Synthesis of phosphonamidate peptides by Staudinger reactions of silylated phosphinic acids and esters†‡§
Ina
Wilkening
,
Giuseppe del
Signore
and
Christian P. R.
Hackenberger
*
Freie Universität Berlin, Institut für Chemie und Biochemie, Takustr. 3, 14195 Berlin, Germany. E-mail: hackenbe@chemie.fu-berlin.de; Fax: +49 30 838 52551; Tel: +49 30 838 52451
First published on 7th September 2010
Abstract
The Staudinger reaction of unprotected azido-peptides with silylated phosphinic acids and esters on the solid support offers a straightforward acid-free entry to different phosphonamidate peptide esters or acids under mild conditions in high purity and yield.
Phosphon- and phosphoramidates are of great interest in various areas of modern organic chemistry, including applications in asymmetric catalysis1–4 as well as bioorganic and pharmaceutical research.5–12 Phosphoramidates have recently received attention for bioconjugation in chemoselective protein modification strategies, which yield phosphoramidate peptides 1.13,14 In contrast to the rather stable phosphoramidates, phosphonamidates are known to be more prone towards P(
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–NH bond cleavage under acidic conditions, in particular when a free OH group is present at the phosphorous atom.11,15,16 Nevertheless, phosphonamidate peptides 2 and 3, in which an amide bond is replaced by P(O)–OR,NH (2) or P(O)–OH,NH (3), have found considerable attention as promising protease inhibitors. Upon incorporation into a peptide backbone phosphonamidates can be regarded as analogues of the transition state during peptide bond cleavage by proteases and thereby mimic the natural substrate (Scheme 1).5,8,11,17–20 This potency makes phosphonamidate peptides 2 and 3 interesting synthetic targets for the development of protocols to probe different proteases5–12 in the elucidation of a variety of biological processes and as targets for drug discovery for the treatment of diseases like HIV6,10 and others.
O)–NH bond cleavage under acidic conditions, in particular when a free OH group is present at the phosphorous atom.11,15,16 Nevertheless, phosphonamidate peptides 2 and 3, in which an amide bond is replaced by P(O)–OR,NH (2) or P(O)–OH,NH (3), have found considerable attention as promising protease inhibitors. Upon incorporation into a peptide backbone phosphonamidates can be regarded as analogues of the transition state during peptide bond cleavage by proteases and thereby mimic the natural substrate (Scheme 1).5,8,11,17–20 This potency makes phosphonamidate peptides 2 and 3 interesting synthetic targets for the development of protocols to probe different proteases5–12 in the elucidation of a variety of biological processes and as targets for drug discovery for the treatment of diseases like HIV6,10 and others.
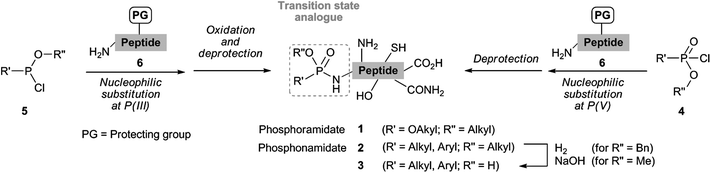 | ||
| Scheme 1 Phosphonamidate synthesis. | ||
Although several synthetic pathways to phosphonamidate peptides 2 and 3 have already been developed, they still face limitations due to the acid lability of the P(O)–NH bond. Most of the synthetic protocols are based on nucleophilic substitution at P(v), in which phosphonochloridates 4 are reacted with side-chain protected amino acids or peptides 6 under rather harsh reaction conditions (Scheme 1).21–23 Extensive purification steps caused by side product formation and subsequent removal of the commonly acid-labile peptide side chain protecting groups after the P(O)–NH bond formation are therefore particularly challenging. Alternatively, nucleophilic substitution of P(III) phosphonochloridites 5 with peptides 6 followed by oxidation can be performed, however with respect to the same limitation of the final protecting group removal of interfering nucleophilic side chains.24 Consequently, only few reports of peptides 3 with a free OH at phosphorus have been reported, which utilize an additional basic hydrolysis8,10,16,18 or hydrogenation step to convert ester 2 into acid 3 (Scheme 1).
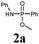
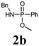
In order to stress the importance of a direct and acid-free synthetic route to phosphonamidate peptides 2 and 3, we first conducted stability tests of phosphonamidate esters with a phenyl (2a) and a benzyl (2b) substituent at nitrogen under standard TFA-deprotection conditions. These studies showed that both compounds are cleaved in TFA within a short period of time. Compounds containing an alkyl residue at nitrogen showed even higher instability due to the electron-donating influence.
Based on these results, we decided to develop a convenient synthetic route under acid-free conditions to both phosphonamidate peptides 2 and 3 (Scheme 2). We reasoned that the Staudinger reaction25 between azides26 and trivalent phosphorus species in the presence of water would be suitable even for peptides 3, since the reaction is mild and does not require the protection of nucleophilic side chains, as demonstrated recently in the bioorthogonal functionalization of whole proteins.13,14
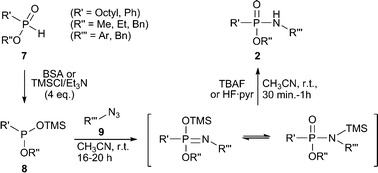 | ||
| Scheme 2 Silylation and Staudinger reaction of phosphinic esters 7. | ||
In order to access phosphonamidates, a phosphonite has to be used in the Staudinger reaction with azides. Since the synthesis of phosphonites is difficult due to their high tendency to oxidize, we propose to use phosphinic esters (H-phosphinates) 7 as starting materials. These compounds are air stable and can be converted by silylation into the trivalent silylated phosphonite species 8in situ, which can be directly used in the subsequent Staudinger reaction with azides 9 to deliver phosphonamidates 2. Previously, the silylation of 7 was already applied for the synthesis of phosphinic peptides12 and for modified nucleotides27–29 but to the best of our knowledge not demonstrated for unprotected peptides 2 or 3.
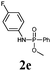
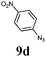
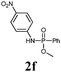
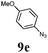
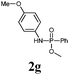
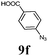
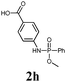
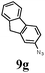
The first goal of our strategy was to find efficient silylating conditions.27–30 Both, TMSCl and BSA (bistrimethylsilyl acetamide), showed a quantitative conversion of the methyl phenylphosphinate (7a) in CH3CN. After the addition of phenyl azide (9a), the reaction was stirred for 16 h at room temperature before desilylation was performed with TBAF to form phosphonamidate 2a in excellent 91% yield (Table 1, entry 1). Alternatively, deprotection with sodium hydroxide solution (1 N) led to similar yields (entry 2). Afterwards, different substituted azides 9 were employed and in all cases the desired products 2 could be obtained in good to high yields after column chromatography (entries 3–10). In case of para-azido benzoic acid 9f HF·pyridine was favoured over TBAF for the desilylation, because the phosphonamidate 2h precipitated from the reaction mixture under these conditions (entry 9). To further probe another silylated alkyl phosphonite 8, which is expected to be more sensitive towards oxidation, methyl octylphosphinate 7d (for preparation see ESI‡) was silylated and reacted with 9a. The corresponding phosphonamidate 2j was formed in a moderate yield (30%), in which hydrolysis byproducts complicated the isolation by column chromatography.
| Entry | Azide 9 | Phosphinic esters 7 | Silylating reagent | Desilylation reagent | Yield (%) | Product 2 |
|---|---|---|---|---|---|---|
| a Conditions: (1) 1 equiv. 9, 1 equiv. 7, 4–6 equiv. BSA or TMSCl/Et3N, CH3CN, r.t., 16–20 h; (2) TBAF or HF·pyr, r.t., 30 min.–1 h, r.t. See also ESI. | ||||||
| 1 |
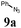
|
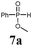
|
BSA | TBAF | 91 |
|
| 2 | 9a | 7a | BSA | NaOH (1 N) | 95 | 2a |
| 3 |
|
7a | BSA | TBAF | 54 |
|
| 4 | 9a |
|
Et3N/TMSCl | TBAF | 82 |
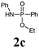
|
| 5 | 9a |
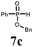
|
BSA | TBAF | 80 |
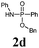
|
| 6 |
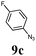
|
7a | BSA | TBAF | 61 |
|
| 7 |
|
7a | BSA | TBAF | 72 |
|
| 8 |
|
7a | BSA | TBAF | 76 |
|
| 9 |
|
7a | BSA | HF·pyridine | 86 |
|
| 10 |
|
7a | BSA | TBAF | 90 |
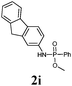
|
| 11 | 9a |
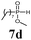
|
BSA | TBAF | 30 |
|
After the synthesis of small phosphonamidates 2a–j in solution, we next attempted to transfer the synthetic route to solid supported aryl azido peptides (Scheme 3), in which we used the salt-free BSA silylation condition. The advantages of such a solid supported process include the easy removal of reagents and additives and the possibility to combine this route with standard Fmoc-based solid-phase peptide synthesis (SPPS).
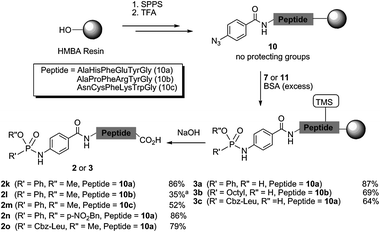 | ||
| Scheme 3 Synthesis of phosphonamidate peptides 2 and 3. Conversions were determined by UV measurement at 280 nm. For conditions and LC-MS analysis see Fig. 1 and ESI.‡aIsolated yield after HPLC purification. | ||
For this purpose, a base-labile commercially available HMBA resin was chosen as the solid support, which enables after SPPS a side chain deprotection by TFA to deliver an immobilized unprotected azido peptide 10. This allows a Staudinger reaction with silyl-phosphonites 8 on the solid support before the peptide is desilylated and cleaved from the resin by basic treatment, thereby evading acidic conditions after the P(O)–NH bond formation (Scheme 3).
Three model peptides 10a–c with para-azido benzoic acid at the N-terminus and different functional groups in the side chains were synthesized, to which an excess of BSA and three different aryl and alkyl phosphinic esters 7 were added (Scheme 3 and ESI‡). In addition to the formation of silyl-phosphonites 8 the peptide side chains are assumed to be also silylated during the reaction. The reaction was then allowed to proceed at room temperature under slight agitation. Finally, the phosphonamidate peptides 2k–o were cleaved with 1 N NaOH from the solid support. For all peptides, LC-MS analysis showed full consumption of the azide and good to high conversions into the corresponding phosphonamidate peptides as determined by UV (see Scheme 3, Fig. 1 and ESI‡).
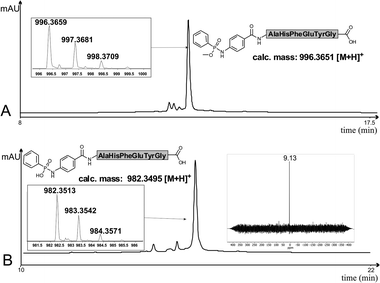 | ||
| Fig. 1 (A) HRMS analysis and HPLC profile of crude 2k; (B) HRMS analysis, 31P-NMR and HPLC profile of crude 3a. For conditions see ESI.‡ | ||
After this successful synthesis of peptide esters 2 we additionally probed the synthesis of the very labile phosphonamidate acid peptides 3. Three phosphinic acids 11a–c were synthesized according to literature protocols (see ESI‡) and applied in an analogous silylation and Staudinger reaction sequence with immobilized peptide 10a (Scheme 3). We were delighted to find that peptides 3a–c were formed as major products. However, small amounts (7–25%) of the corresponding amino peptides appeared in the LC-MS traces, which are presumably formed by P(O)–NH bond cleavage during the HPLC measurements. The high conversion to 3a was further validated by 31P-NMR measurement of the crude peptide 3a in which only one phosphonamidate species was present. Addition of phenylphosphonic acid to the NMR sample indicated that the previous NMR signal did not result from the expected phosphorous cleavage product of 3a.
Finally, we attempted to access a phosphonamidate acid peptide 3 by light cleavage of the nitro benzyl ester 2n. This procedure would allow the synthesis of a relatively stable phosphonamidate peptide, which could be converted to the more labile peptide 3 during biological applications. After ten minutes of UV irradiation concomitant P(O)–NH bond cleavage occurred in addition to the formation of 3a (see ESI‡). Currently, we are further optimizing this procedure by the synthesis of electron rich nitro-benzyl phosphinates which allows milder irradiation conditions.
In conclusion, we have demonstrated that the Staudinger reaction of silylated phosphinic acids and esters with different aryl azides in solution as well as on the solid support represents an efficient and straightforward acid-free protocol for the synthesis of various phosphonamidates including sensitive phosphonamidate acid peptides 3. The target compounds were obtained in very good to excellent yields in solution and in high purities on the solid support without the need of acidic protecting group manipulations. The synthesis of phosphonamidate peptides from alkyl azido peptides for protease inhibition studies as well as other phosphinic acid derivatives for analogous Staudinger reactions is currently under investigation.
The authors acknowledge financial support from the German Science Foundation (Emmy-Noether program, HA 4468/2-1), the SFB 765, the Max-Buchner Stiftung and the Fonds der chemischen Industrie (FCI).
Notes and references
- P. Jiao, D. Nakashima and H. Yamamoto, Angew. Chem., Int. Ed., 2008, 47, 2411 CrossRef CAS.
- D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626 CrossRef CAS.
- M. Rueping, B. J. Nachtsheim, S. A. Moreth and M. Bolte, Angew. Chem., Int. Ed., 2008, 47, 593 CrossRef CAS.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS.
- P. A. Bartlett and C. K. Marlowe, Biochemistry, 1983, 22, 4618 CrossRef CAS.
- N. P. Camp, P. C. D. Hawkins, P. B. Hitchcock and D. Gani, Bioorg. Med. Chem. Lett., 1992, 2, 1047 CrossRef CAS.
- R. E. Galardy, Biochemistry, 1982, 21, 5777 CrossRef CAS.
- N. E. Jacobsen and P. A. Bartlett, J. Am. Chem. Soc., 1981, 103, 654 CrossRef CAS.
- A. P. Kaplan and P. A. Bartlett, Biochemistry, 1991, 30, 8165 CrossRef CAS.
- D. A. Mc Leod, R. I. Brinkworth, J. A. Ashley, K. D. Janda and P. Wirsching, Bioorg. Med. Chem. Lett., 1991, 1, 653 CrossRef CAS.
- K. A. Mookhtiar, C. K. Marlowe, P. A. Bartlett and H. E. Van Wart, Biochemistry, 1987, 26, 1962 CrossRef CAS.
- A. Yiotakis, D. Georgiadis, M. Matziari, A. Makaritis and V. Dive, Curr. Org. Chem., 2004, 8, 1135 CrossRef CAS.
- (a) R. Serwa, I. Wilkening, G. Del Signore, M. Muehlberg, I. Claussnitzer, C. Weise, M. Gerrits and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2009, 48, 8234 CrossRef CAS; (b) R. Serwa, P. Majkut, B. Horstman, J.-M. Swiecicki, M. Gerrits, E. Krause and C. P. R. Hackenberger, Chem. Sci., 2010 10.1039/c0sc00324g; (c) D. M. M. Jaradat, H. Hamouda and C. P. R. Hackenberger, Eur. J. Org. Chem., 2010 DOI:10.1002/ejoc.201000627.
- V. Boehrsch, R. Serwa, P. Majkut, E. Krause and C. P. R. Hackenberger, Chem. Commun., 2010, 46, 3176 RSC.
- P. de Medina, L. S. Ingrassia and M. E. Mulliez, J. Org. Chem., 2003, 68, 8424 CrossRef CAS.
- A. Mucha, J. Grembecka, T. Cierpicki and P. Kafarski, Eur. J. Org. Chem., 2003, 4797 CrossRef CAS.
- J. L. Radkiewicz, M. A. McAllister, E. Goldstein and K. N. Houk, J. Org. Chem., 1998, 63, 1419 CrossRef CAS.
- J. Grembecka, A. Mucha, T. Cierpicki and P. Kafarski, J. Med. Chem., 2003, 46, 2641 CrossRef CAS.
- H. M. Holden, D. E. Tronrud, A. F. Monzingo, L. H. Weaver and B. W. Matthews, Biochemistry, 1987, 26, 8542 CrossRef CAS.
- M. Izquierdomartin and R. L. Stein, J. Am. Chem. Soc., 1992, 114, 1527 CrossRef CAS.
- R. Hirschmann, K. M. Yager, C. M. Taylor, J. Witherington, P. A. Sprengeler, B. W. Phillips, W. Moore and A. B. Smith, J. Am. Chem. Soc., 1997, 119, 8177 CrossRef CAS.
- W. P. Malachowski and J. K. Coward, J. Org. Chem., 1994, 59, 7616 CrossRef.
- A. Mucha, P. Kafarski, F. Plenat and H. J. Cristau, Tetrahedron, 1994, 50, 12743 CrossRef CAS.
- M. D. Fernandez, C. P. Vlaar, H. Fan, Y. H. Liu, F. R. Fronczek and R. P. Hammer, J. Org. Chem., 1995, 60, 7390 CrossRef.
- Y. G. Gololobov and L. F. Kasukhin, Tetrahedron, 1992, 48, 1353 CrossRef CAS.
- S. Braese, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef.
- T. Kline, M. S. Trent, C. M. Stead, M. S. Lee, M. C. Sousa, H. B. Felise, H. V. Nguyen and S. I. Miller, Bioorg. Med. Chem. Lett., 2008, 18, 1507 CrossRef CAS.
- R. A. Fairhurst, S. P. Collingwood and D. Lambert, Synlett, 2001, 473 CAS.
- R. A. Fairhurst, S. P. Collingwood, D. Lambert and E. Wissler, Synlett, 2002, 763 CrossRef CAS.
- H. Vorbrüggen, Silcon-mediated Transformations of Functional groups, Wiley-VCH, Weinheim, 2004 Search PubMed.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: General synthetic procedures, synthesis and characterisation of compounds. See DOI: 10.1039/c0cc02472d |
| § This paper is dedicated to Prof. Helmut Vorbrüggen on the occasion of his 80th birthday. |
| This journal is © The Royal Society of Chemistry 2011 |