjee, M. Swanson, B. C. Roy, X. Jia, M. K. Haldar, S. Malik and D. K. Srivastava, J. Am. Chem. Soc., 2004, 126, 10875–10883 CrossRef CAS.
- M. A. Blaskovich, Q. Lin, F. L. Delarue, J. Sun, H. S. Park, D. Coppola, A. D. Hamilton and S. M. Sebti, Nat. Biotechnol., 2000, 18, 1065–1070 CrossRef CAS.
- Y. Wei, G. L. McLendon, A. D. Hamilton, M. A. Case, C. B. Purring, Q. Lin, H. S. Park, C. S. Lee and T. N. Yu, Chem. Commun., 2001, 1580–1581 RSC.
- H. Bayraktar, P. S. Ghosh, V. M. Rotello and M. J. Knapp, Chem. Commun., 2006, 1390–1392 RSC.
- H. Azuma, Y. Yoshida, D. Paul, S. Shinoda, H. Tsukube and T. Nagasaki, Org. Biomol. Chem., 2009, 7, 1700–1704 RSC.
- H. S. Park, Q. Lin and A. D. Hamilton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5105–5109 CrossRef CAS.
- A. M. Fazal, B. C. Roy, S. Sun, S. Malik and K. Rogers, J. Am. Chem. Soc., 2001, 123, 6283–6290 CrossRef CAS.
- J. Muldoon, A. E. Ashcroft and A. J. Wilson, Chem.–Eur. J., 2010, 16, 100–103 CrossRef CAS.
- H. Takashima, S. Shinkai and I. Hamachi, Chem. Commun., 1999, 2345–2346 RSC.
- J. Ohkanda, R. Satoh and N. Kato, Chem. Commun., 2009, 6949–6951 RSC.
- M. R. Ghadiri, C. Soares and C. Choi, J. Am. Chem. Soc., 1992, 114, 825–831 CrossRef.
- S. Sakai, Y. Shigemasa and T. Sasaki, Bull. Chem. Soc. Jpn., 1999, 72, 1313–1319 CrossRef CAS.
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777–2796 CrossRef CAS.
- N. C. A. Baker, N. McGaughey, N. C. Fletcher, A. V. Chernikov, P. N. Horton and M. B. Hursthouse, Dalton Trans., 2009, 965–972 RSC.
- E. A. P. Armstrong, R. T. Brown, M. S. Sekwale, N. C. Fletcher, X.-Q. Gong and P. Hu, Inorg. Chem., 2004, 43, 1714–1722 CrossRef CAS.
- N. C. Fletcher, C. Martin and H. J. Abrahams, New J. Chem., 2007, 31, 1407–1411 RSC.
- N. C. Fletcher, M. Nieuwenhuyzen and S. Rainey, J. Chem. Soc., Dalton Trans., 2001, 2641–2648 RSC.
- H. Pelletier and J. Kraut, Science, 1992, 258, 1748–1755 CrossRef CAS.
- A. Juris, S. Barigelletti, S. Campagna, V. Balzani, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- E. Mochan and P. Nicholls, Biochim. Biophys. Acta, Bioenerg., 1972, 267, 309–319 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedures, characterization and fluorescence binding studies. See DOI: 10.1039/c0cc04754f |
| This journal is © The Royal Society of Chemistry 2011 |