Improved Cope-type hydroamination reactivity of hydrazine derivatives†‡
Francis
Loiseau
,
Christian
Clavette
,
Michaël
Raymond
,
Jean-Grégoire
Roveda
,
Alishya
Burrell
and
André M.
Beauchemin
*
Centre for Catalysis Research and Innovation, Department of Chemistry, University of Ottawa, 10 Marie-Curie, Ottawa, ON K1N 6N5, Canada. E-mail: andre.beauchemin@uottawa.ca; Fax: +1 613-562-5170; Tel: +1 613-562-5800, ext. 2245
First published on 6th October 2010
Abstract
A systematic investigation on the metal-free, Cope-type hydroamination reactivity of hydrazides and analogues is reported. Optimization of the hydrazide structure resulted in more facile intramolecular reactivity and enabled intermolecular reactions of alkenes, thus providing a direct approach to polysubstituted hydrazides.
Efficient C–N bond-forming reactions continue to emerge from efforts to synthesize diverse nitrogen-containing functional groups from simple building blocks. Hydroamination, the formal addition of N–H bonds across an unsaturated carbon–carbon π bond, represents a highly desirable and versatile strategy to form C–N bonds. To overcome the high activation energy associated with such reactivity, hydroamination efforts have mostly focused on transition metal catalysis,1 including some recent developments in hydrohydrazination.2
The availability of monosubstituted hydrazine derivatives has stimulated intense research and led to applications in agriculture (pesticides), polymer chemistry, photographic products and pharmaceuticals (both as synthetic intermediates and end products).3 Specifically, hydrazides (N-acylhydrazines) have been used in the synthesis of heterocycles, dyestuffs, polymers, and in peptidomimetics (azapeptides),4 agriculture (e.g.daminozide, a plant growth regulator) and pharmaceuticals [e.g. isoniacid (tuberculosis), isocarboxazid (antidepressant), atazanavir (antiretroviral)].3c With most applications featuring monosubstituted hydrazines and hydrazides, broadly applicable methods to access di- and tri-substituted hydrazines are in particular need. In this context, uses of hydrazines in the hydroamination of alkenes and alkynes (hydrohydrazination) are only emerging.2
As part of our interest in metal-freeamination methods, we recently extended the scope of the thermal, concerted, Cope-type hydroamination5 reactivity of hydroxylamines to intermolecular reactions of alkenes, alkynes and allenes (Scheme 1).6 We also recently reported our preliminary results on related reactivity of hydrazines and hydrazides.7 Herein, we disclose a systematic evaluation of the reactivity of hydrazine derivatives, leading to increased reactivity and applicability in intramolecular systems, and enabling intermolecular alkene hydrohydrazidation.
 | ||
| Scheme 1 Cope-type hydroamination of alkenes. | ||
In contrast to hydroxylamines, hydrazine derivatives are remarkably thermally stable. Hydrazides are also typically crystalline and bench stable, and the electron-withdrawing group can facilitate the proton transfer step from the ammonium ylide intermediate.6a,b,7 Speculating that optimizing the structure of the hydrazide group would result in a more facile hydroamination event (through stabilization of the developing charges present in the transition state) and stabilize the dipole intermediate, we embarked on a systematic investigation of related hydrazine derivatives (Table 1).

| Entry | EWG | R1 | Temp/°C | Product | Yieldb (%) |
|---|---|---|---|---|---|
| a Conditions: heated in PhCF3 (0.05 M), in sealed tubes (18–40 h) or in a microwave reactor (10–16 h).b Isolated yield.c NMR yield using an internal standard.d Obtained as a mixture of diastereoisomers (see ESI2). | |||||
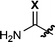
|
|||||
| 1 | X = O (1a) | H | 150 | 2a | 50 |
| 2 | X = S (1b) | H | 100 | 2b | 86 |
|
|||||
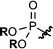
| 3 | R = Et (1c) | H | 120 | 2c | >98c |
| 4 | R = Ph (1d) | H | 110 | 2d | >98c |

|
|||||
| 5 | R = t-Bu (1e) | H | 150 | 2e | 66 |
| 6 | R = 2-pyridyl (1f) | H | 170 | 2f | 64 |
| 7 | R = Ph (1g) | H | 120 | 2g | 93 |
| 8 | R = Ph (1h) | Me | 120 | 2h | 98d |

|
|||||
| 9 | R = H (1i) | Me | 90 | 2i | 90d |
| 10 | R = NO2 (1j) | Me | 70 | 2j | 88d |
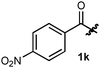
| 11 |
|
Me | 90 | 2k | 91d |
| 12 |

|
H | 95 | 2l | 81 |
The data shown in Table 1 show the generality of the approach and allow a comparison of the relative reactivity of semicarbazides, thiosemicarbazides, phosphohydrazides and hydrazides in the formation of the pyrrolidine ring system (Table 1, entries 1–7). Due to side reactions observed with semicarbazides8 and phosphohydrazides9 at higher temperatures, benzoic hydrazides were selected for further optimization. Gratifyingly, increased reactivity was observed for substrates possessing hydrogen-bonding and electron-withdrawing substituents (entries 9–12). While both hydrazides 1j and 1l resulted in encouraging reactivity at 70 °C,10 the increased solubility of hydrazide 1l in organic solvents led us to explore the reactivity of 3,5-bis(trifluoromethyl)benzoic hydrazides in more challenging intra- and intermolecular reactions.
Thus, we next sought to investigate the cyclization of several substrates to access pyrrolidine and piperidine ring systems (Table 2). The reactivity of the simpler benzoic hydrazides7a (4a–e) is also presented to allow comparison.

| Entry | Substrate | Temp/°C | Product | Yieldb (%) | |
|---|---|---|---|---|---|
| a Conditions: heated in PhCF3 (0.05 M), in a microwave reactor (10–24 h).b Isolated yield.c NMR yield using an internal standard.d Obtained as a mixture of diastereoisomers (see ESI2). | |||||
| 1 | 3a | R1 = R2 = R3 = H n = 1 | 95 | 5a | 81 |
| 2 | 4a | 120 | 6a | 93 | |
| 3 | 3b | R1 = Me, R2 = R3 = H n = 1 | 95 | 5b | 85c |
| 4 | 4b | 120 | 6b | 98c | |
| 5 | 3c | R1 = R2 = H, R3 = Me n = 1 | 150 | 5c | 91 |
| 6 | 4c | 175 | 6c | 75 | |
| 7 | 3d | R1 = R2 = R3 = H n = 2 | 175 | 5d | 82 |
| 8 | 4d | 200 | 6d | 90 | |
| 9 | 3e | R1 = R3 = H, R2 = Et n = 2 | 195 | 5e | 53 |
| 10 | 4e | 220 | 6e | 51d |
As shown in Table 2, the efficiency of the cyclizations to simple 5- and 6-membered rings (5a–e) was comparable to that of the simple benzhydrazide derivatives (6a–e), with the hydroamination proceeding at lower temperatures. While only a modest increase in reactivity was observed, the effect of the improvement was most noticeable with substrates with distal alkene substituents (entries 5–6 and 9–10). Such disubstituted alkenes typically afford lower yields of the cyclised products due to a more challenging hydroamination event and competing side reactions.11 In such systems, modified hydrazides resulted in a marked improvement over reactions obtained using benzoic hydrazides.
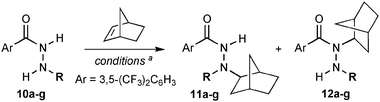
While intramolecular hydroamination reactivity is possible through various catalyzed and metal-free approaches, intermolecular processes are more challenging (especially for alkenes).12 With optimized reagents, we revisited previously unsuccessful attempts to achieve a metal-free intermolecular alkene hydrohydrazidation simply upon heating. The lead result obtained is shown in eqn (1).
 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶1 ratio. The two products are likely formed from a common ammonium ylide intermediate (A, see Scheme 2),13 with the expected major product 11a arising from a proton transfer and the minor product 12a forming through a competing [1,2]-shift of the norbornyl group.14 Such [1,2]-shifts are related to the Stevens' rearrangement and usually occur through a diradical mechanism.15 The scope of this intermolecular reactivity was explored with several hydrazides, as shown in Table 3.
∶1 ratio. The two products are likely formed from a common ammonium ylide intermediate (A, see Scheme 2),13 with the expected major product 11a arising from a proton transfer and the minor product 12a forming through a competing [1,2]-shift of the norbornyl group.14 Such [1,2]-shifts are related to the Stevens' rearrangement and usually occur through a diradical mechanism.15 The scope of this intermolecular reactivity was explored with several hydrazides, as shown in Table 3.
 | ||
| Scheme 2 Intermolecular hydrohydrazidation: divergent reactivity from the ammonium ylide intermediate. | ||
| Entry | Hydrazide (R) | Reaction time/h | Products | Yieldb (%)/ratio (11∶2) |
|---|---|---|---|---|
| a Conditions: heated in PhCF3 (2 M), 160 °C, sealed tube, 17–40 h.b Isolated yields. | ||||
| 1 | Bn | 40 | 11a + 12a | 81 (3.1∶1) |
| 2 | Me | 17 | 11b + 12b | 85 (4.3∶1) |
| 3 | i-Pr | 17 | 11c + 12c | 74 (1.7∶1) |
| 4 | i-Bu | 17 | 11d + 12d | 73 (2.7∶1) |
| 5 | c-C6H11 | 40 | 11e + 12e | 87 (3.2∶1) |
| 6 | (CH2)2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
17 | 11f + 12f | 87 (3.7∶1) |
| 7 | (CH2)3OBn | 17 | 11g + 12g | 86 (3.3∶1) |
Encouragingly, the hydroamination of norbornene proved efficient with several alkylhydrazides, providing the hydroamination products in combined yields ranging from 74–87% (Table 3, entries 1–7). The presence of alkene and benzyl ether functionalities on the hydrazide was also well tolerated (entries 6 and 7). In all cases the expected hydroamination product 11 was favored over rearrangement product 12, with the ratio of products showing little dependence on the size of the hydrazide substituent (R). This observation indicates that proton transfer of the ammonium ylide intermediate is more facile than migration of the alkyl substituents. Importantly, no rearrangement product derived from [1,2]-shift of the R substituent was detected, highlighting the preference for the norbornyl substituent to migrate over several alkyl groups.
In summary, we have performed a systematic investigation of the hydroamination reactivity of hydrazides and related compounds, showing its generality in simple intramolecular systems. More reactive benzoic hydrazides were identified, and 3,5-bis(trifluoromethyl)benzhydrazides proved more efficient in several cyclizations and enabled intermolecular hydrohydrazidations. Extensions of this work to access more substituted ammonium ylides and to enable alkyne hydroamination are in progress and will be reported in due course.
We thank the University of Ottawa, the Canadian Foundation for Innovation, the Ontario Ministry of Research and Innovation (Early Researcher Award to A.M.B.), and NSERC for their support. Scholarships to F.L. (FQRNT), C.C. (NSERC CREATE) and M.R. (NSERC USRA) are also acknowledged. We also thank Ms Roxanne Clément (CCRI) for assistance in preliminary high throughput screening experiments directed at intermolecular reactivity.
Notes and references
- For selected reviews, see: (a) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef and reviews cited therein; (b) I. Aillaud, J. Collin, J. Hannedouche and E. Schulz, Dalton Trans., 2007, 5105 RSC; (c) K. C. Hultzsch, Adv. Synth. Catal., 2005, 347, 367 CrossRef CAS; (d) M. Nobis and B. Drieβen-Hölscher, Angew. Chem., Int. Ed., 2001, 40, 3983 CrossRef CAS; (e) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef.
- For selected examples of metal-catalyzed hydrohydrazidations, see: (a) J. Waser and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 5676 CrossRef CAS; (b) J. Waser and E. M. Carreira, Angew. Chem., Int. Ed., 2004, 43, 4099 CrossRef CAS; (c) J. Waser, B. Gaspar, H. Nambu and E. M. Carreira, J. Am. Chem. Soc., 2006, 128, 11693 CrossRef CAS; (d) K. Alex, A. Tillack, N. Schwarz and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 2304 CrossRef CAS and references cited therein (e) Y. Li, Y. Shi and A. L. Odom, J. Am. Chem. Soc., 2004, 126, 1794 CrossRef CAS; (f) C. Cao, Y. Shi and A. L. Odom, Org. Lett., 2002, 4, 2853 CrossRef CAS; (g) J. M. Hoover, A. DiPasquale, J. M. Mayer and F. E. Michael, J. Am. Chem. Soc., 2010, 132, 5043 CrossRef CAS; (h) S. L. Dabb and B. A. Messerle, Dalton Trans., 2008, 6368 RSC; (i) A. M. Johns, Z. Liu and J. F. Hartwig, Angew. Chem., Int. Ed., 2007, 46, 7259 CrossRef CAS; (j) S. Banerjee, E. Barnea and A. L. Odom, Organometallics, 2008, 27, 1005 CrossRef CAS; (k) K. Alex, A. Tillack, N. Schwarz and M. Beller, Org. Lett., 2008, 10, 2377 CrossRef CAS.
- For reviews on the uses of hydrazines and hydrazides: (a) E. F. Rothgery, Kirk-Othmer Encyclopedia Chemical Technology, John Wiley & Sons, New York, 5th edn, 2004, vol. 13, pp. 562–607 Search PubMed; (b) U. Ragnarsson, Chem. Soc. Rev., 2001, 30, 205 RSC; (c) E. Licandro and D. Perdicchia, Eur. J. Org. Chem., 2004, 665 CrossRef CAS.
- For a review, see: J. Gante, Synthesis, 1989, 405 Search PubMed.
- For an excellent review of the hydroamination reactivity of hydroxylamines, see: N. J. Cooper and D. W. Knight, Tetrahedron, 2004, 60, 243 Search PubMed.
- (a) A. M. Beauchemin, J. Moran, M.-E. Lebrun, C. Séguin, E. Dimitrijevic, L. Zhang and S. I. Gorelsky, Angew. Chem., Int. Ed., 2008, 47, 1410 CrossRef CAS; (b) J. Moran, S. I. Gorelsky, E. Dimitrijevic, M.-E. Lebrun, A.-C. Bédard, C. Séguin and A. M. Beauchemin, J. Am. Chem. Soc., 2008, 130, 17893 CrossRef CAS; (c) J. Bourgeois, I. Dion, P. H. Cebrowski, F. Loiseau, A.-C. Bédard and A. M. Beauchemin, J. Am. Chem. Soc., 2009, 131, 874 CrossRef CAS; (d) J. Moran, J. Y. Pfeiffer, S. I. Gorelsky and A. M. Beauchemin, Org. Lett., 2009, 11, 1895 CrossRef CAS.
- (a) J.-G. Roveda, C. Clavette, A. D. Hunt, S. I. Gorelsky, C. J. Whipp and A. M. Beauchemin, J. Am. Chem. Soc., 2009, 131, 8740 CrossRef CAS; (b) P. H. Cebrowski, J.-G. Roveda, J. Moran, S. I. Gorelsky and A. M. Beauchemin, Chem. Commun., 2008, 492 RSC.
- Heating semicarbazides at higher temperatures (200 °C) results in the formation of aminoisocyanate intermediates, which can lead to alkene cycloadducts: see ref. 7a.
- An aza-Cope elimination process of an ammonium ylide formed viaproton transfer is suspected at higher temperatures. For related reactivity, see: D. G. Morris, B. W. Smith and R. J. Wood, J. Chem. Soc., Chem. Commun., 1968, 1134 Search PubMed.
- See the ESI‡ for details.
- Two (slow) competing side reactions have been observed with hydrazides at high temperatures: aza-Cope elimination (see ref. 9) and formation of the parent imine through elimination of 3,5-bis(trifluoromethyl)benzamide.
- For general reviews on alkene hydroamination, see ref. 1a and e. Even in intramolecular cases, reactivity is usually only general for 5-membered cyclizations, and is diminished by alkene substitution. The near thermoneutral nature of alkene hydroamination provides an additional challenge for intermolecular hydroaminations: A. M. Johns, N. Sakai, A. Ridder and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9306 Search PubMed.
- Such dipoles are also called aminimides. For a seminal review, see: W. J. McKillip, E. A. Sedor, B. M. Culbertson and S. Wawzonek, Chem. Rev., 1973, 73, 255 Search PubMed.
- The structure of 12 was confirmed through analysis of the products resulting from the cleavage (SmI2) of the N–N bond. See ESI‡.
- For a tutorial review, see: (a) J. B. Sweeney, Chem. Soc. Rev., 2009, 38, 1027 RSC. For the rearrangement of hydrazinium ylides see: (b) S. Wawzonek and E. Yeakey, J. Am. Chem. Soc., 1960, 82, 5718 CrossRef CAS; (c) H. P. Benecke and J. H. Wikel, Tetrahedron Lett., 1971, 12, 3479 CrossRef; (d) K. Chantrapromma, W. D. Ollis and I. O. Sutherland, J. Chem. Soc., Perkin Trans. 1, 1983, 1029 RSC.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, optimization data for Table 3 and spectroscopic characterization for all new products. See DOI: 10.1039/c0cc02403a |
| This journal is © The Royal Society of Chemistry 2011 |